 Qiaoyu Li
Qiaoyu Li Yun Feng
Yun Feng Xuechen Zhu2,3,4*
Xuechen Zhu2,3,4*- 1Department of Ophthalmology, Peking University First Hospital, Beijing, China
- 2Department of Pediatrics, Peking University First Hospital, Beijing, China
- 3Department of Human Anatomy, Histology and Embryology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing, China
- 4Neuroscience Research Institute, Peking University, Beijing, China
Model organisms have played a pivotal role in ophthalmic research, providing essential platforms to investigate eye development, regeneration mechanisms, and disease pathology. Recent advancements in gene editing technologies and experimental methodologies have enabled the successful simulation of various human eye diseases, including glaucoma, retinal degeneration, and corneal disorders in model systems. These models have significantly advanced the understanding of the molecular and cellular mechanisms underlying ocular diseases and facilitated the screening and validation of potential therapeutic agents. Xenopus laevis (X. laevis) has emerged as an ideal system for developmental biology research due to its rapid embryonic development, transparent embryos, and ease of observation and manipulation. Its fully sequenced genome allows precise genetic modifications, including gene knockout, knock-in, and expression regulation studies. In ophthalmic research, X. laevis is widely used for studying eye development, disease modeling, and ocular structure. Its accessible embryonic stages and well-characterized eye development make it a valuable model for retinal disease investigations. This review systematically summarizes the applications, construction methods, and research significance of X. laevis models in eye development, disease modeling, and drug screening. It provides an in-depth perspective on the utility of X. laevis in foundational ophthalmic research, offering insights to guide future studies.
1 Introduction
In ophthalmic basic research, model organisms have provided a crucial experimental platform for exploring eye development, regenerative mechanisms, and disease pathologies. In recent years, researchers have leveraged gene-editing technologies and innovative experimental methods to successfully replicate various human ocular diseases, including glaucoma, retinal degeneration, and corneal pathologies, in model animals. These models not only facilitate a deeper understanding of the molecular and cellular mechanisms underlying ophthalmic diseases but also serve as essential tools for screening and validating potential therapeutic drugs.
With its fully sequenced genome, Xenopus enables studies involving gene knockout, gene knock-in, and gene expression regulation. Due to its rapid embryonic development and transparent embryos, X. laevis provides a simple, cost-effective, and highly observable system readily amenable to manipulation. In ophthalmic research, particularly in studies of eye development, ocular disease models, and ocular structure, the transparency of its embryos and the ease of manipulating eye development make it an optimal model for rapidly investigating disease mechanisms. X. laevis is therefore not merely a technical convenience but a powerful complementary model for dissecting fundamental biological mechanisms in a high-throughput and accessible manner.
This review provides a systematic overview of the applications, construction methodologies, and research significance of X. laevis models in studies of eye development, disease modeling, and drug screening. It offers a comprehensive retrospective and in-depth insights into the use of Xenopus in ophthalmic research, aiming to inspire future directions in the field.
2 Model organisms in ophthalmic disease research
Of the models used to study visual field specification and human ocular pathologies (Figure 1), the mouse remains the premier mammalian model due to its 99% gene conservation rate with humans and well-annotated mutant lines that facilitate the study of systemic gene impacts (1–3). However, its significant limitation for studying fundamental developmental concepts is the extreme difficulty or impossibility of administering interventions during early fetal stages. In contrast, Xenopus laevis has emerged as an ideal complementary model and powerful tool for rapid, cost-effective mechanistic discovery, offering a unique combination of advantages not fully replicated in other systems.
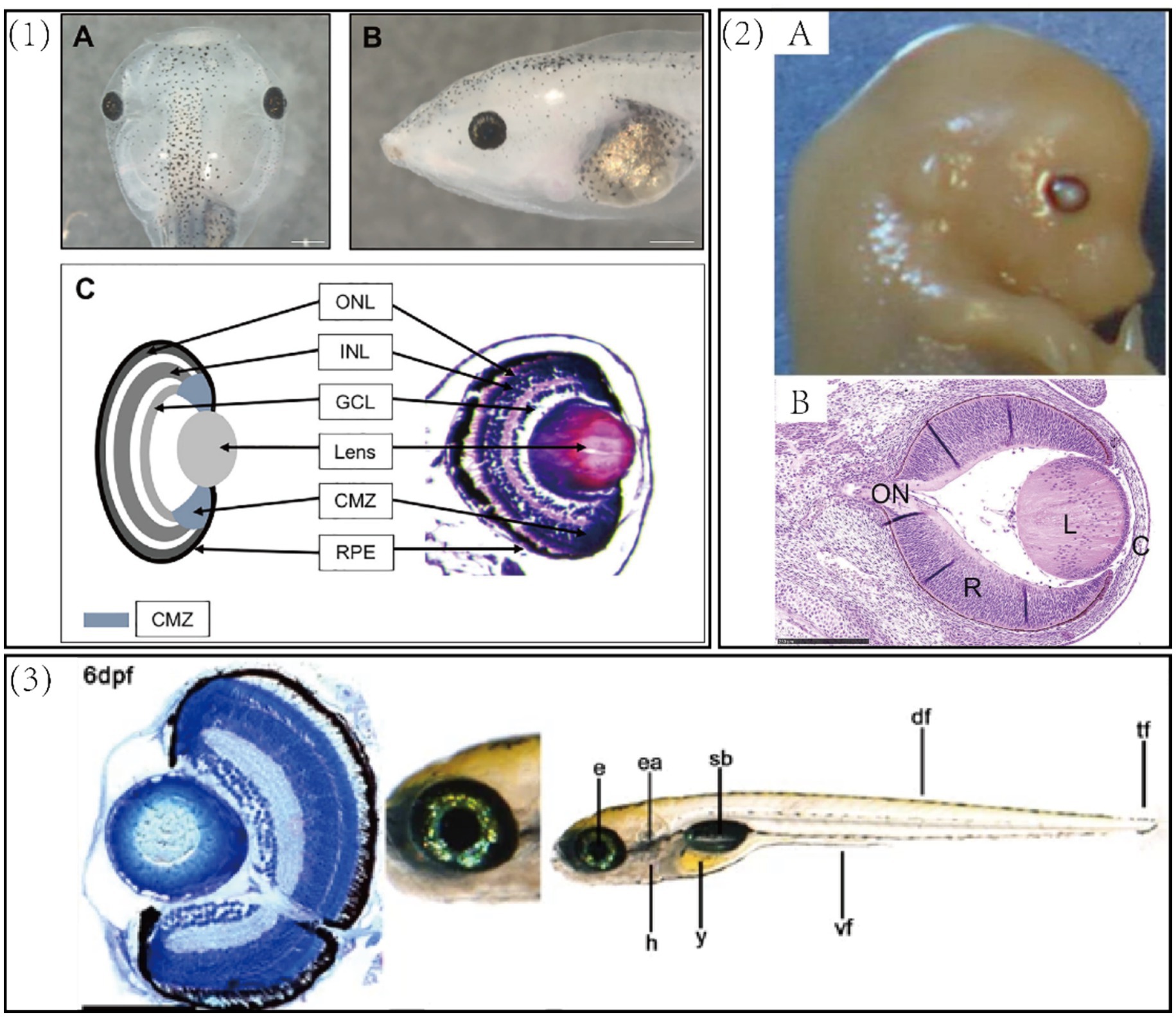
Figure 1. (1) Xenopus laevis tadpole. (A) A dorsal view of a 6-day old swimming tadpole. (B) A side view of the same tadpole. (C) A schematic (left) and transverse section (right) of the tadpole retina. ONL, outer nuclear layer—rod and cone photoreceptors; INL, inner nuclear layer—bipolar, horizontal, and amacrine cells; GCL, ganglion cell layer—retinal ganglion cells and amacrine cells; CMZ, ciliary margin zone—containing retinal stem cells, blue regions at the periphery of the lens. The outer layer covering the eye is the outer corneal epithelium and the layer directly adjacent to the lens is the inner corneal layer. Scale bar = 500 μm (111). (2) (A) Gross morphology of mouse eyeball. (B) Cross-sectional view of the eye (H&E staining). Bar = 250 um. C, cornea; L, lens; ON, optic nerve; R, retina (112). (3) Gross morphology and cross-sectional staining of zebrafish eye. e, eye; ea., ear; h, heart; y, yolk sac; sb, swimbladder, df, dorsal fin; tf, tail fin. Scale bars = 100 μm (113).
While other models such as zebrafish and chicken are also employed, they lack the specific combination of features that make X. laevis particularly special for mechanistic research. Zebrafish share high genetic conservation and transparent embryos but possess duplicated genes that can complicate phenotypic analysis (4). As an amphibian, X. laevis exhibits closer parallels to mammals than to zebrafish in key physiological processes, such as regulating osmotic pressure and undergoing metamorphosis (5). While zebrafish share high genetic conservation and transparent embryos ideal for live imaging, but their relatively small size limits the scale and ease of microsurgical manipulations that are possible in the much larger and more robust X. laevis embryos. The chicken embryo allows for direct observation but is limited by its evolutionary distance and genetic tools (6, 7). Drosophila is powerful for genetic screens but has limited utility for human ocular diseases due to vast physiological differences (8).
The core special of X. laevis lies in its ability to seamlessly integrate powerful genetic manipulation with embryonic accessibility at any developmental stage—a capability rare in other vertebrate models. As a tetrapod, it shares 79% genetic similarity with humans and comparable principles of eye development (9–12). Crucially, its embryos can be easily manipulated and induced at any stage, which is essential for validating developmental concepts in a living system. Specifically: (1) Their large size and transparency facilitate microinjection and single-cell transplantation to study tissue interactions, offering a strong self-control model at the 2- or 4-cell stage (10). (2) The Founder generation 0 (F0) reverse genetics approach enables phenotypic analysis in transgenic larvae without time-consuming breeding, allowing for rapid gain- and loss-of-function studies and the swift generation of transgenic models (9, 13). (3) Researchers can further integrate multi-omics approaches with XenBase resources to decipher developmental transcriptional networks.
It is important to note that gene duplicates in Xenopus may require careful interpretation of phenotypic severity due to potential compensatory mechanisms, and Morpholino use warrants controls (e.g., p53 co-injection) to minimize off-target effects (14). Nonetheless, for research aimed at rapidly and cost-effectively decrypting mechanisms, screening pathogenic genetic variants, and validating developmental hypotheses, the unique combination X. laevis offers—high physiological relevance, exceptional experimental accessibility, and speed—establishes it as a particularly powerful and efficient model system (Table 1).
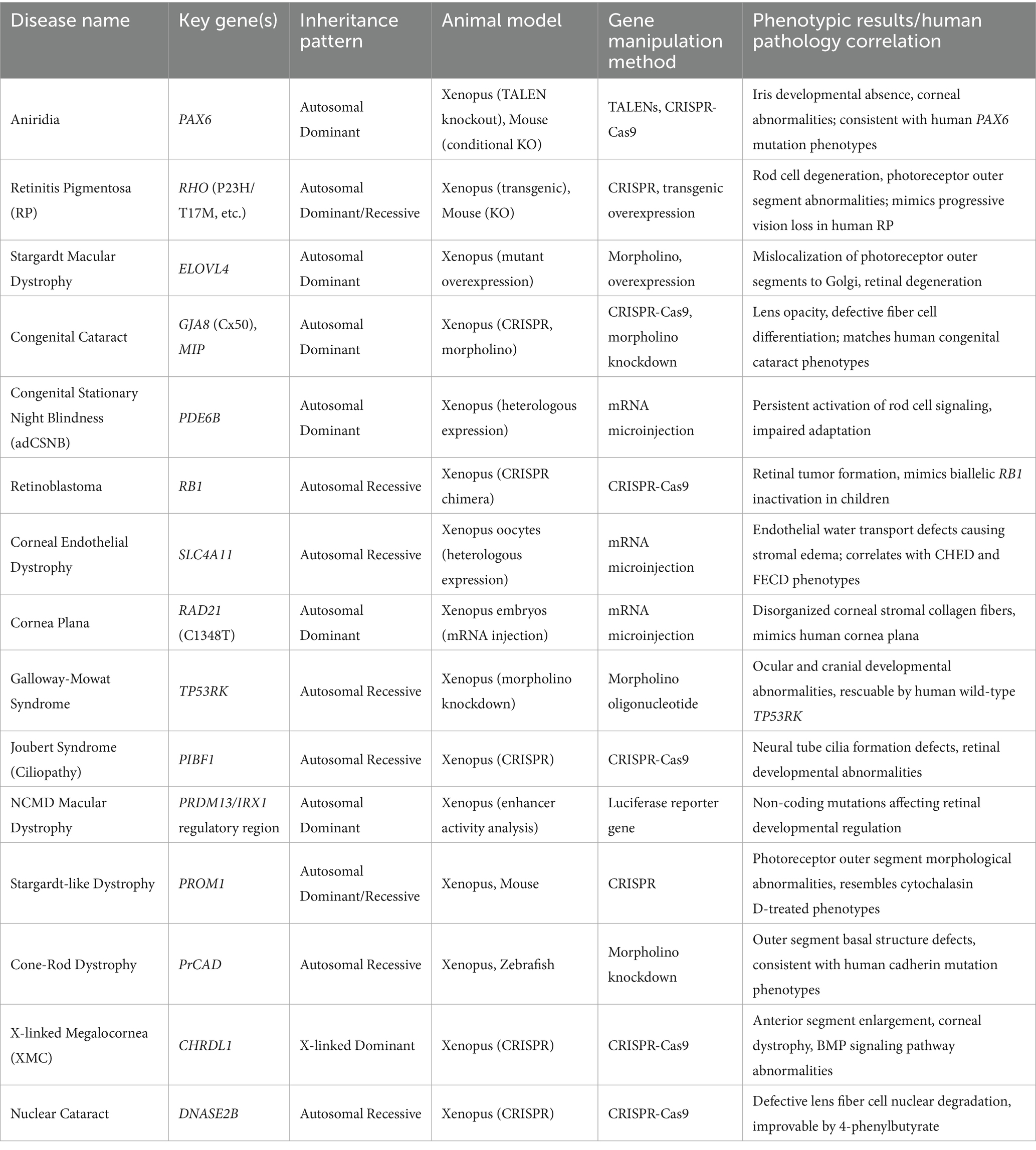
Table 1. Overview of diseases and genes.
3 Current research directions
3.1 Ocular development and eye defects
Previous research has highlighted that embryonic intervention studies are essential for defining the timing of ocular induction, elucidating early regulatory factors in eye development, and clarifying the molecular pathways underlying ocular malformations (10, 15, 16). In X. laevis, ectopic several eye-field transcription factors (EFTFs) require Otx2 to induce the eye field, revealing Otx2’s permissive role in early eye specification. By transplanting fluorescently labeled tissue expressing EFTFs and Otx2 into various regions of host embryos, researchers observed organized, functional eye-like structures, indicating that these factors alone can stimulate and coordinate eye morphogenesis (16–18). In the early stages of embryogenesis (neural plate stage, approximately 12 h post-fertilization), the Pax6 gene is first expressed, serving as the “master regulator” that initiates eye field formation (19). Shortly thereafter, Rx1 expression begins, co-regulating the early development of the ocular region alongside Pax6 (16). Six3 also plays a crucial role at this stage, cooperating with Pax6 and Rx1 to further define and refine the formation and differentiation of the eye field (20). As development progresses to the optic vesicle formation stage (around 24 h post-fertilization), fibroblast growth factor (FGF) signaling becomes active, promoting the extension of the optic vesicle and the formation of the optic cup (21). Concurrently, bone morphogenetic protein (BMP) signaling operates through distinct pathways to ensure the optic cup’s correct shape and positioning (22). Additionally, Sonic Hedgehog (Shh) signaling maintains symmetry and proper morphology in the eye field by modulating cell proliferation and differentiation (23).
Mutations in Pax6 lead to ocular malformations in both vertebrates and invertebrates, with PAX6 mutations in humans causing aniridia (24). In Xenopus, embryos injected with transcription activator-like effector nucleases (TALENs) targeting pax6a and pax6b in over 85% of cells displayed severe disruptions in eye formation and various ocular malformations, including microphthalmia and tissue deficiencies (25). In partial loss-of-function Pax6 mutants, early eye development appears relatively normal, but by the tadpole stage, the iris shows developmental deficiencies similar to the typical phenotype of aniridia in human PAX6 mutation carriers (26). These mutant tadpoles also exhibit other ocular anomalies, such as cataracts and corneal defects, which are frequently observed in human aniridia patients (27).
3.2 Retinal development and disease model
3.2.1 Retinal development model
The neural retina is composed of six distinct cell types—ganglion cells, amacrine cells, horizontal cells, photoreceptors, bipolar cells, and Müller glial cells—that are arranged in a stereotypical layered structure conserved across all vertebrates (12). X. laevis is a well-established model for studying neurodevelopment, particularly retinal cell fate determination (28–31). During Xenopus eye development, eye field specification is a critical step that governs the conversion of neuroectodermal cells into retinal cells. Initially, at the blastula stage, most animal pole cells have the potential to form retinal cells. By stage 12.5, when gastrulation is almost complete and the neural plate is forming, cells in the anterior region of the neural plate (the eye field) will differentiate into retinal tissue and eyes if cultured in a neutral environment or transplanted to the ventral side of an embryo. Hours before stage 12.5, cells transplanted from the same area do not form retina or eyes. This series of molecular changes, occurring within hours, enables neuroectodermal cells in frogs to specialize into retinal cells, a process termed eye field specification. Studying this stage can enhance understanding of the molecular mechanisms behind eye field specialization, thus providing an important model system for retinal development and regeneration research (14). Following optic cup formation (~48 h), the Notch pathway coordinates progenitor proliferation and differentiation (e.g., via Atoh7 and Ngn2) to generate retinal cell diversity (32, 33). Beyond the major molecular players in eye field specification, key regulators like KDM7A and Rax/Rx1 dynamically control this process (34). Regulated by Rax/Rx1, KDM7A is expressed early in Xenopus development and is temporally and spatially regulated in the central nervous system and eye (35). KDM7A expression is dynamic during embryonic development; overexpression disrupts late retinal development, hindering ganglion cell development and promoting horizontal cell formation, potentially contributing to the molecular mechanism regulating the spatiotemporal generation of retinal neuron subtypes (36).
3.2.1.1 Key proteins in retinal specialization
Opsins can detect light and perform imaging or non-imaging tasks, yet little is known about the retinal expression cells, developmental onset, and photoactivation of neural opsin proteins (37). Neural opsins (opn5, 6a, 6b, 8) exhibit stage-specific expression: opn5 and opn8 initiate at stage 37/38, coinciding with retinal circuit activation. Once the retinal circuitry connects to the brain, opn5 mRNA localizes in multiple retinal cell types, including bipolar cells (~70–75%), amacrine cells (~10%), and retinal ganglion cells (~20%), while opn8 is present in amacrine cells (~70%) and ganglion cells (~30%). Opn6a and opn6b mRNA emerge in newly formed photoreceptors by stage 35 and co-localize in rods and cones at stage 37/38. In the mature larval retina (stage 43/44), opn6a and opn6b mRNA preferentially localize in rods and cones, respectively, with newly formed photoreceptors bordering the proliferative ciliary marginal zone co-expressing both genes. The majority of retinal ganglion cells expressing neural protease show c-fos expression under light stimulation, and more than half of intermediate neurons expressing neural protease do so as well (38).
Post-translational modification is critical for the proteins involved in eye field specification, with presenilin (PS)/γ-secretase acting as the catalytic component of γ-secretase to cleave single-pass transmembrane proteins essential for development, such as Notch, netrin receptor DCC, cadherins, drebrin-A, and EphB2 receptors. Studies have shown that inhibiting PS expression in post-synaptic tectal neurons in the Xenopus tadpole retinotectal circuit diminishes visual avoidance behavior and weakens synaptic transmission, significantly reducing NMDA receptor (NMDAR) and AMPA receptor (AMPAR)-mediated currents. Further research indicates that expressing the C-terminal fragment of the EphB2 receptor rescues the NMDAR-mediated response reduction; this fragment, typically cleaved by PS/γ-secretase, is known to upregulate synaptic NMDAR, suggesting that normal PS function is essential for the proper formation and potentiation of retinotectal synapses via EphB2 cleavage (39).
3.2.1.2 Key protein mutations affecting retinal and neurodevelopment
Recent studies have shown that the absence of certain key proteins can lead to severe retinal neurodevelopmental abnormalities, even if their role in eye field specification remains unclear. Nitric Oxide Synthase Interacting Protein (Nosip), associated with various human diseases, is essential for the development of neural precursor tissues, such as the eye and neural crest cells. Research indicates that Nosip gene expression occurs in the developing eye system and neural crest cells of X. laevis, and Nosip inhibition results in severe eye formation defects in both mice and Xenopus. In Nosip-deficient Xenopus embryos, retinal layering and dorsal-ventral patterning of the retina are disrupted. Marker gene analyses (e.g., rax, pax6, and otx2) reveal that induction and differentiation of the eye field are hindered. Nosip deficiency also impairs cranial cartilage structure, inhibits neural crest cell induction and migration, and affects downstream factors of retinoic acid signaling, such as foxc1 (40). The Fezzin family member Nedd4 Binding Protein 3 (N4BP3) is expressed in the neuroectoderm of Xenopus embryos, including the eye, brain, and neural crest cells. Knockdown of N4BP3 in anterior neuroectoderm leads to severe developmental defects in eye, brain, and neural crest-derived cranial cartilage structures. N4BP3 deficiency significantly reduces the expression of eye and brain-specific marker genes, decreases neural crest cell migration, and impacts cell apoptosis and proliferation, suggesting that N4BP3 is essential for early anterior neural development in vertebrates, consistent with findings linking human N4BP3 gene disruption to neurodevelopmental disorders (41).
3.2.2 Retinal disease models
3.2.2.1 Retinitis pigmentosa
Retinitis pigmentosa (RP) is a common hereditary retinal dystrophy characterized by the gradual degeneration of rod cells, followed by the non-cell-autonomous death of cone cells, progressive vision loss, and eventual blindness (42). Over 200 mutations in 50 different genes are associated with this disease, but there is currently no cure (43, 44). Rhodopsin, a cilia-specific G-protein-coupled receptor (GPCR), is transported with high fidelity to the outer segment (OS) of vertebrate rod cells; disruption in rhodopsin trafficking results in photoreceptor apoptosis and RP-associated blindness (45, 46). Mutations in the rhodopsin gene account for approximately 10% of RP cases worldwide. These mutations can cause endoplasmic reticulum (ER) retention and cell death, mislocalization, constitutive activity, or impaired protein–protein interactions. Using a rhodopsin-based gene-editing model in Xenopus, researchers discovered that loss of rhodopsin function leads to significant rod cell degeneration, displaying ultrastructural defects in the outer and inner segments, including vesiculation in the OS and rapid phagocytosis by retinal pigment epithelium (RPE), with occasional cell death, followed by morphological deterioration of cone cells (47, 48). Expression of the autosomal dominant rhodopsin mutation Ter349Glu in Xenopus revealed that this mutation, which adds 51 amino acids to the C-terminus of rhodopsin, causes mislocalization, OS developmental abnormalities, and disk formation defects (49). Studies in Xenopus expressing truncated rhodopsin mutants identified the CCGKN motif (amino acids 322–326) as critical for localization fidelity; additional signals in this region promote mislocalization. While the VXPX motif offsets mislocalization signals, enhancing ciliary targeting and transport (50).
The P23H mutation in rhodopsin is a major cause of autosomal dominant RP. Expression of bovine P23H rhodopsin in Xenopus showed that light-induced degeneration of P23H rods occurs in at least two stages: first, impaired phototransduction, followed by morphological changes (51). The P23H mutation triggers light-dependent degeneration through autophagy: cells degrade misfolded rhodopsin or secretory pathway debris, ultimately leading to apoptosis (48, 52). Visualizing autophagic structures in transgenic Xenopus rod cells expressing the dual-fluorescent autophagy marker mRFP-eGFP-LC3 revealed autophagosome maturation and degradation over 28 h, with more autophagosomes in rods expressing misfolded RHO P23H under light-induced autophagy enhancement (53). Expression of glycosylation-deficient rhodopsin mutants (T4K and T17M) in Xenopus explored light-exacerbated retinal degeneration mechanisms, finding that additional disulfide bonds increased thermal stability and reduced retinal degeneration. These mutants exhibited reduced toxicity when lacking chromophore binding sites or when vitamin A was absent in the diet; however, active conformation of these mutants, even in darkness, led to retinal degeneration, suggesting that vitamin A supplementation may be ineffective or harmful in glycosylation-deficient RP genotypes (54). Studying the effects of valproic acid (VPA) in Xenopus models for RP revealed varied outcomes depending on rhodopsin mutation. VPA improved degeneration associated with P23H rhodopsin, promoted rhodopsin clearance, and restored visual function. However, under light exposure, VPA exacerbated degeneration in the T17M mutant and showed adverse effects in T4K and Q344ter mutants, with outcomes similar to histone deacetylase (HDAC) inhibitors, suggesting effects of VPA was linked to autophagy regulation (55).
Rhodopsin is precisely trafficked to the OS of rod cells and transport defects cause autosomal dominant RP (46). A rhodopsin-photoactivated GFP-1D4 (RHO-paGFP-1D4) model was introduced in Xenopus to monitor rhodopsin trafficking in live cells, demonstrating that this model mirrors rhodopsin function and localization both in vitro and in vivo, enabling simpler and more comprehensive studies of rhodopsin and GPCR trafficking (56). In vivo studies in Xenopus identified that TBC1D32 is essential in retinal development and RPE differentiation, with deficiencies leading to disrupted cilia elongation, apical tight junction breakdown, loss of function, and an epithelial-mesenchymal transition-like phenotype, affecting photoreceptor differentiation and OS transport (57). FAM161A, a widely conserved microtubule-associated protein, has been linked to hereditary RP-associated blindness. Studies in Xenopus show that FAM161A has significant homology with Xenopus Tpx2, indicating a role in microtubule binding similar to Tpx2, laying a foundation for a molecular model of the FAM161A-microtubule complex (58).
3.2.2.2 Genetic macular degeneration
North Carolina Macular Dystrophy (NCMD), a rare autosomal dominant disorder, arises from noncoding single nucleotide variants (SNVs) near PRDM13 or duplications overlapping DNase I hypersensitive sites (near PRDM13/IRX1) (59–61). Researchers mapped interactions between PRDM13 and IRX1 promoters and identified 18 candidate cis-regulatory elements (cCREs), examining their activity via luciferase and Xenopus enhancer assays (59). Stargardt macular dystrophy, caused by mutations in ELOVL4, leads to macular degeneration and early-onset blindness. Overexpressing murine ELOVL4 mutants in Xenopus rods showed that lack of the ER retention di-lysine motif led to mislocalization to Golgi and post-Golgi compartments, rather than the inner segment, altering photoreceptor structure and function and leading to retinal degeneration (62). Mutations in prominin-1 (prom1) have also been implicated in autosomal dominant Stargardt-like macular dystrophy, autosomal recessive retinitis pigmentosa, and cone-rod dystrophy (63). Prom1 expresses in the retinal outer segment (ROS) base, ROS patches, and cone outer segments (COS) in Xenopus and is also localized at the ciliary tips of multiciliated skin cells (64, 65). Mutations in the photoreceptor cadherin (PrCAD) lead to autosomal recessive cone-rod dystrophy (66). PrCAD is expressed in the retina and pineal photoreceptors and is localized to the ROS base in X. laevis, Xenopus tropicalis, and zebrafish retinas, with additional immunoreactivity observed at the ROS plasma membrane and the leading edge of COS disks (67). The mechanisms underlying outer segment (OS) malformations in prom1 and prCAD mutants remain unclear. Previous studies have shown that treating X. laevis eye cups with cytochalasin D induces OS malformations resembling those observed in prom1 mutants (68). Exploring the application of Xenopus model organisms may provide deeper insights into the processes of OS disk morphogenesis and the underlying mechanisms of prom1- and prCAD-related retinal degenerations (64).
3.2.2.3 Color blindness and retinal tumors
Autosomal dominant congenital stationary night blindness (adCSNB) is caused by mutations in genes involved in the rod phototransduction cascade, including rhodopsin (RHO), transducin alpha subunit (GNAT1), and cGMP phosphodiesterase 6 beta subunit (PDE6B) (69, 70). In Xenopus, truncated PDE6B variants (PDE6β1–313, PDE6β1–314 fs50) localize to the phototransduction compartment, retain cGMP binding (non-catalytic), and constitutively activate the cascade by disrupting Pγ regulation, ultimately impairing rod adaptation (69). Complete color blindness links to CNGB3 mutations (e.g., F525N, T383fsX) that heighten photoreceptor susceptibility to cell death. Patch-clamp recordings in Xenopus oocytes expressing Wild Type or mutant channels revealed that these mutations enhance affinity for the activator CPT-cGMP, leading to cytotoxicity, particularly in the F525N variant, mitigated by CNG channel blockers or removal of extracellular calcium (71).
3.2.2.4 Retinoblastoma and autosomal dominant vitreoretinal degeneration
Childhood eye tumors, such as retinoblastoma, are caused by biallelic inactivation of the retinoblastoma 1 (RB1) gene (72). By developing rb1/rbl1 chimeric mutants using CRISPR/Cas9 in Xenopus embryos, researchers rapidly established a retinoblastoma model (73). Snowflake vitreoretinal degeneration, an autosomal dominant disorder, is linked to the KCNJ13 gene encoding the Kir7.1 inwardly rectifying potassium channel. The R162W mutation associated with this disease disrupts functional channel formation. Xenopus oocyte expression of mutant and wild-type Kir7.1 channels showed that the R162W mutation inhibits Kir7.1 activity by potentially altering phosphatidylinositol 4,5-bisphosphate (PIP2) gating (74).
3.3 Corneal development and disease model
3.3.1 Corneal development in Xenopus laevis
The structural and developmental characteristics of the X. laevis cornea closely parallel those of humans (75). The development of the X. laevis cornea begins at stage 25 with the differentiation of a simple embryonic epidermis that overlies the developing optic vesicle. Around stage 30, after the detachment of the lens placode, cranial neural crest cells invade the space between the lens and embryonic epidermis, forming the corneal endothelium. At stage 41, a second wave of migrating cells containing presumptive corneal stromal cells infiltrates the stroma, leading to the formation of inner and outer corneal layers. These layers converge at a central stromal attraction center, which organizes radial fiber alignment. Post-stage 48, secondary stromal keratocytes migrate individually to the center, establishing the stromal layers. By stage 60, the stroma is extensively populated with collagen lamellae and keratocytes, and the stromal attraction center disappears. During early metamorphosis, the embryonic epithelium gradually transitions into adult corneal epithelium, characterized by microvilli coverage. By stage 62, the epithelium thickens, accompanied by extensive apoptosis in the epithelial cells, coinciding with eyelid opening. Post-metamorphosis, the adult frog cornea reaches its mature structure, comprising three cellular layers (epithelium, stroma, and endothelium) and two acellular layers (Bowman’s layer and Descemet’s membrane) (11, 76). Following initial maturation, the X. laevis cornea, particularly the stroma, continues to thicken and expand throughout the animal’s lifespan. In adult frogs, a p63-positive, wave-like structure at the limbus is observed, indicating corneal stem cell (CESC) activity. Proliferation assays reveal active division in basal corneal epithelial cells and limited proliferative activity in stromal and endothelial cells, suggesting ongoing corneal maintenance (11).
The larval corneal epithelium in X. laevis consists of three distinct layers: an outer epithelial layer, a basal epithelial layer, and a deeper fibrous layer containing major sensory nerve trunks. Basal epithelial cells express multiple pluripotency markers, including sox2, p63, c-myc, and klf4, indicating stem-like properties (77, 78). Notably, p63 expression is confined to all basal epithelial cells, whereas c-myc is predominantly localized to a subset of basal epithelial cells and adjacent stromal tissue at the corneal periphery. Furthermore, sox2 is expressed throughout the outer and basal epithelial cells, with higher intensity in a unique subpopulation of multinucleated or lobulated cells often located at the corneal periphery. Proliferation assays using thymidine analog EdU indicate that cell proliferation is concentrated in the basal epithelial layer and contributes to regeneration. Results reveal upregulation of pluripotency markers such as sox2, p63, and oct60 during early lens regeneration, while immunostaining indicates a significant increase in sox2-expressing cells throughout the basal corneal epithelium within 4 h post-lens removal (78).
3.3.2 Corneal disorders
3.3.2.1 Hereditary endothelial dystrophies
SLC4A11 is a member of the SLC4 bicarbonate transporter family expressed in corneal endothelial cells. Mutations in SLC4A11 disrupt endothelial fluid transport, leading to dystrophies like congenital hereditary endothelial dystrophy (CHED) and Fuchs endothelial corneal dystrophy (FECD), but the precise pathogenic mechanisms are unresolved. SLC4A11 encodes the bicarbonate transporter-related protein BTR1, and its mutations are associated with diseases such as CHED and Harboyan syndrome, characterized by vision and hearing loss. SLC4A11-mediated water transport is unique in its solute flux independence, inhibition by classic SLC4 inhibitors, and inactivity in CHED2 mutants (R125H). Studies in Xenopus oocytes and HEK293 cells demonstrate that SLC4A11 facilitates water flux at rates approximately half that of aquaporins (79). Recent findings suggest that mSlc4a11 functions as an H(+)/OH(−) channel independent of co-transport with other ions, identifying OH(−) as its likely substrate. Moreover, mSlc4a11 activity is enhanced by extracellular and intracellular alkalinization (80–82). Using Xenopus as a model system, researchers have traced cellular lineages, examining the division patterns and fate of CESCs during development, homeostasis, and wound healing (83). Notably, a histologically confirmed Bowman’s layer is present in Xenopus laevis, which makes Xenopus a particularly relevant model for investigating a broader spectrum of human corneal pathologies, including those that primarily affect the epithelial-stromal interface (11).
3.3.2.2 Peripheral corneal sclerocornea
The RAD21 (R450C) variant has been identified in a family with peripheral corneal sclerocornea. Microinjection of this rad21 variant mRNA into Xenopus embryos disrupts the organization of stromal collagen fibers. Cells carrying the heterozygous rad21 variant exhibit altered chromatin conformation and expression of cell adhesion genes, impairing neural crest migration and significantly reducing neural crest-derived periocular mesenchyme in the stromal region. These findings elucidate a mechanism by which the RAD21 (R450C) variant causes stromal defects (84).
3.3.2.3 Limbal stem cell deficiency
LSCD, a debilitating condition caused by CESC damage or loss, has been modeled in Xenopus using transient psoralen-AMT treatment followed by UV exposure (PUV) (85). This model recapitulates LSCD features, including pigment invasion, corneal opacity, and neovascularization. PUV-induced depletion of p63-expressing basal epithelial cells promotes mitotic activity in residual cells, eventually restoring corneal transparency. This model provides a platform for understanding LSCD mechanisms and therapeutic strategies (86).
3.3.2.4 Corneal stiffness disorders
Disordered stromal collagen fibers characterize corneal stiffening diseases (87). The RAD21(C1348T) variant is associated with such phenotypes, as shown by injection of wild-type and mutant rad21 mRNA into Xenopus embryos. Mutant embryos exhibit disrupted stromal collagen fibers and reduced fiber diameter, which can be rescued by wild-type rad21 overexpression. These findings highlight RAD21’s critical role in collagen organization and corneal development (88).
3.3.2.5 Corneoscleral dysplasia
Corneoscleral dysplasia, marked by an indistinct corneoscleral boundary, involves RAD21 mutations that disrupt neural crest cell migration through upregulation of PCDHGC3 and WNT9B (89, 90). In Xenopus, rad21 knockdown increases pcdhgc3 and wnt9b expression, impairing neural crest differentiation. Suppressing wnt9b rescues migration defects, suggesting that rad21 regulates corneoscleral boundary formation via WNT signaling modulation (91).
3.3.2.6 X-linked megalocornea
Mutations in Chordin-Like 1 (CHRDL1) cause XMC, characterized by anterior segment enlargement, mosaic corneal dystrophy, premature cataracts, and glaucoma (92, 93). A novel CHRDL1 frameshift mutation (c.807_808delTC) has been identified, leading to loss of function. Using Xenopus as a model, researchers demonstrated that chrdl1 deficiency causes XMC-like phenotypes, linked to disrupted BMP signaling. Knockdown of chrdl1 in Xenopus mimics human XMC, with reduced BMP receptor 1A and altered pSMAD1/5 phosphorylation. These findings implicate impaired BMP antagonism in XMC pathogenesis (94).
3.4 Lens development and disease model
3.4.1 Lens development
In X. laevis, lens development can be categorized into five major stages, each defined by specific cellular and molecular events: In the early stages of development, the lens ectoderm thickens to form the lens placode, which begins to invaginate. By stage 27, the invaginated cell mass forms the lens vesicle primordium. At stage 32, the lens becomes polarized, forming a lens vesicle that separates from the sensory ectoderm. By stage 38, cells in the posterior portion of the lens vesicle elongate to form primary lens fibers, while the anterior cells develop into the epithelial layer. At stage 41, nuclear degradation begins, and primary fiber cells lose their nuclei. Between stages 44 and 48, secondary fiber cells differentiate in the equatorial region, forming concentric layers around the primary fibers, ultimately achieving a structure resembling the mature lens (76, 95).
3.4.2 Congenital cataracts
Dozens of genes associated with congenital hereditary cataracts have been identified using animal models. These genes primarily encode lens proteins, connexins, membrane proteins, extracellular matrix components, cytoskeletal proteins, and transcription factors such as FOXE3, HSF4, MAF, and PITX3 (96). Injection of low doses of morpholino oligonucleotides in X. laevis tailbud stages induces a subtle lens phenotype resembling cataracts (97). Knockout of mammalian homologs of cataract-associated genes, such as TMEM114, CHRLD1, SIPAL3, or CELF1, results in ocular phenotypes loosely associated with cataracts (98, 99). Inactivation of three genes in X. laevis via CRISPR/Cas9 revealed lens-specific defects. Mutants of the gap junction protein and nuclease gja8 exhibited lens opacity, nuclear degradation, and fiber cell organization defects, while dnase2b mutants had normal external morphology but impaired fiber cell differentiation. The potential therapeutic effect of the chemical chaperone 4-phenylbutyrate was demonstrated by improving vision in gja8 mutant tadpoles (96). Connexin 50 (Cx50) is one of the most commonly mutated genes associated with congenital cataracts. The Cx50T39R mutation exhibits dominant effects in X. laevis, resulting in significantly enhanced hemichannel currents, altered voltage gating, and induced cell death. All-atom molecular dynamics simulations revealed that the R39 substitution stabilized an open-state conformation of the N-terminal domain, leading to lens cytotoxicity and cataract formation (100). Mutations in RNA granule component TDRD7 cause pediatric cataracts. In X. laevis, unilateral knockdown of Hspb1 at stage 42 resulted in defects in eye and lens development, which were rescued by co-injection of mouse Hspb1 mRNA (101). In a four-generation Chinese family with congenital cataracts, a novel missense mutation in the MIP gene, c.572C > G (p. P191R), was identified. Swelling assays in X. laevis oocytes showed that the P191R mutation reduced oocyte swelling rates by impairing MIP protein trafficking and decreasing its membrane localization, leading to cataract formation (102).
Channel Protein-Related Mutations: Membrane transporter monocarboxylate transporter 12 (MCT12), also known as creatine transporter 2 (CRT2), is expressed in the lens and associated with cataracts. Studies in X. laevis oocytes and human HEK293T cells revealed that multiple variants of the SLC16A12 gene (p. Ser158Pro, p. Gly205Val, p. Pro395Gln, and p. Ser453Arg) significantly impaired creatine transport. Specifically, the p. Gly205Val and p. Ser453Arg variants failed to localize to the oocyte membrane, suggesting defective protein interactions during transporter processing (103). In autosomal dominant congenital cataract pedigrees, mutations in genes encoding various soluble and membrane proteins have been linked to lamellar cataracts. A novel mutation in the MIP gene (c.494G > A) caused substitution of a highly conserved glycine with aspartic acid (G165D) in aquaporin-0 (AQP0). Functional analysis of Xenopus oocytes expressing AQP0-G165D showed that the mutation disrupted AQP0 trafficking and abolished water channel function. These findings highlight the essential role of AQP0 in maintaining lens transparency and underscore how mutations in conserved residues of aquaporins impact their function (104).
3.5 Others
3.5.1 Glaucoma
Using X. laevis oocytes as a modification and expression system, MβCD-induced currents were validated in oocytes expressing TRPV4. Further studies established the relationship between mechanical strain, free membrane cholesterol, the actin cytoskeleton, and stretch-activated transient receptor potential vanilloid isoform 4 (TRPV4) channels in human trabecular meshwork (TM) cells. These findings demonstrate that membrane cholesterol regulates the transduction of trabecular mechanical signals. This suggests that diet, cholesterol metabolism, and mechanical stress may influence conventional outflow pathways and intraocular pressure in glaucoma and diabetes, partially through TM mechanosensing (105).
3.5.2 Galloway-Mowat syndrome
GAMOS is a rare disease characterized by early-onset nephrotic syndrome and microcephaly, often accompanied by various neurological features. Most cases are caused by pathogenic variants in genes encoding components of the KEOPS complex, including OSGEP, TP53RK, TPRKB, and LAGE3. Computational analyses revealed that three core members of the KEOPS complex—Osgep, Tp53rk, and Tprkb—are highly conserved across species, including X. laevis. RT-PCR and whole-mount in situ hybridization studies investigated the spatial and temporal expression patterns of osgep, tp53rk, and tprkb during early development in X. laevis. Results showed that these genes are expressed during early embryogenesis and are enriched in tissues and organs affected by GAMOS, including the developing eye (106). In a morpholino-mediated tp53rk knockdown model in X. laevis, depletion of endogenous Tp53rk led to abnormal eye and head development. This abnormal phenotype was rescued by expressing human wild-type TP53RK, but not by the c.163C > G mutant or another previously described GAMOS-associated mutant, c.125G > A (p. Gly42Asp) (107).
3.5.3 Nuclear envelopathies
Mutations in nuclear envelope proteins are associated with various human diseases (nuclear envelopathies), particularly mutations in inner nuclear membrane proteins emerin and MAN1, which are linked to X-linked Emery-Dreifuss muscular dystrophy. Both proteins contain a highly conserved LEM domain known to interact with multiple transcription factors. Morpholino-mediated knockdown experiments in X. laevis showed that MAN1 is critical for the development of tissues, including the eye. The effects of MAN1 knockdown could be compensated by ectopic expression of emerin, restoring normal organogenesis. This highlights the essential role of MAN1 in regulating genes critical for organ development and tissue homeostasis, supporting the necessity of LEM proteins in early embryonic development. Furthermore, MAN1 deficiency may contribute to muscle- and retina-specific diseases (108).
3.5.4 Ciliopathies
Regulatory Factor X (RFX) transcription factors are implicated in the pathogenesis of ciliopathies, severe human developmental disorders such as retinal degeneration. Studies on the RFX7 gene in X. laevis revealed its expression in the eye, and knockdown of RFX7 in Xenopus embryos led to defects in neural tube ciliogenesis and neural tube closure failure. RFX7 regulates the expression of RFX4, a gene required for neural tube ciliogenesis. Ectopic expression of Foxj1, a key regulator of motile ciliogenesis, inhibited RFX4 expression but did not affect RFX7 expression (109). Using X. laevis as a model, researchers rapidly evaluated the effects of novel compound heterozygous variants (p. Y503C and p. Q485*) in the centriole gene PIBF1, identified in Joubert Syndrome (JS) patients. The study indicated that JS results from a combination of amorphic and hypomorphic PIBF1 alleles (110).
4 Discussion
The Xenopus system has emerged as an indispensable model for investigating ocular development and disease pathogenesis. Its unique embryonic transparency and experimental tractability have enabled precise characterization of Pax6-mediated eye field specification during early neurulation, revealing conserved genetic networks underlying vertebrate eye morphogenesis. Subsequent studies of corneal development have delineated the spatiotemporal dynamics of neural crest cell migration, providing mechanistic insights into stromal organization and endothelial differentiation. For disease modeling, CRISPR/Cas9 and TALEN approaches have successfully recapitulated human ocular pathologies including aniridia (Pax6 mutations), retinitis pigmentosa (RHO variants), and congenital cataracts (GJA8 defects). Particularly noteworthy is the light-inducible RHO P23H model, which has provided unprecedented live visualization of photoreceptor degeneration dynamics through autophagy marker labeling.
While offering substantial advantages for developmental studies, the Xenopus system presents several constraints for clinical ophthalmology applications. The absence of macular anatomy precludes modeling of age-related macular degeneration, and the lack of intraocular pressure regulation mechanisms limits glaucoma research. Most disease models exhibit accelerated pathology compared to human chronic conditions - for instance, retinal degeneration typically progresses over days rather than years. Additional translational challenges include: (1) inability to perform standard visual function assessments (e.g., electroretinography, visual field testing); (2) limited capacity to study late-stage disease complications; and (3) absence of accessory ocular structures (eyelids, lacrimal apparatus) essential for investigating ocular surface disorders. (4) The species’ ancestral genome duplication event introduces functional redundancy that may obscure genotype–phenotype correlations, while its lack of macular specialization limits modeling of human macular pathologies.
Several strategic developments could enhance the model’s clinical utility. Establishing standardized visual behavior paradigms would facilitate functional assessment of therapeutic interventions. A tiered “Xenopus-to-mammal” validation platform could optimize drug discovery pipelines, leveraging Xenopus for rapid candidate screening followed by mammalian models for comprehensive evaluation. Particularly promising is the exploration of Xenopus’s remarkable regenerative capacity, which may reveal novel neuroprotective mechanisms applicable to retinal degenerative diseases.
5 Conclusion
In recent years, substantial progress has been made in studies related to eye development and associated diseases. Key breakthroughs include the identification of transcription factors and signaling pathway molecules that regulate the development of the lens, retina, and other ocular tissues. These findings have elucidated the genetic mechanisms underlying cataracts, glaucoma, and retinal degenerative diseases. However, the intricate gene regulatory networks governing eye development remain poorly understood, and the goal of organ-level eye regeneration remains a distant challenge. The X. laevis model, with its transparent embryogenesis, experimental flexibility, and efficiency in gene function studies, provides a unique platform for research in eye development. Integrating cutting-edge technologies with the Xenopus model offers promising opportunities to further uncover the molecular and genetic basis of eye development and congenital hereditary diseases.
Author contributions
QL: Writing – original draft, Writing – review & editing. YF: Writing – review & editing, Conceptualization. XZ: Conceptualization, Writing – review & editing, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Supported by Beijing Natural Science Foundation. ID: Z240020 Clinical Medicine Plus X - Young Scholars Project of Peking University, the Fundamental Research Funds for the Central Universities. ID: PKU2024LCXQ005.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yan, W, Laboulaye, MA, Tran, NM, Whitney, IE, Benhar, I, and Sanes, JR. Mouse retinal cell atlas: molecular identification of over sixty amacrine cell types. J Neurosci. (2020) 40:5177–95. doi: 10.1523/JNEUROSCI.0471-20.2020
2. Mai, S, Zhu, X, Wan, EYC, Wu, S, Yonathan, JN, Wang, J, et al. Postnatal eye size in mice is controlled by SREBP2-mediated transcriptional repression of Lrp2 and Bmp2. Development. (2022) 149:dev200633. doi: 10.1242/dev.200633
3. Marcos, S, González-Lázaro, M, Beccari, L, Carramolino, L, Martin-Bermejo, MJ, Amarie, O, et al. Meis1 coordinates a network of genes implicated in eye development and microphthalmia. Development. (2015) 142:3009–20. doi: 10.1242/dev.122176
4. Ghyselinck, NB, and Duester, G. Retinoic acid signaling pathways. Development. (2019) 146:13. doi: 10.1242/dev.167502
5. Carotenuto, R, Pallotta, MM, Tussellino, M, and Fogliano, C. Xenopus laevis (Daudin, 1802) as a model organism for bioscience: a historic review and perspective. Biology. (2023) 12:890. doi: 10.3390/biology12060890
6. Fernández-Garre, P. Fate map of the chicken neural plate at stage 4. Development. (2002) 129:2807–22. doi: 10.1242/dev.129.12.2807
7. Hayashi, M, Ninomiya, Y, Hayashi, K, Linsenmayer, TF, Olsen, BR, and Trelstad, RL. Secretion of collagen types I and II by epithelial and endothelial cells in the developing chick cornea demonstrated by in situ hybridization and immunohistochemistry. Development. (1988) 103:27–36.
8. Teeters, G, Weasner, BM, Ordway, AJ, Weasner, BP, and Kumar, JP. Control of fate specification within the dorsal head of Drosophila melanogaster. Development. (2024) 151:dev199885. doi: 10.1242/dev.199885
9. Khokha, MK. Xenopus white papers and resources: folding functional genomics and genetics into the frog. Genesis. (2012) 50:133–42. doi: 10.1002/dvg.22015
10. Harland, RM, and Grainger, RM. Xenopus research: metamorphosed by genetics and genomics. Trends Genet. (2011) 27:507–15. doi: 10.1016/j.tig.2011.08.003
11. Hu, W, Haamedi, N, Lee, J, Kinoshita, T, and Ohnuma, SI. The structure and development of Xenopus laevis cornea. Exp Eye Res. (2013) 116:109–28. doi: 10.1016/j.exer.2013.07.021
12. Papadogiannis, V, Hockman, D, Mercurio, S, Ramsay, C, Hintze, M, Patthey, C, et al. Evolution of the expression and regulation of the nuclear hormone receptor ERR gene family in the chordate lineage. Dev Biol. (2023) 504:12–24. doi: 10.1016/j.ydbio.2023.09.003
13. Lei, Y, Guo, X, Liu, Y, Cao, Y, Deng, Y, Chen, X, et al. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs). Proc Natl Acad Sci. (2012) 109:17484–9. doi: 10.1073/pnas.1215421109
14. McDonough, MJ. Dissection, culture, and analysis of Xenopus laevis embryonic retinal tissue. J Vis Exp. (2012) 70:4377. doi: 10.3791/4377
15. Chow, RL, and Lang, RA. Early eye development in vertebrates. Annu Rev Cell Dev Biol. (2001) 17:255–96. doi: 10.1146/annurev.cellbio.17.1.255
16. Zuber, ME, Gestri, G, Viczian, AS, Barsacchi, G, and Harris, WA. Specification of the vertebrate eye by a network of eye field transcription factors. Development. (2003) 130:5155–67. doi: 10.1242/dev.00723
17. Danno, H, Michiue, T, Hitachi, K, Yukita, A, Ishiura, S, and Asashima, M. Molecular links among the causative genes for ocular malformation: Otx2 and Sox2 coregulate rax expression. Proc Natl Acad Sci USA. (2008) 105:5408–13. doi: 10.1073/pnas.0710954105
18. Zuber, ME. Eye field transcriptome characterization identifies disease-related and novel genes regulated during early eye formation. Invest Ophthalmol Vis Sci. (2023) 64:2834–4.
19. Walther, C, and Gruss, P. Pax-6, a murine paired box gene, is expressed in the developing CNS. Development. (1991) 113:1435–49.
20. Oliver, G, Mailhos, A, Wehr, R, Copeland, NG, Jenkins, NA, and Gruss, P. Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development. (1995) 121:4045–55.
21. Song, MR, and Ghosh, A. FGF2-induced chromatin remodeling regulates CNTF-mediated gene expression and astrocyte differentiation. Nat Neurosci. (2004) 7:229–35. doi: 10.1038/nn1192
22. Mack, R, and Gozani, O. Functional epigenomics: chromatin complexity untangled. Nat Struct Mol Biol. (2023) 30:1403–5. doi: 10.1038/s41594-023-01088-3
23. Ekker, SC, Ungar, AR, Greenstein, P, von Kessler, DP, Porter, JA, Moon, RT, et al. Patterning activities of vertebrate hedgehog proteins in the developing eye and brain. Curr Biol. (1995) 5:944–55.
24. Sannan, N. S., Genetic insights into the role of PAX6 in ocular development. Vancouver: University of British Columbia Library (2018).
25. Suzuki, KT, Isoyama, Y, Kashiwagi, K, Sakuma, T, Ochiai, H, Sakamoto, N, et al. High efficiency TALENs enable F0 functional analysis by targeted gene disruption in Xenopus laevis embryos. Biol Open. (2013) 2:448–52. doi: 10.1242/bio.20133855
26. Grainger, RM, Lauderdale, JD, Collins, JL, Trout, KL, McCullen Krantz, S, Wolfe, SS, et al. Report on the 2021 aniridia North America symposium on PAX6, aniridia, and beyond. Ocul Surf. (2023) 29:423–31. doi: 10.1016/j.jtos.2023.05.010
27. Nakayama, T, Fisher, M, Nakajima, K, Odeleye, AO, Zimmerman, KB, Fish, MB, et al. Xenopus pax6 mutants affect eye development and other organ systems, and have phenotypic similarities to human aniridia patients. Dev Biol. (2015) 408:328–44. doi: 10.1016/j.ydbio.2015.02.012
28. Feller, MB, and Sun, YH. Introduction to special issue on retinal development. Dev Neurobiol. (2011) 71:1131–2. doi: 10.1002/dneu.20952
29. Sanes, JR, and Zipursky, SL. Design principles of insect and vertebrate visual systems. Neuron. (2010) 66:15–36. doi: 10.1016/j.neuron.2010.01.018
30. Phipps, LS. Model systems for regeneration: Xenopus. Development. (2020) 147:6. doi: 10.1242/dev.180844
31. Kha, CX, Guerin, DJ, and Tseng, KA. Using the Xenopus developmental eye regrowth system to distinguish the role of developmental versus regenerative mechanisms. Front Physiol. (2019) 10:502. doi: 10.3389/fphys.2019.00502
32. Brown, NL, Patel, S, Brzezinski, J, and Glaser, T. Math5 is required for retinal ganglion cell and optic nerve formation. Development. (2001) 128:2497–508. doi: 10.1242/dev.128.13.2497
33. Li, X, Egervari, G, Wang, Y, Berger, SL, and Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol. (2018) 19:563–78. doi: 10.1038/s41580-018-0029-7
34. Meng, Z, Liu, Y, Wang, J, Fan, H, Fang, H, Li, S, et al. Histone demethylase KDM7A is required for stem cell maintenance and apoptosis inhibition in breast cancer. J Cell Physiol. (2020) 235:932–43. doi: 10.1002/jcp.29008
35. Bailey, TJ, E H, H, Zhang, L, Shah, R, Mathers, PH, and Jamrich, M. Regulation of development by Rx genes. Int J Dev Biol. (2004) 48:761–70. doi: 10.1387/ijdb.041878tb
36. Martini, D, Digregorio, M, Voto, IAP, Morabito, G, Degl’Innocenti, A, Giudetti, G, et al. Kdm7a expression is spatiotemporally regulated in developing Xenopus laevis embryos, and its overexpression influences late retinal development. Dev Dyn. (2024) 253:508–18. doi: 10.1002/dvdy.670
37. Schmidt, TM, and Kofuji, P. Novel insights into non-image forming visual processing in the retina. Cell Sci. (2008) 5:77–83.
38. Man, LLH, Storey, SS, Bertolesi, GE, and McFarlane, S. Cell-type expression and activation by light of neuropsins in the developing and mature Xenopus retina. Front Cell Neurosci. (2023) 17:1266945. doi: 10.3389/fncel.2023.1266945
39. Liu, Z, Thakar, A, Santoro, SW, and Pratt, KG. Presenilin regulates retinotectal synapse formation through EphB2 receptor processing. Dev Neurobiol. (2018) 78:1171–90. doi: 10.1002/dneu.22638
40. Flach, H, Krieg, J, Hoffmeister, M, Dietmann, P, Reusch, A, Wischmann, L, et al. Nosip functions during vertebrate eye and cranial cartilage development. Dev Dyn. (2018) 247:1070–82. doi: 10.1002/dvdy.24659
41. Kiem, LM, Dietmann, P, Linnemann, A, Schmeisser, MJ, and Kühl, SJ. The Nedd4 binding protein 3 is required for anterior neural development in Xenopus laevis. Dev Biol. (2017) 423:66–76. doi: 10.1016/j.ydbio.2017.01.009
42. Song, D-J, Bao, XL, Fan, B, and Li, GY. Mechanism of cone degeneration in retinitis pigmentosa. Cell Mol Neurobiol. (2023) 43:1037–48. doi: 10.1007/s10571-022-01243-2
43. Daiger, SP, Sullivan, LS, and Bowne, SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. (2013) 84:132–41. doi: 10.1111/cge.12203
44. Wang, AL, Knight, DK, Vu, TTT, and Mehta, MC. Retinitis pigmentosa: review of current treatment. Int Ophthalmol Clin. (2019) 59:263–80. doi: 10.1097/IIO.0000000000000256
45. Saito, M. Recent advances in the understanding of cilia mechanisms and their applications as therapeutic targets. Front Mol Biosci. (2023):10. doi: 10.3389/fmolb.2023.1232188
46. Xu, J, Zhao, C, and Kang, Y. The formation and renewal of photoreceptor outer segments. Cells. (2024) 13:1357. doi: 10.3390/cells13161357
47. Parain, K, Lourdel, S, Donval, A, Chesneau, A, Borday, C, Bronchain, O, et al. CRISPR/Cas9-mediated models of retinitis pigmentosa reveal differential proliferative response of Müller cells between Xenopus laevis and Xenopus tropicalis. Cells. (2022) 11:807. doi: 10.3390/cells11050807
48. Bogéa, TH, Wen, RH, and Moritz, OL. Light induces ultrastructural changes in rod outer and inner segments, including autophagy, in a transgenic Xenopus laevis P23H rhodopsin model of retinitis pigmentosa. Invest Ophthalmol Vis Sci. (2015) 56:7947–55. doi: 10.1167/iovs.15-16799
49. Hollingsworth, TJ, and Gross, AK. The severe autosomal dominant retinitis pigmentosa rhodopsin mutant Ter349Glu mislocalizes and induces rapid rod cell death. J Biol Chem. (2013) 288:29047–55. doi: 10.1074/jbc.M113.495184
50. Lodowski, KH, Lee, R, Ropelewski, P, Nemet, I, Tian, G, and Imanishi, Y. Signals governing the trafficking and mistrafficking of a ciliary GPCR, rhodopsin. J Neurosci. (2013) 33:13621–38. doi: 10.1523/JNEUROSCI.1520-13.2013
51. Bocchero, U, Tam, BM, Chiu, CN, Torre, V, and Moritz, OL. Electrophysiological changes during early steps of retinitis pigmentosa. Invest Ophthalmol Vis Sci. (2019) 60:933–43. doi: 10.1167/iovs.18-25347
52. Newton, F, and Megaw, R. Mechanisms of photoreceptor death in retinitis pigmentosa. Gene. (2020) 11:1120. doi: 10.3390/genes11101120
53. Wen, RH, Stanar, P, Tam, B, and Moritz, OL. Autophagy in Xenopus laevis rod photoreceptors is independently regulated by phototransduction and misfolded RHO(P23H). Autophagy. (2019) 15:1970–89. doi: 10.1080/15548627.2019.1596487
54. Tam, BM, Noorwez, SM, Kaushal, S, Kono, M, and Moritz, OL. Photoactivation-induced instability of rhodopsin mutants T4K and T17M in rod outer segments underlies retinal degeneration in X. laevis transgenic models of retinitis pigmentosa. J Neurosci. (2014) 34:13336–48. doi: 10.1523/JNEUROSCI.1655-14.2014
55. Vent-Schmidt, RYJ, Wen, RH, Zong, Z, Chiu, CN, Tam, BM, May, CG, et al. Opposing effects of valproic acid treatment mediated by histone deacetylase inhibitor activity in four Transgenic X. laevis models of retinitis pigmentosa. J Neurosci. (2017) 37:1039–54. doi: 10.1523/JNEUROSCI.1647-16.2016
56. Sammons, JD, and Gross, AK. Biochemical analysis of a rhodopsin photoactivatable GFP fusion as a model of G-protein coupled receptor transport. Vis Res. (2013) 93:43–8. doi: 10.1016/j.visres.2013.10.008
57. Bocquet, B. TBC1D32 variants disrupt retinal ciliogenesis and cause retinitis pigmentosa. JCI. Insight. (2023) 8:e169426. doi: 10.1172/jci.insight.169426
58. Levine, TP. Structural bioinformatics predicts that the retinitis Pigmentosa-28 protein of unknown function FAM161A is a homologue of the microtubule nucleation factor Tpx2. F1000Res. (2020) 9:1052. doi: 10.12688/f1000research.25870.1
59. Van de Sompele, S. Multi-omics approach dissects cis-regulatory mechanisms underlying North Carolina macular dystrophy, a retinal enhanceropathy. Am J Hum Genet. (2022) 109:2029–48. doi: 10.1016/j.ajhg.2022.09.013
60. Cipriani, V, Silva, RS, Arno, G, Pontikos, N, Kalhoro, A, Valeina, S, et al. Duplication events downstream of IRX1 cause North Carolina macular dystrophy at the MCDR3 locus. Sci Rep. (2017) 7:7512. doi: 10.1038/s41598-017-06387-6
61. Small, KW, DeLuca, AP, Whitmore, SS, Rosenberg, T, Silva-Garcia, R, Udar, N, et al. North Carolina macular dystrophy is caused by dysregulation of the retinal transcription factor PRDM13. Ophthalmology. (2016) 123:9–18. doi: 10.1016/j.ophtha.2015.10.006
62. Agbaga, MP, Tam, BM, Wong, JS, Yang, LL, Anderson, RE, and Moritz, OL. Mutant ELOVL4 that causes autosomal dominant stargardt-3 macular dystrophy is misrouted to rod outer segment disks. Invest Ophthalmol Vis Sci. (2014) 55:3669–80. doi: 10.1167/iovs.13-13099
63. Permanyer, J, Navarro, R, Friedman, J, Pomares, E, Castro-Navarro, J, Marfany, G, et al. Autosomal recessive retinitis pigmentosa with early macular affectation caused by premature truncation in PROM1. Invest Ophthalmol Vis Sci. (2010) 51:2656–63. doi: 10.1167/iovs.09-4857
64. Han, Z, Anderson, DW, and Papermaster, DS. Prominin-1 localizes to the open rims of outer segment lamellae in Xenopus laevis rod and cone photoreceptors. Invest Ophthalmol Vis Sci. (2012) 53:361–73. doi: 10.1167/iovs.11-8635
65. Abd El-Aziz, MM. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. (2008) 40:1285–7. doi: 10.1038/ng.241
66. Henderson, RH. Biallelic mutation of protocadherin-21 (PCDH21) causes retinal degeneration in humans. Mol Vis. (2010) 16:46–52.
67. Yang, LL. Characterization of protocadherin-21 in photoreceptor disk synthesis. Vancouver: University of British Columbia Library (2012).
68. Williams, DS, Linberg, KA, Vaughan, DK, Fariss, RN, and Fisher, SK. Disruption of microfilament organization and deregulation of disk membrane morphogenesis by cytochalasin D in rod and cone photoreceptors. J Comp Neurol. (1988) 272:161–76.
69. Manes, G, Cheguru, P, Majumder, A, Bocquet, B, Sénéchal, A, Artemyev, NO, et al. A truncated form of rod photoreceptor PDE6 β-subunit causes autosomal dominant congenital stationary night blindness by interfering with the inhibitory activity of the γ-subunit. PLoS One. (2014) 9:e95768. doi: 10.1371/journal.pone.0095768
70. Szabo, V, Kreienkamp, HJ, Rosenberg, T, and Gal, A. P.Gln200Glu, a putative constitutively active mutant of rod α-transducin (GNAT1) in autosomal dominant congenital stationary night blindness. Hum Mutat. (2007) 28:741–2. doi: 10.1002/humu.9499
71. Liu, C, Sherpa, T, and Varnum, MD. Disease-associated mutations in CNGB3 promote cytotoxicity in photoreceptor-derived cells. Mol Vis. (2013) 19:1268–81. doi: 10.1167/13.9.1268
72. Soliman, SE. Genetics and molecular diagnostics in retinoblastoma—an update. The Asia-Pacific. J Ophthalmol. (2017) 6:197–207. doi: 10.22608/APO.201711
73. Naert, T, Colpaert, R, van Nieuwenhuysen, T, Dimitrakopoulou, D, Leoen, J, Haustraete, J, et al. CRISPR/Cas9 mediated knockout of rb1 and rbl1 leads to rapid and penetrant retinoblastoma development in Xenopus tropicalis. Sci Rep. (2016) 6:35264. doi: 10.1038/srep35264
74. Zhang, W, Zhang, X, Wang, H, Sharma, AK, Edwards, AO, and Hughes, BA. Characterization of the R162W Kir7.1 mutation associated with snowflake vitreoretinopathy. Am J Phys Cell Phys. (2013) 304:C440–9. doi: 10.1152/ajpcell.00363.2012
75. Gautam, P, Hamashima, K, Chen, Y, Zeng, Y, Makovoz, B, Parikh, BH, et al. Multi-species single-cell transcriptomic analysis of ocular compartment regulons. Nat Commun. (2021) 12:5675. doi: 10.1038/s41467-021-25968-8
76. Hamilton, PW. New insights into cornea-lens regeneration in Xenopus laevis: The role of Wnt/beta-catenin signaling and the regenerative capacity of the limbal region. Doctoral dissertation, University of Illinois at Urbana-Champaign (2016).
77. Sonam, S. Cellular and molecular characterization of the corneal epithelium in Xenopus frogs. Doctoral dissertation, University of Illinois at Urbana-Champaign (2021).
78. Perry, KJ, Thomas, AG, and Henry, JJ. Expression of pluripotency factors in larval epithelia of the frog Xenopus: evidence for the presence of cornea epithelial stem cells. Dev Biol. (2013) 374:281–94. doi: 10.1016/j.ydbio.2012.12.005
79. Vilas, GL, Loganathan, SK, Liu, J, Riau, AK, Young, JD, Mehta, JS, et al. Transmembrane water-flux through SLC4A11: a route defective in genetic corneal diseases. Hum Mol Genet. (2013) 22:4579–90. doi: 10.1093/hmg/ddt307
80. Myers, EJ, Marshall, A, Jennings, ML, and Parker, MD. Mouse Slc4a11 expressed in Xenopus oocytes is an ideally selective H+/OH- conductance pathway that is stimulated by rises in intracellular and extracellular pH. Am J Phys Cell Phys. (2016) 311:C945–c959. doi: 10.1152/ajpcell.00259.2016
81. Quade, BN, Marshall, A, and Parker, MD. pH dependence of the Slc4a11-mediated H(+) conductance is influenced by intracellular lysine residues and modified by disease-linked mutations. Am J Phys Cell Phys. (2020) 319:C359–c370. doi: 10.1152/ajpcell.00128.2020
82. Loganathan, SK, Schneider, HP, Morgan, PE, Deitmer, JW, and Casey, JR. Functional assessment of SLC4A11, an integral membrane protein mutated in corneal dystrophies. Am J Phys Cell Phys. (2016) 311:C735–c748. doi: 10.1152/ajpcell.00078.2016
83. Adil, MT, and Henry, JJ. Understanding cornea epithelial stem cells and stem cell deficiency: lessons learned using vertebrate model systems. Genesis. (2021) 59:e23411. doi: 10.1002/dvg.23411
84. Zhang, BN, Liu, Y, Yang, Q, Leung, PY, Wang, C, Wong, TCB, et al. rad21 is involved in corneal stroma development by regulating neural crest migration. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21207807
85. Adil, MT. Understanding cornea stem cells during homeostasis and wound healing in Xenopus. US: University of Illinois at Urbana-Champaign (2021) 21:7807.
86. Adil, MT, Simons, CM, Sonam, S, and Henry, JJ. Understanding cornea homeostasis and wound healing using a novel model of stem cell deficiency in Xenopus. Exp Eye Res. (2019) 187:107767. doi: 10.1016/j.exer.2019.107767
87. Formisano, N, van der Putten, C, Grant, R, Sahin, G, Truckenmüller, RK, Bouten, CVC, et al. Mechanical properties of bioengineered corneal stroma. Adv Healthc Mater. (2021) 10:e2100972. doi: 10.1002/adhm.202100972
88. Zhang, BN, Wong, TCB, Yip, YWY, Liu, Z, Wang, C, Wong, JSC, et al. A sclerocornea-associated RAD21 variant induces corneal stroma disorganization. Exp Eye Res. (2019) 185:107687. doi: 10.1016/j.exer.2019.06.001
89. Sehu, KW, and Lee, WR. Ophthalmic pathology: An illustrated guide for clinicians John Wiley & Sons (2012).
90. Klintworth, GK. Genetic disorders of the cornea. Garner and Klintworth’s Pathobiol Ocular Dis. (2008):683–740. doi: 10.3109/9781420020977-37
91. Liu, H, Qi, B, Liu, G, Duan, H, Li, Z, Shi, Z, et al. RAD21 deficiency drives corneal to scleral differentiation fate switching via upregulating WNT9B. iScience. (2024) 27:109875. doi: 10.1016/j.isci.2024.109875
93. Arce-Gonzalez, R, Chacon-Camacho, OF, Navas-Perez, A, Gonzalez-Gonzalez, MC, Martinez-Aguilar, A, and Zenteno, JC. Novel CHRDL1 mutation causing X-linked megalocornea in a family with mild anterior segment manifestations in carrier females. Ophthalmic Genet. (2022) 43:224–9. doi: 10.1080/13816810.2021.2002917
94. Pfirrmann, T, Emmerich, D, Ruokonen, P, Quandt, D, Buchen, R, Fischer-Zirnsak, B, et al. Molecular mechanism of CHRDL1-mediated X-linked megalocornea in humans and in Xenopus model. Hum Mol Genet. (2015) 24:3119–32. doi: 10.1093/hmg/ddv063
95. Brahma, SK, and Mcdevitt, DS. Ontogeny and localization of the lens crystallins in Xenopus laevis lens regeneration. Development. (1974) 32:783–94.
96. Viet, J, Reboutier, D, Hardy, S, Lachke, SA, Paillard, L, and Gautier-Courteille, C. Modeling ocular lens disease in Xenopus. Dev Dyn. (2020) 249:610–21. doi: 10.1002/dvdy.147
97. Huynh, MH, Zhu, SJ, Kollara, A, Brown, T, Winklbauer, R, and Ringuette, M. Knockdown of SPARC leads to decreased cell-cell adhesion and lens cataracts during post-gastrula development in Xenopus laevis. Dev Genes Evol. (2011) 220:315–27. doi: 10.1007/s00427-010-0349-x
98. Rothe, M, Kanwal, N, Dietmann, P, Seigfried, FA, Hempel, A, Schütz, D, et al. An Epha4/Sipa1l3/Wnt pathway regulates eye development and lens maturation. Development. (2017) 144:321–33. doi: 10.1242/dev.147462
99. Siddam, AD, Gautier-Courteille, C, Perez-Campos, L, Anand, D, Kakrana, A, Dang, CA, et al. The RNA-binding protein Celf1 post-transcriptionally regulates p27Kip1 and Dnase2b to control fiber cell nuclear degradation in lens development. PLoS Genet. (2018) 14:e1007278. doi: 10.1371/journal.pgen.1007278
100. Tong, JJ, Khan, U, Haddad, BG, Minogue, PJ, Beyer, EC, Berthoud, VM, et al. Molecular mechanisms underlying enhanced hemichannel function of a cataract-associated Cx50 mutant. Biophys J. (2021) 120:5644–56. doi: 10.1016/j.bpj.2021.11.004
101. Barnum, CE, al Saai, S, Patel, SD, Cheng, C, Anand, D, Xu, X, et al. The Tudor-domain protein TDRD7, mutated in congenital cataract, controls the heat shock protein HSPB1 (HSP27) and lens fiber cell morphology. Hum Mol Genet. (2020) 29:2076–97. doi: 10.1093/hmg/ddaa096
102. Yuan, C, Han, T, Su, P, Liu, M, Zhou, X, Zhang, D, et al. A novel MIP mutation in a Chinese family with congenital cataract. Ophthalmic Genet. (2018) 39:473–6. doi: 10.1080/13816810.2018.1484930
103. Stäubli, A, Capatina, N, Fuhrer, Y, Munier, FL, Labs, S, Schorderet, DF, et al. Abnormal creatine transport of mutations in monocarboxylate transporter 12 (MCT12) found in patients with age-related cataract can be partially rescued by exogenous chaperone CD147. Hum Mol Genet. (2017) 26:4203–14. doi: 10.1093/hmg/ddx310
104. Senthil Kumar, G, Kyle, JW, Minogue, PJ, Dinesh Kumar, K, Vasantha, K, Berthoud, VM, et al. An MIP/AQP0 mutation with impaired trafficking and function underlies an autosomal dominant congenital lamellar cataract. Exp Eye Res. (2013) 110:136–41. doi: 10.1016/j.exer.2012.10.010
105. Lakk, M, Hoffmann, GF, Gorusupudi, A, Enyong, E, Lin, A, Bernstein, PS, et al. Membrane cholesterol regulates TRPV4 function, cytoskeletal expression, and the cellular response to tension. J Lipid Res. (2021) 62:100145. doi: 10.1016/j.jlr.2021.100145
106. Treimer, E, Niedermayer, K, Schumann, S, Zenker, M, Schmeisser, MJ, and Kühl, SJ. Galloway-Mowat syndrome: new insights from bioinformatics and expression during Xenopus embryogenesis. Gene Expr Patterns. (2021) 42:119215. doi: 10.1016/j.gep.2021.119215
107. Treimer, E, Kalayci, T, Schumann, S, Suer, I, Greco, S, Schanze, D, et al. Functional characterization of a novel TP53RK mutation identified in a family with Galloway-Mowat syndrome. Hum Mutat. (2022) 43:1866–71. doi: 10.1002/humu.24472
108. Reil, M, and Dabauvalle, MC. Essential roles of LEM-domain protein MAN1 during organogenesis in Xenopus laevis and overlapping functions of emerin. Eur J Cell Biol. (2013) 92:280–94. doi: 10.1016/j.ejcb.2013.10.008
109. Manojlovic, Z, Earwood, R, Kato, A, Stefanovic, B, and Kato, Y. RFX7 is required for the formation of cilia in the neural tube. Mech Dev. (2014) 132:28–37. doi: 10.1016/j.mod.2014.02.001
110. Ott, T, Kaufmann, L, Granzow, M, Hinderhofer, K, Bartram, CR, Theiß, S, et al. The frog Xenopus as a model to study Joubert syndrome: the case of a human patient with compound heterozygous variants in PIBF1. Front Physiol. (2019) 10:134. doi: 10.3389/fphys.2019.00134
111. Tseng, AS. Seeing the future: using Xenopus to understand eye regeneration. Genesis. (2017) 55:e23003. doi: 10.1002/dvg.23003
112. Graw, J. Mouse models for microphthalmia, anophthalmia and cataracts. Hum Genet. (2019) 138:1007–18. doi: 10.1007/s00439-019-01995-w
Keywords: Xenopus laevis, eye, development model, disease model, ophthalmology
Citation: Li Q, Feng Y and Zhu X (2025) Systematic review: Xenopus laevis as a model for ophthalmic development and disease research. Front. Med. 12:1545958. doi: 10.3389/fmed.2025.1545958
Edited by:
Rong Li, Regeneron Pharmaceuticals, Inc., United StatesReviewed by:
Yuntian Xue, Align Technology, Inc., United StatesChao Ma, Tongren Polytechnic University, China
Copyright © 2025 Li, Feng and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun Feng, c3VwZXJqdW5lQHNpbmEuY29t; Xuechen Zhu, emh1eGNAYmptdS5lZHUuY24=