Abstract
Past studies on common mutant aldosterone-producing adenomas (APAs) had found genotype–phenotype correlations associated with histological appearance. Most of these studies did not perform CYP11B2-guided sequencing of APAs or sequencing for all the currently known aldosterone-driver genes. Hence, misinterpretation of the genotype–phenotype correlations could have occurred. Herein, we aimed to identify the genotype–phenotype correlations associated with the histopathology of the different mutant APAs utilizing CYP11B2-guided sequencing. A total of 33 APAs with confirmed aldosterone-driver mutation (17 KCNJ5 mutant APAs, 8 ATP1A1 mutant APAs, 6 CACNA1D mutant APAs, and 2 CTNNB1 mutant APAs) were immunohistochemically stained using H&E, CYP17A1, CYP11B2, KCNJ5, Ki67, β-catenin, and LHCGR antibody. Interestingly, APAs with a p.Thr41Ala CTNNB1 mutation also harbored a p.Val1373Met CACNA1D mutation. The CTNNB1 double mutant APAs had less expression of CYP17A1 and larger quantities of spironolactone bodies than a single mutant APA with a p.Ser45Phe CTNNB1 mutation. However, both CTNNB1 mutant APAs displayed diffuse active β-catenin expression with prominent nuclear staining that reflects the constitutive activation of the Wnt/β-catenin signaling pathway (p = 0.016 compared to other genotypes) but no significant increase in LHCGR. KCNJ5 mutant APAs displayed distinct existence of atypical cells (6 of the 17 KCNJ5 mutant APAs), whereas CACNA1D mutant APAs had frequent presentations of spironolactone bodies (4 of the 6 CACNA1D mutant APAs), and ATP1A1 mutant APAs had significantly higher Ki67 score than KCNJ5 mutant APAs (p = 0.020). The results of this study support the notion that CYP11B2-guided sequencing of all currently known aldosterone-driver genes can fine-tune existing genotype–phenotype correlations in histopathological profiles.
1 Introduction
Primary aldosteronism (PA), also known as Conn’s syndrome, is one of the most common and potentially curable forms of secondary hypertension, also referred to as endocrine hypertension (1). PA can be caused by the autonomous aldosterone production in one or both adrenal glands, leading to elevated aldosterone levels despite suppressed renin activity (2). In nearly half of PA cases, the identification of a unilateral aldosterone-producing adenoma (APA) or aldosterone-producing nodule (APN) offers the opportunity for a complete cure of hypertension through the removal of the autonomous aldosterone-producing lesion (3). Studies on excised tissue have identified gain-of-function somatic mutations as the cause of autonomous aldosterone production (4–8). The most frequent mutations were found in the genes KCNJ5, CACNA1D, and ATP1A1 (8–11). In addition to the more common aldosterone-driver mutations, past studies have also identified recurring aldosterone-driver somatic mutations in the genes ATP2B3, CTNNB1 (with or without GNA11/GNAQ), CACNA1H, CLCN2, CADM1, and SLC30A1 in APAs (12–18).
The majority of the aldosterone-driver genes encode for ion channels or pumps (KCNJ5, CACNA1D, CACNA1H, ATP1A1, ATP2B3, and SLC30A1) that are involved in regulating intracellular ion homeostasis and plasma membrane potential (9). In general, mutations of these genes lead to the depolarization of the cell membrane due to the impairment of ion transport that activates voltage-gated Ca2+ channels. This alteration consequently increases intracellular calcium levels and promotes the transcription of aldosterone synthase (CYP11B2), leading to excessive aldosterone biosynthesis. Thus, adrenal immunohistochemistry (IHC) for CYP11B2 could detect cells that can synthesize aldosterone, enabling non-functional nodules and adenomas co-existing with those that synthesize aldosterone to be distinguished. This has only been made possible in the last decade, with the successful development of a specific monoclonal antibody for CYP11B2 that can differentiate from the highly homologous CYP11B1 (19). To note, CYP11B2-guided sequencing has a higher detection rate of aldosterone-driver mutations than non-CYP11B2-guided sequencing (20).
Past studies on the excised tissue of mutant APAs have found genotype–phenotype correlations associated with the morphologic appearance (4–6, 8, 21). However, many of these studies did not perform CYP11B2-guided sequencing or sequencing for all the currently known aldosterone-driver genes (most performed was KCNJ5 genotyping). Thus, one could postulate that past studies that had not performed CYP11B2-guided sequencing or performed genotyping of all aldosterone-driver genes could have misinterpreted the genotype–phenotype correlations observed.
Herein, this study aimed to identify the genotype–phenotype correlations associated with the histology of APAs with different mutations utilizing CYP11B2-guided sequencing. Correlation with histological features was performed using IHC staining of CYP17A1 and KCNJ5. CYP17A1 is usually not present in the ZG as its main physiological expression is in the zona fasciculata (ZF) and zona reticularis (ZR) in the adrenal cortex where it converts progesterone to 17α-(OH) progesterone to produce glucocorticoids, or pregnenolone to 17α-(OH) pregnenolone to produce sex steroid precursors (22). KCNJ5 is expressed more in the ZG, though its exact role in the adrenal gland is yet to be defined (23). The proliferation marker Ki67 was used to estimate the proliferation rate of the mutant adenoma. Furthermore, β-catenin nuclear expression was utilized as a measure of constitutive activation of Wnt/β-catenin signaling, and LHCGR expression was measured as CTNNB1. Mutant APAs that harbored a GNA11 or GNAQ aldosterone-driver mutation have been reported to have increased LHCGR expression and onset of disease during high periods of HCG (4, 17, 24).
2 Methods
2.1 Sample acquisition
Adrenal tissues were collected from 66 Czech patients with unilateral PA who underwent adrenalectomy at the University Hospital Hradec Kralove. All the patients gave written informed consent. The study was approved by the University Hospital Hradec Kralove Ethics Committee (201504 S22P) and the National University of Malaysia Research Ethics Committee (FF-2015-092). Case detection and subtype identification were in accordance with local guidelines described in detail previously (25, 26). Confirmation and lateralization of PA were performed as detailed in Supplementary material.
2.2 Genotyping of somatic mutations in APAs
APA lesion was identified in formalin-fixed paraffin-embedded (FFPE) adrenal sections of unilateral PA patients through positive immunohistochemistry (IHC) staining of CYP11B2. CYP11B2 IHC-guided DNA extraction of adenoma tissue was then performed on 3–5 serial sections of 10 μm thickness using a commercial DNA extraction kit to ensure the extraction of APA tissue that expressed aldosterone synthase. Genotype of the APA DNA was determined through targeted sequencing of aldosterone-driver mutation hotspots as previously reported (8, 27) or through commercial targeted sequencing of aldosterone-driver genes (KCNJ5, ATP1A1, ATP2B3, CACNA1D, and CTNNB1) using the DNBSEQ platform powered by combinatorial Probe-Anchor Synthesis (cPAS) and improved DNA Nanoballs (DNB) technology (BGI Genomics Co., Ltd., Hong Kong). The cPAS chemistry works by linking a fluorescent probe to a DNA anchor on the DNB, followed by high-resolution digital imaging. Sequencing-derived raw image files were processed by a DNBSEQ base-calling software for base-calling with default parameters, and the sequence data of each individual was generated as paired-end reads, which were defined as “raw data” and stored in FASTQ format.
A total of 33 APAs had a known aldosterone-driver mutation in the aldosterone-driver genes KCNJ5, ATP1A1, CACNA1D, and CTNNB1, with sufficient material for further immunohistochemical analysis. A total of 24 had been characterized previously (8, 27), of which 3 samples initially classified as wild-type by targeted sequencing of aldosterone-driver mutation hotspots were, in this study, re-classified as having a known aldosterone-driver mutation based on targeted sequencing of aldosterone-driver genes using the DNBSEQ platform. Mutations identified through the DNBSEQ platforms were then annotated and filtered using Polymorphism Phenotyping v2 (PolyPhen-2 v2.2.2 build r394) (28) and Sorting Intolerant From Tolerant (SIFT v1.03) (29). The combined prediction results were considered evidence supporting a deleterious effect of a variant. Variants were also characterized using the Mutation Assessor (30), the Mutation Taster (31), and the Combined Annotation Dependent Depletion (CADD) score (32).
2.3 Immunohistochemistry (IHC) staining
IHC staining was performed on FFPE tissue sections using the detection system EnVision FLEX+, Mouse, High pH (Dako, Denmark) according to the manufacturer’s recommendations. IHC staining for CYP11B2 was performed on all samples, and then sections that had a positive nodule with CYP11B2 were stained with other antibodies (Supplementary Table S1). IHC staining of CYP11B2 and β-catenin was performed using selective mouse monoclonal antibodies, whereas staining of CYP17A1, KCNJ5, and LHCGR was performed using rabbit polyclonal antibodies. The antibodies for CYP11B2 and CYP17A1 were provided as a gift by Prof Celso Gomez-Sanchez (University of Mississippi Medical Center, USA). Staining of Ki67 was performed using a commercial ready-to-use mouse monoclonal antibody (Clone MIB-1, Catalog No. IR62661, Dako, Denmark). Details of IHC staining protocols are described in Supplementary Table S1. Positive control tissues were stained with every batch, ensuring staining was specific and selective in the IHC experiments (Supplementary Figure S1).
Scoring of IHC staining for CYP11B2, CYP17A1, KCNJ5, and LHCGR was performed genotype-blinded, with a score of 0 representing 0% expression to 10 representing 100% expression (further details of the score are provided in Supplementary Table S2; Supplementary Figures S2–S6). As for β-catenin immunostaining, the level of nuclear staining was determined by scoring as detailed in Supplementary Table S3. Intense Ki67 nuclei staining of representative fields of the APAs’ histology was photographed and quantified using Image J with a previously published semi-automated image analysis of high-contrast tissue areas method (detailed in Supplementary material; Supplementary Figure S7) (27, 33). The percentage of atypical cells and spironolactone bodies was determined from three representative images of the APAs’ histology using hematoxylin and eosin-stained (H&E) sections.
2.4 Statistical analysis
All data are presented as mean±SEM for the indicated number of experiments (n) unless specified otherwise as median (IQR). Statistical analysis was performed using standard statistical software, and the statistical significance was set at a p-value of <0.05. Assessment of the normality of data was analyzed using the Shapiro–Wilk test, in which comparisons of normally distributed data were performed using the independent t-test. Non-normally distributed data and ordinal data were compared using the Mann–Whitney U-test. Comparison of categorical data was analyzed using Fisher’s exact test.
3 Results
3.1 CYP11B2-guided sequencing of APAs
Of the 33 APA samples with sufficient material for genotype–phenotype analysis, 17 APAs had an aldosterone-driver mutation in KCNJ5, 8 APAs in ATP1A1, 6 APAs in CACNA1D, and 2 APAs in CTNNB1. Of particular interest, one CTNNB1 mutant APAs harbored two known aldosterone-driver mutations, the p.Thr41Ala CTNNB1 mutation and the p.Val1373Met CACNA1D, which was confirmed by Sanger Sequencing (Figure 1A). The variants replaced an A with a G (c.121A > G) in CTNNB1, resulting in the p.Thr41Ala substitution, and replaced a G with an A (c.4117G > A) in CACNA1D, resulting in the p.Val1373Met substitution. These variants were somatic, only present in the CYP11B2-positive APA tissue but not in the adjacent normal adrenal (Figure 1A). The SIFT score of both mutations was close to 0 (a SIFT score of 0–0.05 predicts an intolerable mutation that can affect protein function), whereas PolyPhen-2 was close to 1 (which also predicts a deleterious variant) (Supplementary Table S4). Similarly, Mutation Assessor with an FI score greater than 2.00 (CTNNB1; 2.68 (M) and CACNA1D; 3.62 (H)) and Mutation Taster scores closer to 1 (CTNNB1; 1 (D) and CACNA1D; 1 (D)) predicted both these mutations as ‘deleterious’ (Supplementary Table S5). These values support the possibility that these CACNA1D and CTNNB1 mutations are both likely pathogenic for APA pathology.
Figure 1

(A) DNA of patient 1 with mutations CTNNB1 T41A and CACNA1D V1373M, and patient 2 with mutation CTNNB1 S45F. Mutations were found in the adenoma but not the adjacent adrenal gland. APA, aldosterone-producing adenoma. (B) Immunohistochemical characterization of APAs harboring a CTNNB1 and CACNA1D pathogenic mutation or (C) just a CTNNB1 pathogenic mutation. Scale bars, 2 mm. (D) Comparison of β-catenin, LHCGR and Ki67 IHC staining of CTNNB1 and CACNA1D double mutant APA and (E)CTNNB1 single mutant APA. Orange arrows indicate nuclear staining.* CTNNB1 and CACNA1D.
Clinical characteristics of patients’ pre-adrenalectomy for each genotype are compared in Table 1. Although there was no significant difference in gender by genotype, the majority of female PA patients (7 of 9) harbored a KCNJ5 mutant APAs. This high prevalence mutation also significantly occurred in younger patients, in contrast to the other somatic mutations that were frequently detected in older male patients (p = 0.002; Table 1). Moreover, patients with KCNJ5 mutant APAs (n = 17) were on a significantly lower number of antihypertensive medications compared to other mutants (n = 16; 3 ± 0.4 vs. 5 ± 0.3; p = 0.016), indicating less resistant hypertension, and having a more pronounced suppression of renin (plasma active renin 2.9 ± 0.1 vs. 3.5 ± 0.3, p = 0.013). Other clinical characteristics listed in Table 1 were not significantly different between patients with or without a KCNJ5 aldosterone-driver mutation. To note, in this cohort, the significant age difference was mainly driven by ATP1A1 mutation, whereas the difference in plasma active renin was mainly driven by CACNA1D mutation (Table 1).
Table 1
| Characteristics | KCNJ5 (n = 17) | ATP1A1 (n = 8) | CACNA1D (n = 6) | CTNNB1 double mutants (n = 1) | CTNNB1 single mutant (n = 1) |
p-value KCNJ5 vs. others |
p-value KCNJ5 vs. ATP1A1 | p-value KCNJ5 vs. CACNA1D | p-value ATP1A1 vs. CACNA1D |
|---|---|---|---|---|---|---|---|---|---|
| Sex (male:female) | 10:7 | 7:1 | 6:0 | 1:0 | 0:1 | 0.118 | 0.205 | 0.124 | 1.000 |
| Age | 43 ± 2 | 59 ± 3 | 48 ± 5 | 53 | 62 | 0.002 | 0.000 | 0.317 | 0.038 |
| SBP, mmHg | 142 ± 5 | 147 ± 11 | 141 ± 9 | 130 | 128 | 0.934 | 0.603 | 0.933 | 0.687 |
| DBP, mmHg | 88 ± 3 | 86 ± 6 | 91 ± 7 | 86 | 88 | 0.946 | 0.669 | 0.657 | 0.575 |
| Number of AH meds | 3 ± 0.4 | 4 ± 0.5 | 5 ± 0.6 | 5 | 5 | 0.016 | 0.133 | 0.056 | 0.586 |
| Serum Na, mmol/L | 140 ± 0.6 | 140 ± 0.8 | 141 ± 1.6 | 143 | 142 | 0.667 | 0.936 | 0.877 | 0.942 |
| Serum K, mmol/L | 3.7 ± 0.1 | 3.8 ± 0.2 | 4.0 ± 0.3 | 3.7 | 3.9 | 0.345 | 0.655 | 0.300 | 0.524 |
| Serum aldosterone, pmol/La | 1,153 ± 132 | 1828 ± 739 | 886 ± 177 | 520 | 470 | 0.358 | 0.884 | 0.293 | 0.302 |
| Plasma active renin, ng/Lb | 2.9 ± 0.1 | 3.5 ± 0.5 | 3.7 ± 0.4 | 3.6 | 3.2 | 0.013 | 0.168 | 0.011 | 0.335 |
Clinical characteristics of patients pre-adrenalectomy.
Data are shown as mean±SEM. Statistics on continuous variables were performed with the independent t-test for normally distributed data, and the Mann–Whitney U-test for non-normally distributed data. A comparison of sex between genotypes was performed using Fisher’s exact test. DBP, diastolic blood pressure; SBP, systolic blood pressure; AH meds, antihypertensive medications; Na, sodium; K, potassium; NS, not significant (p-value > 0.05). Statistical comparison of the clinical characteristics for CACNA1D did not include patients who had CTNNB1 and CACNA1D mutants.
Serum aldosterone values in ng/dL were converted to pmol/L using a conversion factor of 27.74 (44).
1 plasma active renin data from a patient with KCNJ5 mutation was excluded from the analysis as the value is affected by spironolactone due to the patient having severe hypertension complicated by aortic dissection, and treatment withdrawal is not possible.
3.2 Clinical presentation of the double mutant APA
The CACNA1D/CTNNB1 double mutant APAs were adrenalectomized from a 53-year-old male patient. He had been referred for resistant arterial hypertension and a history of hypokalemia (serum potassium of 3.7 mmol/L, Table 1). A screening test for PA was performed when the patient was on verapamil and doxazosin with potassium supplementation. The aldosterone–renin ratio measured then was 80. The saline infusion confirmatory test was positive, with serum aldosterone measuring 200 pmoL/L at the end of infusion after sodium loading. AVS was then performed, and unilateral PA was diagnosed. A computed tomography scan was interpreted as adrenal hyperplasia, although histopathological analysis of the excised adrenal identified a 10-mm nodule (Figure 1B). Post-adrenalectomy of the affected adrenal gland, the blood pressure was controlled (124/84 mmHg) on doxazosin 4 mg, amlodipine 5 mg, and telmisartan 80 mg (Supplementary Table S6).
For comparison, APAs detected to have a single mutation of CTNNB1 (p.S45F) were used. The variant replaced a C with a T (c.134C > T) as shown in Figure 1A. Mutant APAs were adrenalectomized from a 62-year-old woman. She also had resistant arterial hypertension and a history of hypokalemia. A screening test performed while the patient was on verapamil and doxazosin, with potassium supplementation, measured an aldosterone–renin ratio of 223. A saline infusion confirmatory test measured serum aldosterone of 1,260 pmoL/L at the end of infusion (after sodium loading). AVS was performed, and unilateral PA was diagnosed. Like double mutant APAs, the histopathological analysis identified an 11-mm nodule, although the computed tomography scan reported adrenal hyperplasia (Figure 1C). She underwent laparoscopic adrenalectomy of the affected adrenal, which improved her blood pressure (110/66 mmHg), though she still requires some antihypertensive medications (amlodipine 5 mg and telmisartan 80 mg) (Supplementary Table S6).
3.3 Histopathologic features of APAs somatic mutation
The histopathologic features of each variant were identified using H&E staining and immunohistochemical detection of CYP17A1, CYP11B2, KCNJ5, Ki67, β-catenin, and LHCGR (Supplementary Figures S8, S9). All the mutant APAs exhibited moderate to high expression of CYP11B2, with significantly higher IHC scores in ATP1A1 and CACNA1D mutant APAs than KCNJ5 mutant APAs (7 [6–9] and 8 [6–9] vs. 6 [5–6]; p < 0.05; Table 2). In contrast to the KCNJ5 mutant APAs, ATP1A1, CACNA1D, and CTNNB1 mutant APAs had a significantly lower score of CYP17A1 expression (2 [1–4] vs. 7 [6–7]; p ≤ 0.001), with higher score of KCNJ5 (9 [8–10] and 9 [8–9] vs. 4 [4–6]; p < 0.001; Table 2). Furthermore, the non-KCNJ5 mutant APAs had a significantly higher Ki67 score than KCNJ5 mutant APAs, which was mainly driven by ATP1A1 mutation (0.061 ± 0.007 vs. 0.042 ± 0.005; p = 0.019; Table 2). Yet, the average adenoma size, based on the diameter, was significantly larger in KCNJ5 mutant APAs compared to non-KCNJ5-mutant adenomas (13 ± 0.8 vs. 9 ± 0.9; p = 0.001; Table 2). To note, CACNA1D mutant APAs had a significantly lower β-catenin IHC score than KCNJ5 mutant APAs (2 [1–2] vs. 2 [2–3]; p = 0.026; Table 2). However, this lower score in the CACNA1D mutant APAs represents less nuclear staining rather than lower cytoplasmic expression of β-catenin compared to KCNJ5 mutant APAs. The positive nuclear β-catenin staining indicates that the Wnt/β-catenin signaling was active in these APAs. Whereas for the LHCGR, no significant difference was seen between groups (LHCGR score of KCNJ5 mutant APAs: 2 (1–4), n = 7; ATP1A1 mutant APAs: 3 (2–4), n = 3; CACNA1D mutant APAs: 3 (2–3), n = 3; CTNNB1 double mutant APAs: 4, n = 1; CTNNB1 single mutant APAs: 4, n = 1); however, the CTNNB1 double and single mutant APAs did have the highest score.
Table 2
| Characteristics | KCNJ5 (n = 17) | ATP1A1 (n = 8) | CACNA1D (n = 6) | CTNNB1 double mutants (n = 1) | CTNNB1 single mutant (n = 1) | p-value KCNJ5 vs. others | p-value KCNJ5 vs.ATP1A1 | p-value KCNJ5 vs.CACNA1D | p-value ATP1A1 vs.CACNA1D |
|---|---|---|---|---|---|---|---|---|---|
| CYP11B2 score | 6 (5–6) | 7 (6–9) | 8 (6–9) | 4 | 4 | 0.005 | 0.001 | 0.023 | 0.895 |
| CYP17A1score | 7 (6–7) | 2 (1–4) | 2 (1–4) | 1 | 3 | 0.000 | 0.000 | 0.001 | 0.838 |
| KCNJ5 score | 4 (4–6) | 9 (8–10) | 9 (8–9) | 7 | 8 | 0.000 | 0.000 | 0.000 | 0.946 |
| Ki67 scorea | 0.042 ± 0.005 | 0.063 ± 0.008 | 0.051 ± 0.014 | 0.080 | 0.084 | 0.019 | 0.020 | 0.438 | 0.400 |
| β-catenin scorea | 2 (2–3) | 2 (1–3) | 2 (1–2) | 4 | 4 | 0.220 | 0.163 | 0.026 | 0.266 |
| Size of the adenoma, mm | 13 ± 0.8 | 9 ± 1.5 | 8 ± 1.2 | 10 | 11 | 0.001 | 0.013 | 0.002 | 0.521 |
| Atypical cells (Absent:Present) | 6:11 | 8:0 | 6:0 | 1:0 | 1:0 | 0.000 | 0.003 | 0.014 | ND |
| Spironolactone bodies (Absent:Present) | 17:0 | 8:0 | 2:4 | 0:1 | 0:1 | 0.007 | ND | 0.002 | 0.015 |
Comparison of cellular histochemistry by the APA genotype.
Normal variables are shown as mean±SEM, and non-normal variables are shown as median (25%–75%). Statistics were performed with the independent sample t-test, Mann–Whitney U-test, and Fisher’s exact test. ND, Not detected.
KCNJ5 had two missing data for Ki67 score, ATP1A1 had one missing data for Ki67 score, CACNA1D had one missing data for Ki67 score, and two missing data for β-catenin score.
Comparing the CTNNB1 mutant APAs, both the double mutant and single mutant APAs exhibited comparable features in H&E staining, as well as similar expression of CYP11B2, KCNJ5, Ki67, β-catenin, and LHCGR (Table 2, Figures 1B–E, and Supplementary Figure S10). In both APAs harboring CTNNB1 mutations, the β-catenin was diffusely expressed, demonstrating both cytoplasmic and strong nuclear expression, compared to the other mutant APAs that displayed weak nuclear and cytoplasmic β-catenin expression (4 vs. 2 [2–3]; p = 0.016; Table 2 and Supplementary Figure S10). To note, CTNNB1 and CACNA1D double mutant APAs did have a lower CYP17A1 score than single CTNNB1 mutant APAs (1 vs. 3, Table 2 and Figures 1D,E).
Another significant observation from the immunohistochemistry analysis is the histopathologic findings of spironolactone bodies and atypical cells (Table 2; Supplementary Table S7). The spironolactone bodies, detected by H&E staining as round, laminated cytoplasmic inclusions surrounded by a clear halo, were often found in cells with a low cytoplasmic to nucleus ratio, similar to those of the zona glomerulosa of the normal adrenal cortex (Figure 2A). These spironolactone bodies were observed frequently in CACNA1D and CTNNB1 mutant APAs (Table 2; Supplementary Table S7). Interestingly, compared to CTNNB1 single mutant APAs, spironolactone bodies were found in larger quantities in APAs with both CTNNB1 and CACNA1D double mutations (Figure 2A). Conversely, atypical cells with large nuclei and prominent nucleoli were found only in KCNJ5 mutant APAs, while other mutant APAs had monomorphic bland nuclei (Table 2 and Figure 2A). To note, ZG with positive CYP11B2 staining was detectable in the adrenals adjacent to ATP1A1 mutant APAs (n = 2) and KCNJ5 mutant APAs (n = 4; Supplementary Figure S11). The ZG region was determined based on the lack of CYP17A1 expression and intense KCNJ5 pericapsular immunohistochemical positivity. Interestingly, ZG hyperplasia had a trend to be thicker in KCNJ5 mutant APAs (n = 8; 0.32 ± 0.03 mm) compared to other genotypes (n = 15; 0.28 ± 0.02 mm; p = 0.137, Figure 2B).
Figure 2
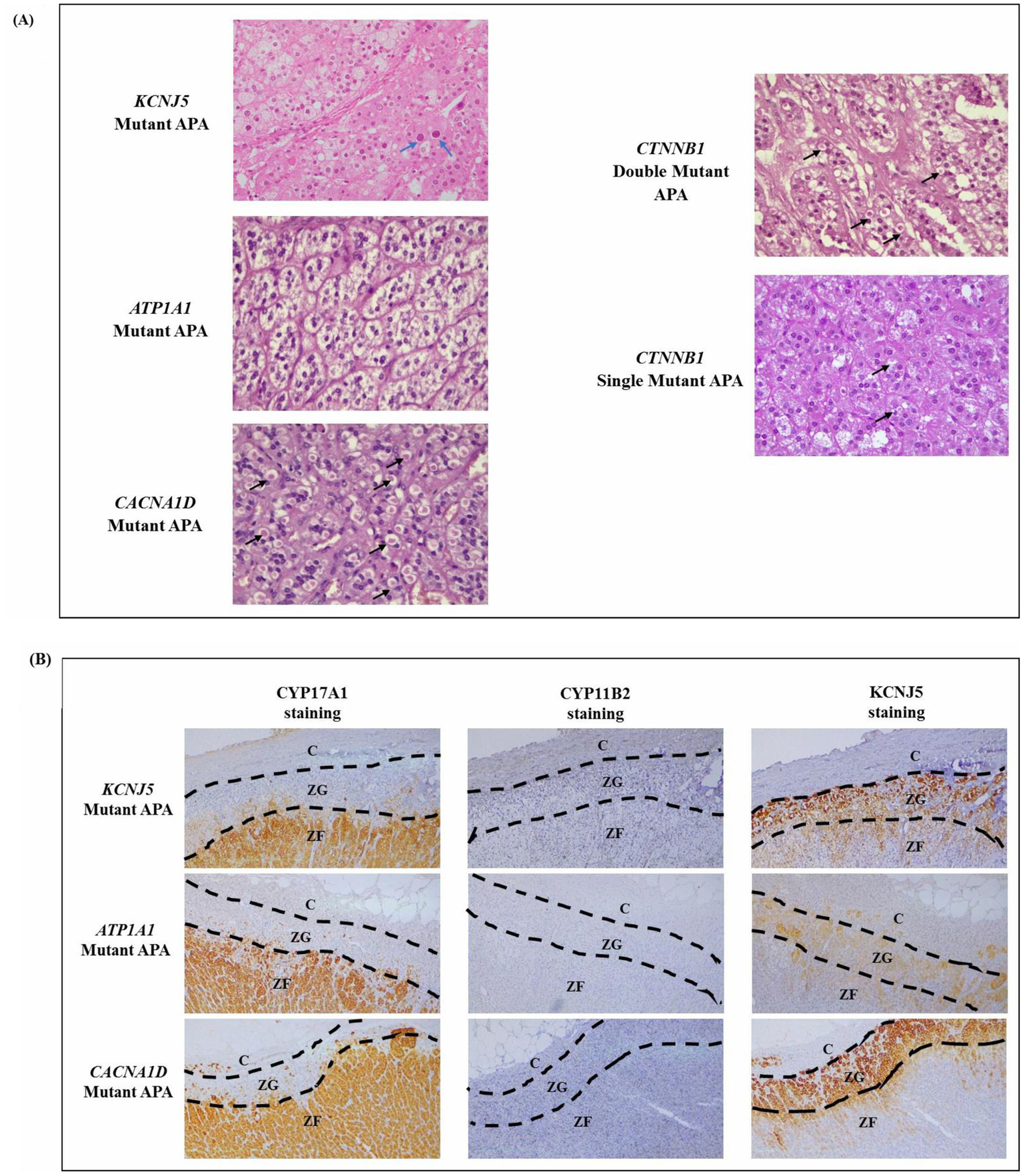
(A) Representative H&E staining of mutant APAs. Black arrows point to spironolactone bodies, and blue arrows point to atypical nuclei. (B) Zona glomerulosa hyperplasia as determined by a lack of CYP17A1 IHC staining and intense KCNJ5 IHC staining.
4 Discussion
Herein, two APAs with pathogenic CTNNB1 mutations were found, with one also harboring a pathogenic CACNA1D mutation (p.V1373M). Somatic mutations in CTNNB1 have been scarcely identified in PA patients, suggesting a low prevalence (4, 15). The majority of APAs with CTNNB1 pathogenic mutations occur mutually exclusive to other aldosterone-driver somatic mutations, though there were rare reports of CTNNB1 mutations existing with the co-driver mutations in GNA11 and GNAQ that activated aldosterone production (17, 20, 34). This is different from the finding of micronodules harboring a pathogenic aldosterone-driving mutation in a separate gene from that of APAs in the same adrenal (20, 34). The rarity of APAs with double pathogenic mutations (in this case, CTNNB1 and CACNA1D) makes their immunohistochemical and biochemical phenotype of interest. The identified variants in CTNNB1, p.T41A, and CACNA1D, p.V1373M, have both been previously found separately in APAs (4, 7, 35). Supporting further its pathogenic nature, several algorithm tools (SIFT, PolyPhen-2, Mutation Assessor, and MutationTaster) identified both missense variants as damaging with functional consequences. Moreover, the sequencing result of the patient’s germline DNA confirmed these variants to be somatic. Therefore, the coexistence of these variants should be considered the presence of two independent concurrent driver mutations harbored by APAs.
This study characterized the histopathologic spectrum of identified somatic mutations in APAs using H&E staining and CYP17A1, CYP11B2, KCNJ5, Ki67, β-catenin, and LHCGR IHC. When comparing the CTNNB1 mutant APAs, both single and double mutant APAs had a seemingly similar histopathologic phenotype, except that CTNNB1 single mutant APAs had a higher expression of CYP17A1 compared to CTNNB1 double mutant APAs. This is in line with previous reports, which showed that APAs harboring the CTNNB1 mutation could display heterogeneous CYP11B2 and CYP11B1 expression (15).
CTNNB1 expression is normally restricted to the ZG in the adrenal gland (36). Nuclear expression of CTNNB1 reflects the constitutive activation of the Wnt/β-catenin signaling pathway (37). During constitutive activation of Wnt/β-catenin signaling in the ZG, translocation of β-catenin to the nucleus is thought to increase expression of nuclear receptors NURR1 and NUR77, responsible for increased transcription of downstream targets including T-cell factor/lymphoid enhancer factor (TCF/LEF-1), CYP21, the Angiotensin I receptor (ATR1), and CYP11B2 (24, 38, 39). Activation of the associated Wnt/β-catenin pathway has also been reported to lead to a functional block in the ability of the ZG cells to transdifferentiate into ZF cells, leading to hyperplasia (40). In this study, APAs harboring CTNNB1 mutations (both single and double) displayed diffuse active β-catenin expression higher than other mutant APAs with prominent nuclear staining. The finding of these CTNNB1 mutant APAs along with the high expression of β-catenin, both in the nucleus and the cytoplasm, highlights the central role of the Wnt/β-catenin signaling pathway in the development of APAs. Thus, targeting this pathway may be an important approach in the treatment of unilateral PA.
Constitutive activation of the Wnt signaling pathway in ZG-like adenomatous cells has also been postulated to lead to dedifferentiation toward their common adrenal–gonadal precursor cell type and lead to aberrant expression of gonadal receptor LHCGR and/or GnRHR (4). However, both single- and double-mutant APAs did not exhibit any significant difference in the expression of LHCGR, unlike the previous study that reported LHCGR was upregulated more than 10-fold in double-mutant APAs harboring CTNNB1 and GNA11/GNAQ mutations (17). Nevertheless, ATP1A1 and CACNA1D mutant APAs did have low expression of β-catenin and LHCGR IHC staining, along with low CYP17A1 and high CYP11B2 and KCNJ5 expression, as previously documented (27).
Clinically, both patients with a CTNNB1 and CACNA1D double mutant APAs and the single CTNNB1 mutant APAs have not achieved complete post-adrenalectomy resolution of hypertension. This contrasts with the previous study that found 10 patients with double mutant APAs of CTNNB1 and GNA11/Q to be completely cured after adrenalectomy (17). However, both patients harboring the CTNNB1 mutations were >50 years old, and thus, age-related essential hypertension cannot be ruled out for the post-adrenalectomy residual hypertension seen. APAs carrying CTNNB1 mutations have been previously reported to have a higher possibility of residual hypertension than other mutant APAs, albeit these were most likely single CTNNB1 mutant APAs (41). Concurringly, in Wu et al.’s study, the eight CTNNB1 mutant APAs found most frequently occurred in females of older age with relatively large adrenal lesions similar to our patient with a single mutant APA.
In summary, the results of this study support the suggestion that CYP11B2-guided sequencing of all the currently known aldosterone-driver genes fine-tunes current genotype–phenotype relationships identified in PA patients with APAs. In addition, this study identified a rare CTNNB1 mutant APA with a likely pathogenic CACNA1D mutation. Although there were many overlaps in histopathologic features between the single and double CTNNB1 mutant APAs, CTNNB1 APAs with a CACNA1D mutation did have a distinct profile with lower expression of CYP17A1 and larger quantities of spironolactone bodies. Furthermore, KCNJ5 mutant APAs were found once again to have a lower Ki67 proliferation index (27). This study found that the finding was mainly driven by the high expression of Ki67 in ATP1A1 mutant APAs. Interestingly, although both patients with CACNA1D and ATP1A1 mutations were on a high number of antihypertensive medications, which suggested that spironolactone was most likely part of their drug management, spironolactone bodies, which are believed to be the result of treatment with spironolactone (42) were only found in CACNA1D mutant APAs (and CTNNB1 mutant APAs). Although the significance of the spironolactone bodies remains unclear, it is postulated that these structures represent a compensatory attempt on the part of the cell to produce more mineralocorticoid or storage of the steroids, as the spironolactone bodies are probably derived from the endoplasmic reticulum, which is considered capable of storing steroids (42, 43). Other clinical parameters of patients harboring an ATP1A1 or CACNA1D mutation are quite closely matched (except for age); thus, the existence of spironolactone bodies only in the CACNA1D mutant APAs but not in ATP1A1 mutant APAs warrants further investigation. Nevertheless, despite the study’s capability to fine-tune the current genotype–phenotype histopathology profiles by utilizing CYP11B2-guided sequencing, a larger sample size is required to support the significant observations associated with the genotypes. Moreover, functional characterization of the mechanism for the variants in double mutant APAs is also needed to confirm the pathogenicity of the variants.
Statements
Data availability statement
The contributions presented in the study are all publicly available. Original sequencing data can be found at PRJNA1269091 (SRA - NCBI) while previously reported sequencing data is published as cited in main article (DOI: 10.1161/HYPERTENSIONAHA.117.09057). The raw data supporting the conclusions of this article can be made available upon request, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by University Hospital Hradec Kralove Ethics Committee (201504 S22P) and the National University of Malaysia Research Ethics Committee (FF-2015-092). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
FP: Conceptualization, Formal analysis, Visualization, Writing – original draft, Investigation, Methodology. MM: Methodology, Validation, Writing – review & editing. GT: Methodology, Validation, Writing – review & editing. AR: Resources, Validation, Writing – review & editing. JC: Resources, Validation, Writing – review & editing. MS: Resources, Validation, Writing – review & editing. EA: Conceptualization, Methodology, Project administration, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. FP’s study was supported by the UK-MY Joint Partnership on Non-communicable Diseases 2019 program (NEWTON-MRC/2020/002) awarded to EA.
Acknowledgments
We thank C. E. Gomez-Sanchez (University of Mississippi Medical Center, Jackson, MS, USA) for his kind gift of his custom-made CYP11B2 antibody. The Endocrine Unit Laboratory of the National University of Malaysia (UKM) Medical Centre, as well as Nur Fatin Syazreen (UKM), provided assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1569619/full#supplementary-material
References
1.
Rossi GP . Primary aldosteronism: JACC state-of-the-art review. J Am Coll Cardiol (2019) 74:2799–811. doi: 10.1016/j.jacc.2019.09.057
2.
Omata K Tomlins SA Rainey WE . Aldosterone-producing cell clusters in normal and pathological states. Horm Metab Res. (2017) 49:951–6. doi: 10.1055/s-0043-122394
3.
Williams TA Gomez-Sanchez CE Rainey WE Giordano TJ Lam AK Marker A et al . International histopathology consensus for unilateral primary aldosteronism. J Clin Endocrinol Metab. (2021) 106:42–54. doi: 10.1210/clinem/dgaa484
4.
Teo AE Garg S Haris Shaikh L Zhou J Karet Frankl FE Gurnell M et al . Pregnancy, primary aldosteronism, and adrenal CTNNB1 mutations. N Engl J Med. (2015) 373:1429–36. doi: 10.1056/NEJMoa1504869
5.
Monticone S Castellano I Versace K Lucatello B Veglio F Gomez-Sanchez CE et al . Immunohistochemical, genetic and clinical characterization of sporadic aldosterone-producing adenomas. Mol Cell Endocrinol. (2015) 411:146–54. doi: 10.1016/j.mce.2015.04.022
6.
Kitamoto T Suematsu S Yamazaki Y Nakamura Y Sasano H Matsuzawa Y et al . Clinical and steroidogenic characteristics of aldosterone-producing adenomas with ATPase or CACNA1D gene mutations. J Clin Endocrinol Metabol. (2016) 101:494–503. doi: 10.1210/jc.2015-3284
7.
Åkerström T Willenberg HS Cupisti K Ip J Backman S Moser A et al . Novel somatic mutations and distinct molecular signature in aldosterone-producing adenomas. Endocr Relat Cancer. (2015) 22:735–44. doi: 10.1530/ERC-15-0321
8.
Azizan EAB Poulsen H Tuluc P Zhou J Clausen MV Lieb A et al . Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat Genet. (2013) 45:1055–60. doi: 10.1038/ng.2716
9.
Fernandes-Rosa FL Boulkroun S Zennaro MC . Genetic and genomic mechanisms of primary aldosteronism. Trends Mol Med. (2020) 26:819–32. doi: 10.1016/j.molmed.2020.05.005
10.
Beuschlein F Boulkroun S Osswald A Wieland T Nielsen HN Lichtenauer UD et al . Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet. (2013) 45:440–4. doi: 10.1038/ng.2550
11.
Choi M Scholl UI Yue P Björklund P Zhao B Nelson-Williams C et al . K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. (2011) 331:768–72. doi: 10.1126/science.1198785
12.
Scholl UI Stölting G Schewe J Thiel A Tan H Nelson-Williams C et al . CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet. (2018) 50:349–54. doi: 10.1038/s41588-018-0048-5
13.
Scholl UI Stölting G Nelson-Williams C Vichot AA Choi M Loring E et al . Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. eLife. (2015) 4:e06315. doi: 10.7554/eLife.06315
14.
Fernandes-Rosa FL Daniil G Orozco IJ Göppner C El Zein R Jain V et al . A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet. (2018) 50:355–61. doi: 10.1038/s41588-018-0053-8
15.
Åkerström T Maharjan R Sven Willenberg H Cupisti K Ip J Moser A et al . Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci Rep. (2016) 6:19546. doi: 10.1038/srep19546
16.
Williams TA Monticone S Mulatero P . KCNJ5 mutations are the Most frequent genetic alteration in primary aldosteronism. Hypertension. (2015) 65:507–9. doi: 10.1161/HYPERTENSIONAHA.114.04636
17.
Zhou J Azizan EAB Cabrera CP Fernandes-Rosa FL Boulkroun S Argentesi G et al . Somatic mutations of GNA11 and GNAQ in CTNNB1-mutant aldosterone-producing adenomas presenting in puberty, pregnancy or menopause. Nat Genet. (2021) 53:1360–72. doi: 10.1038/s41588-021-00906-y
18.
Rege J Bandulik S Nanba K Kosmann C Blinder AR Plain A et al . Somatic SLC30A1 mutations altering zinc transporter ZnT1 cause aldosterone-producing adenomas and primary aldosteronism. Nat Genet. (2023) 55:1623–31. doi: 10.1038/s41588-023-01498-5
19.
Gomez-Sanchez CE Qi X Velarde-Miranda C Plonczynski MW Parker CR Rainey W et al . Development of monoclonal antibodies against human CYP11B1 and CYP11B2. Mol Cell Endocrinol. (2014) 383:111–7. doi: 10.1016/j.mce.2013.11.022
20.
De Sousa K Boulkroun S Baron S Nanba K Wack M Rainey WE et al . Genetic, cellular, and molecular heterogeneity in adrenals with aldosterone-producing adenoma. Hypertension. (2020) 75:1034–44. doi: 10.1161/HYPERTENSIONAHA.119.14177
21.
Mohideen SK Mustangin M Kamaruddin NA Muhammad R Jamal ARA Sukor N et al . Prevalence and histopathological characteristics of KCNJ5 mutant aldosterone-producing adenomas in a multi-ethnic Malaysian cohort. Front Endocrinol (Lausanne). (2019) 10:666. doi: 10.3389/fendo.2019.00666
22.
Yadav R Petrunak EM Estrada DF Scott EE . Structural insights into the function of steroidogenic cytochrome P450 17A1. Mol Cell Endocrinol. (2017) 441:68–75. doi: 10.1016/j.mce.2016.08.035
23.
Chen AX Nishimoto K Nanba K Rainey WE . Potassium channels related to primary aldosteronism: expression similarities and differences between human and rat adrenals. Mol Cell Endocrinol. (2015) 417:141–8. doi: 10.1016/j.mce.2015.09.011
24.
Berthon A Drelon C Ragazzon B Boulkroun S Tissier F Amar L et al . WNT/β-catenin signalling is activated in aldosterone-producing adenomas and controls aldosterone production. Hum Mol Genet. (2014) 23:889–905. doi: 10.1093/hmg/ddt484
25.
Ceral J Solar M Krajina A Ballon M Suba P Cap J . Adrenal venous sampling in primary aldosteronism: a low dilution of adrenal venous blood is crucial for a correct interpretation of the results. Eur J Endocrinol. (2010) 162:101–7. doi: 10.1530/EJE-09-0217
26.
Solar M Malirova E Ballon M Pelouch R Ceral J . Confirmatory testing in primary aldosteronism: extensive medication switching is not needed in all patients. Eur J Endocrinol. (2012) 166:679–86. doi: 10.1530/EJE-11-0914
27.
Tan GC Negro G Pinggera A Tizen Laim NMS Mohamed Rose I Ceral J et al . Aldosterone-producing adenomas: histopathology-genotype correlation and identification of a novel CACNA1D mutation. Hypertension. (2017) 70:129–36. doi: 10.1161/HYPERTENSIONAHA.117.09057
28.
Adzhubei IA Schmidt S Peshkin L Ramensky VE Gerasimova A Bork P et al . A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
29.
Ng PC Henikoff S . SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1093/nar/gkg509
30.
Reva B Antipin Y Sander C . Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. (2011) 39:e118. doi: 10.1093/nar/gkr407
31.
Datta N . Unleashing the power of MutationTaster2 and MutationTaster2021: the machine learning approach to genetic variant analysis. Rev Cuba Inform Méd. (2023) 15:614. Available at: https://www.researchgate.net/publication/237020041_Semiautomated_Image_Analysis_of_High_Contrast_Tissue_Areas_Using_HueSaturation_Brightness_Based_Color_Filtering
32.
Kircher M Witten DM Jain P O'roak BJ Cooper GM Shendure J . A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. (2014) 46:310–5. doi: 10.1038/ng.2892
33.
Mezei T Szakács M Dénes L Jung J Egyed-Zsigmond I . Semiautomated image analysis of high contrast tissue areas using hue/saturation/brightness based color filtering. Acta Med Marisiensis. (2011) 57:679–84. Available at: https://scholar.google.com/scholar_lookup?hl=en&volume=15&publication_year=2023&pages=614&journal=Revista+Cubana+de+Inform%C3%A1tica+M%C3%A9dica&issue=2&author=N.+Datta&title=Unleashing+the+Power+of+MutationTaster2+and+MutationTaster2021%3A+The+Machine+Learning+Approach+to+Genetic+Variant+Analysis
34.
Omata K Satoh F Morimoto R Ito S Yamazaki Y Nakamura Y et al . Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension. (2018) 72:874–80. doi: 10.1161/HYPERTENSIONAHA.118.11086
35.
Gaujoux S Grabar S Fassnacht M Ragazzon B Launay P Libé R et al . β-Catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin Cancer Res. (2011) 17:328–36. doi: 10.1158/1078-0432.CCR-10-2006
36.
Boulkroun S Samson-Couterie B Golib-Dzib JF Amar L Plouin PF Sibony M et al . Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology. (2011) 152:4753–63. doi: 10.1210/en.2011-1205
37.
Tissier F Cavard C Groussin L Perlemoine K Fumey G Hagneré A-M et al . Mutations of β-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. (2005) 65:7622–7. doi: 10.1158/0008-5472.CAN-05-0593
38.
Brunner E Peter O Schweizer L Basler K . Pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the wingless signal in Drosophila. Nature. (1997) 385:829–33. doi: 10.1038/385829a0
39.
Van de Wetering M Cavallo R Dooijes D van Beest M van Es J Loureiro J et al . Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. (1997) 88:789–99. doi: 10.1016/s0092-8674(00)81925-x
40.
Pignatti E Leng S Yuchi Y Borges KS Guagliardo NA Shah MS et al . Beta-catenin causes adrenal hyperplasia by blocking zonal transdifferentiation. Cell Rep. (2020) 31:107524. doi: 10.1016/j.celrep.2020.107524
41.
Wu V-C Wang S-M Chueh S-CJ Yang S-Y Huang K-H Lin Y-H et al . The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci Rep. (2017) 7:39121. doi: 10.1038/srep39121
42.
Davis DA Medline NM . Spironolactone (Aldactone) bodies: concentric lamellar formations in the adrenal cortices of patients treated with spironolactone. Am J Clin Pathol. (1970) 54:22–32. doi: 10.1093/ajcp/54.1.22
43.
Hsu S-M Raine L Martin HF . Spironolactone bodies: an immunoperoxidase study with biochemical correlation. Am J Clin Pathol. (1981) 75:92–5. doi: 10.1093/ajcp/75.1.92
44.
Funder JW Carey RM Mantero F Murad MH Reincke M Shibata H et al . The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metabol. (2016) 101:1889–916. doi: 10.1210/jc.2015-4061
Summary
Keywords
aldosterone-producing adenomas, CYP11B2-guided sequencing, aldosterone-driver mutations, genotype–phenotype correlations, immunohistochemistry analysis
Citation
Pauzi FA, Mustangin M, Tan GC, Ryska A, Ceral J, Solar M and Azizan EA (2025) Histopathological spectrum of common aldosterone-driver gene mutations in aldosterone-producing adenomas. Front. Med. 12:1569619. doi: 10.3389/fmed.2025.1569619
Received
01 February 2025
Accepted
17 April 2025
Published
10 June 2025
Volume
12 - 2025
Edited by
Piotr Glinicki, Centre of Postgraduate Medical Education, Poland
Reviewed by
Juilee Rege, University of Michigan, United States
Felipe Freitas-Castro, Faculdade de Medicina da Universidade de São Paulo, Brazil
Updates
Copyright
© 2025 Pauzi, Mustangin, Tan, Ryska, Ceral, Solar and Azizan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Aisha Azizan, elena.azizan@ukm.edu.my
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.