 Thomas Schmitt†
Thomas Schmitt† Jens Waschke*†
Jens Waschke*†- Ludwig-Maximilian-Universität München, Anatomische Anstalt, Lehrstuhl Anatomie I – Vegetative Anatomie, Munich, Germany
Pemphigus is a severe autoimmune disease impairing barrier functions of epidermis and mucosa. Autoantibodies primarily target the desmosomal adhesion molecules desmoglein (Dsg) 1 and Dsg 3 and induce loss of desmosomal adhesion. Strikingly, autoantibody profiles in pemphigus correlate with clinical phenotypes. Mucosal-dominant pemphigus vulgaris (PV) is characterised by autoantibodies (PV-IgG) against Dsg3 whereas epidermal blistering in PV and pemphigus foliaceus (PF) is associated with autoantibodies against Dsg1. Therapy in pemphigus is evolving towards specific suppression of autoantibody formation and autoantibody depletion. Nevertheless, during the acute phase and relapses of the disease additional treatment options to stabilise desmosomes and thereby rescue keratinocyte adhesion would be beneficial. Therefore, the mechanisms by which autoantibodies interfere with adhesion of desmosomes need to be characterised in detail. Besides direct inhibition of Dsg adhesion, autoantibodies engage signalling pathways interfering with different steps of desmosome turn-over. With this respect, recent data indicate that autoantibodies induce separate signalling responses in keratinocytes via specific signalling complexes organised by Dsg1 and Dsg3 which transfer the signal of autoantibody binding into the cell. This hypothesis may also explain the different clinical pemphigus phenotypes.
Introduction
Pemphigus is a severe autoimmune blistering skin disease disrupting desmosomes and thereby affecting the epidermis of the skin and the mucosa (1, 2). Desmosomes are important epithelial cell-cell contacts providing high mechanical stability (3–5). Disruption of desmosomes causes the cells to detach from each other, leading to acantholysis. The desmosomal transmembrane cadherins comprise desmogleins (Dsg) 1–4 and desmocollins (Dsc) 1–3 (3). They are connected to the plaque proteins plakoglobin (PG), plakophilin (PKP) 1–3, and desmoplakin (DP). The plaque anchors the desmosomes to the keratin filament cytoskeleton (6–11). The composition of desmosomes varies within tissues and different layers of the epidermis which is relevant since Dsg isoforms differ in their function to regulate signalling pathways in pemphigus. Dsg2, which is ubiquitously expressed throughout other epithelia, can only be found in the basal layer of neonatal epidermis but is absent in adult epidermis outside of hair follicles (12–14). In contrast, Dsg1 and Dsc1 expression increase whereas the amount of Dsg3 and Dsc3 decrease towards superficial epidermal layers with Dsg3 being absent in the stratum granulosum. Dsg4 on the other hand is missing from basal cells, described to be only found in the granular layer and hair follicles (13). Starting from the granular layer, corneodesmosine starts additionally linking cadherins together in the extracellular space (13). This shift in composition marks the stepwise maturation of the adaptable desmosomes towards stable corneodesmosomes found in the cornified layer. In mucosal epithelium, the situation is slightly different. Dsg2 is found in the basal layers only (15). Dsg1 is less predominant and particularly absent in the basal layer of mucosa whereas Dsg3 is present in all layers (15). Dsg4 can also be found in all layers above the basal layer. In contrast, Dsc1 is not present in oral mucosa while Dsc2 and 3 are present in all layers (3). The expression patterns of desmosomal cadherins in the mucosa are thus more homogeneous compared to epidermis. In neonatal mouse skin, it was shown, that the more ubiquitous distribution of Dsg3 without a distinct enrichtment of Dsg1 in the upper epidermal layers (16).
All pemphigus variants are rather rare and here we focus on the two main variants of the disease pemphigus vulgaris (PV) and pemphigus foliaceus (PF). Incidence and prevalence of PV vary profoundly in different geographical regions as well as ethnicities. In Germany, the prevalence for PV in 2014 was 94.8 per one million population (pmp) (17). It was shown that some genetic variations particularly of the HLA class II (18–24) but also non-HLA genes (23, 25–37), including DSG3 (38) play a role. Especially, different expression of the transcription factor ST18, which alters signalling pathways including ERK and is associated with apoptosis, was found tocorrelate with increased prevalence in of PV (22, 32, 39, 40). Of course, environmental risk factors might be of importance as well.
For PF, the prevalence was 10.01 pmp (17), with an estimated incidence rate of <1 pmp in the USA and Europe. PF can manifest sporadically, usually at a middle age. It was shown, that genetic-HLA (41–45) and non-HLA markers (28, 46) are relevant as well. However, there are also more frequent endemic PF variants, e.g., in some subtropical areas of Brazil with incidence rates as high as 3–5% and an onset often at a relatively young age (47–52). Other regions such as Tunisia (53) or Colombia (54) show similar variants.
In most cases, PV initially shows mucosal erosions sometimes spreading to the oesophagus, the airways, the anogenital mucosa (2, 55) and in rare cases even the conjunctiva (56) (Figure 1). In about half of the cases after mucosal erosions additional epidermal lesions develop (57). In contrast, PF affects the more superficial layers of the epidermis only and is less severe (2, 58, 59), however a spread to deeper layers as well as the mucosa and thus progression to PV is possible (60).
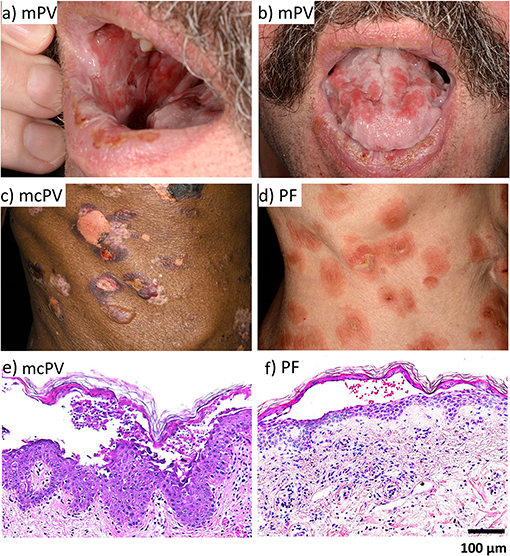
Figure 1. The clinical phenotype and histology of PV and PF. (a) mPV. (b) mPV. (c) mcPV. (d) PF. (e) mcPV. (f) PF.
For diagnosis of pemphigus, the specific histology with separation at the suprabasal layer for PV and at the granular layer for PF is important (3, 61) (Figure 1). The second specific diagnostic marker are the circulating auto-antibodies against Dsg1 and Dsg3 and in some cases against Dsc3 (57, 61–66), Dsc1 (65), or Dsc2 (66, 67) as well as the accumulation of auto-antibodies in perilesional skin (55, 57). Auto-antibody titers often correlate with disease severity (57, 61, 68, 69).
Pemphigus is a very severe disease with a mortality of around 71% if untreated. Secondary infections due to the disruption of the skin barrier are the most severe problem (58, 70). All treatment strategies aim to reduce the levels of circulating pathogenic autoantibodies. With the introduction of systemic corticosteroid therapy for unspecific immunosuppression, the mortality rate was significantly improved to about 21% (71). However, especially under long-time therapy, these medications cause many side effects including increased infection tendency, osteoporosis, low blood sugar, weight gain arterial hypertonia, cataracts, and glaucoma. These side effects can be reduced or delayed by combination therapy with other immune-suppressive agents or antibodies such as Rituximab, which depletes pathogenic B cells, to reduce the steroid dose (72–75). Current combination therapies reduce the mortality to about 5–10% with death mostly resulting from adverse drug side effects (76). These agents usually show less severe but nonetheless considerable side effects including liver toxicity, anaemia, neutropenia, increased infection risk, cold-like symptoms, increased thirst and urination or in some rare worst cases neurological symptoms reaching from sight loss to impaired movement or speech. Patients might also develop an insensitivity to many of these agents (73, 77).
Novel therapy options are for example intravenous injection of high doses of IgG of healthy donors (78, 79), competitively blocking Fc-receptors of inflammatory cells, reducing B-cell response and inducing autoantibody catabolism (80). Another successful concept is plasmapheresis, where the patient's pathogenic autoantibodies are removed (76). In recent years, plasmapheresis is replaced by immunoapheresis, depleting the patient's own autoantibodies by protein A immunoadsorbtion and thus causing less side-effects (81, 82). However, all these therapies have their own drawbacks, including the high costs (83), availability of donor IgG (84) and the need for regular long treatment sessions. Currently the treatment with corticosteroids in combination with immune suppressive agents and Rituximab is still the standard method (72, 85, 86). Furthermore, it is known that not all patients respond to these therapy concepts.
There are some experimental therapy approaches including humanised anti-CD20 antibodies other than Rituximab, designed to be more specific, more potent and/or less immunogenic (87–89). Antibodies directed against targets other than CD20 are also in development (90–92). A similar approach is targeting B-cell signalling and crosstalk with specific inhibitors (93, 94). Furthermore, IgE signalling via IL-4 on Th2 cells which are involved in pemphigus pathology are a novel target (95–97). Targeting Abl-thyrosine kinase-mediated extravasation of autoantibodies into the skin seems to be a promising approach, too (98–100). Another target impacting the autoantibody function are Fc receptors, which prevent IgG degradation and increase autoantibody halflife (101–103). Another approach is to restore Dsg tolerance (104, 105) by transferring Foxp3-expressing regulatory T cells which were shown to be markedly reduced in pemphigus (95). A different emerging option is the therapy with chimeric antigen receptor T-cells (CAR-T)/chimeric autoantibody T-cells (CAA-T) which target a specific antigen and kill the target cell. They could thus deplete autoreactive B-cells (106–108).
For all therapy approaches to reduce autoantibody formation one limitation is that time is required until patients benefit. Therefore, for the acute phase of disease as well as for patients suffering from relapses additional therapeutic options directly stabilising keratinocyte desmosomal adhesion would fulfil an unmet medical need. Directly interfering with keratinocyte signalling to stabilise desmosomes and to induce desmosome assembly could thus be a very promising therapeutic target.
The Role of Autoantibodies
Pathogenic autoantibodies in pemphigus are mostly directed against Dsg1 and Dsg3. Antibodies against other desmosomal and non-desmosomal proteins are also found. However, whilst it was shown that anti-Dsg1 and anti-Dsg3 antibodies are pathogenic, the role of most other antibodies for the pathology of pemphigus is not clear (2, 58, 105–112). One rare exception are the autoantibodie against Dsc isoforms such as Dsc3 which induce a pathology similar to anti-Dsg antibodies (57, 61–67). Nevertheless, it is likely that some of these auto-antibodies may cause additive effects which affect the clinical phenotype (109). To address this possibility, the so-called “multiple hit hypothesis” postulates that for the actual disease to occur the cumulative effects of several of these antibodies against desmosomal and non-desmosomal antigens are needed (110, 111). However, AK23, which is a Dsg3-specific antibody from an active mouse model, and monoclonal autoantibodies targeting Dsg1 isolated from PF patients have been shown to induce pemphigus-typical acantholysis (112, 113). This demonstrates that autoantibodies against Dsg molecules are sufficient to cause pemphigus, at least under experimental conditions.
Pathogenic pemphigus autoantibodies targeting Dsg1 and Dsg3 are mostly directed against EC1 and 2, while antibodies directed for example against EC5 are reported to be non-pathogenic (23, 114). It is theorised that pathogenic pemphigus autoantibodies can evolve from the non-pathogenic antibodies via epitope spreading. It is known that non-pathogenic Dsg1 autoantibodies are increased in unaffected individuals of high incidence endemic populations (23, 45, 115, 116). In endemic PF, the subtype of IgG usually was found to be IgG4 whereas antibodies in unaffected individuals were almost exclusively IgG2 (117). However, non-pathogenic and pathogenic antibodies often share the same light and heavy chains (114), providing credence to this theory (23, 95, 114). Epitope spreading from Dsg3 to Dsg1 is described in the relatively frequent transformation from mucosal-dominant (anti-Dsg3-IgG only) to mucocutaneous PV (anti-Dsg1- and anti-Dsg3-IgG). The same is true for a less frequent transformation from PF (anti-Dsg1-IgG only) to PV (anti-Dsg1- and anti-Dsg3-IgG) (58, 70). Similarly, autoantibodies against plaque proteins or Dsc isoforms also support this hyposthesis (95). A further piece of evidence is the observation that the introduction of a single Dsg3-reactive T-cell was sufficient to induce the expression of polyclonal anti-Dsg3 IgG in mice in vivo (118).
The importance of these most prevalent pemphigus autoantigens is demonstrated further by models to deplete the expression of the corresponding proteins. It was shown that knockout of Dsg3 in mice alone is sufficient to cause a pemphigus-like phenotype (119–121). Similar results were observed for deletion of Dsc3 (122), the autoantigen of atypical pemphigus (123). Similar results were observed in an active Dsc3 mouse model producing autoantibodies against Dsc3 in vivo (124). In contrast, Dsg1 K.O. mice show a complete abrasion of the superficial epidermis comparable to the histology in PF during normal birth and are not viable (125).
Pathogenesis Induced by Autoantibodies Targeting Dsg1 and Dsg3 as Revealed by Experimental Models
Under experimental conditions, autoantibodies against Dsg1 and Dsg3 have been demonstrated to disturb the amount and localisation of the respective desmosomal cadherins as well as to induce reorganisation of the cytoskeleton. anti-Dsg3-IgGs cause reorganisation and depletion of Dsg3 from the cell surface (126, 127). In the skin of PV patients less Dsg3 compared to healthy individuals was detectable (128). Both PV-IgG (126, 129) and the monoclonal antibody AK23 reduced membranous Dsg3 in vitro (130) in human skin ex vivo (127) and in neonatal mice in vivo (127). Starting at a reduction by about 50%, cell adhesion in vitro was significantly reduced (130). For mice injected with PF-IgG a reduction of Dsg1 by about 30% was enough to cause blistering (128). In line with this, blister formation in neonatal mice was inhibited under conditions which blocked the depletion of Dsg3 (127). The internalisation of Dsg3 occurs via endocytosis in a complex with PG. Following the internalisation, the proteins are degraded in the lysosome (126).
Despite the well-established importance of Dsg3 depletion in pemphigus pathogenesis it is yet unclear if this is an early or late mechanism (3). Furthermore, it was shown that different Dsg3 fractions exist which behave differently in terms of depletion (129–132). This is important since localisation of desmosomal cadherins is not limited to the desmosomes (133, 134). Rather, extradesmosomal Dsg molecules (135–137) linked to the actin cytoskeleton (138) are reported as well. It was observed that the extradesmosomal Dsg3 is depleted relatively rapidly following exposure to pemphigus autoantibodies whereas it takes much longer until the Dsg3 integrated in desmosomes is notably impacted by pathogenic IgG (129–131, 139). Since it was observed that acantholysis is actually initiated in the spaces between the desmosomes (140, 141) while keratinocytes stay connected punctually via the desmosomes (142) which are disrupted later (143–145), it is hypothesised that the exradesmosomal Dsg3 complexes represent desmosome precursors, which are later integrated into desmosomes (135, 146). This process could possibly be disrupted by pathogenic autoantibodies leading to endocytosis and degradation of these precursors (129, 135, 139). Since desmosomes are dependent on a dynamic equilibrium and are constantly remodelled under high molecular fluctuation (147, 148) this would prevent desmosome formation and cause disassembly of existing desmosomes (135, 149). A different hypothesis proposes a membrane-receptor-like function for non-desmosomal Dsg3 (59, 144, 150) suggesting that extradesmosomal and desmosomal cadherins may have distinct roles.
Together with the disorganisation of the desmosome a retraction of keratin filaments from the cell surface occurs (151–153). During this process, keratin filaments detach from the desmosomes (142, 143, 145, 154) and concentrate around the nucleus (141, 153–156). It was shown that the retraction of keratin filaments starts even before the internalisation of Dsg3 and correlates with the time the cell dissociation starts. Keratin filaments show a clear association with Dsg3 reorganisation at the membrane, preceding endocytosis (153) and turnover after PV-IgG treatment (151). This mechanism is thus proposed to play an important role in pemphigus pathogenesis and especially the time of onset before desmosome disassembly suggest keratin filament retraction to be a primary pathogenic mechanism (153). The actin cytoskeleton was also shown to be affected by PV-IgG which may interfere with desmosome assembly (129, 157).
Mechanisms Causing Acantholysis in Pemphigus
Meanwhile it is widely accepted that the mechanisms underlying loss of cell adhesion in pemphigus comprise both direct inhibition of Dsg interaction by bound autoantibodies and signalling mechanisms regulating keratinocyte adhesion triggered by autoantibody binding (158). In the pioneering studies on pathologic mechanisms direct inhibition of Dsg3 adhesion by steric hindrance was very suggestive to be causal for acantholysis (159) whereas signalling such as PLC-induced Ca2+ release (156, 160) was regarded more as secondary bystander effect.
Meanwhile, there are some studies available on steric hindrance of Dsg1 and Dsg3 adhesion, which for technical reasons are mostly carried out under cell-free conditions in vitro using bead assays or single-molecule atomic force microscopy (161) but rarely were performed in cultured keratinocytes (Table 1). However, studies using in vivo models or human organ culture are lacking which makes it hard to decide whether effects of autoantibodies on recombinant extracellular domains of Dsg1 and Dsg3 can be transferred to the situation in intact skin where desmosomes are formed in a complex multi-step process. In contrast, there is ample evidence for the role of signalling pathways in the regulation of desmosomal adhesion in PV from studies not only performed in cultured cells but also using in vivo mouse models and ex-vivo human epidermis whereas data for PF are still limited (Table 2).

Table 1. Studies on direct inhibition of Dsg1 and Dsg3 interaction in pemphigus.
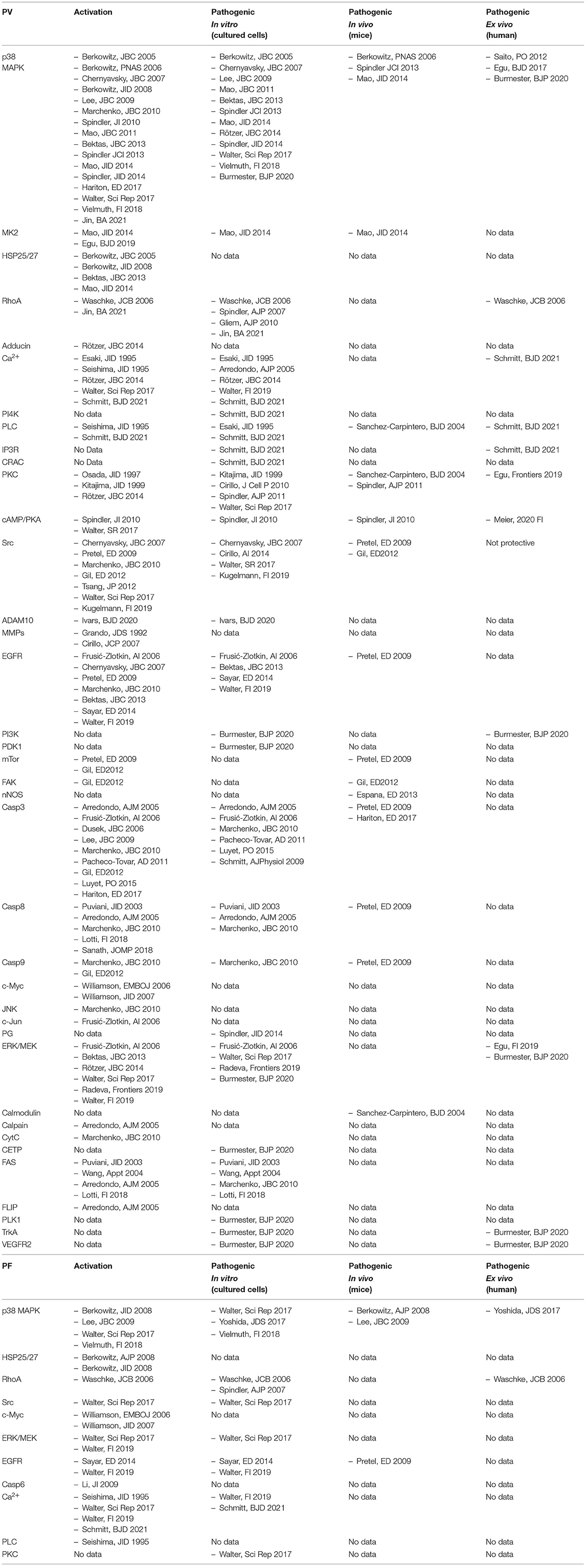
Table 2. Studies on signalling pathways in pemphigus.
Direct Inhibition of Desmoglein Adhesion vs. Cellular Signalling
The cadherins were reported to undergo homo- as well as heterophilic Ca2+-dependent interactions via their extracellular domains (3). The outermost extracellular domain EC1 domain seems to be most important for Dsg adhesion (1, 27, 162–164). As discussed above, the pathogenic autoantibodies found in pemphigus mostly bind to this domain (27, 162, 165, 166). At first glance, it appears obvious that the binding of a large protein like an antibody to this region would cause steric hindrance by directly inhibiting the Dsg binding and thus cell adhesion. For Dsg3 it was possible to show the occurrence of such a direct inhibition by PV-IgG in vitro, both under cell-free conditions and for adhesion to the cell-surface (151, 167) (Table 1). A PV-mouse-model did also show possible indications of direct inhibition of Dsg3 adhesion (165). The finding that the monoclonal anti-Dsg3-antibody AK23, which causes a PV-like phenotype in mice (112), also causes direct inhibition of homophilic Dsg3 interaction under cell-free conditions supports this hypothesis (152, 168). Homophilic Dsc3 and heterophilic Dsc3-Dsg1 were reported to be important for adhesion in keratinocytes as well. Direct inhibition of these interactions greatly reduced the adhesive strength (64). However, another study reported heterophilic interactions between Dsg1 and Dsc1 and Dsg3 and Dsc3 to be the fundamental adhesive unit of desmosomes. In a cell-free assay, mixtures of Dsg1 and Dsc1 or Dsg3 and Dsc3 ectodomains formed large aggregates while Dsg1 or Dsg3 as well as Dsc1 or Dsc3 alone only formed minor to no aggregates. Autoantibodies where effective to directly inhibit binding of Dsg1 to Dsc1 and Dsg3 to Dsc3 beads (169). Direct inhibition has thus to be considered to play a role in PV pathology (150, 167, 170). However, direct inhibition alone is not adequate to cause a complete loss of keratinocyte adhesion. This is highlighted by the fact that impairing cellular signalling but not direct inhibition by incubating the cells at 4 °C did prevent PV-IgG induced loss of cohesion (126). Moreover, inhibition of p38MAPK rescued cell adhesion although direct inhibition of Dsg3 binding by PV-IgG was still detectable which demonstrates that modulation of autoantibody-induced signalling is sufficient to outbalace effects of steric hindrance (137). For PF, the intracellular signalling seems to be even more important since no direct inhibition of Dsg1 interaction was observed in several studies (151, 152, 168, 171).
Over time, several signalling pathways have been identified to be essential for the pathology of pemphigus (59, 152, 171–173) (Table 2). The focus of current research is to integrate all the different pathways and to define their role in pemphigus pathogenesis. Especially, the chronology of many steps is not clear at present.
The Pathology of Patients' Lesions Identifies the Underlying Mechanisms Interfering With Desmosome Turn-Over
Microscopic evaluation of skin lesions in pemphigus revealed that the pathogenic mechanisms involved cannot be simple. Rather, the different clinical phenotypes in PV and PF are defined by characteristic histology and ultrastructural alterations of desmosomes, which only can be explained by severe impairment of desmosome turn-over, have been established as hallmarks of the disease.
In PV, deep epidermal blisters and mucosal erosions typically separate supra-basal keratinocytes in the blister roof from tomb-stone-like cells of the basal layer in the blister bottom (174, 175) (Figure 1). In contrast, superficial erosions in PF tear of granular and apical spinous cell layers. Since Dsg3 is the predominant desmosomal cadherin in oral mucosa whereas Dsg1 is sparsely expressed, it is conceivable why in PF mucosal erosions usually are absent. Similarly, because in the granular layer Dsg1 and Dsc1 are the main desmosomal cadherins expressed, superficial blistering in PF can be explained by the Dsg compensation hypothesis (176). Supporting this, it was reported, that in neonatal mouse skin, the more ubiquitous distribution of Dsg3 without a distinct enrichtment of Dsg1 in the upper epidermal layers provided a protective effect against PF-IgG (16). However, the morphology of epidermal lesions in PV does not reflect the distribution patterns of Dsg1 and Dsg3 which in human skin broadly overlap. Thus, desmoglein compensation cannot explain a sharp supra-basal cleavage plain as found in PV (3, 150). Also, morphologic alterations typical for apoptosis usually are not present on histologic or ultrastructural levels in patients' lessons demonstrating that apoptotic cell death is not a major cause for acantholysis (154, 177, 178).
It has been shown that Dsg3 clustering in the absence of epidermal blistering correlates with the presence of autoantibodies against Dsg3 in patients with mucosal-dominant PV (149, 178, 179). Similarly, inter-desmosomal widening in PV and PF was present in unaffected epidermis suggesting that these events are not sufficient to cause skin blistering. Rather, epidermal blistering occurred when autoantibodies against Dsg1 were present in PV and PF, the number of desmosomes was reduced, desmosomes were smaller in size and keratin filament retraction from desmosomal plaques was identified by electron microscopy (145, 178, 180). Split desmosomes with partial or complete separation of the two desmosomal plaques from neighbouring cells and double-membrane structures, reflecting engulfment of membrane domains, were detected also. Thus, the best strategy to evaluate the relevance of the different mechanisms causing pemphigus pathology is to correlate mechanisms with the morphologic hallmarks of the disease. Direct inhibition of desmoglein binding by steric hindrance through bound autoantibodies would cause splitting of desmosomes but no alterations of desmosome ultrastructure. Indeed, mechanical shear has been demonstrated to cause split desmosomes (181) which therefore may be caused by steric hindrance. Since split desmosomes mostly were found to be smaller and displayed altered insertion of keratin filaments (15, 178, 181, 182), splitting of desmosomes most likely is a consequence of altered desmosome composition. Moreover, most desmosomes in ex vivo human skin models of pemphigus are not split but rather smaller and characterised by less opaque plaques, less dense desmoglea in the desmosomal intercellular space and shorter keratin filaments attached (15, 182). Taken together, the ultrastructural alterations can be explained only by impaired desmosome turn-over (178, 183).
Therefore, the major goal is to decipher which mechanisms shown to be induced by autoantibodies interfere with the distinct phases of desmosome assembly and dissasembly. The different steps of desmosome assembly have been characterised in detail (184). First, Dsg and Dsc molecules together with PG are transported to cell borders with preformed adherens junctions along microtubules and via kinesin. At the membrane, desmosomal cadherins associate with lipid rafts via Flotillin-1 and 2 which are required for proper desmosome assembly (185, 186). Here, intermediate junctions composed of Dsgs and adherens junction components including E-cadherin, PG and β-catenin are assembled in Src-dependent manner (138, 187, 188). In a next step, Dsg molecules are transferred to DP coupled to keratin filaments (9, 189). Actin binding proteins such as adducing, which is regulated via RhoA/Rho kinase, and cortactin are important for directed targeting of Dsg molecules during desmosome assembly (190–193). The assembly of Dsgs with DP is controlled by PKP3 in a Rap1-dependent manner (194), the latter of which is a GTPase activated by cAMP and EPAC1. Inside of desmosomes, Dsg isoforms including Dsg1 and Dsg3 in a Ca2+-dependent and most likely homo- and heterophilic manner interact with Dsc1 and Dsc3 via their extracellular domains (64, 168, 169, 171, 188, 195).
Hyperadhesion as a State of Increased Desmosome Stability
Finally, the desmosomal components are locked inside of desmosomes by signalling mechanisms which control the anchorage of DP to keratin filaments and thereby induce a hyperadhesive state (196). This indicates that once a desmosome is established it is remarkably stable but the protein exchange of desmosomal components is regulated on the molecular level resulting in low half-life of desmosomal components (147, 196, 197). This paradigm allows desmosome disassembly when the net exchange by molecules diffusing out of desmosomes outbalances the assembly process as described above in processes like wound healing and migration (198–200). Also, this explains why uptake of entire desmosomes in double-membrane structures as observed under pathologic conditions in pemphigus ex vivo models is rarely observed in intact epidermis (182).
Interestingly, under hyperadhesive conditions the binding properties of desmogleins change dramatically. For instance, in hyperdahesive keratinocytes oligomerization of Dsg3 but not Dsg1 is enhanced and the Ca2+-dependency of desmosome integrity is abolished (196, 198, 201–203). Rather, if trans-interaction between Dsg3 molecules was maintained desmosomes were preserved under Ca2+-depletion although the ordered array of the Dsg3 extracellular domains in desmosomes was lost (196). This indicates that Ca2+-dependency of Dsg3 interaction may get lost when adhesion molecules and plaque proteins in desmosomes are locked by cytoskeletal anchorage. Moreover, since it was shown that Dsg1 interaction can be rescued after re-substitution of Ca2+ whereas Dsg3 cadherin order was not re-established (171, 196, 204), it is also possible that the roles of Dsg1 and Dsg3 in hyperadhesion are different. This can be concluded from the observation that in intact human epidermis, in which the majority of desmosomes is assumed to be hyperadhesive (198, 201, 202, 205), immunostaining of Dsg1 but not of Dsg3 was drastically altered by Ca2+ depletion indicating that Dsg3 in the epidermis indeed loses its Ca2+ dependency (206). In line with this, the molecular binding strength of Dsg3 but not of Dsg1 was increased when keratinocytes were hyperadhesive.
As the underlying molecular mechanism regulating hyperadhesion, PKC-mediated phosphorylation of DP under control of PKP1 has been shown to result in decreased cytoskeletal anchorage and thus to revert the hyperadhesive state in order to allow desmosome reorganisation (196, 197, 199–201, 207, 208). To allow this adaption, PKC was shown to be sequestered to desmosomes via the adaptor-protein RACK1 in DP-dependent manner and is regulated by keratins (170, 208, 209). However, since in cultured cardiomyocytes and cardiac slice cultures besides inhibition of PKC a hyperadhesive state can also be induced by PKA activation and p38MAPK inhibition, it is likely that other signalling mechanisms involved in the assembly of the desmosomal plaque and its anchorage to keratin filaments are also important for hyperadhesion (210–212). Other targets besides DP appear to be relevant as well because hyperadhesion can be abolished when expression of desmosomal components such as Dsg3 or PKP1 and PKP3 is disturbed (203, 213), which is also similar to cardiomyocytes where depletion of Dsg2, PG, or PKP2 abrogated hyperadhesion (214).
An open question, which also has relevance to identify the central molecular mechanisms impairing desmosome turn-over in pemphigus remains: whether the hyper-adhesive state can be recognised on the level of desmosome ultrastructure. It has been proposed that a dense midline in the desmosome intercellular space where extracellular domains of neighbouring cells trans-interact would be a hallmark of hyperadhesion (198). Indeed, in intact epidermis most desmosomes exhibit dense midlines which is not the case in the vicinity to epidermal blisters or where the desmosome ultrastructure is altered in ex vivo human pemphigus skin models (182, 215). However, in cultured keratinocytes under hyperadhesive conditions desmosomes were lacking midlines completely (196). This indicates that the hyperadhesive state cannot be concluded for a specific desmosome from its ultrastructure in cultured cells. On the other hand it can be assumed that in the epidermis desmosomes with altered morphology and lacking midlines most likely are not hyperadhesive and thus PKC is likely to participate in pemphigus pathology by reverting hyperadhesion of desmosomes.
Autoantibodies Against Dsg1 AND Dsg3 Induce Specific Signalling Pathways in Pemphigus
As summarised above a vast set of signalling pathways has been associated with pemphigus pathogenesis (Figures 2–5 and Table 2). Before we try to allocate the different mechanisms to the steps of desmosome assembly and disassembly, we introduce the concept that the signalling pathways activated by a specific autoantibody are defined by the signalling complexes organised by Dsg1 and Dsg3. It was an important observation that signalling responses in keratinocytes are not a consequence of loss of cell adhesion but rather signalling molecules are associated with Dsg isoforms. This was shown first for Dsg3, which was found to form a complex with activated p38MAPK, a process enhanced by pemphigus autoantibodies (216). This adhesion receptor was regulated in adhesion-dependent manner because reduced Dsg3 adhesion by specific autoantibodies or peptides or tryptophan caused p38MAPK activation whereas stabilisation of Dsg3 binding by cross-linking peptides reduced p38MAPK activity (216, 217). This process is regulated by PG because depletion of PG also caused p38MAPK activation. In contrast, depletion of DP or PKP1 and PKP3, which also cause a profound loss of keratinocyte adhesion, did not enhance p38MAPK activity (203, 218, 219).

Figure 2. Schematic depiction of p38MAPK-dependent signalling in pemphigus (grey RhoA and cAMP steps represent potential rescue pathways).
The signalling complexes organised by Dsg molecules appear to be both isoform- and cell type-specific because Dsg2 in contrast to Dsg3 and Dsg1 does not associate with p38MAPK (14, 220, 221). Nevertheless, in enterocytes Dsg2 regulates p38MAPK activity (222). In contrast, Dsg2 has been shown to bind and to control EGFR in enterocytes in fashion similar to Dsg3 in keratinocytes (187, 223).
All of this led us to propose that Dsg molecules form cell-type specific signalling complexes which serve as adhesion-dependent signalling receptors and regulate complex functions such as keratinocyte migration and wound healing (59, 224). Most recently, we observed that Dsg1 also forms signalling complexes which are at least in part different to Dsg3. The majority of signalling molecules involved in pemphigus pathogenesis analysed so far including p38MAPK, PLC, and PKC were associated with both Dsg1 and Dsg3. However, PI4K acting upstream of the PLC/Ca2+/PKC pathway was found exclusively to interact with Dsg1 (220). The fact that p38MAPK binds to both Dsg1 and Dsg3 is compatible with the observation that blister formation in vivo caused by PV-IgG and PF-IgG is blocked by inhibition of p38MAPK (225, 226) and that both PV-IgG and PF-IgG reduced activity of RhoA in p38MAPK-dependent manner (127, 227).
Moreover, these findings are in line with the observation that autoantibody fractions including antibodies targeting Dsg1 in PV and PF induce a different signalling response compared to antibodies against Dsg3. Most striking, PLC activation and Ca2+ influx was found only when Dsg1 autoantibodies were present in PV-IgG and PF-IgG fractions but not induced by mucosal-dominant PV-IgG or AK23 (152, 220, 228). Similarly, ERK activation was detectable after incubation with PF-IgG and PV-IgG only when Dsg1 antibodies were present and similar to Ca2+ influx activation was preserved in Dsg3-deficient keratinocytes. In contrast, EGFR activation was found to be dependent on Src and caused by AK23 and mucosal-dominant PV-IgG and thus in the absence of autoantibodies against Dsg1 (152, 228). Similarly, Src- and ADAM10-dependent EGFR activation has been found in response to PV-IgG including autoantibodies against Dsg1/3 whereas blister formation with additional autoantibodies against Dsc2/3 was ADAM10-independent (66).
For PKC it is not clear whether the different isoforms are regulated via Dsg1 or Dsg3. PKC was found to associate with both Dsg1 and Dsg3 and inhibition of PKC ameliorated loss of cell adhesion in response to PF-IgG and PV-IgG independent of the presence of autoantibodies against Dsg1 (152, 220). Also, PKC was found to be involved in Dsg3 depletion in vivo (127). On the other hand, at least for Ca2+-dependent PKC isoforms activation via autoantibodies against Dsg1 can be assumed (220).
Taken together, the studies on composition of signalling complexes on one hand and evaluation of signalling pathways on the other hand suggest that besides p38MAPK activation autoantibodies against Dsg3 in PV cause SRC-dependent EGFR activation whereas autoantibodies against Dsg1 in PV and PF activate both ERK and the PI4K/PLC/Ca2+ pathway. The hypothesis that the signalling pattern orchestrated by Dsg1 and Dsg3 in pemphigus are to some extent different also helps to explain why blister formation in pemphigus requires antibodies against Dsg1. In this line of thoughts, some pathologic effects such as clustering and depletion of Dsg3 molecules in unaffected skin of mucosal-dominant PV patients (149, 179) can be induced by signalling pathways such as p38MAPK, Src/EGFR and PKC. However, clustering of Dsg1 as well as shrinkage and loss of desmosomes which finally cause epidermal blistering (149, 179, 182) require ERK and PLC/Ca2+ signalling in addition to p38MAPK. This third line of evidence that the relative contribution of various signalling pathways for skin blistering in pemphigus is different comes from studies in human skin ex vivo where inhibition of p38MAPK, ERK, and PLC activity as well as of Ca2+ influx were sufficient to abrogate skin blistering whereas inhibition of Src and PKC was not (182, 191, 215, 220, 229).
Another important aspect is that inhibition of p38MAPK is effective to abolish PV-IgG-induced epidermal blistering in ex vivo human skin models whereas antibodies against Dsg3 such as AK23 or in mucosal-dominant PV-IgG fractions are not sufficient to induce skin blistering, although they activate p38MAPK (152, 182). This suggests that p38MAPK for epidermal blistering is required but not sufficient. In a mucosa model, inhibition of p38MAPK did not block blistering although AK23 in mucosa activated p38MAPK indicating that the relevance of signalling pathways in pemphigus is also tissue-specific (15). All these results indicate that the clinical phenotype of PV with mucosal erosions and deep epidermal blistering on one hand and PF characterised by superficial acantholysis on the other hand are unlikely to be caused by different expression pattern of Dsg3 and Dsg1 alone as suggested by the Dsg compensation hypothesis. Rather, the mechanisms regulating cell adhesion in mucosa and epidermal layers appear to be different and to be regulated by Dsg3 and Dsg1 (3, 150).
Finally, it has to be noted that the observation of Dsg3 and Dsg1 to regulate keratinocyte adhesion and function via different mechanisms is also in line with the different phenotypes of mouse models deficient for these molecules. Dsg3-deficient mice besides hair-loss display rather mild erosions in the skin with PV-typical histology, which have been observed to spontaneously heal within several days, but also suffer from eye involvement in conjunctiva and cornea (56, 119, 224). The phenotype may be explained by the observation that in both affected and unaffected skin p38MAPK activity was up-regulated, which was found to be required for wound healing (224). This shows that Dsg3 supports epidermal integrity at least in part via regulation of signalling pathways controlling cell adhesion. In contrast, Dsg1-deficient mice lose their superficial epidermis during birth and all die within 24 h due to complete loss of epidermal barrier properties (125). Because TJ morphology was altered in heterozygous mice, it is likely that the function of Dsg1 via Erbin to control EGFR-mediated TJ assembly in the superficial epidermis is involved also (230–232) (Figures 2–5).
The Central Role of p38MAPK to Regulate Desmosome Turn-Over
For some signalling pathways the role in pemphigus pathogenesis and the mechanisms underlying loss of desmosomal adhesion became clearer during the last two decades.
An important molecule contributing to pathogenesis of pemphigus is p38MAPK, a protein involved mostly in the reaction to external stressors, including oxidative (233, 234), thermal and chemical stress (234, 235) but can also be activated by growth factors (236). Upon activation by phosphorylation at threonine residue 180 and tyrosine 182 (225, 237–239) p38MAPK activates other kinases further downstream and finally impacts cell growth, differentiation (240), migration as well as wound healing (224) possibly also modulating gene expression (235) and was also reported to directly phosphorylate keratins in keratinocytes and enterocytes (241–243).
p38MAPK is activated by PV-IgG (225, 237) as well by PF -IgG (226) in keratinocytes in vitro and in mouse skin in vivo. p38MAPK is associated with both intra- as well as extradesmosomal Dsg3 (187) and is phosphorylated in a protein complex with Dsg3 and PG (216). p38MAPK then via MAPKAPK2 (56, 244) activates HSP27 (225, 237, 245). In skin biopsies of PV and PF patients, activation of p38MAPK (238), MAKAPK2 (244) as well as HSP27 (238) were confirmed also. HSP27 regulates both actin (236) as well as intermediary filament (246) dynamic restructuring. Moreover, p38MAPK is associated with the keratin cytoskeleton (151, 161, 216, 237).
To delineate the role of p38MAPK in pemphigus pathogenesis it was shown that specific inhibition of p38MAPK or its downstream target MAPKAPK2 are sufficient to reduce loss of adhesion in keratinocytes in vitro (221, 247), depletion of both extra- and intradesmosomal Dsg3 (132, 233, 244) from both the soluble and insoluble fractions (132) and the activation of and reorganisation of the cytoskeleton by HSP27 (237) upon PV-IgG treatment. In murine skin in vivo, p38MAPK inhibition was sufficient to prevent skin blistering and activation of HSP25, the murine analogue to human HSP27, in the skin (225, 226) upon PV- or PF-IgG treatment. In human skin ex-vivo, inhibition of p38MAPK ameliorated the pathogenic effects of PV-IgG, including skin blistering, Dsg3 fragmentation, reduction of desmosome number and size, membrane detachment and keratin retraction (182). In contrast, inhibition of p38MAPK was protective against Dsg1 clustering but did not prevent acantholysis induced by PF-IgG (113).
However, it is currently unclear how exactly p38MAPK is activated after antibody binding to Dsg1 and Dsg3. One study reported activation with two peaks in activity at different points in time, which was different in vivo vs. in vitro as well as between PV- and PF-IgG. It was proposed that both peaks would most likely cause different cellular responses (248). In other experiments, different peaking patterns were reported (249) or experiments were only performed at specific time points (233) making it impossible to directly compare many of these results (Figures 2, 5).
Rho A/Adducin-Mediated Desmosome Assembly
One pathway by which p38MAPK could deplete Dsg3 from intra- and extradesmosomal pools is interference with RhoA/adducing-mediated assembly of desmosomes. Reorganisation of the actin cytoskeleton was observed upon treatment with both PV-IgG and PF-IgG (171, 237). RhoA was shown to induce reorganisation of the actin cytoskeleton (157) and is inactivated following PV-IgG and PF-IgG treatment (227). This mechanism appears to be important in pemphigus because pharmacologic activation of RhoA inhibited epidermal blistering in response to PV-IgG and PF-IgG in human skin ex vivo and inactivation of Rho-GTPases induced epidermal splitting similar to autoantibodies (227, 250). It was demonstrated that inhibition of p38MAPK abolished RhoA inactivation after PV-IgG treatment identifying RhoA as another downstream target of p38MAPK (127, 227). PKP2 was shown to prevent the localisation of RhoA to cellular interfaces having similar effects as RhoA inactivation (190). RhoA activity was furthermore linked to possible apoptotic signalling in pemphigus (251). The effects of RhoA inactivation include reduced incorporation of DP into the desmosomal plaque (190), reduced anchorage of the desmosomes to the keratin cytoskeleton (227) and depletion of Dsg3 and Dsg1 finally leading to loss of cell adhesion (250, 252–254).
All these effects can be explained by the fact that RhoA via Rho kinase regulates the actin binding protein adducin (192). Adducin is essential for cell adhesion in keratinocytes by controlling the incorporation of Dsg3 into desmosomes (193). Other actin binding proteins such as cortactin, which have been shown to be involved in pemphigus pathogenesis, also bind to extra-desmosomal Dsg3 and thus may participate in desmosome assembly (191). Thus, it is possible that inhibition of desmosome assembly via p38MAPK-mediated inactivation of Rho family GTPases and actin binding proteins is a general paradigm in pemphigus pathogenesis (Figures 2, 5).
Plakoglobin Acts as Signal Transducer and Transcription Factor in Pemphigus
As described above, PG is essential for the assembly of desmosomes (255–257) and cellular adhesion (8, 256, 258) e.g., as part of the Dsg3-PG complex (187). This complex is later incorporated into the desmosomes and becomes insoluble via interaction with Dsg1 (259). It was shown, that PV-IgG impairs the distribution of PG (260–262) and its association with Dsg3 (263). As described this is a result of PG endocytosis (218, 260). It was shown that the composition of the desmosomes and cellular adhesion and keratin filament integrity is largely dependent on PG (218, 261). Loss of PG activated p38MAPK as discussed above (218). However, PG was reported to contribute to both desmosomes and adherents junctions (255, 256) but is also found free at the cell membrane as well as in the cytosole (130) and even in the nucleus (218) of keratinocytes. It is thus most likely displaying an even broader range of functions. The localisation of PG is regulated by phosphorylation via GSK3β and thus inhibitors of this pathway were shown to be protective against PV-IgG-induced skin blistering in vivo (264). By its relocalization to the nucleus, PG also regulates gene expression in pemphigus patients. This role is distinct from its function as a plaque protein (218). PG increases expression of urokinase-type plasminogen-activator receptor (uPAR) at the cell membrane. uPAR was described to be involved in wound healing and cellular reorganisation at the cell membrane (262). Nuclear PG also increased expression of the proto-oncogene c-Myc, which ultimately leads to hyperproliferation, reduced cell adhesion and eventually skin blistering in pemphigus patients (264, 265). This increase of nuclear c-Myc is characteristic for PV and is not observed for other skin diseases including PF, again highlighting that for both variants of pemphigus different signalling pathways are activated. It was observed that c-Myc activity is already increased very early in the pathogenesis and does correlate with disease severity (265). Very recently, inhibitors against the signalling cascade PI3K/PDK1/Akt, which regulate GSK3β, have been shown to be protective against skin blistering in vivo and ex vivo human skin and also inhibition of the down-stream molecule mTOR was shown to protective in mice (229, 266). Therefore, it is possible that this signalling pathway regulates desmosome turn-over also by mechanisms different from PG (Figures 3, 5).

Figure 3. Schematic depiction of EGFR-dependent signalling pathways (purple lines indicate diffusion of extracellular second messengers).
The Src/ADAM10/EGFR Receptor Pathway in Pemphigus
Src activated rapidly and in a Dsg3-dependent manner (267) after PV-IgG treatment (249). Src activity was reported to cause loss of Dsg3 from the membrane, phosphorylation of PKP3, thereby destabilising the desmosomal plaque (268) and to induce keratin retraction (249). These changes cause loss of adhesion in vitro (191, 268) and skin blistering in mice in vivo (66, 172, 191, 266). In peri-lesional skin of pemphigus patients an increased Src activity was observed. In contrast to that, in lesional skin its activity was notably reduced, indicating an activation of Src before akantholysis (267). However, in human skin Src inhibition was not completely effective to prevent acantholysis caused by PV-IgG fractions including higher anti-Dsg1 IgG concentrations. Because Src was no longer activated after 24 h and Src inhibition was only protective during timeframes when Src was active (191), it can be assumed that Src plays a less central role in pemphigus pathogenesis and likely contributes during the initial phase only when the extradesmosomal Dsg3 is depleted. In addition to its pathogenic role, Src is also essential for the Ca2+-dependent incorporation of Dsg3 into desmosomes by stabilising the complex of E-cadherin together with Dsg3, potentially via cortactin (187, 191). Thus, Src seems to play an interesting dual role by stimulating desmosome formation under basal conditions but causing desmosome disassembly under pathogenic conditions.
One target molecule of Src in pemphigus pathogenesis appears to be EGFR which was shown to interact with (8, 269) and to be activated via Src (228, 249, 270). EGFR inhibition was shown to be protective against effects induced by PV-IgG and PF-IgG in vitro and in vivo (228, 271–273). EGFR is an important central molecule of a complex network of signalling pathways (269) and can be activated by ligands like the EGF or TGF but also in a ligand-independent manner via phosphorylation by p38MAPK and formation of intracellular signalling complexes (269, 270, 272, 274). EGFR was shown to regulate cellular adhesion and inhibiting it's basal activity promotes localisation of PG and DP to the desmosomal plaque and strengthens cellular adhesion (272, 275). Furthermore, it was demonstrated to be involved in pemphigus signalling (249, 272, 273) but not to be crucial because EGFR-independent loss of keratinocyte adhesion after incubation with PV-IgG has been observed as well (217). EGFR activity leads to endocytosis of Dsg3 and PG causing loss of adhesion (272, 273).
EGFR downstream signalling is broad and the exact mechanisms involved in acantholysis are not perfectly clear but may involve PI3K and ERK which recently were associated with pemphigus pathogenesis (152, 215, 229). PI3K, acting via PDK1/Akt/mTOR/GSK3β on nuclear localisation of PG may cause hyper-proliferation and loss of adhesion (269, 276, 277). Similarly, the Ras/Raf/MEK/ERK pathway is involved in the regulation of cell proliferation vs. differentiation and causes loss of adhesion as well (228, 230, 269, 278–281). Another possible target is JNK1 impacting cell proliferation and migration (269, 282, 283).
Besides direct activation of EGFR by phosphorylation at Y845, Src is also involved in the release of activating EGFR ligands in response to pemphigus autoantibodies (66, 228). In this context, metalloproteinases (MMPs) are involved which can cleave cellular proteins and thus convert proteins to active forms, release second messengers or cleave and inactive proteins. They are well-known to be involved in the pathology of bullous pemphigoid (284). It was furthermore reported that in PV and bullous pemphigoid levels of serin proteinases activity were increased in patients' serum and blister exudates whereas MMP inhibitors were reduced (285). MMP9 was reported to be significantly upregulated in PV model in vivo and in vitro (286). Recently, ADAM10 was reported to be directly involved in pemphigus pathogenesis (66). ADAM10 is activated downstream of Src thereby modulating EGFR activation via release of EGF and Betacellulin (BTC). Interestingly, the activation of ADAM10 is dependent on the autoantibody profile and restricted to antibodies targeting Dsg1 and Dsg3. In contrast, additional autoantibodies against Dsc2 and 3 triggered a more severe and earlier acantholysis independent of ADAM10 (66) (Figures 3, 5).
Dsg1 Regulates EGFR Signalling Towards ERK Which May Be Involved in Pemphigus
Dsg1 is a very important antagonist of EGFR signalling. EGFR promotes cell migration and proliferation in the basal layer of the epidermis. In contrast, Dsg1 expression in superficial epidermis shifts the gears more towards keratinocyte differentiation. This antagonistic role is a result of at least one shared pathway. While EGFR is known to activate ERK1/2, Dsg1 is a known suppressor of this signalling pathway (215, 228, 230, 279–281, 287). This suppression is mediated via the Dsg1-Erbin-SHOC2-complex which interferes with EGFR-induced activation of MEK1/2 and ERK1/2 (228, 279–281). Therefore, this pathway represents a very important crossing point between Dsg3- and Dsg1-dependent signalling and a protective role of Dsg1 against signalling induced by anti-Dsg3 autoantibodies in pemphigus can explain why in mucosal PV the epidermis is not affected although Dsg3 distribution is severely altered (149, 178, 179). In this line of thoughts, anti-Dsg1 antibodies in PV would shut off the inherent suppression on ERK via Erbin-SHOC2 and thereby activate ERK in the absence of EGFR activation as observed following incubation with PV-IgG and PF-IgG (152, 228). This hypothesis is also in line with the inefficiency to prevent loss of adhesion by EGFR suppression in presence of higher concentrations of anti-Dsg1 autoantibodies, which releases the suppression of Erk1/2 signalling (217) (Figures 3, 5).
Dsg1-Mediated Ca2+-Signalling is Important For Epidermal Blistering in Pemphigus
Relatively early in pemphigus research, PV-IgG treatment was demonstrated to induce activation of PLC and Ca2+-influx via IP3R (156, 160). More recently, direct interaction of DSG1 and PI4K (220, 288, 289), an upstream kinase of PLC (290), was reported. For PLC, an interaction with both Dsg1 and Dsg3 was shown (220). Activated PLC generates the second messengers IP3 and DAG (156, 172) which leads to a release of Ca2+ from the endoplasmic reticulum (ER) to the cytoplasm (291). Stim1 and Orai1 are two ubiquitously expressed proteins, which together form the so-called CRAC (Ca2+-release-activated-channel). Stim 1 is located at the membrane of the ER (292) where it serves as a sensor for the Ca2+ concentration. Upon low Ca2+, Stim1 contacts Orai1, located at the plasma membrane, and both together from a Ca2+-specific channel causing Ca2+ influx and replenishing the Ca2+ stores of the ER (293–298). In line with this, inhibition of Ca2+ signalling via inhibition of either PI4K, PLC, IP3R or CRAC was sufficient to ameliorate pathogenic effects of pemphigus autoantibodies in vitro and inhibition of PLC and IP3R abrogated skin blistering in human skin ex vivo (220).
DAG and Ca2+ released by these mechanisms are activators of PKC (195, 299), causing its redistribution to cell contacts (200, 299). PV-IgG was demonstrated to induce PKC activation as well (127, 152, 172, 208, 300). The exact mechanism of action of PKC is yet unknown, however, it was observed that PKC interacts with the keratin filaments (161, 208, 301) and is regulated by PKP1 (207). PKC was furthermore shown to phosphorylate DP which causes destabilisation of the desmosomal plaque (197, 208, 209). Inhibition of PKC reduces the PV-IgG-induced depletion of Dsg3 (127), keratin retraction (208) and loss of cell adhesion (127, 152) in vitro and in vivo (127, 172) and skin blistering in mice in vivo (127, 172). PKC thus clearly plays a role in pemphigus pathogenesis. However, since it was reported that inhibition of PKC was not sufficient to prevent skin blistering in human skin ex vivo (215), PKC is probably not the only downstream target of the PLC/Ca2+ pathway. Furthermore, it needs to be noted that there are three types of PKCs with various isoforms, activated by different sets of second messengers. More specific, DAG and cytoplasmic Ca2+ activate Ca2+-dependent PKC (cPKC), DAG activates novel PKC (nPKC) and translocation activates atypical PKC (aPKC) isoforms, respectively (299). At least cPKCs and nPKCs are downstream targets of PLC and thus may be activated in response to autoantibodies in pemphigus (Figures 4, 5).

Figure 4. Schematic depiction of specifically Dsg1-dependent signalling mechanisms (grey RhoA complex represents a potential rescue pathway). The specific target of Erk1/2-Elk1 is not known.
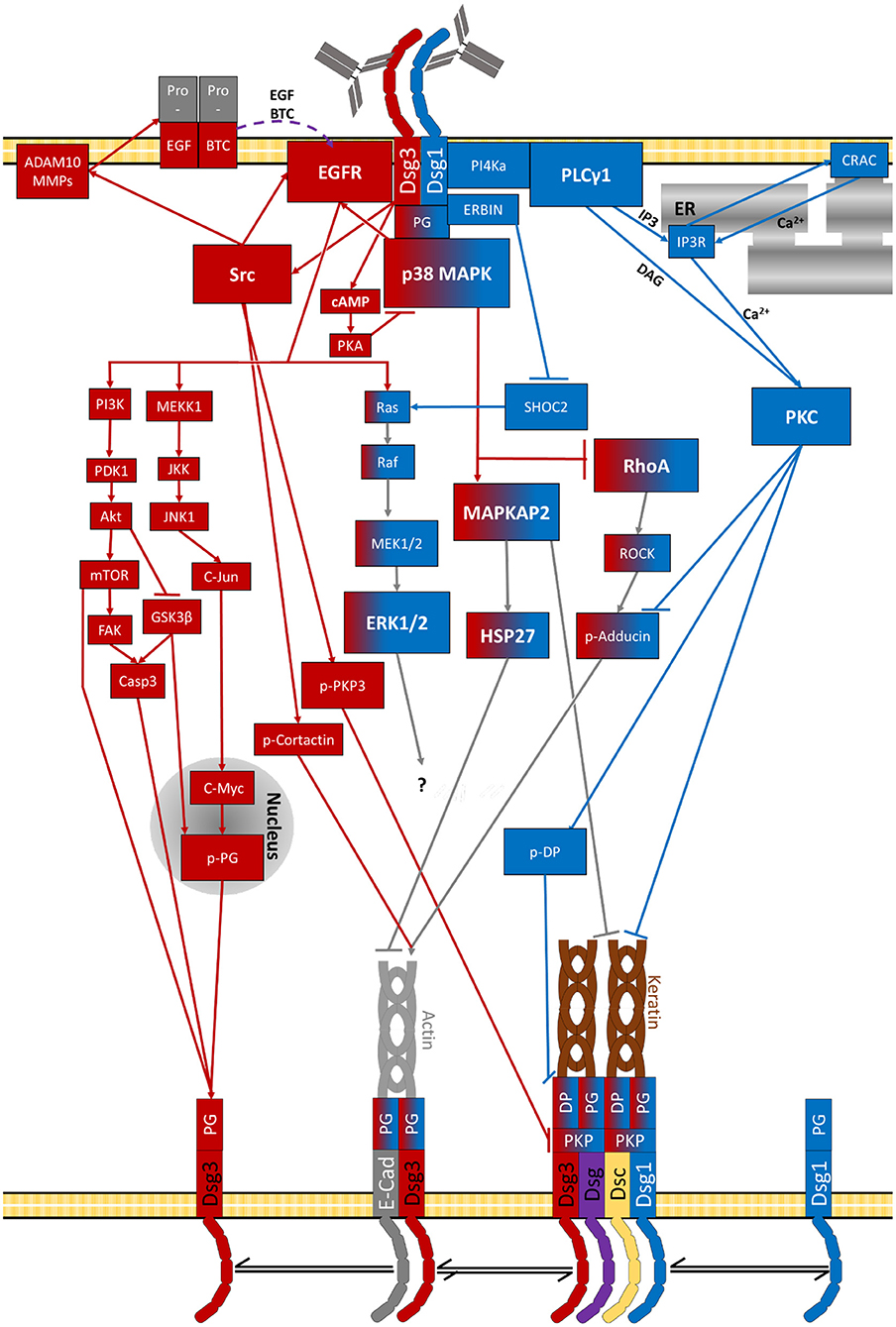
Figure 5. Signalling pathways activated by pemphigus-IgG (purple lines indicate diffusion of extracellular second messengers). The specific target of Erk1/2-Elk1 is not known.
Pre-Apoptotic Caspase Signalling Reducing Dsg1 and Dsg3 in Pemphigus
Several receptor-induced signalling cascades can initiate signalling associated with apoptosis. The caspase protein family plays a central role for this signalling. Caspases cleave certain cellular proteins (154) causing morphologic changes such as condensation of the nucleus, fragmentation of the DNA, cell shrinkage (302) and degradation of the cytoskeleton (303, 304). Amongst the proteins which are cleaved by caspases are the desmosomal cadherins and the plaque proteins causing degradation of desmosomes and cytoskeleton (305). The possibility that these apoptotic pathways can be active in pemphigus was demonstrated in several studies (79, 154, 302, 306–310). Apoptosis can occur in the skin (307, 309, 311, 312) as well as the mucosa (308) of pemphigus patients. However, in acantholytic areas apoptotic cell death is not typically observed (154, 177, 178).
It was shown that PV-IgG increases the activity of pro-apoptotic and decreases that of anti-apoptotic proteins in keratinocytes in vitro (309, 311, 313) and mice in vivo (266, 306, 307). Caspase3 seems to be the most relevant in this context (306, 307, 314). Caspase signalling may be activated by PV-IgG in several ways. Increased amounts of soluble Fas receptor ligands were found in pemphigus patients' sera and in mice after injection of PV-IgG. These induce activation of caspase8 which causes cleavage of Dsg molecules. Inhibition of Fas receptors and depletion of soluble Fas ligand were also shown to be protective in keratinocytes in vitro and mouse skin in vivo (309, 315). Activation via caspase8 was reported as well (316). In some patients anti-mitochondrial autoantibodies were found and it was shown that they can penetrate into mitochondria. These antibodies were reported to cause JNK activation and via caspases may induce degradation of Dsg3 (317, 318). Similarly, Dsg1 was also identified as a caspase3 target (319). The activation via c-Jun is also supported by the observation that inhibition of p38MAPK signalling, which causes c-Jun activation via EGFR, was sufficient to block caspase3 activation after PV-IgG treatment (248). Furthermore, EGFR was reported to induce caspase signalling and apoptosis (273). Thus, signalling by antibodies against Dsg3 may use this Src/EGFR signalling axis to activate caspases via PI3K/PDK1/Akt/GSK3β and nNOS/mTOR/FAK suggestive because inhibitors against this pathway have been shown to be protective against skin blistering (229, 266, 320, 321).
Moreover, caspase signalling could also be induced via the Ca2+ pathway. In this context, a mechanism was described which links the mitochondrial Ca2+ specific channel VDAC1 to IP3R via so-called mitochondria-associated membranes (MAM). The resulting Ca2+ flux was reported to lead to mitochondrial swelling and induction of apoptosis-related signals (322–326). Caspase3 activity can further be linked to p38MAPK, c-Myc (318), and RhoA activity (251). In line with this, several studies demonstrated that activation of p38MAPK, cell dissociation, and keratin retraction in keratinocytes in vitro (318) and skin blistering in mice in vivo (79, 266, 306, 307, 318) were significantly ameliorated under pharmacological inhibition of caspase activity.
However, the relevance of apoptosis for the pathology of pemphigus is controversly discussed and often attributed to be a secondary effect caused by acantholysis (111, 154). It was demonstrated that acantholysis takes place independent of apoptosis in keratinocytes in vitro (154, 318, 327, 328) as well as in pemphigus patient skin (154, 177, 318, 328). One group reported a slight increase of caspase3 in vitro and in vivo before the appearance of lesions but without any other sings of apoptosis (318). Taken together, all studies indicate a possible role of caspases for acantholysis independent of apoptotic cell death (306, 318). Since it was already found that caspases also influence several processes including proliferation, differentiation and cell cycle regulation (329), it can be concluded that caspases activate secondary pathways which contribute to skin blistering (154, 302). The fact that both Dsg1 and Dsg3 can be cleaved by caspase3 also would provide a mechanism how apoptotic signalling is directly interfering with desmosome turnover (Figures 3, 5).
Rescue Pathways in Pemphigus Such as Dsg2 and cAMP Upregulation
Most interestingly, autoantibodies in pemphigus not only cause pathogenic effects leading to loss of keratinocyte adhesion but also induce rescue pathways. One compensatory mechanism is up-regulation of Dsg2 expression in PV patients' lesions (330). It was demonstrated that forced expression of Dsg2 in the upper skin layers of transgenic mice was protective against PF-IgG-induced skin lesions (331). Moreover, in cultured keratinocytes it was shown that expression of Dsg2 (221) similar to Dsg1 (332) was protective against anti-Dsg3-antibody-induced loss of adhesion. Because Dsg2 can undergo heterophilic interaction with Dsg3, which is less sensitive to direct inhibition of autoantibodies targeting Dsg3, it is possible that Dsg2 up-regulation is supporting to maintain cell adhesion in PV (333).
Another protective pathway is dependent on the second messenger cAMP. Increased levels of cAMP were observed after incubation with PV-IgG and a pharmacological increase in cAMP blocked the effects of PV-IgG in vitro and skin blistering in mice in vivo (334). It was demonstrated that cAMP was protective against activation of p38MAPK which may involve PKA activation (334). This mechanism may be related to a phenomenon referred to as positive adhesiotropy by which adrenergic signalling in the heart strengthens cardiomyocyte adhesion via PKA-mediated PG phosphorylation (210, 212, 214). Recently, a case of a pemphigus patient was reported who was treated with Apremilast, a phosphodiesterase-4 inhibitor which enhances intracellular cAMP levels. Treatment ameliorated the symptoms and led to a decrease in autoantibody titers (335). Thus, the protective effect was attributed to changes in immune cell signalling rather than modulation of keratinocyte signalling.
Most recently it was reported, that mechanical stimulation also plays a very important role for the resistance against pemphigus-IgG-induced effects. Stretching of cells reportedly induced activation of RohA, strengthening the cortical acting network and its crosslinking, leading to increased cell contractility. This was shown to reduce pemphigus IgG induced effects, indicating, that activation of RhoA in pemphigus might represent an insufficient rescue pathway (336) (Figures 2, 5).
Conclusion: How to Elucidate a Feasible Approach to Stabilise Desmosomal Adhesion for Pemphigus Therapy?
From this review it can be concluded that some signalling pathways such as p38MAPK, ERK and PLC/Ca2+ appear to be more relevant for skin blistering in pemphigus than others. This is supported by a recent study using an unbiased approach by testing a library of 141 small molecule inhibitors in different experimental models of pemphigus which found that inhibition of ERK and p38MAPK signalling was effective to ameliorate PV-IgG-induced loss of cell adhesion (229).
On the other hand, inhibition of p38MAPK in clinical studies was not beneficial for pemphigus patients (271). This maybe explained in part by the fact that inhibition of a central signalling pathway involved in many different functions (234) is associated with severe side effects. It can be envisaged that the situation is not different for approaches modulating ERK or PLC/Ca2+ signalling indicating that at least systemic therapy using inhibitors of these pathways are most likely not feasible. Nevertheless, since topical application of Dsg-crosslinking peptides or drugs to increase cAMP were effective in pemphigus mouse models in vivo (216, 334) it may also be possible to reduce side-effects by site-specific application of modulators of signalling pathways in the future. Therefore, it is very encouraging that topical application of selumetinib to inhibit MEK1, which is the kinase up-stream of ERK1/2, was protective in the human skin organ culture model (229).
Moreover, identification of signalling mechanisms critical for pemphigus pathology allows to focus on the molecular targets of the associated signalling molecules. Because PKC downstream of PLC/Ca2+ has been shown to regulate desmosome stability via phosphorylation of DP, the modulation of DP cytoskeletal anchorage may be a promising target for therapy. Similarly, given that p38MAPK induces pathogenic effects via inhibition of RhoA, which via Rho kinase and adducin phosphorylation is important for desmosome assembly (190, 192, 193, 227), therapeutic approaches to stimulate desmosome assembly should be characterised in more detail. Finally, since the small inhibitor library helped to identify a role in keratinocyte adhesion regulation for VEGFR2, TrkA, PI3K, and PDK1 (Table 2), new candidates for treatment options have been elucidated as well (229). Therefore, we believe that studies on agents for additional treatment of patients suffering from the acute phase of pemphigus or facing relapses are of high importance in the future.
Author Contributions
TS: concept, original draft, figure design, and revision. JW: concept and revision. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the DFG FOR2497 (PEGASUS) grant to JW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We cordially thank Prof. Dr. Flaig and Prof. Dr. Gerd Plewig (Dermatology Department, LMU Munich) as well as Prof. Dr. Thomás Cunha and Prof. Dr. Michael Hertl (Dermatology Department, University of Marburg) for contribution of images showing clinics and histology of pemphigus.
References
1. Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, et al. Pemphigus. Nat Rev Dis Primers. (2017) 3:17026. doi: 10.1038/nrdp.2017.26
2. Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet. (2019) 394:882–94. doi: 10.1016/S0140-6736(19)31778-7
3. Waschke J. The desmosome and pemphigus. Histochem Cell Biol. (2008) 130:21–54. doi: 10.1007/s00418-008-0420-0
4. Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol. (2004) 5:271–81. doi: 10.1038/nrm1356
5. Price AJ, Cost A-L, Ungewiß H, Waschke J, Dunn AR, Grashoff C. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat Commun. (2018) 9:7523. doi: 10.1038/s41467-018-07523-0
6. Hatzfeld M, Haffner C, Schulze K, Vinzens U. The function of plakophilin 1 in desmosome assembly and actin filament organization. J Cell Biol. (2000) 149:209–22. doi: 10.1083/jcb.149.1.209
7. Bonné S, Gilbert B, Hatzfeld M, Chen X, Green KJ, van Roy F. Defining desmosomal plakophilin-3 interactions. J Cell Biol. (2003) 161:403–16. doi: 10.1083/jcb.200303036
8. Cirillo N, Prime SS. Desmosomal interactome in keratinocytes: a systems biology approach leading to an understanding of the pathogenesis of skin disease. Cell Mol Life Sci. (2009) 66:3517–33. doi: 10.1007/s00018-009-0139-7
9. Godsel LM, Hsieh SN, Amargo EV, Bass AE, Pascoe-McGillicuddy LT, Huen AC, et al. Desmoplakin assembly dynamics in four dimensions: multiple phases differentially regulated by intermediate filaments and actin. J Cell Biol. (2005) 171:1045–59. doi: 10.1083/jcb.200510038
10. Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and beta -catenin signaling. J Biol Chem. (2002) 277:10512–22. doi: 10.1074/jbc.M108765200
11. Smith EA, Fuchs E. Defining the interactions between intermediate filaments and desmosomes. J Cell Biol. (1998) 141:1229–41. doi: 10.1083/jcb.141.5.1229
12. Wu H, Stanley JR, Cotsarelis G. Desmoglein isotype expression in the hair follicle and its cysts correlates with type of keratinization and degree of differentiation. J Invest Dermatol. (2003) 120:1052–7. doi: 10.1046/j.1523-1747.2003.12234.x
13. Berika M, Garrod D. Desmosomal adhesion in vivo. Cell Commun Adhes. (2014) 21:65–75. doi: 10.3109/15419061.2013.876018
14. Hartlieb E, Kempf B, Partilla M, Vigh B, Spindler V, Waschke J. Desmoglein 2 is less important than desmoglein 3 for keratinocyte cohesion. PLoS ONE. (2013) 8:e53739. doi: 10.1371/journal.pone.0053739
15. Egu DT, Sigmund AM, Schmidt E, Spindler V, Walter E, Waschke J, et al. New ex vivo human oral mucosa model reveals that p38MAPK inhibition is not effective in preventing autoantibody-induced mucosal blistering in pemphigus. Br J Dermatol. (2019) 182:987–94. doi: 10.1111/bjd.18237
16. Wu H, Wang ZH, Yan A, Lyle S, Fakharzadeh S, Wahl JK, et al. Protection against pemphigus foliaceus by desmoglein 3 in neonates. N Engl J Med. (2000) 343:31–5. doi: 10.1056/NEJM200007063430105
17. Hübner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Invest Dermatol. (2016) 136:2495–8. doi: 10.1016/j.jid.2016.07.013
18. Yan L, Wang J-M, Zeng K. Association between HLA-DRB1 polymorphisms and pemphigus vulgaris: a meta-analysis. Br J Dermatol. (2012) 167:768–77. doi: 10.1111/j.1365-2133.2012.11040.x
19. Zivanovic D, Bojic S, Medenica L, Andric Z, Popadic D. Human leukocyte antigen class II (DRB1 and DQB1) alleles and haplotypes frequencies in patients with pemphigus vulgaris among the Serbian population. HLA. (2016) 87:367–74. doi: 10.1111/tan.12796
20. Ahmed AR, Yunis EJ, Khatri K, Wagner R, Notani G, Awdeh Z, et al. Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. Proc Natl Acad Sci USA. (1990) 87:7658–62. doi: 10.1073/pnas.87.19.7658
21. Shams S, Amirzargar AA, Yousefi M, Rezaei N, Solgi G, Khosravi F, et al. HLA class II (DRB, DQA1 and DQB1) allele and haplotype frequencies in the patients with pemphigus vulgaris. J Clin Immunol. (2009) 29:175–9. doi: 10.1007/s10875-008-9244-x
22. Vodo D, Sarig O, Sprecher E. The genetics of pemphigus vulgaris. Front Med. (2018) 5:226. doi: 10.3389/fmed.2018.00226
23. Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. (2018) 66:255–70. doi: 10.1007/s12026-018-8986-7
24. Lee CW, Yang HY, Kim SC, Jung JH, Hwang JJ. HLA class II allele associations in Korean patients with pemphigus. Dermatology. (1998) 197:349–52. doi: 10.1159/000018030
25. Gibson WT, Walter MA, Ahmed AR, Alper CA, Cox DW. The immunoglobulin heavy chain and disease association: application to pemphigus vulgaris. Hum Genet. (1994) 94:675–83. doi: 10.1007/BF00206963
26. Zitouni M, Martel P, Ben Ayed M, Raux G, Gilbert D, Joly P, et al. Pemphigus is not associated with allotypic markers of immunoglobulin kappa. Genes Immun. (2002) 3:50–2. doi: 10.1038/sj.gene.6363817
27. Di Zenzo G, Di Lullo G, Corti D, Calabresi V, Sinistro A, Vanzetta F, et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J Clin Invest. (2012) 122:3781–90. doi: 10.1172/JCI64413
28. Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. (2008) 128:939–48. doi: 10.1038/sj.jid.5701132
29. Collins AM, Sewell WA, Edwards MR. Immunoglobulin gene rearrangement, repertoire diversity, and the allergic response. Pharmacol Ther. (2003) 100:157–70. doi: 10.1016/j.pharmthera.2003.07.002
30. Yamagami J, Kacir S, Ishii K, Payne AS, Siegel DL, Stanley JR. Antibodies to the desmoglein 1 precursor proprotein but not to the mature cell surface protein cloned from individuals without pemphigus. J Immunol. (2009) 183:5615–21. doi: 10.4049/jimmunol.0901691
31. Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. (2005) 115:888–99. doi: 10.1172/JCI24185
32. Sarig O, Bercovici S, Zoller L, Goldberg I, Indelman M, Nahum S, et al. Population-specific association between a polymorphic variant in ST18, encoding a pro-apoptotic molecule, and pemphigus vulgaris. J Invest Dermatol. (2012) 132:1798–805. doi: 10.1038/jid.2012.46
33. Javor J, Chmurova N, Parnicka Z, Ferencik S, Grosse-Wilde H, Buc M, et al. TNF-alpha and IL-10 gene polymorphisms show a weak association with pemphigus vulgaris in the Slovak population. J Eur Acad Dermatol Venereol. (2010) 24:65–8. doi: 10.1111/j.1468-3083.2009.03260.x
34. Mosaad YM, Fathy H, Fawzy Z, El-Saied MA. Tumor necrosis factor-α−308 GA and interleukin-6−174 GC promoter polymorphisms and pemphigus. Hum Immunol. (2012) 73:560–5. doi: 10.1016/j.humimm.2012.02.001
35. Eberhard Y, Burgos E, Gagliardi J, Vullo CM, Borosky A, Pesoa S, et al. Cytokine polymorphisms in patients with pemphigus. Arch Dermatol Res. (2005) 296:309–13. doi: 10.1007/s00403-004-0528-6
36. Slomov E, Loewenthal R, Korostishevsky M, Goldberg I, Brenner S, Gazit E. Pemphigus vulgaris is associated with the transporter associated with antigen processing (TAP) system. Hum Immunol. (2005) 66:1213–22. doi: 10.1016/j.humimm.2005.11.004
37. Tanasilovic S, Popadic S, Medenica L, Popadic D. Pemphigus vulgaris and pemphigus foliaceus determined by CD86 and CTLA4 polymorphisms. Clin Dermatol. (2017) 35:236–41. doi: 10.1016/j.clindermatol.2016.05.021
38. Capon F, Bharkhada J, Cochrane NE, Mortimer NJ, Setterfield JF, Reynaert S, et al. Evidence of an association between desmoglein 3 haplotypes and pemphigus vulgaris. Br J Dermatol. (2006) 154:67–71. doi: 10.1111/j.1365-2133.2005.06882.x
39. Assaf S, Malki L, Mayer T, Mohamad J, Peled A, Pavlovsky M, et al. ST18 affects cell-cell adhesion in pemphigus vulgaris in a tumor necrosis factor-α-dependent fashion. Br J Dermatol. (2020) 2020:19679. doi: 10.1111/bjd.19679
40. Radeva MY, Walter E, Stach RA, Yazdi AS, Schlegel N, Sarig O, et al. ST18 enhances PV-IgG-induced loss of keratinocyte cohesion in parallel to increased ERK activation. Front Immunol. (2019) 10:770. doi: 10.3389/fimmu.2019.00770
41. Sena Nogueira Maehara L de, De-Souza-Santana FC, Porro AM, Marcos EV, Ura S, Nolte IM, et al. HLA class II alleles of susceptibility and protection in Brazilian and Dutch pemphigus foliaceus. Br J Dermatol. (2018) 178:e212–4. doi: 10.1111/bjd.16022
42. Lombardi ML, Mercuro O, Ruocco V, Lo Schiavo A, Lombari V, Guerrera V, et al. Common human leukocyte antigen alleles in pemphigus vulgaris and pemphigus foliaceus Italian patients. J Invest Dermatol. (1999) 113:107–10. doi: 10.1046/j.1523-1747.1999.00626.x
43. Loiseau P, Lecleach L, Prost C, Lepage V, Busson M, Bastuji-Garin S, et al. HLA class II polymorphism contributes to specify desmoglein derived peptides in pemphigus vulgaris and pemphigus foliaceus. J Autoimmun. (2000) 15:67–73. doi: 10.1006/jaut.2000.0388
44. del Mar Sáez-de-Ocariz M, Vega-Memije ME, Zúñiga J, Salgado N, Ruíz J, Balbuena A, et al. HLA-DRB1*0101 is associated with foliaceous pemphigus in Mexicans. Int J Dermatol. (2005) 44:350. doi: 10.1111/j.1365-4632.2005.02038.x
45. Abida O, Zitouni M, Kallel-Sellami M, Mahfoudh N, Kammoun A, Ben Ayed M, et al. Tunisian endemic pemphigus foliaceus is associated with the HLA-DR3 gene: anti-desmoglein 1 antibody-positive healthy subjects bear protective alleles. Br J Dermatol. (2009) 161:522–7. doi: 10.1111/j.1365-2133.2009.09207.x
46. Ben Jmaa M, Abida O, Bahloul E, Toumi A, Khlif S, Fakhfakh R, et al. Role of FOXP3 gene polymorphism in the susceptibility to Tunisian endemic Pemphigus Foliaceus. Immunol Lett. (2017) 184:105–11. doi: 10.1016/j.imlet.2017.02.005
47. Hans-Filho G, Aoki V, Bittner NR, Bittner GC. Fogo selvagem: endemic pemphigus foliaceus. An Bras Dermatol. (2018) 93:638–50. doi: 10.1590/abd1806-4841.20188235
48. Diaz LA, Sampaio SA, Rivitti EA, Martins CR, Cunha PR, Lombardi C, et al. Endemic pemphigus foliaceus (fogo selvagem). I Clinical features and immunopathology. J Am Acad Dermatol. (1989) 20:657–69. doi: 10.1016/S0190-9622(89)70079-7
49. Empinotti JC, Aoki V, Filgueira A, Sampaio SA, Rivitti EA, Sanches JA, et al. Clinical and serological follow-up studies of endemic pemphigus foliaceus (fogo selvagem) in Western Parana, Brazil (2001-2002). Br J Dermatol. (2006) 155:446–50. doi: 10.1111/j.1365-2133.2006.07302.x
50. Aoki V, Rivitti EA, Diaz LA. Update on fogo selvagem, an endemic form of pemphigus foliaceus. J Dermatol. (2015) 42:18–26. doi: 10.1111/1346-8138.12675
51. Diaz LA, Arteaga LA, Hilario-Vargas J, Valenzuela JG, Li N, Warren S, et al. Anti-desmoglein-1 antibodies in onchocerciasis, leishmaniasis and Chagas disease suggest a possible etiological link to Fogo selvagem. J Invest Dermatol. (2004) 123:1045–51. doi: 10.1111/j.0022-202X.2004.23438.x
52. Culton DA, Qian Y, Li N, Rubenstein D, Aoki V, Filhio GH, et al. Advances in pemphigus and its endemic pemphigus foliaceus (Fogo Selvagem) phenotype: a paradigm of human autoimmunity. J Autoimmun. (2008) 31:311–24. doi: 10.1016/j.jaut.2008.08.003
53. Bastuji-Garin S, Souissi R, Blum L, Turki H, Nouira R, Jomaa B, et al. Comparative epidemiology of pemphigus in Tunisia and France: unusual incidence of pemphigus foliaceus in young Tunisian women. J Invest Dermatol. (1995) 104:302–5. doi: 10.1111/1523-1747.ep12612836
54. Robledo MA, Prada S, Jaramillo D, Leon W. South American pemphigus foliaceus: study of an epidemic in El Bagre and Nechi, Colombia 1982 to 1986. Br J Dermatol. (1988) 118:737–44. doi: 10.1111/j.1365-2133.1988.tb02590.x
55. Kneisel A, Hertl M. Autoimmune bullous skin diseases. Part 1: clinical manifestations. J Dtsch Dermatol Ges. (2011) 9:844–56. doi: 10.1111/j.1610-0387.2011.07793.x
56. Vielmuth F, Rötzer V, Hartlieb E, Hirneiß C, Waschke J, Spindler V. Pemphigus autoantibodies induce blistering in human conjunctiva. Invest Ophthalmol Vis Sci. (2016) 57:4442–9. doi: 10.1167/iovs.16-19582
57. Schmidt E, Goebeler M, Hertl M, Sárdy M, Sitaru C, Eming R, et al. S2k-Leitlinie zur Diagnostik des Pemphigus vulgaris/foliaceus und des bullösen Pemphigoids. J Deutschen Dermatologischen Gesellschaft. (2015) 13:713–27. doi: 10.1111/ddg.40_12612
58. Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. (2011) 29:432–6. doi: 10.1016/j.clindermatol.2011.01.013
59. Waschke J, Spindler V. Desmosomes and extradesmosomal adhesive signaling contacts in pemphigus. Med Res Rev. (2014) 34:1127–45. doi: 10.1002/med.21310
60. Pigozzi B, Peserico A, Schiesari L, Alaibac M. Pemphigus foliaceus evolving into pemphigus vulgaris: a probable example of ‘intermolecular epitope spreading’ confirmed by enzyme-linked immunosorbent assay study. J Eur Acad Dermatol Venerol. (2008) 22:242–4. doi: 10.1111/j.1468-3083.2007.02298.x
61. Kneisel A, Hertl M. Autoimmune bullous skin diseases. Part 2: diagnosis and therapy. J Dtsch Dermatol Ges. (2011) 9:927–47. doi: 10.1111/j.1610-0387.2011.07809.x
62. Mao X, Nagler AR, Farber SA, Choi EJ, Jackson LH, Leiferman KM, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am J Pathol. (2010) 177:2724–30. doi: 10.2353/ajpath.2010.100483
63. Rafei D, Müller R, Ishii N, Llamazares M, Hashimoto T, Hertl M, et al. IgG autoantibodies against desmocollin 3 in pemphigus sera induce loss of keratinocyte adhesion. Am J Pathol. (2011) 178:718–23. doi: 10.1016/j.ajpath.2010.10.016
64. Spindler V, Heupel W-M, Efthymiadis A, Schmidt E, Eming R, Rankl C, et al. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J Biol Chem. (2009) 284:30556–64. doi: 10.1074/jbc.M109.024810
65. Müller R, Heber B, Hashimoto T, Messer G, Müllegger R, Niedermeier A, et al. Autoantibodies against desmocollins in European patients with pemphigus. Clin Exp Dermatol. (2009) 34:3241. doi: 10.1111/j.1365-2230.2009.03241.x
66. Ivars M, España A, Alzuguren P, Pelacho B, Lasarte JJ, López-Zabalza MJ. The involvement of ADAM10 in acantholysis in mucocutaneous pemphigus vulgaris depends on the autoantibody profile of each patient. Br J Dermatol. (2020) 182:1194–204. doi: 10.1111/bjd.18382
67. Geller S, Gat A, Zeeli T, Hafner A, Eming R, Hertl M, et al. The expanding spectrum of IgA pemphigus: a case report and review of the literature. Br J Dermatol. (2014) 171:650–6. doi: 10.1111/bjd.12940
68. Lenz P, Amagai M, Volc-Platzer B, Stingl G, Kirnbauer R. Desmoglein 3-ELISA: a pemphigus vulgaris-specific diagnostic tool. Arch Dermatol. (1999) 135:143–8. doi: 10.1001/archderm.135.2.143
69. Ishii K, Amagai M, Hall RP, Hashimoto T, Takayanagi A, Gamou S, et al. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. (1997) 159:2010–7.
70. Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. (2016) 11:175–97. doi: 10.1146/annurev-pathol-012615-044313
71. Carson PJ, Hameed A, Ahmed AR. Influence of treatment on the clinical course of pemphigus vulgaris. J Am Acad Dermatol. (1996) 34:645–52. doi: 10.1016/S0190-9622(96)80066-1
72. Eming R. Pemphigus. Modellerkrankung für zielgerichtete Therapien Hautarzt. (2015) 66:574–82. doi: 10.1007/s00105-015-3656-3
73. Hooten JN, Hall RP, Cardones AR. Updates on the management of autoimmune blistering diseases. Skin Therapy Lett. (2014) 19:1–6.
74. Herrmann G, Hunzelmann N, Engert A. Treatment of pemphigus vulgaris with anti-CD20 monoclonal antibody (rituximab). Br J Dermatol. (2003) 148:602–3. doi: 10.1046/j.1365-2133.2003.05209_10.x
75. Müller R, Hunzelmann N, Baur V, Siebenhaar G, Wenzel E, Eming R, et al. Targeted immunotherapy with rituximab leads to a transient alteration of the IgG autoantibody profile in pemphigus vulgaris. Dermatol Res Pract. (2010) 2010:321950. doi: 10.1155/2010/321950
76. Mazzi G, Raineri A, Zanolli FA, Da Ponte C, de Roia D, Santarossa L, et al. Plasmapheresis therapy in pemphigus vulgaris and bullous pemphigoid. Transfus Apher Sci. (2003) 28:13–8. doi: 10.1016/S1473-0502(02)00095-2
77. Lunardon L, Payne AS. Inhibitory human antichimeric antibodies to rituximab in a patient with pemphigus. J Allergy Clin Immunol. (2012) 130:800–3. doi: 10.1016/j.jaci.2012.03.022
78. Eming R, Sticherling M, Hofmann SC, Hunzelmann N, Kern JS, Kramer H, et al. S2k-Leitlinie zur Therapie des Pemphigus vulgaris/foliaceus und des bullösen Pemphigoid. J Deutschen Dermatologischen Gesellschaft. (2015) 13:833–45. doi: 10.1111/ddg.140_12606
79. Arredondo J, Chernyavsky AI, Karaouni A, Grando SA. Novel mechanisms of target cell death and survival and of therapeutic action of IVIg in pemphigus. Am J Pathol. (2005) 167:1531–44. doi: 10.1016/S0002-9440(10)61239-4
80. Getsios S, Waschke J, Borradori L, Hertl M, Müller EJ. From cell signaling to novel therapeutic concepts: international pemphigus meeting on advances in pemphigus research and therapy. J Invest Dermatol. (2010) 130:1764–8. doi: 10.1038/jid.2010.111
81. Shimanovich I, Herzog S, Schmidt E, Opitz A, Klinker E, Bröcker E-B, et al. Improved protocol for treatment of pemphigus vulgaris with protein A immunoadsorption. Clin Exp Dermatol. (2006) 31:768–74. doi: 10.1111/j.1365-2230.2006.02220.x
82. Schmidt E, Klinker E, Opitz A, Herzog S, Sitaru C, Goebeler M, et al. Protein A immunoadsorption: a novel and effective adjuvant treatment of severe pemphigus. Br J Dermatol. (2003) 148:1222–9. doi: 10.1046/j.1365-2133.2003.05302.x
83. Baum S, Scope A, Barzilai A, Azizi E, Trau H. The role of IVIg treatment in severe pemphigus vulgaris. J Eur Acad Dermatol Venerol. (2006) 20:548–52. doi: 10.1111/j.1468-3083.2006.01540.x
84. Bayry J, Kazatchkine MD, Kaveri SV. Erratum: shortage of human intravenous immunoglobulin—reasons and possible solutions. Nat Rev Neurol. (2008) 4:405. doi: 10.1038/ncpneuro0856
85. Kridin K, Ahmed AR. The evolving role of rituximab in the treatment of pemphigus vulgaris: a comprehensive state-of-the-art review. Expert Opin Biol Ther. (2021) 21:443–54. doi: 10.1080/14712598.2021.1874915
86. Maho-Vaillant M, Perals C, Golinski M-L, Hébert V, Caillot F, Mignard C, et al. Rituximab and corticosteroid effect on desmoglein-specific B-cells and desmoglein-specific T-follicular-helper-cells in pemphigus. J Invest Dermatol. (2021). doi: 10.1016/j.jid.2021.01.031
87. Du FH, Mills EA, Mao-Draayer Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun Highlights. (2017) 8:12. doi: 10.1007/s13317-017-0100-y
88. Rapp M, Pentland A, Richardson C. Successful treatment of pemphigus vulgaris with ofatumumab. J Drugs Dermatol. (2018) 17:1338–9.
89. Reddy V, Klein C, Isenberg DA, Glennie MJ, Cambridge G, Cragg MS, et al. Obinutuzumab induces superior B-cell cytotoxicity to rituximab in rheumatoid arthritis and systemic lupus erythematosus patient samples. Rheumatology. (2017) 56:1227–37. doi: 10.1093/rheumatology/kex067
90. Stohl W. Inhibition of B cell activating factor (BAFF) in the management of systemic lupus erythematosus (SLE). Expert Rev Clin Immunol. (2017) 13:623–33. doi: 10.1080/1744666X.2017.1291343
91. Musette P, Bouaziz JD. B cell modulation strategies in autoimmune diseases: new concepts. Front Immunol. (2018) 9:622. doi: 10.3389/fimmu.2018.00622
92. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. (2016) 2:16039. doi: 10.1038/nrdp.2016.39
93. Crofford LJ, Nyhoff LE, Sheehan JH, Kendall PL. The role of Bruton's tyrosine kinase in autoimmunity and implications for therapy. Expert Rev Clin Immunol. (2016) 12:763–73. doi: 10.1586/1744666X.2016.1152888
94. Corneth OB, Klein Wolterink RG, Hendriks RW. BTK signaling in B cell differentiation and autoimmunity. Curr Top Microbiol Immunol. (2016) 393:67–105. doi: 10.1007/82_2015_478
95. Amber KT, Staropoli P, Shiman MI, Elgart GW, Hertl M. Autoreactive T cells in the immune pathogenesis of pemphigus vulgaris. Exp Dermatol. (2013) 22:699–704. doi: 10.1111/exd.12229
96. Eming R, Hennerici T, Bäcklund J, Feliciani C, Visconti KC, Willenborg S, et al. Pathogenic IgG antibodies against desmoglein 3 in pemphigus vulgaris are regulated by HLA-DRB1*04:02-restricted T cells. J Immunol. (2014) 193:4391–9. doi: 10.4049/jimmunol.1401081
97. Nagel A, Lang A, Engel D, Podstawa E, Hunzelmann N, de Pita O, et al. Clinical activity of pemphigus vulgaris relates to IgE autoantibodies against desmoglein 3. Clin Immunol. (2010) 134:320–30. doi: 10.1016/j.clim.2009.11.006
98. Chen W-S, Chen Y-J, Yang C-S, Juan C-K. Pemphigus foliaceus associated with imatinib therapy in a patient with gastrointestinal stromal tumor. Int J Dermatol. (2018) 57:e93–5. doi: 10.1111/ijd.14142
99. Sood A, Sinha P, Raman DK, Sinha A. Imatinib-induced IgA pemphigus: subcorneal pustular dermatosis type. Indian Dermatol Online J. (2018) 9:331–3. doi: 10.4103/idoj.IDOJ_331_17
100. Ono S, Egawa G, Nomura T, Kitoh A, Dainichi T, Otsuka A, et al. Abl family tyrosine kinases govern IgG extravasation in the skin in a murine pemphigus model. Nat Commun. (2019) 10:1–13. doi: 10.1038/s41467-019-12232-3
101. Ward ES, Ober RJ. Targeting FcRn to generate antibody-based therapeutics. Trends Pharmacol Sci. (2018) 39:892–904. doi: 10.1016/j.tips.2018.07.007
102. Goebl NA, Babbey CM, Datta-Mannan A, Witcher DR, Wroblewski VJ, Dunn KW. Neonatal Fc receptor mediates internalization of Fc in transfected human endothelial cells. Mol Biol Cell. (2008) 19:5490–505. doi: 10.1091/mbc.e07-02-0101
103. Sesarman A, Vidarsson G, Sitaru C. The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell Mol Life Sci. (2010) 67:2533–50. doi: 10.1007/s00018-010-0318-6
104. Yokoyama T, Matsuda S, Takae Y, Wada N, Nishikawa T, Amagai M, et al. Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int Immunol. (2011) 23:365–73. doi: 10.1093/intimm/dxr020
105. Amber KT, Maglie R, Solimani F, Eming R, Hertl M. Targeted therapies for autoimmune bullous diseases: current status. Drugs. (2018) 78:1527–48. doi: 10.1007/s40265-018-0976-5
106. Vafia K, Groth S, Beckmann T, Hirose M, Dworschak J, Recke A, et al. Pathogenicity of autoantibodies in anti-p200 pemphigoid. PLoS ONE. (2012) 7:e41769. doi: 10.1371/journal.pone.0041769
107. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
108. Wegner A. Chimeric antigen receptor T cells for the treatment of cancer and the future of preclinical models for predicting their toxicities. Immunotherapy. (2017) 9:669–80. doi: 10.2217/imt-2017-0028
109. Kalantari-Dehaghi M, Anhalt GJ, Camilleri MJ, Chernyavsky AI, Chun S, Felgner PL, et al. Pemphigus vulgaris autoantibody profiling by proteomic technique. PLoS ONE. (2013) 8:e57587. doi: 10.1371/journal.pone.0057587
110. Grando SA, Pittelkow MR, Shultz LD, Dmochowski M, Nguyen VT. Pemphigus: an unfolding story. J Investig Dermatol. (2001) 117:990–5. doi: 10.1046/j.0022-202x.2001.01489.x
111. Pan M, Liu X, Zheng J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin Exp Dermatol. (2011) 36:703–7. doi: 10.1111/j.1365-2230.2011.04092.x
112. Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, et al. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. (2003) 170:2170–8. doi: 10.4049/jimmunol.170.4.2170
113. Yoshida K, Ishii K, Shimizu A, Yokouchi M, Amagai M, Shiraishi K, et al. Non-pathogenic pemphigus foliaceus (PF) IgG acts synergistically with a directly pathogenic PF IgG to increase blistering by p38MAPK-dependent desmoglein 1 clustering. J Dermatol Sci. (2017) 85:197–207. doi: 10.1016/j.jdermsci.2016.12.010
114. Di Zenzo G, Amber KT, Sayar BS, Müller EJ, Borradori L. Immune response in pemphigus and beyond: progresses and emerging concepts. Semin Immunopathol. (2016) 38:57–74. doi: 10.1007/s00281-015-0541-1
115. Abida O, Masmoudi A, Rebaï A, Ben Ayed M, Mahfoudh N, Kallel-Sellami M, et al. The familial feature of Tunisian endemic pemphigus foliaceus. Br J Dermatol. (2009) 161:951–3. doi: 10.1111/j.1365-2133.2009.09386.x
116. Joly P, Mokhtar I, Gilbert D, Thomine E, Fazza B, Bardi R, et al. Immunoblot and immunoelectronmicroscopic analysis of endemic Tunisian pemphigus. Br J Dermatol. (1999) 140:44–9. doi: 10.1046/j.1365-2133.1999.02605.x
117. Kallel Sellami M, Ben Ayed M, Mouquet H, Drouot L, Zitouni M, Mokni M, et al. Anti-desmoglein 1 antibodies in Tunisian healthy subjects: arguments for the role of environmental factors in the occurrence of Tunisian pemphigus foliaceus. Clin Exp Immunol. (2004) 137:195–200. doi: 10.1111/j.1365-2249.2004.02493.x
118. Takahashi H, Kuwana M, Amagai M. A single helper T cell clone is sufficient to commit polyclonal naive B cells to produce pathogenic IgG in experimental pemphigus vulgaris. J Immunol. (2009) 182:1740–5. doi: 10.4049/jimmunol.182.3.1740
119. Koch PJ, Mahoney MG, Ishikawa H, Pulkkinen L, Uitto J, Shultz L, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. (1997) 137:1091–102. doi: 10.1083/jcb.137.5.1091
120. Amagai M, Tsunoda K, Suzuki H, Nishifuji K, Koyasu S, Nishikawa T. Use of autoantigen-knockout mice in developing an active autoimmune disease model for pemphigus. J Clin Invest. (2000) 105:625–31. doi: 10.1172/JCI8748
121. Baron S, Hoang A, Vogel H, Attardi LD. Unimpaired skin carcinogenesis in desmoglein 3 knockout mice. PLoS ONE. (2012) 7:50024. doi: 10.1371/journal.pone.0050024
122. Chen J, Den Z, Koch PJ. Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. (2008) 121:2844–9. doi: 10.1242/jcs.031518
123. Kamiya K, Aoyama Y, Wakashima C, Kudo T, Nakajima K, Sano S, et al. Atypical pemphigus with immunoglobulin G autoantibodies against desmoglein 3 and desmocollin 3. J Dermatol. (2016) 43:429–31. doi: 10.1111/1346-8138.13086
124. Lotti R, Atene CG, Marconi A, Di Rocco G, Reggiani Bonetti L, Zanocco Marani T, et al. Development of a desmocollin-3 active mouse model recapitulating human atypical pemphigus. Front Immunol. (2019) 10:1387. doi: 10.3389/fimmu.2019.01387
125. Kugelmann D, Radeva MY, Spindler V, Waschke J. Desmoglein 1 deficiency causes lethal skin blistering. J Invest Dermatol. (2019) 139:1596–9.e2. doi: 10.1016/j.jid.2019.01.002
126. Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, et al. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem. (2006) 281:7623–34. doi: 10.1074/jbc.M512447200
127. Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J. The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca2+-induced differentiation. Am J Pathol. (2011) 179:1905–16. doi: 10.1016/j.ajpath.2011.06.043
128. Shu E, Yamamoto Y, Aoyama Y, Kitajima Y. Intraperitoneal injection of pemphigus vulgaris-IgG into mouse depletes epidermal keratinocytes of desmoglein 3 associated with generation of acantholysis. Arch Dermatol Res. (2007) 299:165–7. doi: 10.1007/s00403-007-0754-9
129. Jennings JM, Tucker DK, Kottke MD, Saito M, Delva E, Hanakawa Y, et al. Desmosome disassembly in response to pemphigus vulgaris IgG occurs in distinct phases and can be reversed by expression of exogenous Dsg3. J Invest Dermatol. (2011) 131:706–18. doi: 10.1038/jid.2010.389
130. Yamamoto Y, Aoyama Y, Shu E, Tsunoda K, Amagai M, Kitajima Y. Anti-desmoglein 3 (Dsg3) monoclonal antibodies deplete desmosomes of Dsg3 and differ in their Dsg3-depleting activities related to pathogenicity. J Biol Chem. (2007) 282:17866–76. doi: 10.1074/jbc.M607963200
131. Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol. (1999) 112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x
132. Jolly PS, Berkowitz P, Bektas M, Lee H-E, Chua M, Diaz LA, et al. p38MAPK signaling and desmoglein-3 internalization are linked events in pemphigus acantholysis. J Biol Chem. (2010) 285:8936–41. doi: 10.1074/jbc.M109.087999
133. Wolff K, Schreiner E. Ultrastructural localization of pemphigus autoantibodies within e epidermis. Nature. (1971) 229:59–61. doi: 10.1038/229059a0
134. Jones JC, Yokoo KM, Goldman RD. A cell surface desmosome-associated component: identification of tissue-specific cell adhesion molecule. Proc Natl Acad Sci USA. (1986) 83:7282–6. doi: 10.1073/pnas.83.19.7282
135. Sato M, Aoyama Y, Kitajima Y. Assembly pathway of desmoglein 3 to desmosomes and its perturbation by pemphigus vulgaris-IgG in cultured keratinocytes, as revealed by time-lapsed labeling immunoelectron microscopy. Lab Invest. (2000) 80:1583–92. doi: 10.1038/labinvest.3780168
136. Bédane C, Prost C, Thomine E, Intrator L, Joly P, Caux F, et al. Binding of autoantibodies is not restricted to desmosomes in pemphigus vulgaris: comparison of 14 cases of pemphigus vulgaris and 10 cases of pemphigus foliaceus studied by western immunoblot and immunoelectron microscopy. Arch Dermatol Res. (1996) 288:343–52. doi: 10.1007/BF02507101
137. Vielmuth F, Waschke J, Spindler V. Loss of desmoglein binding is not sufficient for keratinocyte dissociation in pemphigus. J Invest Dermatol. (2015) 135:3068–77. doi: 10.1038/jid.2015.324
138. Tsang SM, Brown L, Lin K, Liu L, Piper K, O'Toole EA, et al. Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J Pathol. (2012) 227:81–93. doi: 10.1002/path.3982
139. Mao X, Choi EJ, Payne AS. Disruption of desmosome assembly by monovalent human pemphigus vulgaris monoclonal antibodies. J Invest Dermatol. (2009) 129:908–18. doi: 10.1038/jid.2008.339
140. Takahashi Y, Patel HP, Labib RS, Diaz LA, Anhalt GJ. Experimentally induced pemphigus vulgaris in neonatal BALB/c mice: a time-course study of clinical, immunologic, ultrastructural, and cytochemical changes. J Investig Dermatol. (1985) 84:41–6. doi: 10.1111/1523-1747.ep12274679
141. Hu CH, Michel B, Schiltz JR. Epidermal acantholysis induced in vitro by pemphigus autoantibody. An ultrastructural study. Am J Pathol. (1978) 90:345–62.
142. Diercks GF, Pas HH, Jonkman MF. The ultrastructure of acantholysis in pemphigus vulgaris. Br J Dermatol. (2009) 160:460–1. doi: 10.1111/j.1365-2133.2008.08971.x
143. Bystryn J-C, Grando SA. A novel explanation for acantholysis in pemphigus vulgaris: the basal cell shrinkage hypothesis. J Am Acad Dermatol. (2006) 54:513–6. doi: 10.1016/j.jaad.2005.12.003
144. Spindler V, Waschke J. Desmosomal cadherins and signaling: lessons from autoimmune disease. Cell Commun Adhes. (2014) 21:77–84. doi: 10.3109/15419061.2013.877000
145. Wilgram GF, Caulfield JB, Lever WF. An electron microscopic study of acantholysis in pemphigus vulgaris. J Invest Dermatol. (1961) 36:373–82. doi: 10.1038/jid.1961.58
146. Cirillo N, Femiano F, Gombos F, Lanza A. Serum from pemphigus vulgaris reduces desmoglein 3 half-life and perturbs its de novo assembly to desmosomal sites in cultured keratinocytes. FEBS Lett. (2006) 580:3276–81. doi: 10.1016/j.febslet.2006.04.089
147. Windoffer R, Borchert-Stuhlträger M, Leube RE. Desmosomes: interconnected calcium-dependent structures of remarkable stability with significant integral membrane protein turnover. J Cell Sci. (2002) 115:1717–32. doi: 10.1242/jcs.115.8.1717
148. Gloushankova NA, Wakatsuki T, Troyanovsky RB, Elson E, Troyanovsky SM. Continual assembly of desmosomes within stable intercellular contacts of epithelial A-431 cells. Cell Tissue Res. (2003) 314:399–410. doi: 10.1007/s00441-003-0812-3
149. Oktarina DA, van der Wier G, Diercks GF, Jonkman MF, Pas HH. IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein non-assembly depletion hypothesis. Br J Dermatol. (2011) 165:552–62. doi: 10.1111/j.1365-2133.2011.10463.x
150. Spindler V, Waschke J. Pemphigus-a disease of desmosome dysfunction caused by multiple mechanisms. Front Immunol. (2018) 9:136. doi: 10.3389/fimmu.2018.00136
151. Vielmuth F, Walter E, Fuchs M, Radeva MY, Buechau F, Magin TM, et al. Keratins regulate p38MAPK-dependent desmoglein binding properties in pemphigus. Front Immunol. (2018) 9:528. doi: 10.3389/fimmu.2018.00528
152. Walter E, Vielmuth F, Rotkopf L, Sárdy M, Horváth ON, Goebeler M, et al. Different signaling patterns contribute to loss of keratinocyte cohesion dependent on autoantibody profile in pemphigus. Sci Rep. (2017) 7:3579. doi: 10.1038/s41598-017-03697-7
153. Schlögl E, Radeva MY, Vielmuth F, Schinner C, Waschke J, Spindler V. Keratin retraction and desmoglein3 internalization independently contribute to autoantibody-induced cell dissociation in pemphigus vulgaris. Front Immunol. (2018) 9:858. doi: 10.3389/fimmu.2018.00858
154. Schmidt E, Waschke J. Apoptosis in pemphigus. Autoimmun Rev. (2009) 8:533–7. doi: 10.1016/j.autrev.2009.01.011
155. Cirillo N, Femiano F. Dell'Ermo A, Arnese P, Gombos F, Lanza A. A novel method to investigate pemphigus-induced keratinocyte dysmorphisms through living cell immunofluorescence microscopy. Virchows Arch. (2007) 450:683–90. doi: 10.1007/s00428-007-0410-y
156. Esaki C, Seishima M, Yamada T, Osada K, Kitajima Y. Pharmacologic evidence for involvement of phospholipase C in pemphigus IgG-induced inositol 1,4,5-trisphosphate generation, intracellular calcium increase, and plasminogen activator secretion in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol. (1995) 105:329–33. doi: 10.1111/1523-1747.ep12319948
157. Gliem M, Heupel W-M, Spindler V, Harms GS, Waschke J. Actin reorganization contributes to loss of cell adhesion in pemphigus vulgaris. Am J Physiol Cell Physiol. (2010) 299:C606–13. doi: 10.1152/ajpcell.00075.2010
158. Spindler V, Eming R, Schmidt E, Amagai M, Grando S, Jonkman MF, et al. Mechanisms causing loss of keratinocyte cohesion in pemphigus. J Invest Dermatol. (2018) 138:32–7. doi: 10.1016/j.jid.2017.06.022
159. Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. (1991) 67:869–77. doi: 10.1016/0092-8674(91)90360-B
160. Seishima M, Esaki C, Osada K, Mori S, Hashimoto T, Kitajima Y. Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J Invest Dermatol. (1995) 104:33–7. doi: 10.1111/1523-1747.ep12613469
161. Vielmuth F, Spindler V, Waschke J. Atomic force microscopy provides new mechanistic insights into the pathogenesis of pemphigus. Front Immunol. (2018) 9:485. doi: 10.3389/fimmu.2018.00485
162. Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol. (2001) 167:5439–48. doi: 10.4049/jimmunol.167.9.5439
163. Tselepis C, Chidgey M, North A, Garrod D. Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci USA. (1998) 95:8064–9. doi: 10.1073/pnas.95.14.8064
164. Runswick SK, O'Hare MJ, Jones L, Streuli CH, Garrod DR. Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nat Cell Biol. (2001) 3:823–30. doi: 10.1038/ncb0901-823
165. Shimizu A, Ishiko A, Ota T, Tsunoda K, Amagai M, Nishikawa T. IgG binds to desmoglein 3 in desmosomes and causes a desmosomal split without keratin retraction in a pemphigus mouse model. J Investig Dermatol. (2004) 122:1145–53. doi: 10.1111/j.0022-202X.2004.22426.x
166. Shimizu H, Masunaga T, Ishiko A, Kikuchi A, Hashimoto T, Nishikawa T. Pemphigus vulgaris and pemphigus foliaceus sera show an inversely graded binding pattern to extracellular regions of desmosomes in different layers of human epidermis. J Investig Dermatol. (1995) 105:153–9. doi: 10.1111/1523-1747.ep12316695
167. Vielmuth F, Hartlieb E, Kugelmann D, Waschke J, Spindler V. Atomic force microscopy identifies regions of distinct desmoglein 3 adhesive properties on living keratinocytes. Nanomedicine. (2015) 11:511–20. doi: 10.1016/j.nano.2014.10.006
168. Heupel W-M, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol. (2008) 181:1825–34. doi: 10.4049/jimmunol.181.3.1825
169. Ishii K, Yoshida K, Stanley JR, Yamagami J, Amagai M, Ishiko A. Pemphigus vulgaris and foliaceus IgG autoantibodies directly block heterophilic trans-interaction between desmoglein and desmocollin. J Invest Dermatol. (2020). doi: 10.1016/j.jid.2020.02.010
170. Vielmuth F, Wanuske M-T, Radeva MY, Hiermaier M, Kugelmann D, Walter E, et al. Keratins regulate the adhesive properties of desmosomal cadherins through signaling. J Invest Dermatol. (2018) 138:121–31. doi: 10.1016/j.jid.2017.08.033
171. Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. (2005) 115:3157–65. doi: 10.1172/JCI23475
172. Sánchez-Carpintero I, España A, Pelacho B, López Moratalla N, Rubenstein DS, Diaz LA, et al. In vivo blockade of pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br J Dermatol. (2004) 151:565–70. doi: 10.1111/j.1365-2133.2004.06147.x
173. Schmidt E, Spindler V, Eming R, Amagai M, Antonicelli F, Baines JF, et al. Meeting report of the pathogenesis of pemphigus and pemphigoid meeting in Munich, September 2016. J Invest Dermatol. (2017) 137:1199–203. doi: 10.1016/j.jid.2017.01.028
175. Ohata C, Ishii N, Furumura M. Locations of acantholysis in pemphigus vulgaris and pemphigus foliaceus. J Cutan Pathol. (2014) 41:880–9. doi: 10.1111/cup.12384
176. Udey MC, Stanley JR. Pemphigus–diseases of antidesmosomal autoimmunity. JAMA. (1999) 282:572–6. doi: 10.1001/jama.282.6.572
177. Janse IC, van der Wier G, Jonkman MF, Pas HH, Diercks GF. No evidence of apoptotic cells in pemphigus acantholysis. J Invest Dermatol. (2014) 134:2039–41. doi: 10.1038/jid.2014.60
178. Sokol E, Kramer D, Diercks GF, Kuipers J, Jonkman MF, Pas HH, et al. Large-scale electron microscopy maps of patient skin and mucosa provide insight into pathogenesis of blistering diseases. J Invest Dermatol. (2015) 135:1763–70. doi: 10.1038/jid.2015.109
179. van dW, Pas HH, Kramer D, Diercks GF, Jonkman MF. Smaller desmosomes are seen in the skin of pemphigus patients with anti-desmoglein 1 antibodies but not in patients with anti-desmoglein 3 antibodies. J Invest Dermatol. (2014) 134:140. doi: 10.1038/jid.2014.140
180. Wilgram GF, Caulfield JB, Madgic EB. An electron microscopic study of acantholysis and dyskeratosis in pemphigus foliaceus. With a special note on peculiar intracytoplasmic bodies. J Investig Dermatol. (1964) 43:287–99. doi: 10.1038/jid.1964.159
181. Stahley SN, Warren MF, Feldman RJ, Swerlick RA, Mattheyses AL, Kowalczyk AP. Super-resolution microscopy reveals altered desmosomal protein organization in tissue from patients with pemphigus vulgaris. J Invest Dermatol. (2016) 136:59–66. doi: 10.1038/JID.2015.353
182. Egu DT, Walter E, Spindler V, Waschke J. Inhibition of p38MAPK signalling prevents epidermal blistering and alterations of desmosome structure induced by pemphigus autoantibodies in human epidermis. Br J Dermatol. (2017) 177:1612–8. doi: 10.1111/bjd.15721
183. Kitajima Y. 150(th) anniversary series: desmosomes and autoimmune disease, perspective of dynamic desmosome remodeling and its impairments in pemphigus. Cell Commun Adhes. (2014) 21:943397. doi: 10.3109/15419061.2014.943397
184. Nekrasova O, Green KJ. Desmosome assembly and dynamics. Trends Cell Biol. (2013) 23:537–46. doi: 10.1016/j.tcb.2013.06.004
185. Stahley SN, Saito M, Faundez V, Koval M, Mattheyses AL, Kowalczyk AP. Desmosome assembly and disassembly are membrane raft-dependent. PLoS ONE. (2014) 9:e87809. doi: 10.1371/journal.pone.0087809
186. Völlner F, Ali J, Kurrle N, Exner Y, Eming R, Hertl M, et al. Loss of flotillin expression results in weakened desmosomal adhesion and Pemphigus vulgaris-like localisation of desmoglein-3 in human keratinocytes. Sci Rep. (2016) 6:28820. doi: 10.1038/srep28820
187. Rötzer V, Hartlieb E, Vielmuth F, Gliem M, Spindler V, Waschke J. E-cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell Mol Life Sci. (2015) 72:4885–97. doi: 10.1007/s00018-015-1977-0
188. Shafraz O, Rübsam M, Stahley SN, Caldara AL, Kowalczyk AP, Niessen CM, et al. E-cadherin binds to desmoglein to facilitate desmosome assembly. Elife. (2018) 7:37629. doi: 10.7554/eLife.37629
189. Bass-Zubek AE, Hobbs RP, Amargo EV, Garcia NJ, Hsieh SN, Chen X, et al. Plakophilin 2: a critical scaffold for PKC alpha that regulates intercellular junction assembly. J Cell Biol. (2008) 181:jcb.200712133. doi: 10.1083/jcb.200712133
190. Godsel LM, Dubash AD, Bass-Zubek AE, Amargo EV, Klessner JL, Hobbs RP, et al. Plakophilin 2 couples actomyosin remodeling to desmosomal plaque assembly via RhoA. Mol Biol Cell. (2010) 21:2844–59. doi: 10.1091/mbc.e10-02-0131
191. Kugelmann D, Rötzer V, Walter E, Egu DT, Fuchs MT, Vielmuth F, et al. Role of Src and cortactin in pemphigus skin blistering. Front Immunol. (2019) 10:626. doi: 10.3389/fimmu.2019.00626
192. Rötzer V, Breit A, Waschke J, Spindler V. Adducin is required for desmosomal cohesion in keratinocytes. J Biol Chem. (2014) 289:14925–40. doi: 10.1074/jbc.M113.527127
193. Hiermaier M, Kliewe F, Schinner C, Stüdle C, Maly IP, Wanuske MT, et al. The actin-binding protein α-adducin modulates desmosomal turnover and plasticity. J Invest Dermatol. (2020). doi: 10.1101/787846
194. Todorovic V, Koetsier JL, Godsel LM, Green KJ. Plakophilin 3 mediates Rap1-dependent desmosome assembly and adherens junction maturation. Mol Biol Cell. (2014) 25:3749–64. doi: 10.1091/mbc.e14-05-0968
195. Nie Z, Merritt A, Rouhi-Parkouhi M, Tabernero L, Garrod D. Membrane-impermeable cross-linking provides evidence for homophilic, isoform-specific binding of desmosomal cadherins in epithelial cells. J Biol Chem. (2011) 286:2143–54. doi: 10.1074/jbc.M110.192245
196. Bartle EI, Rao TC, Beggs RR, Dean WF, Urner TM, Kowalczyk AP, et al. Protein exchange is reduced in calcium-independent epithelial junctions. J Cell Biol. (2020) 219:6153. doi: 10.1083/jcb.201906153
197. Hobbs RP, Green KJ. Desmoplakin regulates desmosome hyperadhesion. J Invest Dermatol. (2012) 132:482–5. doi: 10.1038/jid.2011.318
198. Garrod D, Kimura TE. Hyper-adhesion: a new concept in cell-cell adhesion. Biochem Soc Trans. (2008) 36:195–201. doi: 10.1042/BST0360195
200. Wallis S, Lloyd S, Wise I, Ireland G, Fleming TP, Garrod D. The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol Biol Cell. (2000) 11:1077–92. doi: 10.1091/mbc.11.3.1077
201. Kimura TE, Merritt AJ, Garrod DR. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol. (2007) 127:775–81. doi: 10.1038/sj.jid.5700643
202. Cirillo N, Lanza A, Prime SS. Induction of hyper-adhesion attenuates autoimmune-induced keratinocyte cell-cell detachment and processing of adhesion molecules via mechanisms that involve PKC. Exp Cell Res. (2010) 316:580–92. doi: 10.1016/j.yexcr.2009.10.005
203. Fuchs M, Foresti M, Radeva MY, Kugelmann D, Keil R, Hatzfeld M, et al. Plakophilin 1 but not plakophilin 3 regulates desmoglein clustering. Cell Mol Life Sci. (2019) 76:3465–76. doi: 10.1007/s00018-019-03083-8
204. Waschke J, Menendez-Castro C, Bruggeman P, Koob R, Amagai M, Gruber HJ, et al. Imaging and force spectroscopy on desmoglein 1 using atomic force microscopy reveal multivalent Ca(2+)-dependent, low-affinity trans-interaction. J Membr Biol. (2007) 216:83–92. doi: 10.1007/s00232-007-9037-9
205. Garrod DR, Berika MY, Bardsley WF, Holmes D, Tabernero L. Hyper-adhesion in desmosomes: its regulation in wound healing and possible relationship to cadherin crystal structure. J Cell Sci. (2005) 118:5743–54. doi: 10.1242/jcs.02700
206. Fuchs M, Sigmund AM, Waschke J, Vielmuth F. Desmosomal hyperadhesion is accompanied with enhanced binding strength of desmoglein 3 molecules. Biophys J. (2020) 119:1489–500. doi: 10.1016/j.bpj.2020.09.008
207. Tucker DK, Stahley SN, Kowalczyk AP. Plakophilin-1 protects keratinocytes from pemphigus vulgaris IgG by forming calcium-independent desmosomes. J Invest Dermatol. (2014) 134:1033–43. doi: 10.1038/jid.2013.401
208. Dehner C, Rötzer V, Waschke J, Spindler V. A desmoplakin point mutation with enhanced keratin association ameliorates pemphigus vulgaris autoantibody-mediated loss of cell cohesion. Am J Pathol. (2014) 184:2528–36. doi: 10.1016/j.ajpath.2014.05.016
209. Kröger C, Loschke F, Schwarz N, Windoffer R, Leube RE, Magin TM. Keratins control intercellular adhesion involving PKC-α-mediated desmoplakin phosphorylation. J Cell Biol. (2013) 201:681–92. doi: 10.1083/jcb.201208162
210. Schinner C, Vielmuth F, Rötzer V, Hiermaier M, Radeva MY, Co TK, et al. Adrenergic signaling strengthens cardiac myocyte cohesion. Circ Res. (2017) 120:1305–17. doi: 10.1161/CIRCRESAHA.116.309631
211. Schinner C, Erber BM, Yeruva S, Waschke J. Regulation of cardiac myocyte cohesion and gap junctions via desmosomal adhesion. Acta Physiol. (2019) 226:e13242. doi: 10.1111/apha.13242
212. Shoykhet M, Trenz S, Kempf E, Williams T, Gerull B, Schinner C, et al. Cardiomyocyte adhesion and hyperadhesion differentially require ERK1/2 and plakoglobin. JCI Insight. (2020) 5:140066. doi: 10.1172/jci.insight.140066
213. Keil R, Rietscher K, Hatzfeld M. Antagonistic regulation of intercellular cohesion by plakophilins 1 and 3. J Invest Dermatol. (2016) 136:2022–9. doi: 10.1016/j.jid.2016.05.124
214. Yeruva S, Kempf E, Egu DT, Flaswinkel H, Kugelmann D, Waschke J. Adrenergic signaling-induced ultrastructural strengthening of intercalated discs via plakoglobin is crucial for positive adhesiotropy in murine cardiomyocytes. Front Physiol. (2020) 11:430. doi: 10.3389/fphys.2020.00430
215. Egu DT, Kugelmann D, Waschke J. Role of PKC and ERK signaling in epidermal blistering and desmosome regulation in pemphigus. Front Immunol. (2019) 10:2883. doi: 10.3389/fimmu.2019.02883
216. Spindler V, Rötzer V, Dehner C, Kempf B, Gliem M, Radeva M, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J Clin Invest. (2013) 123:800–11. doi: 10.1172/JCI60139
217. Heupel W-M, Müller T, Efthymiadis A, Schmidt E, Drenckhahn D, Waschke J. Peptides targeting the desmoglein 3 adhesive interface prevent autoantibody-induced acantholysis in pemphigus. J Biol Chem. (2009) 284:8589–95. doi: 10.1074/jbc.M808813200
218. Spindler V, Dehner C, Hübner S, Waschke J. Plakoglobin but not desmoplakin regulates keratinocyte cohesion via modulation of p38MAPK signaling. J Invest Dermatol. (2014) 134:1655–64. doi: 10.1038/jid.2014.21
219. Wanuske M-T, Brantschen D, Schinner C, Stüdle C, Walter E, Hiermaier M, et al. Clustering of desmosomal cadherins by desmoplakin is essential for cell-cell adhesion. Acta Physiol. (2021) 231:e13609. doi: 10.1111/apha.13609
220. Schmitt T, Egu DT, Walter E, Sigmund AM, Eichkorn R, Yazdi A, et al. Ca2+ signalling is critical for autoantibody-induced blistering of human epidermis in pemphigus. Br J Dermatol. (2021) 2021:bjd.20091. doi: 10.1111/bjd.20091
221. Hartlieb E, Rötzer V, Radeva M, Spindler V, Waschke J. Desmoglein 2 compensates for desmoglein 3 but does not control cell adhesion via regulation of p38 mitogen-activated protein kinase in keratinocytes. J Biol Chem. (2014) 289:17043–53. doi: 10.1074/jbc.M113.489336
222. Ungewiß H, Vielmuth F, Suzuki ST, Maiser A, Harz H, Leonhardt H, et al. Desmoglein 2 regulates the intestinal epithelial barrier via p38 mitogen-activated protein kinase. Sci Rep. (2017) 7:6329. doi: 10.1038/s41598-017-06713-y
223. Ungewiß H, Rötzer V, Meir M, Fey C, Diefenbacher M, Schlegel N, et al. Dsg2 via Src-mediated transactivation shapes EGFR signaling towards cell adhesion. Cell Mol Life Sci. (2018) 75:2869. doi: 10.1007/s00018-018-2869-x
224. Rötzer V, Hartlieb E, Winkler J, Walter E, Schlipp A, Sardy M, et al. Desmoglein 3-dependent signaling regulates keratinocyte migration and wound healing. J Invest Dermatol. (2016) 136:301–10. doi: 10.1038/JID.2015.380
225. Berkowitz P, Hu P, Warren S, Liu Z, Diaz LA, Rubenstein DS. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc Natl Acad Sci USA. (2006) 103:12855–60. doi: 10.1073/pnas.0602973103
226. Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol. (2008) 173:1628–36. doi: 10.2353/ajpath.2008.080391
227. Waschke J, Spindler V, Bruggeman P, Zillikens D, Schmidt G, Drenckhahn D. Inhibition of Rho A activity causes pemphigus skin blistering. J Cell Biol. (2006) 175:721–7. doi: 10.1083/jcb.200605125
228. Walter E, Vielmuth F, Wanuske M-T, Seifert M, Pollmann R, Eming R, et al. Role of Dsg1- and Dsg3-mediated signaling in pemphigus autoantibody-induced loss of keratinocyte cohesion. Front Immunol. (2019) 10:1128. doi: 10.3389/fimmu.2019.01128
229. Burmester IA, Flaswinkel S, Thies C-S, Kasprick A, Kamaguchi M, Bumiller-Bini V, et al. Identification of novel therapeutic targets for blocking acantholysis in pemphigus. Br J Pharmacol. (2020) 177:5114–30. doi: 10.1111/bph.15233
230. Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, et al. Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol. (2009) 185:1243–58. doi: 10.1083/jcb.200809044
231. Rübsam M, Mertz AF, Kubo A, Marg S, Jüngst C, Goranci-Buzhala G, et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat Commun. (2017) 8:1250. doi: 10.1038/s41467-017-01170-7
232. Cohen-Barak E, Godsel LM, Koetsier JL, Hegazy M, Kushnir-Grinbaum D, Hammad H, et al. The role of desmoglein 1 in gap junction turnover revealed through the study of SAM syndrome. J Invest Dermatol. (2020) 140:556–67.e9. doi: 10.1016/j.jid.2019.08.433
233. Mao X, Sano Y, Park JM, Payne AS. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J Biol Chem. (2011) 286:1283–91. doi: 10.1074/jbc.M110.172874
234. Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. (1994) 265:808–11. doi: 10.1126/science.7914033
235. Tan Y, Rouse J, Zhang A, Cariati S, Cohen P, Comb MJ, et al. and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. (1996) 15:4629–42. doi: 10.1002/j.1460-2075.1996.tb00840.x
236. Guay J, Lambert H, Gingras-Breton G, Lavoie JN, Huot J, Landry J. Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J Cell Sci. (1997) 110:357–68. doi: 10.1242/jcs.110.3.357
237. Berkowitz P, Hu P, Liu Z, Diaz LA, Enghild JJ, Chua MP, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. (2005) 280:23778–84. doi: 10.1074/jbc.M501365200
238. Berkowitz P, Diaz LA, Hall RP, Rubenstein DS. Induction of p38MAPK and HSP27 phosphorylation in pemphigus patient skin. J Invest Dermatol. (2008) 128:738–40. doi: 10.1038/sj.jid.5701080
239. Nahas N, Molski TF, Fernandez GA, Sha'afi RI. Tyrosine phosphorylation and activation of a new mitogen-activated protein (MAP)-kinase cascade in human neutrophils stimulated with various agonists. Biochem J. (1996) 318:247–53. doi: 10.1042/bj3180247
240. Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science. (1996) 272:1347–9. doi: 10.1126/science.272.5266.1347
241. Sawant M, Schwarz N, Windoffer R, Magin TM, Krieger J, Mücke N, et al. Threonine 150 phosphorylation of keratin 5 is linked to epidermolysis bullosa simplex and regulates filament assembly and cell viability. J Invest Dermatol. (2018) 138:627–36. doi: 10.1016/j.jid.2017.10.011
242. Meir M, Burkard N, Ungewiß H, Diefenbacher M, Flemming S, Kannapin F, et al. Neurotrophic factor GDNF regulates intestinal barrier function in inflammatory bowel disease. J Clin Invest. (2019) 129:2824–40. doi: 10.1172/JCI120261
243. Wöll S, Windoffer R, Leube RE. p38 MAPK-dependent shaping of the keratin cytoskeleton in cultured cells. J Cell Biol. (2007) 177:795–807. doi: 10.1083/jcb.200703174
244. Mao X, Li H, Sano Y, Gaestel M, Mo Park J, Payne AS, et al. kinase 2 (MK2)-dependent and -independent models of blister formation in pemphigus vulgaris. J Invest Dermatol. (2014) 134:68–76. doi: 10.1038/jid.2013.224
245. Larsen JK, Yamboliev IA, Weber LA, Gerthoffer WT. Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am J Physiol. (1997) 273:L930–40. doi: 10.1152/ajplung.1997.273.5.L930
246. Perng MD, Cairns L, van den IJssel P, Prescott A, Hutcheson AM, Quinlan RA. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci. (1999) 112:2099–112. doi: 10.1242/jcs.112.13.2099
247. Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS ONE. (2012) 7:e50696. doi: 10.1371/journal.pone.0050696
248. Lee HE, Berkowitz P, Jolly PS, Diaz LA, Chua MP, Rubenstein DS. Biphasic activation of p38MAPK suggests that apoptosis is a downstream event in pemphigus acantholysis. J Biol Chem. (2009) 284:12524–32. doi: 10.1074/jbc.M808204200
249. Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J Biol Chem. (2007) 282:13804–12. doi: 10.1074/jbc.M611365200
250. Spindler V, Drenckhahn D, Zillikens D, Waschke J. Pemphigus IgG causes skin splitting in the presence of both desmoglein 1 and desmoglein 3. Am J Pathol. (2007) 171:906–16. doi: 10.2353/ajpath.2007.070028
251. Liang J, Zeng X, Halifu Y, Chen W, Hu F, Wang P, et al. Blocking RhoA/ROCK inhibits the pathogenesis of pemphigus vulgaris by suppressing oxidative stress and apoptosis through TAK1/NOD2-mediated NF-κB pathway. Mol Cell Biochem. (2017) 436:151–8. doi: 10.1007/s11010-017-3086-x
252. Hall A, Nobes CD. Rho GTPases: molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos Trans R Soc Lond B, Biol Sci. (2000) 355:965–70. doi: 10.1098/rstb.2000.0632
253. Lee SH, Dominguez R. Regulation of actin cytoskeleton dynamics in cells. Mol Cells. (2010) 29:311–25. doi: 10.1007/s10059-010-0053-8
254. Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. (2010) 11:633–43. doi: 10.1038/nrm2957
255. Lewis JE, Wahl JK, Sass KM, Jensen PJ, Johnson KR, Wheelock MJ. Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J Cell Biol. (1997) 136:919–34. doi: 10.1083/jcb.136.4.919
256. Yin T, Getsios S, Caldelari R, Godsel LM, Kowalczyk AP, Müller EJ, et al. Mechanisms of plakoglobin-dependent adhesion: desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J Biol Chem. (2005) 280:40355–63. doi: 10.1074/jbc.M506692200
257. Al-Amoudi A, Castaño-Diez D, Devos DP, Russell RB, Johnson GT, Frangakis AS. The three-dimensional molecular structure of the desmosomal plaque. Proc Natl Acad Sci USA. (2011) 108:6480–5. doi: 10.1073/pnas.1019469108
258. Marcozzi C, Burdett ID, Buxton RS, Magee AI. Coexpression of both types of desmosomal cadherin and plakoglobin confers strong intercellular adhesion. J Cell Sci. (1998) 111:495–509. doi: 10.1242/jcs.111.4.495
259. Pasdar M, Li Z, Chlumecky V. Plakoglobin: kinetics of synthesis, phosphorylation, stability, and interactions with desmoglein and E-cadherin. Cell Motil Cytoskeleton. (1995) 32:258–72. doi: 10.1002/cm.970320403
260. Caldelari R, de Bruin A, Baumann D, Suter MM, Bierkamp C, Balmer V, et al. A central role for the armadillo protein plakoglobin in the autoimmune disease pemphigus vulgaris. J Cell Biol. (2001) 153:823–34. doi: 10.1083/jcb.153.4.823
261. de Bruin A, Caldelari R, Williamson L, Suter MM, Hunziker T, Wyder M, et al. Plakoglobin-dependent disruption of the desmosomal plaque in pemphigus vulgaris. Exp Dermatol. (2007) 16:468–75. doi: 10.1111/j.1600-0625.2007.00557.x
262. Lo Muzio L, Pannone G, Staibano S, Mignogna MD, Rubini C, Farronato G, et al. Strict correlation between uPAR and plakoglobin expression in pemphigus vulgaris. J Cutan Pathol. (2002) 29:540–8. doi: 10.1034/j.1600-0560.2002.290906.x
263. Aoyama Y, Owada MK, Kitajima Y. A pathogenic autoantibody, pemphigus vulgaris-IgG, induces phosphorylation of desmoglein 3, and its dissociation from plakoglobin in cultured keratinocytes. Eur J Immunol. (1999) 29:2233–40. doi: 10.1002/(SICI)1521-4141(199907)29:07<2233::AID-IMMU2233>3.0.CO;2-4
264. Williamson L, Raess NA, Caldelari R, Zakher A, de Bruin A, Posthaus H, et al. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. (2006) 25:3298–309. doi: 10.1038/sj.emboj.7601224
265. Williamson L, Hunziker T, Suter MM, Müller EJ. Nuclear c-Myc: a molecular marker for early stage pemphigus vulgaris. J Invest Dermatol. (2007) 127:1549–55. doi: 10.1038/sj.jid.5700735
266. Pretel M, España A, Marquina M, Pelacho B, López-Picazo JM, López-Zabalza MJ. An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp Dermatol. (2009) 18:771–80. doi: 10.1111/j.1600-0625.2009.00893.x
267. Tsang SM, Brown L, Gadmor H, Gammon L, Fortune F, Wheeler A, et al. Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac-1/Cdc42 in the regulation of actin organisation and dynamics. Exp Cell Res. (2012) 318:2269–83. doi: 10.1016/j.yexcr.2012.07.002
268. Cirillo N, AlShwaimi E, McCullough M, Prime SS. Pemphigus vulgaris autoimmune globulin induces Src-dependent tyrosine-phosphorylation of plakophilin 3 and its detachment from desmoglein 3. Autoimmunity. (2014) 47:134–40. doi: 10.3109/08916934.2013.866100
269. Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. (2005) 1:2005.0010. doi: 10.1038/msb4100014
270. Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J Biol Chem. (1999) 274:8335–43. doi: 10.1074/jbc.274.12.8335
271. Sayar BS, Rüegg S, Schmidt E, Sibilia M, Siffert M, Suter MM, et al. EGFR inhibitors erlotinib and lapatinib ameliorate epidermal blistering in pemphigus vulgaris in a non-linear, V-shaped relationship. Exp Dermatol. (2014) 23:33–8. doi: 10.1111/exd.12290
272. Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J Biol Chem. (2013) 288:9447–56. doi: 10.1074/jbc.M112.438010
273. Frusić-Zlotkin M, Raichenberg D, Wang X, David M, Michel B, Milner Y. Apoptotic mechanism in pemphigus autoimmunoglobulins-induced acantholysis–possible involvement of the EGF receptor. Autoimmunity. (2006) 39:563–75. doi: 10.1080/08916930600971836
274. Hackel PO, Zwick E, Prenzel N, Ullrich A. Epidermal growth factor receptors: critical mediators of multiple receptor pathways. Curr Opin Cell Biol. (1999) 11:184–9. doi: 10.1016/S0955-0674(99)80024-6
275. Lorch JH, Klessner J, Park JK, Getsios S, Wu YL, Stack MS, et al. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J Biol Chem. (2004) 279:37191–200. doi: 10.1074/jbc.M405123200
276. Duda P, Akula SM, Abrams SL, Steelman LS, Martelli A, Cocco L, et al. Targeting GSK3 and associated signaling pathways involved in cancer. Cells. (2020) 9:51110. doi: 10.3390/cells9051110
277. Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, van Waes C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: attractive targets for molecular-oriented therapy. Expert Opin Ther Targets. (2011) 15:63–74. doi: 10.1517/14728222.2011.541440
278. Doma E, Rupp C, Baccarini M. EGFR-Ras-Raf signaling in epidermal stem cells: roles in hair follicle development, regeneration, tissue remodeling and epidermal cancers. Int J Mol Sci. (2013) 14:19361–84. doi: 10.3390/ijms141019361
279. Valenzuela-Iglesias A, Burks HE, Arnette CR, Yalamanchili A, Nekrasova O, Godsel LM, et al. Desmoglein 1 regulates invadopodia by suppressing EGFR/Erk signaling in an erbin-dependent manner. Mol Cancer Res. (2019) 17:1195–206. doi: 10.1158/1541-7786.MCR-18-0048
280. Hammers CM, Stanley JR. Desmoglein-1, differentiation, and disease. J Clin Invest. (2013) 123:1419–22. doi: 10.1172/JCI69071
281. Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, et al. Desmoglein-1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Invest. (2013) 123:1556–70. doi: 10.1172/JCI65220
282. Li G, Gustafson-Brown C, Hanks SK, Nason K, Arbeit JM, Pogliano K, et al. c-Jun is essential for organization of the epidermal leading edge. Dev Cell. (2003) 4:865–77. doi: 10.1016/S1534-5807(03)00159-X
283. Clarke N, Arenzana N, Hai T, Minden A, Prywes R. Epidermal growth factor induction of the c-jun promoter by a Rac pathway. Mol Cell Biol. (1998) 18:1065–73. doi: 10.1128/MCB.18.2.1065
284. Hiroyasu S, Turner CT, Richardson KC, Granville DJ. Proteases in pemphigoid diseases. Front Immunol. (2019) 10:1454. doi: 10.3389/fimmu.2019.01454
285. Grando SA. Decompensation in proteinase-inhibitor system and application of proteinase inhibitors in pemphigus and pemphigoid. J Dermatol Sci. (1992) 4:95–7. doi: 10.1016/0923-1811(92)90065-J
286. Cirillo N, Lanza M, Rossiello L, Gombos F, Lanza A. Defining the involvement of proteinases in pemphigus vulgaris: evidence of matrix metalloproteinase-9 overexpression in experimental models of disease. J Cell Physiol. (2007) 212:36–41. doi: 10.1002/jcp.20997
287. Sumigray KD, Lechler T. Cell adhesion in epidermal development and barrier formation. Curr Top Dev Biol. (2015) 112:383–414. doi: 10.1016/bs.ctdb.2014.11.027
288. Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, et al. Architecture of the human interactome defines protein communities and disease networks. Nature. (2017) 545:505–9. doi: 10.1038/nature22366
289. Sowa ME, Bennett EJ, Gygi SP, Harper JW. Defining the human deubiquitinating enzyme interaction landscape. Cell. (2009) 138:389–403. doi: 10.1016/j.cell.2009.04.042
290. Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. (2013) 93:1019–137. doi: 10.1152/physrev.00028.2012
291. Streb H, Irvine RF, Berridge MJ, Schulz I. Release of Ca2+ from a non-mitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature. (1983) 306:67–9. doi: 10.1038/306067a0
292. Hewavitharana T, Deng X, Wang Y, Ritchie MF, Girish GV, Soboloff J, et al. Location and function of STIM1 in the activation of Ca2+ entry signals. J Biol Chem. (2008) 283:26252–62. doi: 10.1074/jbc.M802239200
293. Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. (2012) 13:549–65. doi: 10.1038/nrm3414
294. Putney JW. Capacitative calcium entry revisited. Cell Calcium. (1990) 11:611–24. doi: 10.1016/0143-4160(90)90016-N
295. Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. (1992) 355:353–6. doi: 10.1038/355353a0
296. Nguyen NT, Han W, Cao W-M, Wang Y, Wen S, Huang Y, et al. Store-operated calcium entry mediated by ORAI and STIM. Compr Physiol. (2018) 8:981–1002. doi: 10.1002/cphy.c170031
297. Zhang SL, Yeromin AV, Zhang XH-F, Yu Y, Safrina O, Penna A, et al. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci USA. (2006) 103:9357–62. doi: 10.1073/pnas.0603161103
298. Bird GS, Hwang S-Y, Smyth JT, Fukushima M, Boyles RR, Putney JW. STIM1 is a calcium sensor specialized for digital signaling. Curr Biol. (2009) 19:1724–9. doi: 10.1016/j.cub.2009.08.022
299. Osada K, Seishima M, Kitajima Y. Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J Invest Dermatol. (1997) 108:482–7. doi: 10.1111/1523-1747.ep12289726
300. Kitajima Y, Aoyama Y, Seishima M. Transmembrane signaling for adhesive regulation of desmosomes and hemidesmosomes, and for cell-cell datachment induced by pemphigus IgG in cultured keratinocytes: involvement of protein kinase C. J Investig Dermatol Symp Proc. (1999) 4:137–44. doi: 10.1038/sj.jidsp.5640197
301. Loschke F, Homberg M, Magin TM. Keratin isotypes control desmosome stability and dynamics through PKCα. J Invest Dermatol. (2016) 136:202–13. doi: 10.1038/JID.2015.403
302. Bektas M, Jolly P, Rubenstein DS. Apoptotic pathways in pemphigus. Dermatol Res Pract. (2010) 2010:456841. doi: 10.1155/2010/456841
303. Sokolowski JD, Gamage KK, Heffron DS, Leblanc AC, Deppmann CD, Mandell JW. Caspase-mediated cleavage of actin and tubulin is a common feature and sensitive marker of axonal degeneration in neural development and injury. Acta Neuropathol Commun. (2014) 2:16. doi: 10.1186/2051-5960-2-16
304. Vakifahmetoglu-Norberg H, Norberg E, Perdomo AB, Olsson M, Ciccosanti F, Orrenius S, et al. Caspase-2 promotes cytoskeleton protein degradation during apoptotic cell death. Cell Death Dis. (2013) 4:e940. doi: 10.1038/cddis.2013.463
305. Weiske J, Schöneberg T, Schröder W, Hatzfeld M, Tauber R, Huber O. The fate of desmosomal proteins in apoptotic cells. J Biol Chem. (2001) 276:41175–81. doi: 10.1074/jbc.M105769200
306. Li N, Zhao M, Wang J, Liu Z, Diaz LA. Involvement of the apoptotic mechanism in pemphigus foliaceus autoimmune injury of the skin. J Immunol. (2009) 182:711–7. doi: 10.4049/jimmunol.182.1.711
307. Pacheco-Tovar D, López-Luna A, Herrera-Esparza R, Avalos-Díaz E. The caspase pathway as a possible therapeutic target in experimental pemphigus. Autoimmune Dis. (2011) 2011:563091. doi: 10.4061/2011/563091
308. Sanath AK, Devy AS, Aithal S, Kumar GS, Prasad BG, Pradeep PS. Caspase cascade pathways of apoptosis in oral pemphigus: an immunohistochemical study. J Oral Maxillofac Pathol. (2018) 22:48–53. doi: 10.4103/jomfp.JOMFP_79_17
309. Puviani M, Marconi A, Cozzani E, Pincelli C. Fas ligand in pemphigus sera induces keratinocyte apoptosis through the activation of caspase-8. J Investig Dermatol. (2003) 120:164–7. doi: 10.1046/j.1523-1747.2003.12014.x
310. Sayama K, Yonehara S, Watanabe Y, Miki Y. Expression of Fas antigen on keratinocytes in vivo and induction of apoptosis in cultured keratinocytes. J Investig Dermatol. (1994) 103:330–4. doi: 10.1111/1523-1747.ep12394858
311. Wang X, Brégégère F, Frusić-Zlotkin M, Feinmesser M, Michel B, Milner Y. Possible apoptotic mechanism in epidermal cell acantholysis induced by pemphigus vulgaris autoimmunoglobulins. Apoptosis. (2004) 9:131–43. doi: 10.1023/B:APPT.0000018795.05766.1f
312. Cuevas-Gonzalez JC, Vega-Memíje ME, García-Vázquez FJ, Aguilar-Urbano MA. Detection of apoptosis in pemphigus vulgaris by TUNEL technique. An Bras Dermatol. (2016) 91:296–9. doi: 10.1590/abd1806-4841.20164598
313. Pelacho B, Natal C, España A, Sánchez-Carpintero I, Iraburu MJ, López-Zabalza MJ. Pemphigus vulgaris autoantibodies induce apoptosis in HaCaT keratinocytes. FEBS Lett. (2004) 566:6–10. doi: 10.1016/j.febslet.2004.03.107
314. Hariton WV, Galichet A, Vanden Berghe T, Overmiller AM, Mahoney MG, Declercq W, et al. Feasibility study for clinical application of caspase-3 inhibitors in Pemphigus vulgaris. Exp Dermatol. (2017) 26:1274–7. doi: 10.1111/exd.13458
315. Lotti R, Shu E, Petrachi T, Marconi A, Palazzo E, Quadri M, et al. Soluble Fas ligand is essential for blister formation in pemphigus. Front Immunol. (2018) 9:370. doi: 10.3389/fimmu.2018.00370
316. Lotti R, Marconi A, Truzzi F, Dallaglio K, Gemelli C, Borroni RG, et al. A previously unreported function of β(1)B integrin isoform in caspase-8-dependent integrin-mediated keratinocyte death. J Invest Dermatol. (2010) 130:2569–77. doi: 10.1038/jid.2010.195
317. Marchenko S, Chernyavsky AI, Arredondo J, Gindi V, Grando SA. Antimitochondrial autoantibodies in pemphigus vulgaris: a missing link in disease pathophysiology. J Biol Chem. (2010) 285:3695–704. doi: 10.1074/jbc.M109.081570
318. Luyet C, Schulze K, Sayar BS, Howald D, Müller EJ, Galichet A. Preclinical studies identify non-apoptotic low-level caspase-3 as therapeutic target in pemphigus vulgaris. PLoS ONE. (2015) 10:e0119809. doi: 10.1371/journal.pone.0119809
319. Dusek RL, Getsios S, Chen F, Park JK, Amargo EV, Cryns VL, et al. The differentiation-dependent desmosomal cadherin desmoglein 1 is a novel caspase-3 target that regulates apoptosis in keratinocytes. J Biol Chem. (2006) 281:3614–24. doi: 10.1074/jbc.M508258200
320. Gil MP, Modol T, España A, López-Zabalza MJ. Inhibition of FAK prevents blister formation in the neonatal mouse model of pemphigus vulgaris. Exp Dermatol. (2012) 21:254–9. doi: 10.1111/j.1600-0625.2012.01441.x
321. España A, Mòdol T, Gil MP, López-Zabalza MJ. Neural nitric oxide synthase participates in pemphigus vulgaris acantholysis through upregulation of Rous sarcoma, mammalian target of rapamycin and focal adhesion kinase. Exp Dermatol. (2013) 22:125–30. doi: 10.1111/exd.12088
322. Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, et al. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. (2009) 19:1363–76. doi: 10.1038/cr.2009.98
323. Magrì A, Reina S, de Pinto V. VDAC1 as pharmacological target in cancer and neurodegeneration: focus on its role in apoptosis. Front Chem. (2018) 6:108. doi: 10.3389/fchem.2018.00108
324. Tomasello MF, Guarino F, Reina S, Messina A, de Pinto V. The voltage-dependent anion selective channel 1 (VDAC1) topography in the mitochondrial outer membrane as detected in intact cell. PLoS ONE. (2013) 8:e81522. doi: 10.1371/journal.pone.0081522
325. Shoshan-Barmatz V, Shteinfer-Kuzmine A, Verma A. VDAC1 at the intersection of cell metabolism, apoptosis, and diseases. Biomolecules. (2020) 10:111485. doi: 10.3390/biom10111485
326. Yang Z, Schumaker LM, Egorin MJ, Zuhowski EG, Guo Z, Cullen KJ. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin Cancer Res. (2006) 12:5817–25. doi: 10.1158/1078-0432.CCR-06-1037
327. Farb RM, Dykes R, Lazarus GS. Anti-epidermal-cell-surface pemphigus antibody detaches viable epidermal cells from culture plates by activation of proteinase. Proc Natl Acad Sci USA. (1978) 75:459–63. doi: 10.1073/pnas.75.1.459
328. Schmidt E, Gutberlet J, Siegmund D, Berg D, Wajant H, Waschke J. Apoptosis is not required for acantholysis in pemphigus vulgaris. Am J Physiol Cell Physiol. (2009) 296:C162–72. doi: 10.1152/ajpcell.00161.2008
329. Schwerk C, Schulze-Osthoff K. Non-apoptotic functions of caspases in cellular proliferation and differentiation. Biochem Pharmacol. (2003) 66:1453–8. doi: 10.1016/S0006-2952(03)00497-0
330. Iwatsuki K, Han GW, Fukuti R, Ohtsuka M, Kikuchi S, Akiba H, et al. Internalization of constitutive desmogleins with the subsequent induction of desmoglein 2 in pemphigus lesions. Br J Dermatol. (1999) 140:35–43. doi: 10.1046/j.1365-2133.1999.02604.x
331. Brennan D, Hu Y, Medhat W, Dowling A, Mahoney MG. Superficial dsg2 expression limits epidermal blister formation mediated by pemphigus foliaceus antibodies and exfoliative toxins. Dermatol Res Pract. (2010) 2010:410278. doi: 10.1155/2010/410278
332. Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Investig Dermatol. (2005) 124:939–46. doi: 10.1111/j.0022-202X.2005.23714.x
333. Sigmund AM, Steinert LS, Egu DT, Bayerbach FC, Waschke J, Vielmuth F. Dsg2 upregulation as a rescue mechanism in pemphigus. Front Immunol. (2020) 11:581370. doi: 10.3389/fimmu.2020.581370
334. Spindler V, Vielmuth F, Schmidt E, Rubenstein DS, Waschke J. Protective endogenous cyclic adenosine 5'-monophosphate signaling triggered by pemphigus autoantibodies. J Immunol. (2010) 185:6831–8. doi: 10.4049/jimmunol.1002675
335. Meier K, Holstein J, Solimani F, Waschke J, Ghoreschi K. Case report: apremilast for therapy-resistant pemphigus vulgaris. Front Immunol. (2020) 11:588315. doi: 10.3389/fimmu.2020.588315
Keywords: adhesion, pemphigus, signalling, autoantibodies, autoimmune blistering disease
Citation: Schmitt T and Waschke J (2021) Autoantibody-Specific Signalling in Pemphigus. Front. Med. 8:701809. doi: 10.3389/fmed.2021.701809
Received: 28 April 2021; Accepted: 08 July 2021;
Published: 09 August 2021.
Edited by:
Kyle T. Amber, Rush University, United StatesReviewed by:
Detlef Zillikens, University of Lübeck, GermanyMarian Dmochowski, Poznan University of Medical Sciences, Poland
Copyright © 2021 Schmitt and Waschke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jens Waschke, amVucy53YXNjaGtlQG1lZC51bmktbXVlbmNoZW4uZGU=
†ORCID: Thomas Schmitt orcid.org/0000-0003-3781-7236
Jens Waschke orcid.org/0000-0003-1182-5422