 Minjie Pan
Minjie Pan Xiaojing Zhou
Xiaojing Zhou- 1Department of Respiratory and Critical Care Medicine, Zhoushan Hospital, Zhoushan, China
- 2Department of Neurology, Zhoushan Hospital, Zhoushan, China
Chronic obstructive pulmonary disease (COPD) is a chronic respiratory disease characterized by irreversible airway remodeling and is a global burden on the healthcare system. The World Health Organization predicts it will be the third leading cause of death by 2030. The causes of airway remodeling in COPD are complex. Several elements, such as the lung parenchyma and interstitium, as well as endothelium, mesenchymal cells, and a range of bioactive chemicals, work together to either encourage or impede the alteration of the airway’s structure during the remodeling process. Airway remodeling is an important factor in the irreversible limitation of ventilatory function. To reduce airway remodeling, significant efforts are being directed to find effective therapeutic ways that inhibit airway remodeling. In China, many patients use traditional Chinese medicine (TCM). Some TCM can improve the symptoms and lung function of COPD patients. Here, we describe the molecular mechanisms and key cellular players of airway remodeling in COPD patients and review the Chinese herbal medicines that may effectively inhibit airway remodeling.
1 Introduction
The global recognition of COPD is growing due to its widespread nature, and it poses a risk to one’s life and well-being. A study shows the prevalence of COPD in the United States from 2007 to 2012 was 13.1–14.3% (1). Also, in China, COPD causes a significant financial and medical burden. COPD is typified by an irreversible reduction in lung function and is linked to particulate matter inhalation, as well as genetic, developmental, and social variables (2, 3). There are various mechanisms involved in the pathophysiologic alterations in COPD, such as the development of emphysema and minor airway lesions, which cause an irreversible loss in lung function. The hallmark of COPD is airway remodeling. Under normal physiological conditions, tissue remodeling also occurs, mediating a mild response to inflammation while fostering tissue growth and mending damage. On the other hand, excessive remodeling in pathological situations may result in modifications to pertinent structures, which could impact organ function and complicate follow-up treatment. Structural alterations define airway remodeling and result from dysregulated tissue formation and breakdown. Causes of structural changes include disorders of angiogenic factors, deposition and fibrosis of subepithelial matrix proteins, proliferation and hypertrophy of smooth muscle cells in the airways, epithelial abnormalities, epithelial–mesenchymal transition (EMT), which result in thickening of the airway wall and airflow restriction (4–6). Various cytokines, chemokines, growth factors, and other bioactive substances participate in this process (6–8). Fibrosis in COPD occurs mainly in the airway wall and predominantly in the small airways (6, 9). To better understand airway remodeling in patients with COPD, this article briefly reviews the pathological mechanism of airway remodeling, describing the relationship between oxidative stress and multiple pathophysiologic changes during airway remodeling. TCM has a long history in China and plays a role in the treatment of COPD. In this review, we discuss several important components to highlight the role of TCM in airway remodeling.
2 Definition and evaluation of airway remodeling
Airway remodeling refers to both quantitative and qualitative modifications of the airway wall. Epithelial damage, goblet cell hyperplasia, enlargement of submucosal mucous glands, and mucus hypersecretion can be observed in the airway lumen. Fibroblast activation and proliferation, extracellular matrix (ECM) modifications, angiogenesis and vascular remodeling, and airway smooth muscle (ASM) proliferation and hypertrophy are among the alterations in the subepithelial structure of the airways (9, 10). Airway remodeling can be assessed by non-invasive tools, including high-resolution computed tomography (HRCT), positron emission tomography (PET) scan, single-photon emission computed tomography (SPECT), optical imaging with fluorescence and bioluminescence (11). Several indicators like airway wall area percentage, lumen diameter, lumen thickness, and airway lumen area, can help to evaluate airway remodeling (12–14). Airway MRI has low resolution, however, by inhalation of 129Xe, airway remodeling can also be sensitively measured (15, 16). Optical coherence tomography (OCT) is a non-invasive, high-resolution optical imaging technique. When OCT and bronchoscopy are used together, the airway wall can be seen in detail, and peripheral airway remodeling can be positively identified (17–19).
In recent years, an overlap phenomenon has been observed in asthma and COPD, where symptoms are similar to those of COPD but respond significantly to bronchodilators, called asthma-COPD overlap (ACO). A 10–40% of patients with COPD and 15–35% of patients with asthma may have an ACO (20). A study using low-dose CT analysis to assess proximal airway remodeling, emphysema, and air trapping in patients with ACO found that lung function indices (FEV1, FEV1/FVC, etc.) were significantly lower in patients with ACO than in patients with non-severe asthma, suggesting that the degree of airflow obstruction was more severe in patients with ACO. The degree of emphysema was more severe in patients with ACO than that in patients with non-severe asthma, while was similar to that in patients with mild to moderate COPD (21–23). Using multi-row spiral CT (MDCT) to assess airway wall thickness and airway lumen area in patients with ACO and asthma, researchers found that airway wall thickness was significantly greater in the ACO group compared to the asthma group and healthy controls. Overall, the degree of lung function impairment and airway remodeling in patients with ACO was intermediate between those with asthma and COPD (24).
3 Physiological factors affecting airway remodeling
Chronic obstructive pulmonary disease patients are predominantly in the elderly population. The prevalence of COPD increases with age, and senescence is an independent risk factor for COPD. Structural cells such as airway epithelial cells, ASM cells, and fibroblasts undergo senescence with age, which is characterized by decreased cell proliferation, impaired function, and secretion of senescence-associated secretory factors (SASPs). SASPs can contribute to inflammatory responses, ECM deposition, and proliferation of ASM. Aging also leads to decreased antioxidant capacity and increased oxidative stress, immune dysfunction, and increased inflammatory response, which leads to thickening and reduced elasticity of the airway wall (25). Aging and airway remodeling reinforce each other in a vicious cycle.
Usually, the human respiratory tract is inhabited by a complex mixture of microorganisms. The composition of the lung microbiota is dynamic and plays a crucial role in regulating innate and adaptive lung immunity. It has been found that in normal lung tissue, common bacteria include Prevotella spp, Streptococcus spp, Veronica spp, Aspergillus spp, Proteus spp, Neisseria spp, Clostridium spp, Haemophilus influenzae spp, Rochesteria spp, Actinomyces spp, Streptococcus pepticus spp, Corynebacterium spp, and Pseudomonas spp. Common fungi include Candida spp, Saccharomyces cerevisiae, Penicillium spp, and Dendrobatrachium spp. And viruses such as Cycloviridae, Iridoviridae, Herpesviridae, and Poxviridae are found (26). Compared to other parts of the body, like the gut, the microbial density of normal lung tissue is low, and its composition changes dynamically with time, the environment, and the host’s health status. In patients with COPD, dysbiosis of the lung flora can lead to colonization by pathogens such as Staphylococcus aureus and Gram-negative bacteria. These pathogens can trigger an inflammatory response, leading to the production of harmful metabolites such as lipopolysaccharides (LPS) and short-chain fatty acids (SCFAs), resulting in overactivation of pro-inflammatory Th17 cells and dysfunction of regulatory T-cells (Tregs), leading to lung tissue damage and dysfunction. Lung dysbiosis can also affect the function of immune cells, such as neutrophils and macrophages, as well as the levels of immunomodulatory factors, resulting in a dysregulated immune response that promotes inflammation and tissue damage (27).
4 Oxidative stress promotes airway remodeling
In COPD, Oxidative stress has been found to have a crucial role in the development of airway remodeling (28–30). Mitochondrial dysfunction leads to decreased oxidative phosphorylation, increased Reactive Oxygen Species (ROS) production, and changes in cell metabolism, which are increasingly recognized as essential events in the pathogenesis of a series of lung diseases, including COPD (31, 32).
Mitochondria are the center of oxidative stress in cells and the major ROS source. Mitophagy is a process by which cells selectively degrade damaged mitochondria through an autophagy mechanism, to maintain mitochondrial and intracellular homeostasis (33). The PINK1/Parkin signaling pathway predominantly regulates mitochondrial autophagy. Under physiological conditions, PINK1 is translocated to the outer mitochondrial membrane, where it remains stable. However, when mitochondrial damage occurs, the loss of membrane potential results in the accumulation of PINK1 on the surface of the mitochondria. This accumulation facilitates the recruitment of the Parkin protein, which tags the damaged mitochondria for degradation. Consequently, this process initiates the formation of autophagosomes that ultimately lead to the lysosomal degradation of the impaired mitochondria (34). Mitophagy plays a dual role in the development of lung diseases (35). In a physiological state, mitophagy keeps the population of mitochondria and is crucial in protecting cells against disease (36). Mitophagy has been suggested to play a protective role in the development of COPD in some studies (37–39). The pathological state of mitophagy aggravates the inflammatory reaction. After exposure to cigarette-smoke (CS), mitochondrial autophagy in airway epithelial cells is increased to clear damaged mitochondria, and excessive autophagy contributes to the progression of COPD (40). Mitochondrial protein Nix is a selective autophagy receptor. When cigarette extract stimulated airway epithelial cells, the expression of Nix protein increased. Nix overexpression can enhance mitophagy and aggravate the damage to mitochondria and cells (41). Mitophagy can also promote pulmonary fibrosis, which is related to airway remodeling (42). Cigarette-smoke extract (CSE) up-regulates mitophagy in ASM through ERK1/2 phosphorylation. The imbalance of ASM cell mitophagy induced by CS may be one of the mechanisms contributing to airway remodeling in COPD (43, 44). However, some studies have shown that mitochondrial autophagy is impaired in patients with COPD. Studies have demonstrated that the levels of Parkin are diminished in the lung tissue of COPD patients. Furthermore, silencing PINK1 and Parkin in bronchial epithelial cells results in a reduction of mitophagy and an increase in the production of ROS (37).
Reactive Oxygen Species induce lipid peroxidation and produce toxic products such as malondialdehyde, which can damage proteins by causing protein cross-links and stimulate lung inflammation, leading to alveolar wall destruction (45). CS can activate cells via toll-like receptor 4 (TLR4) while altering the mitochondrial activity of airway epithelium and promoting the formation of free ROS (46). Endoplasmic reticulum stress (ERS) refers to the impairment of the normal function of the endoplasmic reticulum (ER), accumulating unfolded or misfolded proteins and disrupting Ca2+ homeostasis. In COPD patients, ERS promotes oxidative stress and increases ROS production by inhibiting the expression of antioxidant enzymes, promoting the expression of ROS-generating enzymes, and raising the expression of oxidoreductase cytochrome P450 (47).
Oxidative stress causes increased cell proliferation, including ASM, in COPD patients (28, 48, 49). Hypertrophic hyperplasia of ASM is mainly seen in the peripheral remodeled airway (50, 51). When ASM malfunctions, it releases inflammatory mediators. ASM cells have multiple functions, including contraction, proliferation, and migration, termed phenotypic plasticity. This regulation of phenotypic transitions is accomplished through complex coordinated interactions between external stimuli (52). It has been demonstrated that oxidative stress and peroxide encourage ASM to contract and proliferate, encouraging airway remodeling in COPD (53, 54). In addition, lipopolysaccharide, ECM, and growth factors also contribute to the increased ASM proliferative phenotype (55–57). Potassium-calcium channels are present in ASM cells. KCa3.1 and KCa1.1 are two channels with different conductance. KCa3.1 plays a critical role when activated by calcium ions, maintaining cell viability through the exchange of potassium and calcium ions. Freshly isolated human ASM cells express large conductance KCa1.1 channels (58). A loss of KCa1.1 channels and gain in KCa3.1 channels occurs when ASM cells switch from a contractile to a proliferative phenotype. This was observed in platelet-derived growth factor-induced activation of asthmatic human bronchial smooth muscle cells (58, 59). KCa3.1 Blocking may at least partially prevent ASM reconfiguration (59).
Hypertrophic and hyperplastic ASM cells exhibit metabolic changes, including increased glutamine metabolism, impaired fatty acid oxidation (FAO), and reduced ATP levels. Glutamine serves as a precursor in the biosynthesis of glutathione. An elevation in glutamine levels may facilitate the production of the glutathione, an important intracellular antioxidant, thereby augmenting the antioxidant capacity of cells and diminishing the concentration of ROS (48).
5 Airway inflammation and immune response accelerate airway remodeling
T lymphocytes, macrophages and neutrophils are the primary inflammatory cells that infiltrate the airways of COPD patients (9, 60). The neutrophil is increased in sputum and bronchoalveolar lavage fluid (BALF) from patients with COPD, and its recruitment to the airways and parenchyma requires the initial adhesion of E-selectin to endothelial cells. The expression of E-selectin was up-regulated in airway endothelial cells of COPD patients (61). The neutrophil secretes serine endopeptidase, including neutrophil elastase, cathepsin, and protease, as well as MMP-8 and MMP-9, which may contribute to alveolar destruction (62). T lymphocytes and macrophages predominated in the airway wall of COPD patients, with CD8+ T lymphocytes dominating the T lymphocyte phenotype. This is not the same as the asthma patients’ airway CD4+ T lymphocyte infiltration (63).
Patients with COPD had increased T lymphocytes in their lung parenchyma, peripheral blood, and central airways. The rise in CD8+ T cells was larger than in CD4+ T cells (64). Mice lacking B or T cells failed to develop airway remodeling, suggesting the importance of lymphocytes in airway construction (65). Patients with COPD have higher expression levels of CXCR3, a receptor for the chemokines CXCL9/10/11, in their lung T cells. The accumulation of CD4+ T and CD8+ T cells may be facilitated by increased production of CXCL10 in bronchiolar epithelial cells (66). Patients with COPD also have more lymphocytes in their lungs. B-lymphocytes exist as lymphoid follicles in the lung parenchyma and the surrounding airway (64). Decreased expression of CXCL13, a B-cell chemokine, attenuated CS-induced BALF inflammatory cells and partially protected the alveolar wall from destruction, but did not affect the development of airway remodeling (67).
Macrophages appear to play a key role in COPD. COPD patients show higher levels of macrophages and macrophage chemoattractant in their lung parenchyma and airways (68). Based on surface molecules and distribution, macrophages in the lung can be classified as either interstitial macrophages (IM) or alveolar macrophages (AM) (69). AM is found in the lungs’ air spaces, while IM is found in the spaces between the alveoli and blood vessels. While both AM and IM are macrophages with increased phagocytic capacities, AM is regarded to have a more robust capacity (70, 71). When AM encounters IFN-inducible protein-10 and IFN-γ-induced cytokines, they can release MMP-12, accelerating tissue deterioration and elastic degradation (72). Laminin A1 is strongly expressed on AM and is induced by TGF-β, has been reported to be overexpressed in idiopathic pulmonary fibrosis patients (73). AM from smokers with COPD were classified into M1 and M2 phenotypes. Type 2 immune response, also called adaptive immunity, is mediated by M2. In patients with COPD, macrophages tended to differentiate into M2 cells (74). Pathological remodeling of the pulmonary airway can result from an overabundance of type 2 immune responses (73, 75). The development of COPD is closely associated with the imbalance of M1 and M2 in the airways (69). Peroxisome proliferation-activated receptor-γ (PPAR-γ), a nuclear transcription factor essential to AM polarization, is a member of the nuclear hormone receptor superfamily (76). In COPD patients, there is dysregulation of PPAR-γ expression. In COPD mice, stimulation of PPAR-γ reduces airway remodeling and enhances AM’s polarization stability (77). As a result, maintaining the normal structure of the airways requires controlling the balance between M1 and M2 (Figure 2). In addition to the imbalance of macrophages, the metabolic characteristics of AMs change significantly. CS causes Ams and neutrophils to generate a large amount of ROS, leading to mitochondrial dysfunction and increased oxidative stress. And in COPD patients who smoke, this phenomenon persists even after they quit smoke (78). Abnormal iron metabolism in AMs, including reducing iron-chelating proteins, also leads to iron accumulation and increased ROS production (79).
6 Oxidative stress can cause airway remodeling by promoting EMT
An essential part of the airway’s innate immune system is the epithelium. It is the fundamental barrier of the airway. The airway epithelium consists of four cell types: basal cell, ciliated cell, secretory cell, and intermediate cell (80). Several processes help epithelial repair when the healthy airway epithelium is harmed or when an inflammatory response occurs: 1. Migration of epithelial cells to the injury site. This process can be regulated by integrin-mediated activation of transforming growth factor-β (TGF-β) and regulation of the WNT/β-catenin signaling pathway (81, 82). 2. Matrix components are deposited at the site of injury, these include ECM glycoproteins, fibronectin, and vitronectin, as well as basement membrane components such as laminin and collagen IV (81, 83). 3. Mesenchymal stem cells differentiate into epithelial stem cells during the repair process and then rebuild various epithelial cells to ensure the integrity of the airway epithelium (84). The loss of integrity in the airway epithelium results in exacerbated tissue permeability and immunological activation. Airflow blockage and microbiological overgrowth are caused by excessive mucus secretion and poor mucociliary clearance (85).
The pathologic process of epithelial cells changing into mesenchymal cells by phenotypic transformation is known as EMT, and it is a commonly acknowledged mechanism for modifying airway walls. During the EMT process, epithelial cells lose polarity, acquire markers of mesenchymal cells, and can migrate (86, 87). EMT is commonly categorized into three types. Type I EMT is associated with embryogenesis and organ development. Type II EMT is linked to tissue repair and chronic inflammation-associated wound healing. Type III EMT is responsible for malignant epithelial cells acquiring a migratory phenotype (88, 89). Type II EMT primarily affects the small airways, mediating small airway fibrosis (89). CS induces EMT in bronchial epithelial cells in multiple ways, including producing TGF-β1, increasing oxidative stress, phosphorylating ERK1/2, and SMAD2/3 (90–92). Oxidative stress plays a key role in the pathogenesis of CS-induced EMT in the airway epithelium by promoting the expression of EMT-related transcription factors and promoting the degradation of extracellular matrix (47). ROS can affect TGF-β1-induced EMT in A549 cells (93, 94). Also, the Wnt/β-Catenin and NF-κB signaling pathways are involved in airway epithelial EMT in COPD patients (95–97) (Figure 1). Monocytes in the lung can stimulate EMT in lung epithelial cells through the Toll-like receptor 2/matrix metalloproteinases -9(TLR2/MMP-9) axis (98). Heparin-binding EGF-like growth factor (HB-EGF) is the ligand of the EGF super-family and is up-regulated in the sputum and lung tissue in COPD patients. It is thought to induce EMT in bronchial epithelial cells (99, 100). Additionally, EMT is believed to represent a novel source of fibroblasts, promoting ECM deposition by increasing fibroblast accumulation (101). Keratin 15 (KRT15) is a type of keratin that is closely related to the repair and regeneration of tissue cells after injury. KRT15 can inhibit EMT by promoting MMP-9 expression and protecting the alveolar structure, and is a potential target for the treatment of COPD (102, 103).
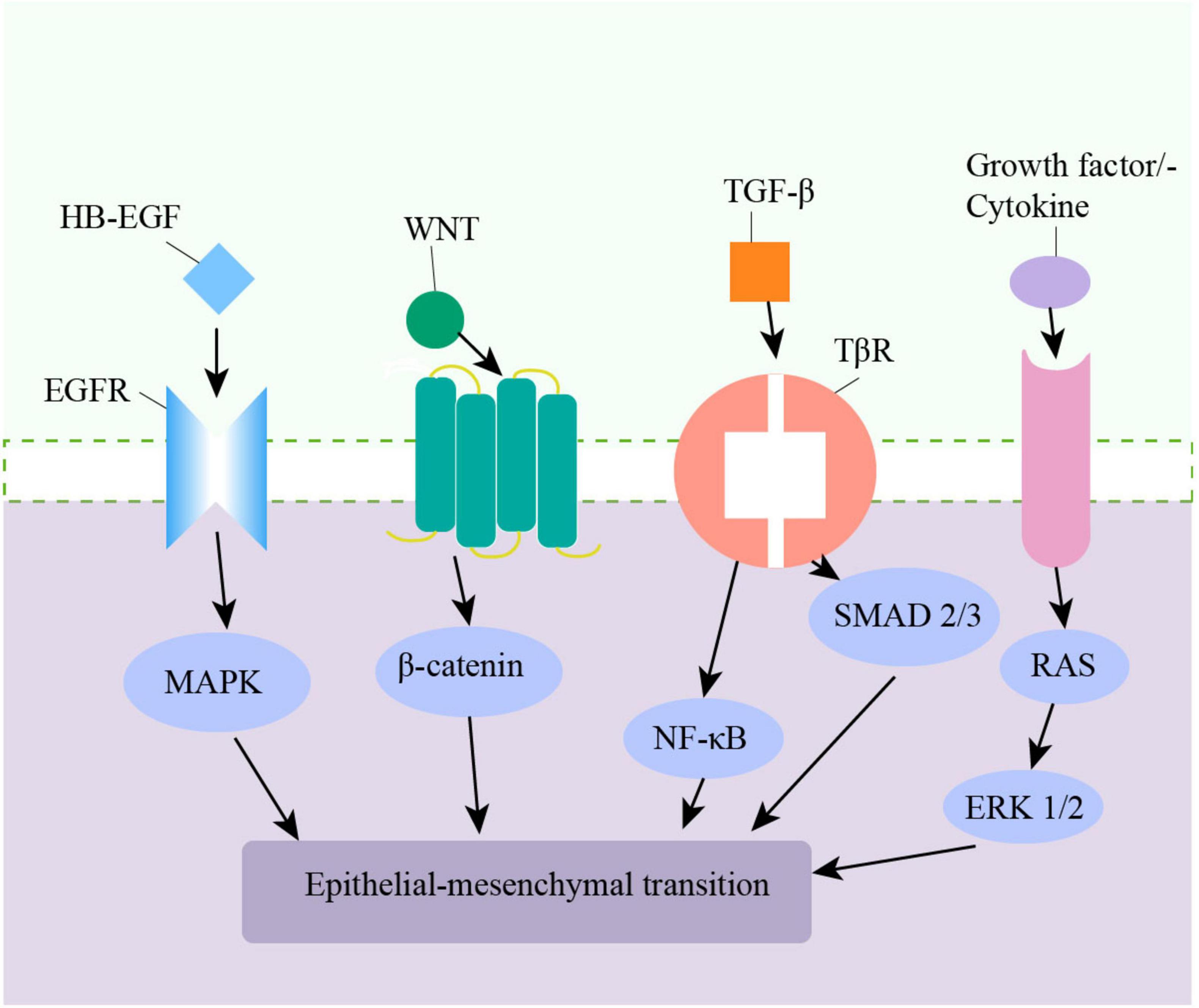
Figure 1. Multiple pathways in the airway lead to epithelial–mesenchymal transition (EMT).
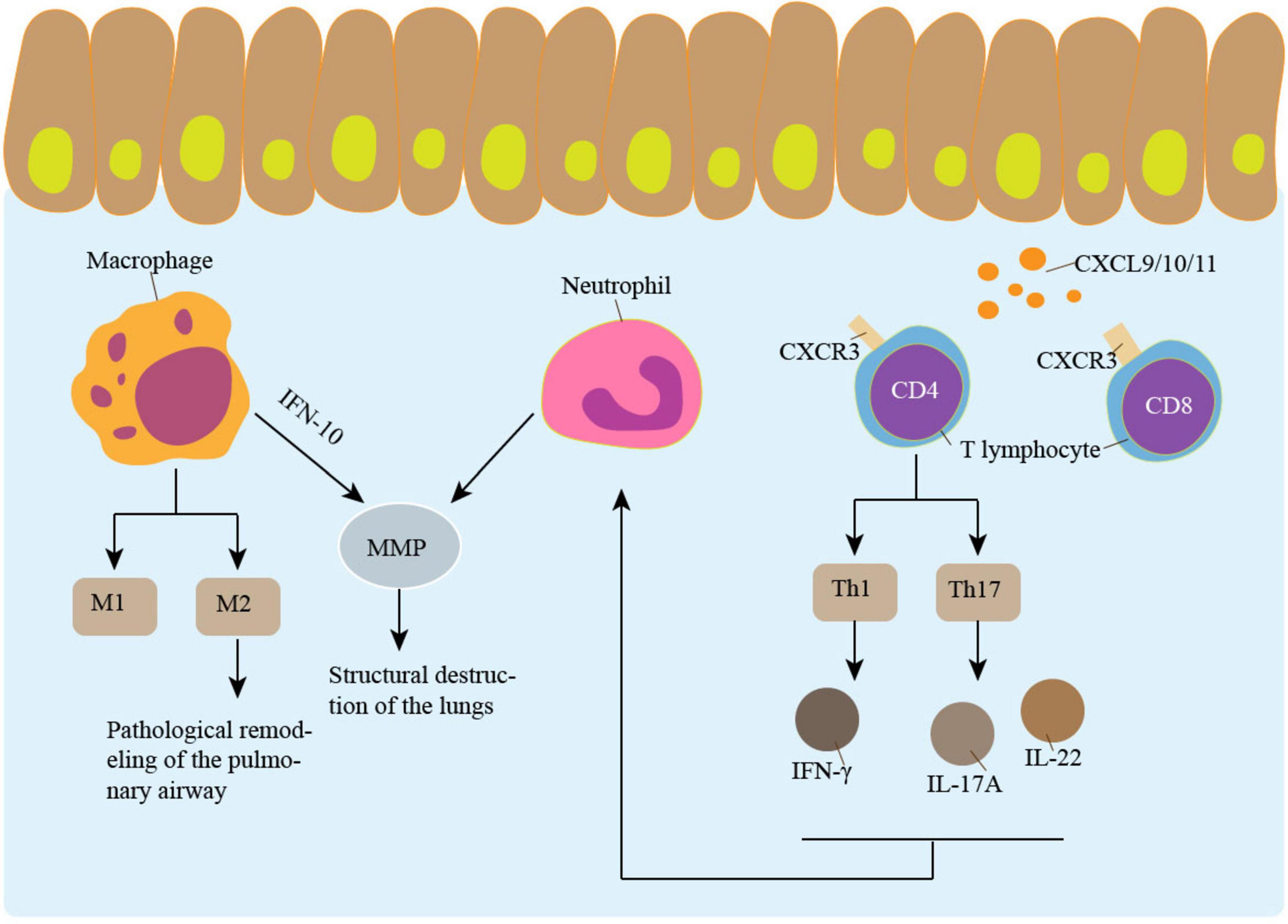
Figure 2. Inflammation promotes airway remodeling. Macrophages, neutrophil and lymphocyte are involved in airway remodeling. M2 type macrophages play an important role in airway remodeling. IL-10-induced MMP secretion by macrophages is involved in the pathogenesis of emphysema. The CXCL9/10/11-CXCR3 Axis recruits CD4 + T cells and CD8 + T cells. CD4 + T cells secrete a variety of cytokines that participate in airway remodeling and promote neutrophil aggregation. Neutrophil increases emphysema through MMP.
6.1 Airway remodeling is closely related to ECM abnormality
One of the consequences of EMT is ECM oversecretion. The ECM regulates tissue development and homeostasis (104). ECM shapes the thickness and elasticity of the airway wall. Abnormal buildup of ECM proteins may result in airway remodeling, for example, increased synthesis and breakdown of ECM (105). The most prevalent components in the ECM of COPD patients include proteoglycan (PGs), glycosaminoglycan (GAGs), collagen, and elastin. These components significantly impact lung tissue’s composition and function, and interact when tissue is injured (106, 107).
The lung’s three main ECM compartments are the alveolar interstitium, the foundation membrane, and the airway lamina propria. The main protein components of the basement membrane are collagen IV and laminin. Fibrillar collagen, elastin, fibronectin, glycosaminoglycans, and proteoglycans are the main components found in the lamina propria and interstitium. Fibroblasts and activated myofibroblasts are the primary ECM-producing cells in the lung. Endogenous proteases, of which matrix metalloproteinases (MMPs) are the predominant kind in the lung, control the destruction of ECM (108). It has been demonstrated that MMPs, such as MMP-1, -2, -7, -9, and -12, destroy a range of matrix elements, including collagen and elastin. Elastin is the main target of disintegration in the process of remodeling. The degradation of elastin is a major cause of emphysema, resulting in a loss of lung tissue elasticity. Collagen V and VI are involved in the formation of the reticular basement membrane, which serves to preserve the shape of the lungs, and their increased expression in the airways of COPD patients may result in airway wall thickening and airflow limitation. Furthermore, chondroitin sulfate demonstrated a considerable increase in BALF during COPD exacerbation, which could be linked to airway remodeling (109). Although the precise function of MMP-12 is uncertain, it is significant since in animal models, small airway remodeling can be inhibited by MMP-12 knockdown or MMP-9/-12 inhibitors (110–113). Oxidative stress can cause changes in ECM expression, and higher levels of oxidative stress may promote collagen buildup. In cardiac fibrosis, elevated levels of oxidative stress have been linked to increased amounts of collagen, and antioxidant therapy has been proven to lower collagen levels (114).
6.2 Transforming growth factor-β is essential for fibrosis
Transforming growth factor-β is a cytokine known to increase ECM production (115). TGF-β1 is a key component in fibrotic conditions and is necessary for developing myofibroblasts. It is produced by mast cells and granulocytes. Elevated ROS levels activate TGF-β1, which stimulates the fibrotic response (116). ROS can stimulate TGF-β1 secretion from airway epithelial cells, release the TGF-β1 receptor from the latency-associated peptide (LAP) complex by dissociation from LAP or activate matrix metalloproteinases (117, 118) TGF-β1 primarily activates cytosolic Smad protein via transmembrane receptor serine/threonine kinase. The activation of the TGF-β1-induced α-SMA promoter and the in vitro expression of α-SMA protein were markedly enhanced by SMAD3 overexpression (119). The expression of α-SMA, a hallmark of mature myofibroblasts, regulates fibroblasts and encourages contraction of the wound edge (120). Myofibroblast apoptosis is reduced in pathological situations, ECM formation persists, and breakdown is impeded, resulting in tissue sclerosis (107). Several studies have indicated that there are more α-SMA positive cells in the bronchi of COPD patients (121, 122). In circulation, the level of α-SMA was also significantly increased in patients with COPD (123). The TGF-β-independent pathway induced by IL-4 and IL-13 can initiate the transformation of resident fibroblasts into myofibroblasts. It has also been demonstrated that IL-4 stimulates the synthesis of collagen, which encourages airway remodeling even more (107).
7 Oxidative stress and necroptosis interact to promote airway remodeling
Cigarette-smoke induces numerous mechanisms of cell death, and the pattern of cell death dictates when inflammatory reactions start. Necroptosis involves greater inflammatory reactions and the release of damage-associated molecular patterns (DAMPs) than apoptosis (124). Necroptosis is a form of programd inflammatory cell death (39). It is characterized by organelle enlargement, plasma membrane disruption, and cytolysis (125). The route involved in necroptosis is mainly dependent on receptor-interacting protein kinase (RIPK) and Mixed lineage kinase domain-like protein (MLKL), which are also the pathway’s primary detecting proteins (126). RIPK3, RIPK1, and MLKL create a multiprotein complex called the necrosome (127). RIPK1 recruits and phosphorylates RIPK3 to form ripoptosome, but this is not absolute, and there is evidence of RIPK1-independent necroptosis. Then phosphorylates MLKL to form necrosomes (128).
Oxidative stress and necroptosis reinforce each other. On the one hand, ROS production is dependent on key proteins of necroptosis, and it has been observed that TNF-induced ROS is suppressed after lowering the level of RIPK1/RIP3 proteins (129), on the other hand, oxidative stress induces necroptosis when sufficient amounts of ROS are present in the cell (130). ROS can activate early growth response gene 1 (Egr-1), which further activates RIPK1 and RIPK3, and initiating the necroptotic pathway. Mitochondria-targeted antioxidants can inhibit the production of mitochondrial ROS and reduce necroptosis (131).
Necroptosis has been linked to immune cell infiltration and has been demonstrated to take place throughout the pathogenesis of COPD (132). Necroptosis mediates the generation of airway mucus and stimulates inflammatory responses at the cellular level (133, 134). Blocking the necroptosis pathway can decrease inflammatory cell infiltration into the airway and lower airway wall thickness (135–137). Necroptosis exacerbates the inflammatory reactions that CS causes in macrophages, and in bone marrow-derived macrophages (BMDM), inhibiting necroptosis reduces the transcriptional levels of inflammatory cytokines that CS induces. The transcription factor NF-κB, which controls the expression of inflammatory genes, is typically made up of a P50/P65 heterodimer, in which P65 regulates NF-κB’s transcriptional activation. Necroptosis controls P65’s phosphorylation state, which controls the transcription of inflammatory genes (138). Through kinase activity, RIPK1 influences cell viability and death. Its activation can result in a highly inflammatory lytic form of cell death or apoptosis dependent on RIPK1 kinase, and blocking RIPK1 kinase can lessen the chance of airway remodeling (139).
8 Exosomes regulate airway remodeling by delivering bioactive substances
Extracellular vesicles (EVs) can be produced by almost all types of cells and are released when cells are activated, damaged, or apoptotic. Due to the complexity of EV origin, vesicle size is the most widely used parameter for distinguishing EV types. EV can be divided into three types according to particle size, the smallest of which is called exosome (140). Exosomes mediate cell-to-cell communication by delivering a variety of substances, including proteins, DNA, and RNA, to targeted cells (141, 142). Exosomes are involved in the development of COPD and airway remodeling (143). Oxidative stress is closely related to the production and function of exosomes. Oxidative stress can affect exosome production and release through multiple pathways, including regulation of lysosomal function and extracellular glutathione levels. It has been suggested that low levels of oxidative stress can promote the release of exosomes, while high levels of oxidative stress can inhibit their release. Oxidative stress can also affect the levels of proteins, lipids, and nucleic acids in exosomes (144). EV isolated from airway epithelial cells is also thought to induce fibroblast differentiation and increase the expression of type 1 collagen and α-SMA. CS stimulates airway epithelial cells to release exosomes, which transport microRNA-21(miR-21) to target cells. MiR-21 regulates the von Hippel-lindau protein (PVHL)/Hypoxia-inducible factors 1 (Hif-1) pathway, and promote fibroblast differentiation and EMT (143). When epithelial cells are exposed to CSE, EV-associated miR-422a is reduced, which causes fibroblasts to produce more type 1 collagen and α-SMA (145). Epithelial cell-derived exosomes also modulate inflammatory responses. The expression of miR-221 and miR-320 in exosomes from epithelial cell stimulates the secretion of pro-inflammatory cytokines, thereby activating AM, which in turn promotes inflammation (146). Immune cells present in the airways can release large amounts of EVs. EVs generated from granulocytes induced by bacterial formylated peptides carry more neutrophil elastase (NE). EV-bound NE cleaves type 1 collagen, causing alveolar expansion and higher airway resistance (147). T lymphocyte derived EVs from patients with COPD can stimulate bronchial epithelial cells to release pro-inflammatory factors and inhibit the synthesis of anti-inflammatory factors, causing inflammatory airway damage (148).
Extracellular vesicle may be a potential tool for the treatment of COPD by its substance-carrying capacity. As mentioned before, MSCs has been suggested to improve lung function in animal models with COPD. For MSCs are pluripotent stem cells that are capable of self-renewal and pluripotent differentiation into osteoblasts, adipocytes and many other cells (149). The MSCs can exploit its therapeutic advantage by releasing EVs. As with bone marrow (BM)-MSC-EVs, the therapeutic effect of the intraperitoneal combination of BM-MSCs and BM-MSC-Exosomes was explored in a study of CS-induced mitochondrial dysfunction in mice with COPD. The combination therapy alleviated COPD through anti-inflammatory and targeting mitochondrial genes, reducing the recruitment of neutrophils, CD4+ lymphocytes and macrophages in the bronchoalveolar lavage. Compared with BM-MSCs alone or their exosomes, the combined use of BM-MSCs and exosomes has a protective effect on the development of COPD (150). Placental Mesenchymal stem cell (PL-MSC)-derived products reduce inflammatory cytokine levels, reduce pulmonary macrophage and neutrophil infiltration, and enhance lung function (151). In a variety of fibrotic diseases, including pulmonary fibrosis, MSC exosomes have been shown to reduce fibroblast differentiation and reduce fibrosis (152).
9 Various herbal medicines may improve airway remodeling through antioxidants
The extract obtained from the whole herb of Polygonum aviculare L exerts anti-inflammatory activity. The ethyl acetate fraction of Polygonum aviculare L. inhibits the contraction of human ASM in mice and humans, and this inhibition is achieved by inhibiting calcium-permeable ion channels. In addition, Polygonum aviculare L. extract contains quercetin, which is also known to inhibit ASM contraction (153). Quercetin is a natural polyphenolic compound with a variety of pharmacological activities such as anti-inflammatory, antioxidant, and antitumor. It has been found that quercetin can enhance antioxidant defense by scavenging free radicals, inhibiting lipid peroxidation, and activating the Nrf2 pathway (154).
Epigallocatechin gallate (EGCG), the most abundant catechin analog in green tea, is a powerful scavenger of oxygen free radicals. It protects cells and DNA from damage. EGCG prevents CSE-induced oxidative stress, reduces ROS production and accumulation in airway epithelial cells, and inhibits the activation of NF-κB and the downstream expression of proinflammatory-type mediators (155).
Resveratrol inhibits pro-inflammatory cytokines and chemokines, and holds promise for treating COPD by reducing oxidative stress, inhibiting ROS, and suppressing inflammatory responses. Sirtuin 1 (SIRT1) and p38 MAPK are considered therapeutic targets for resveratrol (156, 157). In human umbilical vein endothelial cells (HUVECs), resveratrol attenuated CSE-induced endothelial cell apoptosis by inducing autophagy in a Notch1-dependent manner. In human airway epithelial cells, resveratrol attenuated CSE-induced cellular senescence by regulating the miR-34a/SIRT1/NF-κB pathway, suggesting a protective effect on vascular endothelial cells (158, 159).
Polyphenols are a class of natural phenolic compounds widely found in nature, with remarkable biological activities such as antioxidant, anti-inflammatory and anti-tumor. Polyphenols can exert a variety of functions by regulating the NF-κB and MAPK inflammatory pathways, the Nrf2 oxidative stress pathway, and the SIRT1/PGC-1α pathway, thus inhibiting airway inflammation, alleviating oxidative stress, reducing airway mucus hypersecretion, and restoring the balance between proteases and antiproteases (160). Curcumin, a natural polyphenol compound isolated from traditional Chinese medicine such as turmeric, was found to effectively attenuate airway inflammation and airway remodeling in COPD mice and inhibit the proliferation of human bronchial epithelial cells, which may be related to its inhibition of IκBα degradation and Cyclooxygenase-2 (COX-2) expression, suggesting that curcumin may reduce COPD airway remodeling through inhibition of the NF-κB signaling pathway and COX-2 expression. Curcumin can also activate the Nrf2 signaling pathway, which enhances the antioxidant defense of cells (161, 162).
Acute exacerbations are often induced by viral infections (such as influenza) in COPD patients, and the antiviral and anti-inflammatory effects of ZE339 may help to alleviate exacerbations. Ze339 is a CO2-extract from Petasites hybridus. It mainly contains compounds called petasins. ZE339 can not only inhibit viral replication, but also exert anti-inflammatory effects by inhibiting the signal transducer and activator of transcription (STAT) pathway and regulating cytokines. ZE339 may inhibit airway remodeling in COPD patients by inhibiting inflammatory response and cytokine release (163, 164).
10 Conclusion
One crucial aspect of COPD is airway remodeling, which controls the emergence of airflow restriction. The primary characteristics of airway remodeling include epithelial abnormalities, epithelial-mesenchymal transition, fibroblast differentiation and proliferation, and airway smooth muscle hypertrophy and proliferation. Multiple pathophysiological mechanisms are involved in airway remodeling, with oxidative stress being one of the most prominent. Air pollution and cigarettes may promote oxidative stress, leading to ASM proliferation, increased EMT, and influence ECM secretion. Inflammatory cell imbalance and dysfunction in the airways are also linked to elevated oxidative stress. Different oxidation levels influence the generation and release of exosomes that play a role in developing COPD and airway remodeling. In conclusion, better research is needed to understand the link between oxidative stress and airway remodeling. Conventional therapies for COPD consist of bronchodilators, glucocorticoids, antibiotics, and expectorants; nevertheless, they cannot limit airway remodeling successfully. Herbal treatment may improve COPD airway remodeling through antioxidants; further study of Chinese medicine is promising.
Author contributions
MP: writing – original draft. XZ: writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cadham C, Oh H, Han M, Mannino D, Cook S, Meza R, et al. The prevalence and mortality risks of PRISm and COPD in the United States from NHANES 2007-2012. Respir Res. (2024) 25:208. doi: 10.1186/s12931-024-02841-y
2. Christenson S, Smith B, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. (2022) 399:2227–42. doi: 10.1016/S0140-6736(22)00470-6
3. Lareau S, Fahy B, Meek P, Wang A. Chronic obstructive pulmonary disease (COPD). Am J Respir Crit Care Med. (2019) 199:1–2. doi: 10.1164/rccm.1991P1
4. Varricchi G, Ferri S, Pepys J, Poto R, Spadaro G, Nappi E, et al. Biologics and airway remodeling in severe asthma. Allergy. (2022) 77:3538–52. doi: 10.1111/all.15473
5. Bergeron C, Boulet L. Structural changes in airway diseases: characteristics, mechanisms, consequences, and pharmacologic modulation. Chest. (2006) 129:1068–87. doi: 10.1378/chest.129.4.1068
6. Liu G, Philp A, Corte T, Travis M, Schilter H, Hansbro N, et al. Therapeutic targets in lung tissue remodelling and fibrosis. Pharmacol Ther. (2021) 225:107839. doi: 10.1016/j.pharmthera.2021.107839
7. Cao X, Wang Y, Chen Y, Zhao M, Liang L, Yang M, et al. Advances in traditional Chinese medicine for the treatment of chronic obstructive pulmonary disease. J Ethnopharmacol. (2023) 307:116229. doi: 10.1016/j.jep.2023.116229
8. Fehrenbach H, Wagner C, Wegmann M. Airway remodeling in asthma: what really matters. Cell Tissue Res. (2017) 367:551–69. doi: 10.1007/s00441-016-2566-8
9. Jeffery P. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. (2001) 164:S28–38. doi: 10.1164/ajrccm.164.supplement_2.2106061
10. James A, Wenzel S. Clinical relevance of airway remodelling in airway diseases. Eur Respir J. (2007) 30:134–55. doi: 10.1183/09031936.00146905
11. McAnulty R. Models and approaches to understand the role of airway remodelling in disease. Pulm Pharmacol Ther. (2011) 24:478–86. doi: 10.1016/j.pupt.2011.07.005
12. Shimizu K, Hasegawa M, Makita H, Nasuhara Y, Konno S, Nishimura M. Comparison of airway remodelling assessed by computed tomography in asthma and COPD. Respir Med. (2011) 105:1275–83. doi: 10.1016/j.rmed.2011.04.007
13. Nambu A, Zach J, Schroeder J, Jin G, Kim S, Kim Y, et al. Quantitative computed tomography measurements to evaluate airway disease in chronic obstructive pulmonary disease: relationship to physiological measurements, clinical index and visual assessment of airway disease. Eur J Radiol. (2016) 85:2144–51. doi: 10.1016/j.ejrad.2016.09.010
14. Xie H, Zhao Z, Zhang W, Li L. Quantitative analysis of lung function and airway remodeling using ventilation/perfusion single photon emission tomography/computed tomography and HRCT in patients with chronic obstructive pulmonary disease and asthma. Ann Nucl Med. (2023) 37:504–16. doi: 10.1007/s12149-023-01848-7
15. McIntosh M, Kooner H, Eddy R, Jeimy S, Licskai C, Mackenzie C, et al. Asthma control, airway mucus, and 129Xe MRI ventilation after a single benralizumab dose. Chest. (2022) 162:520–33. doi: 10.1016/j.chest.2022.03.003
16. Biederer J. MR imaging of the airways. Br J Radiol. (2023) 96:20220630. doi: 10.1259/bjr.20220630
17. d’Hooghe J, Goorsenberg A, de Bruin D, Roelofs J, Annema J, Bonta P. Optical coherence tomography for identification and quantification of human airway wall layers. PLoS One. (2017) 12:e0184145. doi: 10.1371/journal.pone.0184145
18. Chen Y, Ding M, Guan W, Wang W, Luo W, Zhong C, et al. Validation of human small airway measurements using endobronchial optical coherence tomography. Respir Med. (2015) 109:1446–53. doi: 10.1016/j.rmed.2015.09.006
19. Podoleanu A. Optical coherence tomography. J Microsc. (2012) 247:209–19. doi: 10.1111/j.1365-2818.2012.03619.x
20. João J, Silva Barbosa J, Sales da Silva LL, Fukuzaki S, de Campos EC, Camargo LDN, et al. Effects of plant protease inhibitors (Pep-3-EcTI, Pep-BbKI, and Pep-BrTI) versus corticosteroids on inflammation, remodeling, and oxidative stress in an asthma-COPD (ACO) model. Front Pharmacol. (2024) 15:1282870. doi: 10.3389/fphar.2024.1282870
21. Hardin M, Cho M, McDonald M, Beaty T, Ramsdell J, Bhatt S, et al. The clinical and genetic features of COPD-asthma overlap syndrome. Eur Respir J. (2014) 44:341–50. doi: 10.1183/09031936.00216013
22. Liang J, Xia T, Wu S, Liu S, Guan Y. Application research on asthma-COPD overlap using low-dose CT scan and quantitative analysis. Clin Radiol. (2024) 79:e1473–80. doi: 10.1016/j.crad.2024.09.005
23. Hwang H, Lee S, Seo J, Lee J, Kim N, Lee S, et al. Visual and quantitative assessments of regional xenon-ventilation using dual-energy CT in asthma-chronic obstructive pulmonary disease overlap syndrome: a comparison with chronic obstructive pulmonary disease. Korean J Radiol. (2020) 21:1104–13. doi: 10.3348/kjr.2019.0936
24. Niwa M, Fujisawa T, Karayama M, Furuhashi K, Mori K, Hashimoto D, et al. Differences in airway structural changes assessed by 3-dimensional computed tomography in asthma and asthma-chronic obstructive pulmonary disease overlap. Ann Allergy Asthma Immunol. (2018) 121:704–10.e1. doi: 10.1016/j.anai.2018.08.006.
25. Wang W, Zhou K, Wang L, Qin Q, Liu H, Qin L, et al. Aging in chronic lung disease: will anti-aging therapy be the key to the cure? Eur J Pharmacol. (2024) 980:176846. doi: 10.1016/j.ejphar.2024.176846
26. Stankovic M. Lung microbiota: from healthy lungs to development of chronic obstructive pulmonary disease. Int J Mol Sci. (2025) 26:1403. doi: 10.3390/ijms26041403
27. Wong K, Segal L. Correcting dysbiosis in the lungs of COPD, one pathogen at a time. Cell Host Microbe. (2023) 31:925–7. doi: 10.1016/j.chom.2023.05.019
28. Hiemstra P, van der Does A. Reprogramming of cellular metabolism: driver for airway remodelling in COPD? Eur Respir J. (2017) 50:1702197. doi: 10.1183/13993003.02197-2017
29. Mumby S, Adcock I. Recent evidence from omic analysis for redox signalling and mitochondrial oxidative stress in COPD. J Inflamm (Lond). (2022) 19:10. doi: 10.1186/s12950-022-00308-9
30. Lee I, Yang C. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol. (2012) 84:581–90. doi: 10.1016/j.bcp.2012.05.005
31. Rowlands D. Mitochondria dysfunction: a novel therapeutic target in pathological lung remodeling or bystander? Pharmacol Ther. (2016) 166:96–105. doi: 10.1016/j.pharmthera.2016.06.019
32. Cipollina C, Bruno A, Fasola S, Cristaldi M, Patella B, Inguanta R, et al. Cellular and molecular signatures of oxidative stress in bronchial epithelial cell models injured by cigarette smoke extract. Int J Mol Sci. (2022) 23:1770. doi: 10.3390/ijms23031770
33. Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. (2023) 13:736–66. doi: 10.7150/thno.79876
34. Ai L, de Freitas Germano J, Huang C, Aniag M, Sawaged S, Sin J, et al. Enhanced Parkin-mediated mitophagy mitigates adverse left ventricular remodelling after myocardial infarction: role of PR-364. Eur Heart J. (2025) 46:380–93. doi: 10.1093/eurheartj/ehae782
35. Albano G, Montalbano A, Gagliardo R, Profita M. Autophagy/mitophagy in airway diseases: impact of oxidative stress on epithelial cells. Biomolecules. (2023) 13:1217. doi: 10.3390/biom13081217
36. Youle R, Narendra D. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. (2011) 12:9–14. doi: 10.1038/nrm3028
37. Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy. (2015) 11:547–59. doi: 10.1080/15548627.2015.1017190
38. Vanella L, Li Volti G, Distefano A, Raffaele M, Zingales V, Avola R, et al. A new antioxidant formulation reduces the apoptotic and damaging effect of cigarette smoke extract on human bronchial epithelial cells. Eur Rev Med Pharmacol Sci. (2017) 21:5478–84. doi: 10.26355/eurrev_201712_13938
39. Yan J, Wan P, Choksi S, Liu Z. Necroptosis and tumor progression. Trends Cancer. (2022) 8:21–7. doi: 10.1016/j.trecan.2021.09.003
40. Tulen C, Wang Y, Beentjes D, Jessen P, Ninaber D, Reynaert N, et al. Dysregulated mitochondrial metabolism upon cigarette smoke exposure in various human bronchial epithelial cell models. Dis Model Mech. (2022) 15:dmm049247. doi: 10.1242/dmm.049247
41. Zhang M, Shi R, Zhang Y, Shan H, Zhang Q, Yang X, et al. Nix/BNIP3L-dependent mitophagy accounts for airway epithelial cell injury induced by cigarette smoke. J Cell Physiol. (2019) 234:14210–20. doi: 10.1002/jcp.28117
42. Osborn-Heaford H, Ryan A, Murthy S, Racila A, He C, Sieren J, et al. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J Biol Chem. (2012) 287:3301–12. doi: 10.1074/jbc.M111.308387
43. Fang L, Zhang M, Li J, Zhou L, Tamm M, Roth M. Airway smooth muscle cell mitochondria damage and mitophagy in COPD via ERK1/2 MAPK. Int J Mol Sci. (2022) 23:13987. doi: 10.3390/ijms232213987
44. Barnes P, Baker J, Donnelly L. Autophagy in asthma and chronic obstructive pulmonary disease. Clin Sci (Lond). (2022) 136:733–46. doi: 10.1042/CS20210900
45. Boukhenouna S, Wilson M, Bahmed K, Kosmider B. Reactive oxygen species in chronic obstructive pulmonary disease. Oxid Med Cell Longev. (2018) 2018:5730395. doi: 10.1155/2018/5730395
46. Pace E, Di Vincenzo S, Di Salvo E, Genovese S, Dino P, Sangiorgi C, et al. MiR-21 upregulation increases IL-8 expression and tumorigenesis program in airway epithelial cells exposed to cigarette smoke. J Cell Physiol. (2019) 234:22183–94. doi: 10.1002/jcp.28786
47. Chai H, Yao S, Gao Y, Hu Q, Su W. Developments in the connection between epithelial-mesenchymal transition and endoplasmic reticulum stress (Review). Int J Mol Med. (2025) 56:102. doi: 10.3892/ijmm.2025.5543
48. Michaeloudes C, Kuo C, Haji G, Finch D, Halayko A, Kirkham P, et al. Metabolic re-patterning in COPD airway smooth muscle cells. Eur Respir J. (2017) 50:1700202. doi: 10.1183/13993003.00202-2017
49. Kume H, Yamada R, Sato Y, Togawa R. Airway smooth muscle regulated by oxidative stress in COPD. Antioxidants (Basel). (2023) 12:142. doi: 10.3390/antiox12010142
50. Jones R, Noble P, Elliot J, James A. Airway remodelling in COPD: it’s not asthma! Respirology. (2016) 21:1347–56. doi: 10.1111/resp.12841
51. Roth M. Airway and lung remodelling in chronic pulmonary obstructive disease: a role for muscarinic receptor antagonists? Drugs. (2015) 75:1–8. doi: 10.1007/s40265-014-0319-0
52. Pan S, Conaway S, Deshpande D. Mitochondrial regulation of airway smooth muscle functions in health and pulmonary diseases. Arch Biochem Biophys. (2019) 663:109–19. doi: 10.1016/j.abb.2019.01.002
53. Kojima K, Kume H, Ito S, Oguma T, Shiraki A, Kondo M, et al. Direct effects of hydrogen peroxide on airway smooth muscle tone: roles of Ca2+ influx and Rho-kinase. Eur J Pharmacol. (2007) 556:151–6. doi: 10.1016/j.ejphar.2006.11.007
54. Shiraki A, Kume H, Oguma T, Makino Y, Ito S, Shimokata K, et al. Role of Ca2+ mobilization and Ca2+ sensitization in 8-iso-PGF 2 alpha-induced contraction in airway smooth muscle. Clin Exp Allergy. (2009) 39:236–45. doi: 10.1111/j.1365-2222.2008.03164.x
55. Pera T, Gosens R, Lesterhuis A, Sami R, van der Toorn M, Zaagsma J, et al. Cigarette smoke and lipopolysaccharide induce a proliferative airway smooth muscle phenotype. Respir Res. (2010) 11:48. doi: 10.1186/1465-9921-11-48
56. Dekkers B, Schaafsma D, Nelemans S, Zaagsma J, Meurs H. Extracellular matrix proteins differentially regulate airway smooth muscle phenotype and function. Am J Physiol Lung Cell Mol Physiol. (2007) 292:L1405–13. doi: 10.1152/ajplung.00331.2006
57. Wright D, Trian T, Siddiqui S, Pascoe C, Johnson J, Dekkers B, et al. Phenotype modulation of airway smooth muscle in asthma. Pulm Pharmacol Ther. (2013) 26:42–9. doi: 10.1016/j.pupt.2012.08.005
58. Shepherd M, Duffy S, Harris T, Cruse G, Schuliga M, Brightling C, et al. KCa3.1 Ca2+ activated K+ channels regulate human airway smooth muscle proliferation. Am J Respir Cell Mol Biol. (2007) 37:525–31. doi: 10.1165/rcmb.2006-0358OC
59. Yu Z, Wang Y, Song Y, Lu H, Hou L, Cui Y, et al. Up-regulation of KCa3.1 promotes human airway smooth muscle cell phenotypic modulation. Pharmacol Res. (2013) 77:30–8. doi: 10.1016/j.phrs.2013.09.002
60. Sutherland E, Martin R. Airway inflammation in chronic obstructive pulmonary disease: comparisons with asthma. J Allergy Clin Immunol. (2003) 112:819–27. doi: 10.1016/S0091
61. Di Stefano A, Maestrelli P, Roggeri A, Turato G, Calabro S, Potena A, et al. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am J Respir Crit Care Med. (1994) 149:803–10. doi: 10.1164/ajrccm.149.3.7509705
62. Fahy J, Dickey B. Airway mucus function and dysfunction. N Engl J Med. (2010) 363:2233–47. doi: 10.1056/NEJMra0910061
63. Aoshiba K, Nagai A. Differences in airway remodeling between asthma and chronic obstructive pulmonary disease. Clin Rev Allergy Immunol. (2004) 27:35–43. doi: 10.1385/CRIAI:27:1:035
64. Hogg J, Chu F, Utokaparch S, Woods R, Elliott W, Buzatu L, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. (2004) 350:2645–53. doi: 10.1056/NEJMoa032158
65. Aurora A, Baluk P, Zhang D, Sidhu S, Dolganov G, Basbaum C, et al. Immune complex-dependent remodeling of the airway vasculature in response to a chronic bacterial infection. J Immunol. (2005) 175:6319–26. doi: 10.4049/jimmunol.175.10.6319
66. Saetta M, Mariani M, Panina-Bordignon P, Turato G, Buonsanti C, Baraldo S, et al. Increased expression of the chemokine receptor CXCR3 and its ligand CXCL10 in peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2002) 165:1404–9. doi: 10.1164/rccm.2107139
67. Bracke K, Verhamme F, Seys L, Bantsimba-Malanda C, Cunoosamy D, Herbst R, et al. Role of CXCL13 in cigarette smoke-induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2013) 188:343–55. doi: 10.1164/rccm.201211-2055OC
68. Traves S, Culpitt S, Russell R, Barnes P, Donnelly L. Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax. (2002) 57:590–5. doi: 10.1136/thorax.57.7.590
69. Byrne A, Mathie S, Gregory L, Lloyd C. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. (2015) 70:1189–96. doi: 10.1136/thoraxjnl-2015-207020
70. Kapellos T, Bassler K, Aschenbrenner A, Fujii W, Schultze J. Dysregulated functions of lung macrophage populations in COPD. J Immunol Res. (2018) 2018:2349045. doi: 10.1155/2018/2349045
71. Hoppstädter J, Diesel B, Zarbock R, Breinig T, Monz D, Koch M, et al. Differential cell reaction upon Toll-like receptor 4 and 9 activation in human alveolar and lung interstitial macrophages. Respir Res. (2010) 11:124. doi: 10.1186/1465-9921-11-124
72. Grumelli S, Corry D, Song L, Song L, Green L, Huh J, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. (2004) 1:e0010008. doi: 10.1371/journal.pmed.0010008
73. Puttur F, Gregory L, Lloyd C. Airway macrophages as the guardians of tissue repair in the lung. Immunol Cell Biol. (2019) 97:246–57. doi: 10.1111/imcb.12235
74. da Silva C, de Souza Nogueira J, do Nascimento A, Victoni T, Bártholo T, da Costa C, et al. COPD patients exhibit distinct gene expression, accelerated cellular aging, and bias to M2 macrophages. Int J Mol Sci. (2023) 24:9913. doi: 10.3390/ijms24129913
75. Lloyd C, Snelgrove R. Type 2 immunity: expanding our view. Sci Immunol. (2018) 3:eaat1604. doi: 10.1126/sciimmunol.aat1604
76. Nobs S, Kopf M. PPAR-γ in innate and adaptive lung immunity. J Leukoc Biol. (2018) 104:737–41. doi: 10.1002/JLB.3MR0118-034R
77. He S, Tian R, Zhang X, Yao Q, Chen Q, Liu B, et al. PPARγ inhibits small airway remodeling through mediating the polarization homeostasis of alveolar macrophages in COPD. Clin Immunol. (2023) 250:109293. doi: 10.1016/j.clim.2023.109293
78. Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. (1996) 21:669–81. doi: 10.1016/0891-5849(96)00155-4
79. Ogger P, Byrne A. Macrophage metabolic reprogramming during chronic lung disease. Mucosal Immunol. (2021) 14:282–95. doi: 10.1038/s41385-020-00356-5
80. Crystal R. Airway basal cells. The smoking gun of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2014) 190:1355–62. doi: 10.1164/rccm.201408-1492PP
81. Siddiqui S, Bachert C, Bjermer L, Buchheit K, Castro M, Qin Y, et al. Eosinophils and tissue remodeling: relevance to airway disease. J Allergy Clin Immunol. (2023) 152:841–57. doi: 10.1016/j.jaci.2023.06.005
82. Iosifidis T, Garratt L, Coombe D, Knight D, Stick S, Kicic A. Airway epithelial repair in health and disease: orchestrator or simply a player? Respirology. (2016) 21:438–48. doi: 10.1111/resp.12731
83. Barker T, Engler A. The provisional matrix: setting the stage for tissue repair outcomes. Matrix Biol. (2017) 61:1–4. doi: 10.1016/j.matbio.2017.04.003
84. Fang S, Zhang S, Dai H, Hu X, Li C, Xing Y. The role of pulmonary mesenchymal cells in airway epithelium regeneration during injury repair. Stem Cell Res Ther. (2019) 10:366. doi: 10.1186/s13287-019-1452-1
85. Johansson K, Woodruff P, Ansel K. Regulation of airway immunity by epithelial miRNAs. Immunol Rev. (2021) 304:141–53. doi: 10.1111/imr.13028
86. Bartis D, Mise N, Mahida R, Eickelberg O, Thickett D. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. (2014) 69:760–5. doi: 10.1136/thoraxjnl-2013-204608
87. Sohal S, Walters E. Epithelial mesenchymal transition (EMT) in small airways of COPD patients. Thorax. (2013) 68:783–4. doi: 10.1136/thoraxjnl-2013-203373
88. Sohal S, Mahmood M, Walters E. Clinical significance of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD): potential target for prevention of airway fibrosis and lung cancer. Clin Transl Med. (2014) 3:33. doi: 10.1186/s40169-014-0033-2
89. Mahmood M, Shukla S, Ward C, Walters E. The underappreciated role of epithelial mesenchymal transition in chronic obstructive pulmonary disease and its strong link to lung cancer. Biomolecules. (2021) 11:1394. doi: 10.3390/biom11091394
90. Milara J, Peiró T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. (2013) 68:410–20. doi: 10.1136/thoraxjnl-2012-201761
91. Jiang B, Guan Y, Shen H, Zhang L, Jiang J, Dong X, et al. Akt/PKB signaling regulates cigarette smoke-induced pulmonary epithelial-mesenchymal transition. Lung Cancer. (2018) 122:44–53. doi: 10.1016/j.lungcan.2018.05.019
92. Mahmood M, Reid D, Ward C, Muller H, Knight D, Sohal S, et al. Transforming growth factor (TGF) β1 and Smad signalling pathways: a likely key to EMT-associated COPD pathogenesis. Respirology. (2017) 22:133–40. doi: 10.1111/resp.12882
93. Singh S, Verma S, Kumar S, Ahmad M, Nischal A, Singh S, et al. Evaluation of oxidative stress and antioxidant status in chronic obstructive pulmonary disease. Scand J Immunol. (2017) 85:130–7. doi: 10.1111/sji.12498
94. Hu Y, He K, Wang D, Yuan X, Liu Y, Ji H, et al. TMEPAI regulates EMT in lung cancer cells by modulating the ROS and IRS-1 signaling pathways. Carcinogenesis. (2013) 34:1764–72. doi: 10.1093/carcin/bgt132
95. Eapen M, Sohal SS. WNT/β-catenin pathway: a novel therapeutic target for attenuating airway remodelling and EMT in COPD. EBioMedicine. (2020) 62:103095. doi: 10.1016/j.ebiom.2020.103095
96. Su X, Wu W, Zhu Z, Lin X, Zeng Y. The effects of epithelial-mesenchymal transitions in COPD induced by cigarette smoke: an update. Respir Res. (2022) 23:225. doi: 10.1186/s12931-022-02153-z
97. Su X, Chen J, Lin X, Chen X, Zhu Z, Wu W, et al. FERMT3 mediates cigarette smoke-induced epithelial-mesenchymal transition through Wnt/β-catenin signaling. Respir Res. (2021) 22:286. doi: 10.1186/s12931-021-01881-y
98. Li Y, Lu X, Li W, Shi Z, Du W, Xu H, et al. The circRERE/miR-144-3p/TLR2/MMP9 signaling axis in COPD pulmonary monocytes promotes the EMT of pulmonary epithelial cells. Biochem Biophys Res Commun. (2022) 625:1–8. doi: 10.1016/j.bbrc.2022.07.119
99. Eapen M, Sharma P, Thompson I, Lu W, Myers S, Hansbro P, et al. Heparin-binding epidermal growth factor (HB-EGF) drives EMT in patients with COPD: implications for disease pathogenesis and novel therapies. Lab Invest. (2019) 99:150–7. doi: 10.1038/s41374-018-0146-0
100. Lai T, Li Y, Chen M, Pan G, Wen X, Mai Z, et al. Heparin-binding epidermal growth factor contributes to COPD disease severity by modulating airway fibrosis and pulmonary epithelial-mesenchymal transition. Lab Invest. (2018) 98:1159–69. doi: 10.1038/s41374-018-0049-0
101. Pain M, Bermudez O, Lacoste P, Royer P, Botturi K, Tissot A, et al. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. (2014) 23:118–30. doi: 10.1183/09059180.00004413
102. Zhu W, Han L, Wu Y, Tong L, He L, Wang Q, et al. Keratin 15 protects against cigarette smoke-induced epithelial mesenchymal transformation by MMP-9. Respir Res. (2023) 24:297. doi: 10.1186/s12931-023-02598-w
103. Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell Biol. (2008) 129:705–33. doi: 10.1007/s00418-008-0435-6
104. Theocharis A, Skandalis S, Gialeli C, Karamanos N. Extracellular matrix structure. Adv Drug Deliv Rev. (2016) 97:4–27. doi: 10.1016/j.addr.2015.11.001
105. Burgess J, Ceresa C, Johnson S, Kanabar V, Moir L, Nguyen T, et al. Tissue and matrix influences on airway smooth muscle function. Pulm Pharmacol Ther. (2009) 22:379–87. doi: 10.1016/j.pupt.2008.12.007
106. Stolz D, Leeming D, Kristensen J, Karsdal M, Boersma W, Louis R, et al. Systemic Biomarkers of Collagen and Elastin Turnover Are Associated With Clinically Relevant Outcomes in COPD. Chest. (2017) 151:47–59. doi: 10.1016/j.chest.2016.08.1440
107. Singla A, Reuter S, Taube C, Peters M, Peters K. The molecular mechanisms of remodeling in asthma, COPD and IPF with a special emphasis on the complex role of Wnt5A. Inflamm Res. (2023) 72:577–88. doi: 10.1007/s00011-023-01692-5
108. Brandsma C, Van den Berge M, Hackett T, Brusselle G, Timens W. Recent advances in chronic obstructive pulmonary disease pathogenesis: from disease mechanisms to precision medicine. J Pathol. (2020) 250:624–35. doi: 10.1002/path.5364
109. Karakioulaki M, Papakonstantinou E, Stolz D. Extracellular matrix remodelling in COPD. Eur Respir Rev. (2020) 29:190124. doi: 10.1183/16000617.0124-2019
110. Karnati S, Seimetz M, Kleefeldt F, Sonawane A, Madhusudhan T, Bachhuka A, et al. Chronic obstructive pulmonary disease and the cardiovascular system: vascular repair and regeneration as a therapeutic target. Front Cardiovasc Med. (2021) 8:649512. doi: 10.3389/fcvm.2021.649512
111. Churg A, Zhou S, Wright J. Series matrix metalloproteinases in lung health and disease: matrix metalloproteinases in COPD. Eur Respir J. (2012) 39:197–209. doi: 10.1183/09031936.00121611
112. Churg A, Wang R, Wang X, Onnervik P, Thim K, Wright J. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax. (2007) 62:706–13. doi: 10.1136/thx.2006.068353
113. Elshaw S, Henderson N, Knox A, Watson S, Buttle D, Johnson S. Matrix metalloproteinase expression and activity in human airway smooth muscle cells. Br J Pharmacol. (2004) 142:1318–24. doi: 10.1038/sj.bjp.0705883
114. Martins S, Zilhão R, Thorsteinsdóttir S, Carlos A. Linking oxidative stress and DNA damage to changes in the expression of extracellular matrix components. Front Genet. (2021) 12:673002. doi: 10.3389/fgene.2021.673002
115. Yamauchi K, Inoue H. Airway remodeling in asthma and irreversible airflow limitation-ECM deposition in airway and possible therapy for remodeling-. Allergol Int. (2007) 56:321–9. doi: 10.2332/allergolint.R-07-151
116. Lee J, Song K, Hong J, Shin H, Park E, Baek J, et al. Role of lung ornithine aminotransferase in idiopathic pulmonary fibrosis: regulation of mitochondrial ROS generation and TGF-β1 activity. Exp Mol Med. (2024) 56:478–90. doi: 10.1038/s12276-024-01170-w
117. Thuan D, Zayed H, Eid A, Abou-Saleh H, Nasrallah G, Mangoni A, et al. A potential link between oxidative stress and endothelial-to-mesenchymal transition in systemic sclerosis. Front Immunol. (2018) 9:1985. doi: 10.3389/fimmu.2018.01985
118. Ma M, Shi F, Zhai R, Wang H, Li K, Xu C, et al. TGF-β promote epithelial-mesenchymal transition via NF-κB/NOX4/ROS signal pathway in lung cancer cells. Mol Biol Rep. (2021) 48:2365–75. doi: 10.1007/s11033-021-06268-2
119. Gu L, Zhu Y, Yang X, Guo Z, Xu W, Tian X. Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol Sin. (2007) 28:382–91. doi: 10.1111/j.1745-7254.2007.00468.x
120. Shinde A, Humeres C, Frangogiannis N. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:298–309. doi: 10.1016/j.bbadis.2016.11.006
121. Karvonen H, Lehtonen S, Harju T, Sormunen R, Lappi-Blanco E, Mäkinen J, et al. Myofibroblast expression in airways and alveoli is affected by smoking and COPD. Respir Res. (2013) 14:84. doi: 10.1186/1465-9921-14-84
122. Fei J, Fu L, Cao W, Hu B, Zhao H, Li J. Low Vitamin D status is associated with epithelial-mesenchymal transition in patients with chronic obstructive pulmonary disease. J Immunol. (2019) 203:1428–35. doi: 10.4049/jimmunol.1900229
123. Holm Nielsen S, Willumsen N, Leeming D, Daniels S, Brix S, Karsdal M, et al. Serological assessment of activated fibroblasts by alpha-smooth muscle actin (α-SMA): a noninvasive biomarker of activated fibroblasts in lung disorders. Transl Oncol. (2019) 12:368–74. doi: 10.1016/j.tranon.2018.11.004
124. Chen D, Yu J, Zhang L. Necroptosis: an alternative cell death program defending against cancer. Biochim Biophys Acta. (2016) 1865:228–36. doi: 10.1016/j.bbcan.2016.03.003
125. Ye K, Chen Z, Xu Y. The double-edged functions of necroptosis. Cell Death Dis. (2023) 14:163. doi: 10.1038/s41419-023-05691-6
126. Liu C, Li P, Zheng J, Wang Y, Wu W, Liu X. Role of necroptosis in airflow limitation in chronic obstructive pulmonary disease: focus on small-airway disease and emphysema. Cell Death Discov. (2022) 8:363. doi: 10.1038/s41420-022-01154-7
127. Mizumura K, Maruoka S, Shimizu T, Gon Y. Autophagy, selective autophagy, and necroptosis in COPD. Int J Chron Obstruct Pulmon Dis. (2018) 13:3165–72. doi: 10.2147/COPD.S175830
128. Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. (2015) 25:347–53. doi: 10.1016/j.tcb.2015.01.001
129. Vanlangenakker N, Vanden Berghe T, Bogaert P, Laukens B, Zobel K, Deshayes K, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. (2011) 18:656–65. doi: 10.1038/cdd.2010.138
130. Morgan M, Kim Y, Liu Z. Lipid rafts and oxidative stress-induced cell death. Antioxid Redox Signal. (2007) 9:1471–83. doi: 10.1089/ars.2007.1658
131. Xu F, Luo M, He L, Cao Y, Li W, Ying S, et al. Necroptosis contributes to urban particulate matter-induced airway epithelial injury. Cell Physiol Biochem. (2018) 46:699–712. doi: 10.1159/000488726
132. Wang Y, Su X, Yin Y, Wang Q. Identification and analysis of necroptosis-related genes in COPD by bioinformatics and experimental verification. Biomolecules. (2023) 13:482. doi: 10.3390/biom13030482
133. Wang Y, Zhou J, Xu X, Li Z, Chen H, Ying S, et al. Endoplasmic reticulum chaperone GRP78 mediates cigarette smoke-induced necroptosis and injury in bronchial epithelium. Int J Chron Obstruct Pulmon Dis. (2018) 13:571–81. doi: 10.2147/COPD.S150633
134. Luan G, Zhu Z, Wu K, Yin S. Theaflavin-3,3’-digallate attenuates cigarette smoke extract-induced pulmonary emphysema in mice by suppressing necroptosis. Exp Ther Med. (2022) 23:11. doi: 10.3892/etm.2021.10933
135. Mao K, Luo P, Geng W, Xu J, Liao Y, Zhong H, et al. An integrative transcriptomic and metabolomic study revealed that melatonin plays a protective role in chronic lung inflammation by reducing necroptosis. Front Immunol. (2021) 12:668002. doi: 10.3389/fimmu.2021.668002
136. Lu Z, Van Eeckhoutte H, Liu G, Nair P, Jones B, Gillis C, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2021) 204:667–81. doi: 10.1164/rccm.202009-3442OC
137. Mizumura K, Maruoka S, Gon Y, Choi A, Hashimoto S. The role of necroptosis in pulmonary diseases. Respir Investig. (2016) 54:407–12. doi: 10.1016/j.resinv.2016.03.008
138. Wang Y, Wang X, Wu P, Wang Y, Ren L, Xu A. Necroptosis mediates cigarette smoke-induced inflammatory responses in macrophages. Int J Chron Obstruct Pulmon Dis. (2020) 15:1093–101. doi: 10.2147/COPD.S233506
139. Van Eeckhoutte H, Donovan C, Kim R, Conlon T, Ansari M, Khan H, et al. RIPK1 kinase-dependent inflammation and cell death contribute to the pathogenesis of COPD. Eur Respir J. (2023) 61:2201506. doi: 10.1183/13993003.01506-2022
140. Wu J, Ma Y, Chen Y. Extracellular vesicles and COPD: foe or friend? J Nanobiotechnology. (2023) 21:147. doi: 10.1186/s12951-023-01911-5
141. Verweij F, Balaj L, Boulanger C, Carter D, Compeer E, D’Angelo G, et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat Methods. (2021) 18:1013–26. doi: 10.1038/s41592-021-01206-3
142. Mohammadipoor A, Antebi B, Batchinsky A, Cancio L. Therapeutic potential of products derived from mesenchymal stem/stromal cells in pulmonary disease. Respir Res. (2018) 19:218. doi: 10.1186/s12931-018-0921-x
143. Xu H, Ling M, Xue J, Dai X, Sun Q, Chen C, et al. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics. (2018) 8:5419–33. doi: 10.7150/thno.27876
144. Zhang W, Liu R, Chen Y, Wang M, Du J. Crosstalk between oxidative stress and exosomes. Oxid Med Cell Longev. (2022) 2022:3553617. doi: 10.1155/2022/3553617
145. Dai Z, Lin L, Xu Y, Hu L, Gou S, Xu X. Extracellular vesicle dynamics in COPD: understanding the role of miR-422a, SPP1 and IL-17 A in smoking-related pathology. BMC Pulm Med. (2024) 24:173. doi: 10.1186/s12890-024-02978-y
146. Lee H, Zhang D, Zhu Z, Dela Cruz C, Jin Y. Epithelial cell-derived microvesicles activate macrophages and promote inflammation via microvesicle-containing microRNAs. Sci Rep. (2016) 6:35250. doi: 10.1038/srep35250
147. Genschmer K, Russell D, Lal C, Szul T, Bratcher P, Noerager B, et al. Activated PMN exosomes: pathogenic entities causing matrix destruction and disease in the lung. Cell. (2019) 176:113–26.e15. doi: 10.1016/j.cell.2018.12.002.
148. Qiu Q, Dan X, Yang C, Hardy P, Yang Z, Liu G, et al. Increased airway T lymphocyte microparticles in chronic obstructive pulmonary disease induces airway epithelial injury. Life Sci. (2020) 261:118357. doi: 10.1016/j.lfs.2020.118357
149. El Omar R, Beroud J, Stoltz J, Menu P, Velot E, Decot V. Umbilical cord mesenchymal stem cells: the new gold standard for mesenchymal stem cell-based therapies? Tissue Eng Part B Rev. (2014) 20:523–44. doi: 10.1089/ten.TEB.2013.0664
150. Maremanda K, Sundar I, Rahman I. Protective role of mesenchymal stem cells and mesenchymal stem cell-derived exosomes in cigarette smoke-induced mitochondrial dysfunction in mice. Toxicol Appl Pharmacol. (2019) 385:114788. doi: 10.1016/j.taap.2019.114788
151. Harrell C, Miloradovic D, Sadikot R, Fellabaum C, Markovic B, Miloradovic D, et al. Molecular and cellular mechanisms responsible for beneficial effects of mesenchymal stem cell-derived product Exo-d-MAPPS; in attenuation of chronic airway inflammation. Anal Cell Pathol (Amst). (2020) 2020:3153891. doi: 10.1155/2020/3153891
152. Rajabi H, Konyalilar N, Erkan S, Mortazavi D, Korkunc S, Kayalar O, et al. Emerging role of exosomes in the pathology of chronic obstructive pulmonary diseases; destructive and therapeutic properties. Stem Cell Res Ther. (2022) 13:144. doi: 10.1186/s13287-022-02820-4
153. Luo X, Xue L, Xu H, Zhao Q, Wang Q, She Y, et al. Polygonum aviculare L. extract and quercetin attenuate contraction in airway smooth muscle. Sci Rep. (2018) 8:3114. doi: 10.1038/s41598-018-20409-x
154. Araújo N, de Matos N, Oliveira M, de Souza A, Castro T, Machado-Júnior P, et al. Quercetin improves pulmonary function and prevents emphysema caused by exposure to cigarette smoke in male mice. Antioxidants (Basel). (2022) 11:181. doi: 10.3390/antiox11020181
155. Lakshmi S, Reddy A, Kodidhela L, Varadacharyulu N. Epigallocatechin gallate diminishes cigarette smoke-induced oxidative stress, lipid peroxidation, and inflammation in human bronchial epithelial cells. Life Sci. (2020) 259:118260. doi: 10.1016/j.lfs.2020.118260
156. Knobloch J, Sibbing B, Jungck D, Lin Y, Urban K, Stoelben E, et al. Resveratrol impairs the release of steroid-resistant inflammatory cytokines from human airway smooth muscle cells in chronic obstructive pulmonary disease. J Pharmacol Exp Ther. (2010) 335:788–98. doi: 10.1124/jpet.110.166843
157. Ma B, Li X. Resveratrol extracted from Chinese herbal medicines: a novel therapeutic strategy for lung diseases. Chin Herb Med. (2020) 12:349–58. doi: 10.1016/j.chmed.2020.07.003
158. Zong D, Liu X, Li J, Ouyang R, Long Y, Chen P, et al. Resveratrol attenuates cigarette smoke induced endothelial apoptosis by activating Notch1 signaling mediated autophagy. Respir Res. (2021) 22:22. doi: 10.1186/s12931-021-01620-3
159. Zeng X, Yang X, Liu X. Resveratrol attenuates cigarette smoke extract induced cellular senescence in human airway epithelial cells by regulating the miR-34a/SIRT1/NF-κB pathway. Medicine (Baltimore). (2022) 101:e31944. doi: 10.1097/MD.0000000000031944
160. Zhang Y, Wang L, Zeng J, Shen W. Research advances in polyphenols from Chinese herbal medicine for the prevention and treatment of chronic obstructive pulmonary disease: a review. Naunyn Schmiedebergs Arch Pharmacol. (2025) 1–20. doi: 10.1007/s00210-025-03945-y
161. Yuan J, Liu R, Ma Y, Zhang Z, Xie Z. Curcumin attenuates airway Inflammation and airway remolding by inhibiting NF-κB signaling and COX-2 in cigarette smoke-induced COPD mice. Inflammation. (2018) 41:1804–14. doi: 10.1007/s10753-018-0823-6
162. Reis R, Orak D, Yilmaz D, Cimen H, Sipahi H. Modulation of cigarette smoke extract-induced human bronchial epithelial damage by eucalyptol and curcumin. Hum Exp Toxicol. (2021) 40:1445–62. doi: 10.1177/0960327121997986
163. Jakwerth C, Grass V, Erb A, Pichlmair A, Boonen G, Butterweck V, et al. Inhibition of SARS-CoV-2 infection and replication by Petasites hybridus CO2-extract (Ze 339). Biomed Pharmacother. (2024) 170:115959. doi: 10.1016/j.biopha.2023.115959
Keywords: COPD, airway remodeling, epithelial-mesenchymal transition, inflammation, oxidative stress
Citation: Pan M and Zhou X (2025) Airway remodeling in chronic obstructive pulmonary disease: characteristics and opportunities. Front. Med. 12:1556868. doi: 10.3389/fmed.2025.1556868
Received: 22 January 2025; Accepted: 10 July 2025;
Published: 28 July 2025.
Edited by:
Ulrich Matthias Zissler, University of Applied Sciences Rosenheim, GermanyCopyright © 2025 Pan and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojing Zhou, enhqY2N4MzE1QDE2My5jb20=