 Qianfeng Qin
Qianfeng Qin Cunwei Cao
Cunwei Cao Jiarong Liang1
Jiarong Liang1- 1Department of Dermatology and Venereology, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
- 2Fangchenggang Wanqing Institute of Mycosis Prevention and Control, Fangchenggang, Guangxi, China
- 3Guangxi Key Laboratory of Mycosis Prevention and Treatment, Nanning, Guangxi, China
Background: PASH syndrome is a rare autoinflammatory disorder characterized primarily by pyoderma gangrenosum, acne, and suppurative hidradenitis. It is frequently misdiagnosed or underdiagnosed due to its diverse clinical manifestations. PASH syndrome is considered a polygenic autoinflammatory disease associated with multiple gene variants, among which MEFV variants may play a significant role. This case report describes a PASH syndrome patient with the MEFV variant (NM_000243.3.442G > C; p.Glu148Gln). The E148Q variant appears with high frequency as a homozygote in the gnomAD database, and its pathogenicity remains controversial.
Case presentation: This case report describes a Chinese male patient with PASH syndrome who initially presented with severe acne and suppurative hidradenitis that were unresponsive to conventional therapy, subsequently developing pyoderma gangrenosum, leading to the diagnosis of PASH syndrome. Whole-exome sequencing performed during the diagnostic workup revealed the MEFV gene variant (p.E148Q). Treatment with adalimumab ultimately achieved favorable therapeutic outcomes.
Conclusion: This case report represents the first documentation of a PASH syndrome patient carrying the homozygous E148Q variant. Adalimumab demonstrated good therapeutic efficacy; however, disease recurrence occurred due to poor medication adherence, highlighting the importance of long-term management. The pathogenicity of E148Q alone is relatively weak and likely depends on polygenic accumulation or epigenetic dysregulation. It may contribute to disease pathogenesis through a “variant load” mechanism in conjunction with ethnic differences and other genetic or environmental factors.
1 Introduction
PASH syndrome (pyoderma gangrenosum, acne, and suppurative hidradenitis) is a rare and complex autoinflammatory disease encompassing three main clinical manifestations: pyoderma gangrenosum (PG), acne, and hidradenitis suppurativa (HS). Since Markus Braun-Falco first proposed PASH syndrome in 2011, its diverse clinical presentations have often been overlooked by clinicians, leading to misdiagnosis or delayed diagnosis (1, 2).
Currently, PASH syndrome is considered a polygenic autoinflammatory disease. Multiple gene variants have been associated with this condition, including those in NCSTN, PSENEN, NOD2, PSTPIP1, NLRC4, OTULIN, GJB2 and MEFV (p.I591T) (3, 4). A study investigating genetic variants associated with hidradenitis suppurativa (HS) identified two predominant variants significantly correlated with HS: rs10512572 located on chromosome 17 near the SOX9 gene, and rs17090189 located on chromosome 13 near the KLF5 gene (5), which may be associated with PASH syndrome. Among these, MEFV gene variants are closely associated with familial Mediterranean fever (FMF), which is aa classic monogenic autoinflammatory disease (6). Studies have demonstrated elevated MEFV variant rates in patients with complex HS (including PASH syndrome), potentially playing an important role in autoinflammatory pathogenesis (7). The MEFV (p.E148Q) variant identified in this case has been previously reported in patients with PG and familial Mediterranean fever (FMF), but its role in PASH syndrome remains unclear (8, 9). In previous reports, the genetic status of E148Q includes heterozygous, compound heterozygous, and homozygous states. Additionally, this variant appears with high frequency in homozygous form in the gnomAD database. Its pathogenicity remains controversial: traditionally, scholars have considered E148Q a variant of uncertain significance (10); however, other studies have published different perspectives, with one study showing no statistical differences in symptoms among heterozygous, compound heterozygous, and homozygous patients, concluding that E148Q is a disease-causing variant (9).
This study reports a challenging case of PASH syndrome in which whole exome sequencing detected the MEFV gene variant (p.E148Q). This represents the first reported case of this variant identified in a patient with PASH syndrome.
2 Case presentation
The patient is a 25-year-old Asian male residing in southern China, working as a truck driver, with a 6-year smoking history and no family history of pyoderma gangrenosum (PG), severe acne, hidradenitis suppurativa (HS), or familial Mediterranean fever (FMF). In July 2021, the patient presented to our hospital with erythema, papules, and nodules on the head and face accompanied by pruritus, which gradually spread to the trunk, extremities, and axillae, without fever, abdominal pain, chest pain, or joint pain. The presentation met diagnostic criteria for severe acne and hidradenitis suppurativa. Despite standard treatment, therapeutic response was suboptimal, causing significant psychological distress for the patient. In November 2022, the patient developed extensive erythema and nodules on the left lower extremity, which gradually progressed to ulcerative lesions (Figure 1). Histopathological examination confirmed the diagnosis of pyoderma gangrenosum (Figure 2).
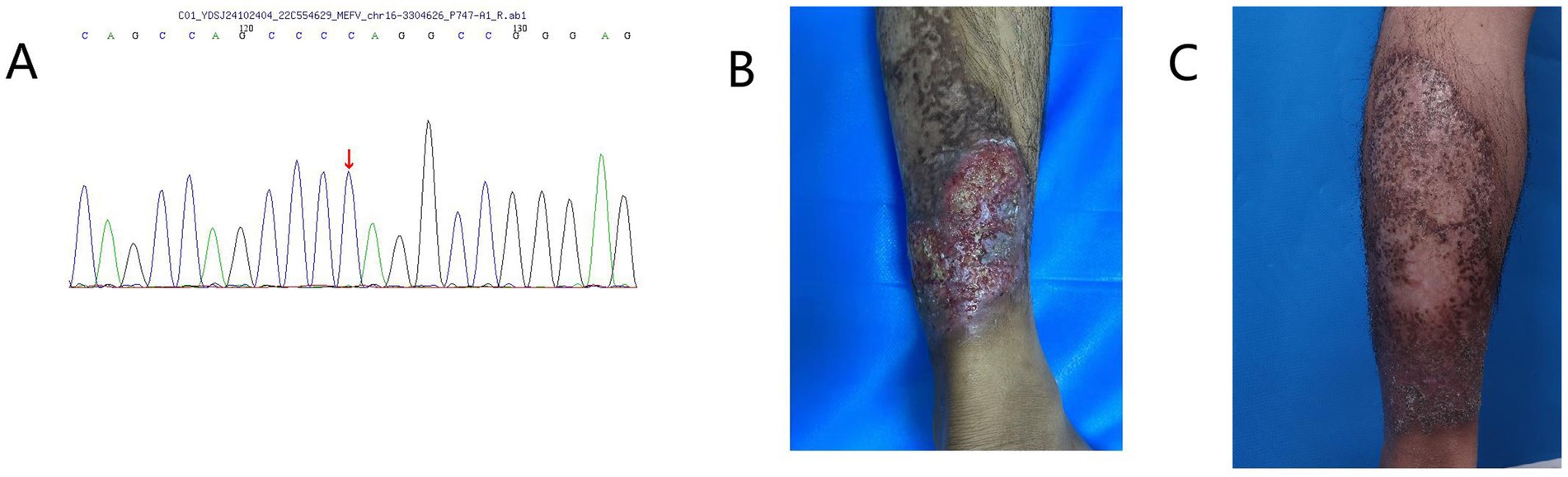
Figure 1. (A) Whole-exome sequencing identified a NM_000243.3.442G > C (p.E148Q) variant of the MEFV gene in the patient. (B) Before adalimumab treatment, the patient had a non-healing ulcerative lesion on the left lower limb. (C) After treatment with adalimumab, the patient’s skin lesions on the left lower limb healed.
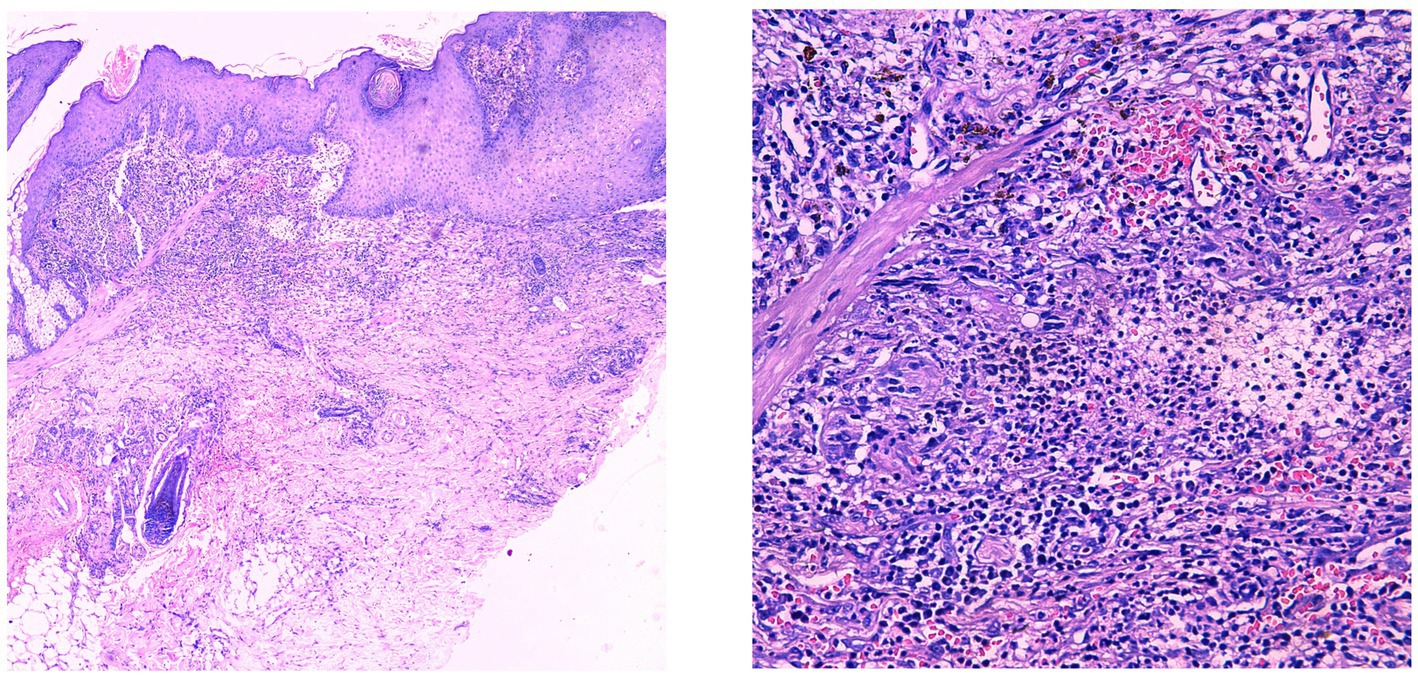
Figure 2. Pseudoepitheliomatous hyperplasia of the epidermis with extensive lymphocytic and neutrophilic infiltration in the superficial to mid-dermis.
Initial treatment with prednisolone acetate resulted in partial ulcer control but incomplete healing. Self-discontinuation of medication led to re-expansion of lesions. Adalimumab therapy initiated in August 2023 resulted in significant improvement of lesions with marked ulcer reduction; however, due to irregular follow-up, disease recurrence occurred after medication discontinuation. The recurrent exacerbations caused the patient extreme physical and psychological distress. Based on medical history and clinical presentation, a final diagnosis of PASH syndrome was established. To exclude other genetic disorders and elucidate the genetic basis of PASH syndrome, we performed whole-exome sequencing and analysis on the patient at no charge. The homozygous MEFV variant (E148Q) was identified (Figure 1), while the aforementioned variants rs10512572 and rs17090189, as well as genetic variants in NCSTN, PSENEN, NOD2, PSTPIP1, NLRC4, OTULIN, GJB2, and MEFV (p.I591T), were not detected. Unfortunately, due to family members being occupied with work, no familial segregation study was conducted. During treatment, recurrent lesions and treatment resistance presented significant clinical management challenges. Disease control was ultimately achieved through a combination regimen of adalimumab with antibiotics and corticosteroids. The patient expressed satisfaction with the current treatment outcomes.
3 Discussion and conclusion
Current literature indicates that PASH syndrome exhibits highly heterogeneous clinical manifestations, with no unified diagnostic and therapeutic guidelines currently available (4). This patient’s therapeutic response to adalimumab aligns with previous reports, suggesting potential efficacy of this medication for PASH syndrome (11). However, poor medication adherence and irregular follow-up led to multiple disease recurrences, highlighting the importance of long-term medication management and ensuring good patient compliance (12).
A recent study found that in FMF patients, the presence of multiple variants may be associated with higher occurrence rates of moderate disease severity (13). Another study found that isolated MEFV variants result in limited IL-1β secretion increase, requiring cooperation with other variants to reach the pathogenic threshold for FMF (14). Both studies support the “variant burden” hypothesis. Therefore, E148Q may act in conjunction with multiple other moderate/high-frequency variants (polygenic risk) rather than as a single potent pathogenic variant. Additionally, there may be many incorrectly diagnosed patients, which together with variant burden contributes to the high frequency phenomenon of 5,896 homozygotes for the MEFV gene c.442G > C (p.Glu148Gln) variant in the gnomAD database.
Recent scholars have discovered that among FMF patients, whether p.E148Q/p.E148Q or p.E148Q/p.E148Q + V726A, Druze individuals comprise a higher proportion compared to other ethnic groups, suggesting a connection between E148Q variant pathogenicity and ethnicity (15). In this PASH syndrome patient, although whole exome sequencing only detected homozygous MEFV (p.E148Q) variant, considering the clinical complexity and the relatively weak pathogenicity of this MEFV variant, there may be undiscovered synergistic variants (such as non-coding variants, epigenetic regulatory abnormalities, or polygenic cumulative effects). Certain exons of some genes (such as NCSTN, PSENEN) may not have been effectively captured due to high GC content or complex structures. Furthermore, smoking may exacerbate the pro-inflammatory effects of E148Q through epigenetic silencing of anti-inflammatory genes (such as IL-10) (16). These factors may explain why the patient ultimately presented with PASH rather than typical FMF. Whether E148Q is the primary driving factor or merely serves as a modifier of inflammatory response requires more comprehensive genomic analysis and functional experiments to elucidate the complete mechanism.
This study has several limitations. First, as a single-case analysis without familial segregation studies, establishing definitive genotype–phenotype associations remains challenging. Second, whole-exome sequencing has inherent limitations, being unable to detect potentially pathogenic variants in non-coding regulatory regions and intronic sequences. Additionally, although we identified the MEFV E148Q variant, and previous studies suggest that MEFV variants may cause neutrophil dysfunction (6) (providing important insights into the pathogenesis of PASH syndrome), our study lacks functional validation to determine whether this variant contributes to disease development through effects on neutrophil function. Future studies should include larger cohorts and multicenter collaborations to address these limitations, while conducting more comprehensive experimental research to clarify the pathogenic mechanisms of the E148Q variant in PASH syndrome.
Data availability statement
The whole-exome sequencing data generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) under BioProject accession number PRJNA1297302.
Ethics statement
Ethical approval was not required for the studies involving humans because case reports typically only require ethics approval if the case study involves some form of intervention that deviates from standard practice. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant(s)/patient(s) for the publication of this case report.
Author contributions
QQ: Writing – original draft, Writing – review & editing. CC: Resources, Supervision, Writing – review & editing. JL: Writing – review & editing. BL: Writing – review & editing. JS: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the Science and Technology Plan of Guangxi Province of China (AB24010134, AD23026325) and National Key Research and Development Program of China (2022YFC2504800).
Acknowledgments
We would like to thank the patient for their participation in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Braun-Falco, M, Kovnerystyy, O, Lohse, P, and Ruzicka, T. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH)--a new autoinflammatory syndrome distinct from PAPA syndrome. J Am Acad Dermatol. (2012) 66:409–15. doi: 10.1016/j.jaad.2010.12.025
2. Yan, A, Gallardo, M, Savu, A, and Kaffenberger, B. Pyoderma gangrenosum, acne, and suppurative hidradenitis (PASH) syndrome: a single-institution case series with a focus on management. Arch Dermatol Res. (2024) 316:397. doi: 10.1007/s00403-024-03125-7
3. Feola, H, Luna Cian, R, Mancinelli, MC, Cordoba, LM, and Crespi, C. PASH syndrome: a novel MEFV mutation variant with excellent response to adalimumab in a refractory case. Int J Dermatol. (2024) 64:183–5. doi: 10.1111/ijd.17330
4. Maronese, CA, Moltrasio, C, and Marzano, AV. Hidradenitis suppurativa-related autoinflammatory syndromes: an updated review on the clinics, genetics, and treatment of pyoderma gangrenosum, acne and suppurative hidradenitis (PASH), pyogenic arthritis, pyoderma gangrenosum, acne and suppurative hidradenitis (PAPASH), synovitis, acne, pustulosis, hyperostosis and osteitis (SAPHO), and rarer forms. Dermatol Clin. (2024) 42:247–65. doi: 10.1016/j.det.2023.12.004
5. Sun, Q, Broadaway, KA, Edmiston, SN, Fajgenbaum, K, Miller-Fleming, T, Westerkam, LL, et al. Genetic variants associated with hidradenitis Suppurativa. JAMA Dermatol. (2023) 159:930–8. doi: 10.1001/jamadermatol.2023.2217
6. Ozen, S, Demirkaya, E, Amaryan, G, Kone-Paut, I, Polat, A, Woo, P, et al. Results from a multicentre international registry of familial Mediterranean fever: impact of environment on the expression of a monogenic disease in children. Ann Rheum Dis. (2014) 73:662–7. doi: 10.1136/annrheumdis-2012-202708
7. Vural, S, Gundogdu, M, Gokpinar Ili, E, Durmaz, CD, Vural, A, Steinmuller-Magin, L, et al. Association of pyrin mutations and autoinflammation with complex phenotype hidradenitis suppurativa: a case-control study. Br J Dermatol. (2019) 180:1459–67. doi: 10.1111/bjd.17466
8. Marzano, AV, Damiani, G, Ceccherini, I, Berti, E, Gattorno, M, and Cugno, M. Autoinflammation in pyoderma gangrenosum and its syndromic form (pyoderma gangrenosum, acne and suppurative hidradenitis). Br J Dermatol. (2017) 176:1588–98. doi: 10.1111/bjd.15226
9. Topaloglu, R, Ozaltin, F, Yilmaz, E, Ozen, S, Balci, B, Besbas, N, et al. E148Q is a disease-causing MEFV mutation: a phenotypic evaluation in patients with familial Mediterranean fever. Ann Rheum Dis. (2005) 64:750–2. doi: 10.1136/ard.2004.026963
10. Van Gijn, ME, Ceccherini, I, Shinar, Y, Carbo, EC, Slofstra, M, Arostegui, JI, et al. New workflow for classification of genetic variants' pathogenicity applied to hereditary recurrent fevers by the international study Group for Systemic Autoinflammatory Diseases (INSAID). J Med Genet. (2018) 55:530–7. doi: 10.1136/jmedgenet-2017-105216
11. Saint-Georges, V, Peternel, S, Kastelan, M, and Brajac, I. Tumor necrosis factor antagonists in the treatment of pyoderma Gangrenosum, acne, and Suppurative hidradenitis (PASH) syndrome. Acta Dermatovenerol Croat. (2018) 26:173–8.
12. Join-Lambert, O, Duchatelet, S, Delage, M, Miskinyte, S, Coignard, H, Lemarchand, N, et al. Remission of refractory pyoderma gangrenosum, severe acne, and hidradenitis suppurativa (PASH) syndrome using targeted antibiotic therapy in 4 patients. J Am Acad Dermatol. (2015) 73:S66–9. doi: 10.1016/j.jaad.2015.07.040
13. Ulu, K, Coskuner, T, Baykal, GO, Yarar, MH, Yigit, RE, Turkmen, S, et al. The enigma of familial Mediterranean fever: phenotypic characterization of patients harbouring variants of uncertain significance. Rheumatology (Oxford). (2025) 64:2902–9. doi: 10.1093/rheumatology/keaf139
14. Omenetti, A, Carta, S, Delfino, L, Martini, A, Gattorno, M, and Rubartelli, A. Increased NLRP3-dependent interleukin 1beta secretion in patients with familial Mediterranean fever: correlation with MEFV genotype. Ann Rheum Dis. (2014) 73:462–9. doi: 10.1136/annrheumdis-2012-202774
15. Orouk Awaad, E, Khoury, L, van Straalen, JW, Miller-Barmak, A, Gazitt, T, Haddad-Haloun, J, et al. E148Q variant: a familial Mediterranean fever-causing mutation or a sequence variant? Eur J Pediatr. (2024) 183:4499–506. doi: 10.1007/s00431-024-05690-5
Keywords: PASH syndrome, pyoderma gangrenosum, acne, suppurative hidradenitis, MEFV variant, E148Q, adalimumab
Citation: Qin Q, Cao C, Liang J, Li B and Su J (2025) Case Report: Diagnostic difficulties, treatment, and association of the MEFV E148Q variant in a patient with PASH syndrome. Front. Med. 12:1557540. doi: 10.3389/fmed.2025.1557540
Edited by:
Laura Crisponi, National Research Council (CNR), ItalyReviewed by:
Quan Sun, University of North Carolina at Chapel Hill, United StatesCristina Mendez-Vidal, Virgen del Rocío University Hospital, Spain
Copyright © 2025 Qin, Cao, Liang, Li and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaguang Su, MTM4NzgxMDY5NjVAMTYzLmNvbQ==