Abstract
Introduction:
Chronic kidney disease (CKD) is a globally prevalent condition characterized by high morbidity and a progressive course that often culminates in end-stage renal disease (ESRD), necessitating dialysis or kidney transplantation. In recent years, genetic factors have received increasing attention in the pathogenesis of CKD, particularly among patients with unexplained renal dysfunction. Genetic screening has emerged as a valuable diagnostic tool. Mutations in the TMEM216 gene, a pathogenic variant associated with ciliopathy, have been implicated in severe renal impairment. This study presents a case analysis that explores the impact of TMEM216 mutations on kidney function and their potential clinical significance.
Case presentation:
We report a case of a 21-year-old male who developed proteinuria at the age of 15 without an apparent cause. Over the subsequent 6 years, his serum creatinine levels gradually increased, ultimately progressing to ESRD, accompanied by complications such as hypertension and secondary hyperparathyroidism. Imaging studies revealed bilateral renal cysts and a congenital bicuspid aortic valve. Whole-exome sequencing identified compound heterozygous mutations in TMEM216 [c.253C > T (p.R85*) and c.143 T > C (p.L48P)], consistent with an autosomal recessive inheritance pattern. Family analysis indicated that each parent carried one of the mutations. The combination of clinical and genetic findings suggests that the patient’s renal insufficiency may be attributed to TMEM216 mutations, highlighting their potential role in the progression of CKD.
Conclusion:
This study presents a severe case of renal dysfunction attributed to mutations in the TMEM216 gene, thereby expanding the clinical phenotypic spectrum associated with this gene. Mutations in ciliopathy-related genes may contribute to proteinuria and renal failure by disrupting the polarization and functionality of renal tubular epithelial cells. For young patients with unexplained CKD, genetic testing can serve as an early diagnostic tool to identify the underlying etiology and facilitate personalized treatment strategies. Future research on TMEM216-related nephropathy should aim to further elucidate its pathogenic mechanisms and explore potential therapeutic targets to enhance patient outcomes and advance precision medicine.
Introduction
Chronic kidney disease (CKD) is a highly prevalent condition worldwide, with a global median prevalence of 9.5% (IQR 5.9–11.7%). According to the Global Burden of Disease (GBD) study, approximately 697.5 million individuals were affected by CKD globally in 2017, and by 2040, CKD is projected to become the fifth leading cause of death worldwide (1, 2). Beyond its high prevalence, CKD imposes a substantial disease burden, as many patients ultimately progress to end-stage renal disease (ESRD), necessitating dialysis or kidney transplantation. The primary etiologies of CKD include diabetes, hypertension, and tubulointerstitial diseases. In addition to these established causes, genetic factors have garnered increasing attention in recent years for their role in the pathogenesis of CKD.
Hereditary kidney diseases represent a heterogeneous group of disorders resulting from gene mutations that impact renal structure and/or function. These conditions typically manifest as progressive kidney dysfunction, which, if left untreated, may lead to CKD or ESRD. Common hereditary kidney disorders include autosomal dominant polycystic kidney disease (ADPKD), Alport syndrome, and various tubulointerstitial nephropathies, each characterized by unique genetic backgrounds and clinical features (3–5).
This case report focuses on a 21-year-old male diagnosed with ESRD. Through genetic testing and an analysis of family history, a diagnosis of hereditary kidney disease due to a mutation in the TMEM216 gene was established. This case underscores the critical importance of genetic screening in patients with unexplained kidney pathology or a family history of renal disease. Early detection and intervention are vital for slowing disease progression and improving clinical outcomes. The identification of the causal mutation in this patient not only confirmed the hereditary nature of the disease but also offered potential insights into the clinical course and therapeutic targeting of genetically driven kidney disorders. Additional literature regarding TMEM216 is summarized in Table 1 (6–13).
Table 1
| Author | Description | Year |
|---|---|---|
| Glass et al. (6) | More than 40 genes, including TMEM216, have been identified as associated with Joubert syndrome. | 1993 |
| Edvardson et al. (7) | TMEM216 has been localized to primary cilia and plasma membranes in ciliated cell lines and primary cells, playing a significant role in the apical migration of centrioles and the subsequent formation of primary cilia. | 2010 |
| AY Wang (8) | The TMEM216 gene is crucial for cell polarity, morphogenesis, and ciliogenesis. | 2011 |
| Huang et al. (9) | Studies in Caenorhabditis elegans revealed that TMEM216 functions as part of the transition zone (TZ) module, contributing to the spectrum of ciliopathy phenotypes. | 2011 |
| Garcia-Gonzalo et al. (11) | Mutations in TMEM216 and other genes encoding ciliary components cause ciliopathies, indicating that dysfunction in the transition zone is a common pathogenic mechanism underlying these disorders. | 2011 |
| Szymanska et al. (10) | TMEM216 is one of the disease-causing genes for Meckel-Gruber syndrome (MKS). | 2012 |
| Gogendeau et al. (12) | Depletion of the TMEM216 protein in Paramecium can induce persistent ciliary shedding. | 2020 |
| Wang et al. (13) | The TMEM216-SUFU-GLI2/GLI3 axis plays an essential role in ciliopathies and aberrant Hedgehog (Hh) signaling induced by TMEM216 deficiency. | 2024 |
Published studies on the role of TMEM216 in ciliopathies.
Case report
A 21-year-old male presented with proteinuria that first manifested 6 years ago without an identifiable cause. At that time, no further medical assessment or treatment was pursued. Four years ago, an evaluation of renal function at an external hospital revealed an elevated serum creatinine level of 170.00 μmol/L. Subsequent follow-up indicated a gradual increase in serum creatinine levels. He intermittently took Haikun Shenxi Capsules (two capsules, three times daily), a traditional Chinese medicine preparation primarily composed of fucoidan sulfate polysaccharide, which is clinically utilized for the treatment of chronic renal failure. Its reported therapeutic effects include improvement of renal blood flow, attenuation of glomerulosclerosis, promotion of renal function recovery, scavenging of reactive oxygen species, and inhibition of lipid peroxidation (14). Four months prior to admission, the patient’s serum creatinine level rose to 421.54 μmol/L, and his hemoglobin level was recorded at 113.00 g/L. Roxadustat was subsequently introduced to address renal anemia. Two weeks before admission, the patient experienced bone pain accompanied by pruritus. Laboratory tests revealed a parathyroid hormone (PTH) level of 627.50 pg./mL, urea 25.75 mmol/L, creatinine 598.51 μmol/L, and estimated glomerular filtration rate (eGFRcr) of 11.06 mL/min/1.73 m2. Serum inorganic phosphorus was 1.55 mmol/L. He was treated with sevelamer and calcitriol, which led to partial relief of pruritus and bone pain. However, renal function deteriorated significantly. Ten days prior to the latest evaluation, serum urea increased to 31.30 mmol/L and creatinine to 946.77 μmol/L, with an eGFRcr of 6.38 mL/min/1.73 m2. Urinalysis revealed “++” proteinuria, with a 24-h urine protein excretion of 1220.67 mg/24 h. PTH level was 253.10 pg./mL (Figure 1). B-mode ultrasonography of the urinary system revealed multiple anechoic lesions in the bilateral renal cortices, consistent with bilateral renal cysts (Figure 2A). The patient had a history of hypertension lasting over 4 years, with maximum readings of 140–150/100–90 mmHg. Initially, blood pressure was managed with valsartan capsules; however, due to suboptimal control, the treatment was switched to benidipine, resulting in improved blood pressure stability. Additionally, the patient had a previous diagnosis of hypothyroidism and was regularly taking levothyroxine at a dosage of half to three-quarters of a tablet daily. Transthoracic color Doppler echocardiography (CDE) revealed a bicuspid aortic valve (type I, R/L), characterized by the fusion of the right and left coronary cusps arranged in a left–right orientation, with a commissural angle of approximately 160°. A fused ridge was noted between the fused leaflets, which opened well but exhibited a visible raphé (Figure 2B). Chest and upper abdominal CT scans no significant abnormalities. During the evaluation, the patient exhibited mildly delayed cognitive development compared to peers. However, no overt dysmorphic features or neurological signs were observed.
Figure 1

Timeline of renal function-related laboratory tests in the patient, demonstrating their relationship with disease progression and treatment. (A) Urea (mmol/L); (B) Serum creatinine (μmol/L); (C) Estimated glomerular filtration rate (eGFR, mL/min/1.73 m2).
Figure 2
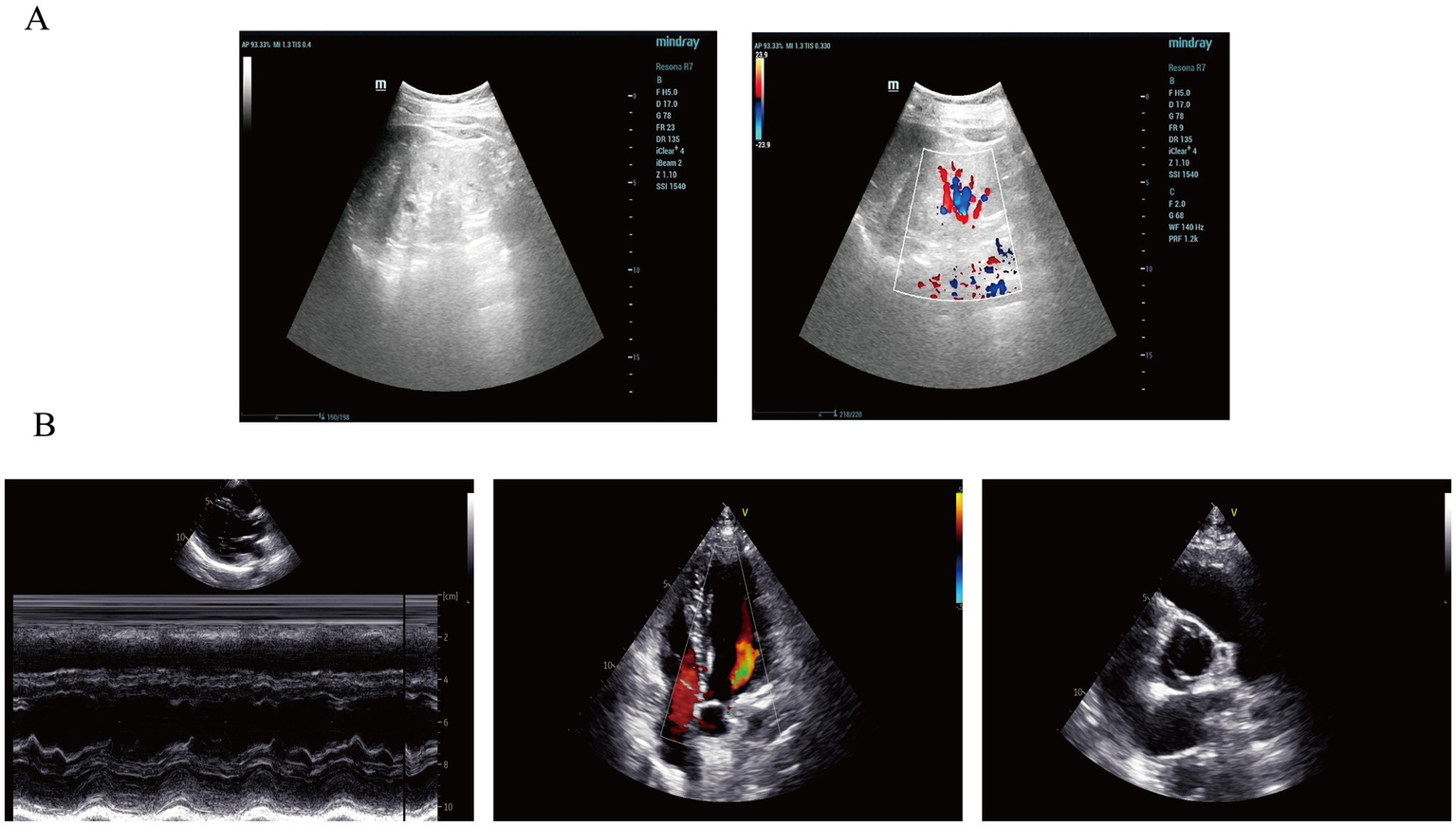
Imaging findings of the patient in this case report. (A) Renal ultrasonography revealed multiple anechoic lesions in the renal cortex. In the right kidney, the largest cyst was located at the lower pole, measuring 0.8 × 0.5 cm, with well-defined margins and good internal echogenicity. In the left kidney, multiple cortical cysts were observed, with the largest measuring 1.0 × 0.8 cm in the mid-region, showing clear margins and good posterior acoustic enhancement. These findings are consistent with bilateral renal cysts. (B) Color Doppler echocardiography (CDE) demonstrated normal sizes of all cardiac chambers, with no thickening of the interventricular septum or left ventricular wall, and preserved wall motion. The aortic valve showed fusion of the left and right coronary cusps, arranged in a right–left orientation with a commissural angle of approximately 160°. A fusion ridge was visible between the fused cusps, with good valve opening and a commissural slit, indicating a congenital bicuspid aortic valve (right–left, Type I). The morphology and structure of the remaining valves appeared normal.
The patient is a young male presenting with mild intellectual developmental difficulties. CDE indicated a congenital bicuspid aortic valve malformation, and a hereditary condition could not be excluded. To investigate the potential genetic etiology underlying the onset and progression of the disease, whole-exome sequencing (WES) was performed on the patient and his parents. Sequencing was conducted on the Illumina NovaSeq 6,000 platform, and raw reads were aligned to the human reference genome (hg38/GRCh38) using Burrows-Wheeler Aligner (BWA). Variant calling was subsequently carried out using the Verita Trekker® variant detection system and GATK, followed by variant annotation and interpretation using ANNOVAR and the Enliven® annotation system (15). According to the guidelines and recommendations from the American College of Medical Genetics and Genomics (ACMG), and after filtering based on inheritance pattern, age of onset, population frequency, and pathogenicity predictions, compound heterozygous variants were identified in the TMEM216 gene of the patient: c.253C > T (p.R85*) and c.143 T > C (p.L48P). The inheritance of these variants was validated by Sanger sequencing. The results showed that both the patient and his father carried the heterozygous c.253C > T (p.R85*) variant, which results in a cytosine (C) to thymine (T) substitution at nucleotide position 253 of the cDNA, creating a premature stop codon at amino acid position 85 (p.R85*). Meanwhile, the patient and his mother both carried the heterozygous c.143 T > C (p.L48P) variant, which leads to a thymine (T) to cytosine (C) substitution at nucleotide position 143, resulting in a leucine (Leu) to proline (Pro) substitution at position 48 of the protein (p.L48P). The c.253C > T (p.R85*) variant has been previously reported as pathogenic in studies related to Meckel syndrome and other ciliopathies, leading to premature stop codon formation. This variant showed co-segregation with the disease phenotype in two affected family members (10). Fulfilling the ACMG criteria of PVS1 + PM3 + PP1 + PM2_Supporting. It has been classified as pathogenic (P) in the ClinVar database and as a disease-causing mutation (DM) in the HGMD database (16, 17). This pathogenic variant is known to cause loss of protein function and is associated with severe ciliopathy phenotypes. In contrast, the c.143 T > C (p.L48P) variant has not been previously reported in the literature, and no functional studies or familial segregation data are available to support its pathogenicity. Based on the PM3 + PM2_Supporting criteria, it was classified as a variant of uncertain significance (VUS) (Figure 3). Interestingly, although the patient’s father also carries the pathogenic c.253C > T (p.R85*) variant in TMEM216, he does not exhibit any clinical renal phenotype, suggesting that the additional c.143 T > C (p.L48P) variant may play a contributory role in the manifestation of renal disease in the patient. The inheritance pattern observed in this family is consistent with autosomal recessive inheritance (Figure 4).
Figure 3

DNA sequencing results of TMEM216 variants in the patient’s family. Sanger sequencing of the TMEM216 gene revealed (A) the patient and his mother both carry the heterozygous variant c.143 T > C (p.L48P), and (B) the patient and his father both carry the heterozygous variant c.253C > T (p.R85*).
Figure 4

Pedigree of the reported family. Filled black symbols indicate individuals affected by hereditary kidney disease. The arrow denotes the proband carrying compound heterozygous mutations in exons 3 and 4 of the TMEM216 gene (clinical details provided in the main text).
Discussion
Hereditary kidney diseases are prevalent across various populations, with multiple common genetic variants have been confirmed to be closely associated with the onset of IgA nephropathy (IgAN), membranous nephropathy, and nephrotic syndrome (18–21). Consequently, the pathogenesis of most kidney diseases is no longer regarded as driven by a single factor but rather results from the combined influence of various genetic and environmental contributors that collectively promote disease initiation and pathological progression. In this report, we present a male patient with hereditary renal insufficiency caused by mutations in the TMEM216 gene, which ultimately led to ESRD. The WES of the patient and his parents revealed a heterozygous nonsense mutation in exon 4 of TMEM216 inherited from the father, and a heterozygous missense mutation in exon 3 inherited from the mother. Both mutations were identified in the patient.
TMEM216 encodes a tetraspan transmembrane protein located in the ciliary transition zone. By interacting with other cilia-associated proteins, TMEM216 plays a role in regulating the formation and localization of primary cilia (22). Primary cilia are critical for kidney organogenesis, particularly in maintaining the differentiation and proliferation of epithelial cells (23). Previous studies have shown that deletion of the ciliary regulatory gene TMEM16A in murine nephrons leads to a reduced number of primary cilia in proximal tubular cells, accompanied by increased proteinuria. This suggests that a reduction in cilia number can disrupt the polarity and function of tubular epithelial cells, thereby impairing the reabsorptive capacity of renal tubules—especially the reabsorption of low-molecular-weight proteins (24, 25).
Studies have demonstrated that knockdown of TMEM216 impairs ciliogenesis in polarized cells and disrupts proper docking of the centrosome at the apical cell surface (26). Mutations in TMEM216 are closely associated with disorders such as Joubert syndrome and Meckel syndrome, which are characterized by multisystem abnormalities including neurodevelopmental defects, renal anomalies, and retinal degeneration (Table 2) (6, 27–31). Among these manifestations, renal abnormalities typically include renal cysts, renal insufficiency, and cystic renal dysplasia. Clinically, patients with Joubert or Meckel syndrome may present with progressive decline in renal function, suggesting that TMEM216 mutations may contribute to renal pathologies in these syndromes by impairing ciliogenesis (32).
Table 2
| System organ classification (SOC) | Joubert syndrome | Meckel syndrome |
|---|---|---|
| Nervous system | Brainstem hypoplasia, vermian hypoplasia of the cerebellum, ataxia, hypotonia, developmental delay, epilepsy, oculomotor disturbances, and hydrocephalus. | Occipital encephalocele, craniorachischisis, partial corpus callosum agenesis, dandy-walker malformation, anencephaly. |
| Ocular | Nystagmus, ocular motility disturbances, retinal dystrophy, optic nerve atrophy, visual impairment, hypertelorism, choroidal retinal defects, optic disc anomalies. | Retinal coloboma, congenital microphthalmia |
| Facial and craniofacial features | Mild craniofacial anomalies | Cleft lip, cleft palate |
| Respiratory system | Neonatal respiratory abnormalities (alternating periods of rapid and slow breathing), apnea. | Pulmonary hypoplasia |
| Renal system | Renal structural abnormalities (such as renal hypoplasia), tubulointerstitial disease, and Renal cysts. | Bilateral cystic dysplasia of the kidneys, kidney enlargement, and corticomedullary differentiation. |
| Hepatobiliary system | Liver fibrosis, cholestasis, and liver dysfunction. | Bile duct proliferation and dilation, hepatic fibrosis, hepatic cysts |
| Skeletal system | Polydactyly and spinal deformities | Polydactyly, bowed and shortened long bones |
| Reproductive system | Undescended testes and external genitalia anomalies | Hypoplasia of male genitalia |
| Psychiatric and behavioral disorders | Autistic-like features, attention deficit, cognitive impairments, inattention, hyperactivity, stereotypies, emotional instability, anxiety, self-harm, aggression, and autism | Intellectual disability, developmental delay |
| General symptoms/systemic | Developmental delay, growth retardation | Growth restriction |
| Other manifestations | Hearing loss (sensorineural), feeding difficulties, gastroesophageal reflux | Cystic hygroma |
Clinical features of Joubert syndrome and Meckel syndrome.
In the present case report, the patient developed proteinuria at the age of 15 without any obvious cause, followed by a gradual increase in serum creatinine over 6 years, ultimately progressing to ESRD accompanied by secondary hyperparathyroidism and other related complications. Ultrasonographic imaging revealed bilateral renal cysts and a congenital bicuspid aortic valve malformation. Whole-exome sequencing identified compound heterozygous mutations in the TMEM216 gene (c.253C > T (p.R85*) and c.143 T > C (p.L48P)). From a clinical perspective, the pathogenesis of renal insufficiency in this patient can be interpreted within the framework of ciliopathy-related mechanisms. Defects in primary cilia impair the ability of renal tubular epithelial cells to sense urinary flow, resulting in dysregulation of electrolyte metabolism, particularly calcium, which contributes to the development of proteinuria and progressive renal dysfunction. Genetically, both the patient and his parents (with no known consanguinity) were found to carry TMEM216 mutations, with the patient exhibiting compound heterozygosity. This finding supports an autosomal recessive inheritance pattern. The TMEM216 mutations likely impair ciliogenesis and ciliary function, leading to secondary injury of the renal tubules and contributing to the onset and progression of CKD.
The current therapeutic principles for TMEM216 mutation–associated renal insufficiency primarily include delaying disease progression, renal replacement therapy (RRT), and reducing the risk of complications. In early-stage patients, cAMP modulators or mTOR inhibitors may be considered to slow the progression of cystic lesions (33), while closely monitoring renal function deterioration. For patients who have progressed to ESRD, peritoneal dialysis (PD) is often the preferred modality, especially in younger individuals or those with significant neurological involvement; hemodialysis serves as an alternative when PD fails or is not tolerated. Kidney transplantation remains the optimal option for improving long-term outcomes, but comprehensive evaluation of hepatic and neurological comorbidities is essential to ensure the feasibility and safety of transplantation (34). In addition, gene therapy approaches such as antisense oligonucleotide (ASO) technology have shown promising results in reducing cyst formation in CEP290-related renal disease models (35) and may represent a potential precision treatment strategy for TMEM216-related kidney disorders in the future. TMEM216 mutations are typically associated with severe renal phenotypes, often accompanied by central nervous system abnormalities or syndromic manifestations. However, most previously reported TMEM216 mutations have rarely presented with isolated renal involvement (7, 26, 36). In our reported case, the patient exhibited primarily renal phenotypes—renal cysts and renal failure—alongside intellectual developmental difficulties, highlighting the possibility that TMEM216 mutations can manifest predominantly as renal disease. For young patients with unexplained CKD, it is necessary to perform exome sequencing or targeted gene testing to identify potential hereditary causes early. Moreover, individuals carrying pathogenic variants should receive genetic counseling and undergo longitudinal follow-up based on their renal function status to guide long-term management. Early diagnosis and personalized interventions can effectively slow disease progression, reduce complications, and improve overall prognosis.
Meanwhile, further investigations into the role of TMEM216 in kidney diseases, particularly in tubular injury, interstitial fibrosis, and other ciliopathies—may provide critical insights into the molecular mechanisms underlying renal pathophysiology and reveal novel therapeutic targets.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LS: Writing – original draft, Writing – review & editing. MX: Methodology, Writing – review & editing. XD: Methodology, Writing – review & editing. XL: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was sponsored by the National Natural Science Foundation of China (82101660), Dalian Science and Technology Innovation Fund Project (2021JJ12SN41) and Liaoning Provincial Department of Education Innovative Talent Cultivation Project (LJ222410161016).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
GBD Chronic Kidney Disease Collaboration . Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2020) 395:709–33. doi: 10.1016/s0140-6736(20)30045-3
2.
Foreman KJ Marquez N Dolgert A Fukutaki K Fullman N McGaughey M et al . Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet. (2018) 392:2052–90. doi: 10.1016/s0140-6736(18)31694-5
3.
Parfrey PS . Hereditary renal diseases. Curr Opin Nephrol Hypertens. (1993) 2:192–200. doi: 10.1097/00041552-199303000-00004
4.
Savige J Lipska-Zietkiewicz BS Watson E Hertz JM Deltas C Mari F et al . Guidelines for genetic testing and management of alport syndrome. Clin J Am Soc Nephrol. (2022) 17:143–54. doi: 10.2215/cjn.04230321
5.
Praga M González E . Acute interstitial nephritis. Kidney Int. (2010) 77:956–61. doi: 10.1038/ki.2010.89
6.
Glass IA Dempsey JC Parisi M Doherty D . Joubert Syndrome In: AdamMPFeldmanJMirzaaGMPagonRAWallaceSEAmemiyaA, editors. GeneReviews(®). Seattle, WA: University of Washington (1993).
7.
Edvardson S Shaag A Zenvirt S Erlich Y Hannon GJ Shanske AL et al . Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. (2010) 86:93–7. doi: 10.1016/j.ajhg.2009.12.007
8.
Wang A . TMEM216 joins its ciliary cousins in ciliopathies. Clin Genet. (2011) 79:45–7. doi: 10.1111/j.1399-0004.2010.01556_2.x
9.
Huang L Szymanska K Jensen VL Janecke AR Innes AM Davis EE et al . TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet. (2011) 89:713–30. doi: 10.1016/j.ajhg.2011.11.005
10.
Szymanska K Berry I Logan CV Cousins SR Lindsay H Jafri H et al . Founder mutations and genotype-phenotype correlations in Meckel-Gruber syndrome and associated ciliopathies. Cilia. (2012) 1:18. doi: 10.1186/2046-2530-1-18
11.
Garcia-Gonzalo FR Corbit KC Sirerol-Piquer MS Ramaswami G Otto EA Noriega TR et al . A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. (2011) 43:776–84. doi: 10.1038/ng.891
12.
Gogendeau D Lemullois M Borgne PL Castelli M Aubusson-Fleury A Arnaiz O et al . MKS-NPHP module proteins control ciliary shedding at the transition zone. PLoS Biol. (2020) 18:e3000640. doi: 10.1371/journal.pbio.3000640
13.
Wang Y Yao H Zhang Y Mu N Lu T Du Z et al . TMEM216 promotes primary ciliogenesis and hedgehog signaling through the SUFU-GLI2/GLI3 axis. Sci Signal. (2024) 17:eabo0465. doi: 10.1126/scisignal.abo0465
14.
Li S Li J Shi F Yang L Ye M . Protection effect of intracellular melanin from Lachnum YM156 and Haikunshenxi capsule combination on adenine-induced chronic renal failure in mice. Medchemcomm. (2017) 8:917–23. doi: 10.1039/c6md00646a
15.
Zhou J Zhou R Feng Q Song X Chen X . Phenotypic and genotypic analysis of a patient with Miyoshi myopathy caused by truncated protein. Gene. (2024) 893:147929. doi: 10.1016/j.gene.2023.147929
16.
Bachmann-Gagescu R Dempsey JC Phelps IG O'Roak BJ Knutzen DM Rue TC et al . Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. (2015) 52:514–22. doi: 10.1136/jmedgenet-2015-103087
17.
Capalbo A Valero RA Jimenez-Almazan J Pardo PM Fabiani M Jiménez D et al . Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: translational research genomic data from 14,125 exomes. PLoS Genet. (2019) 15:e1008409. doi: 10.1371/journal.pgen.1008409
18.
Rasouly HM Groopman EE Heyman-Kantor R Fasel DA Mitrotti A Westland R et al . The burden of candidate pathogenic variants for kidney and genitourinary disorders emerging from exome sequencing. Ann Intern Med. (2019) 170:11–21. doi: 10.7326/m18-1241
19.
Kiryluk K Li Y Scolari F Sanna-Cherchi S Choi M Verbitsky M et al . Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. (2014) 46:1187–96. doi: 10.1038/ng.3118
20.
Xie J Liu L Mladkova N Li Y Ren H Wang W et al . The genetic architecture of membranous nephropathy and its potential to improve non-invasive diagnosis. Nat Commun. (2020) 11:1600. doi: 10.1038/s41467-020-15383-w
21.
Dufek S Cheshire C Levine AP Trompeter RS Issler N Stubbs M et al . Genetic identification of two novel loci associated with steroid-sensitive nephrotic syndrome. J Am Soc Nephrol. (2019) 30:1375–84. doi: 10.1681/asn.2018101054
22.
Liu Y Cao S Yu M Hu H . TMEM216 deletion causes Mislocalization of cone opsin and rhodopsin and photoreceptor degeneration in zebrafish. Invest Ophthalmol Vis Sci. (2020) 61:24. doi: 10.1167/iovs.61.8.24
23.
Patel A . The primary cilium calcium channels and their role in flow sensing. Pflugers Arch. (2015) 467:157–65. doi: 10.1007/s00424-014-1516-0
24.
Ruppersburg CC Hartzell HC . The Ca2+−activated cl- channel ANO1/TMEM16A regulates primary ciliogenesis. Mol Biol Cell. (2014) 25:1793–807. doi: 10.1091/mbc.E13-10-0599
25.
Schenk LK Buchholz B Henke SF Michgehl U Daniel C Amann K et al . Nephron-specific knockout of TMEM16A leads to reduced number of glomeruli and albuminuria. Am J Physiol Renal Physiol. (2018) 315:F1777–f1786. doi: 10.1152/ajprenal.00638.2017
26.
Valente EM Logan CV Mougou-Zerelli S Lee JH Silhavy JL Brancati F et al . Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. (2010) 42:619–25. doi: 10.1038/ng.594
27.
Alexiev BA Lin X Sun CC Brenner DS . Meckel-Gruber syndrome: pathologic manifestations, minimal diagnostic criteria, and differential diagnosis. Arch Pathol Lab Med. (2006) 130:1236–8. doi: 10.5858/2006-130-1236-ms
28.
Gana S Serpieri V Valente EM . Genotype-phenotype correlates in Joubert syndrome: A review. Am J Med Genet C Semin Med Genet. (2022) 190:72–88. doi: 10.1002/ajmg.c.31963
29.
Hartill V Szymanska K Sharif SM Wheway G Johnson CA . Meckel-Gruber syndrome: an update on diagnosis, clinical management, and research advances. Front Pediatr. (2017) 5:244. doi: 10.3389/fped.2017.00244
30.
Ickowicz V Eurin D Maugey-Laulom B Didier F Garel C Gubler MC et al . Meckel-Grüber syndrome: sonography and pathology. Ultrasound Obstet Gynecol. (2006) 27:296–300. doi: 10.1002/uog.2708
31.
Spahiu L Behluli E Grajçevci-Uka V Liehr T Temaj G . Joubert syndrome: molecular basis and treatment. J Mother Child. (2022) 26:118–23. doi: 10.34763/jmotherandchild.20222601.d-22-00034
32.
Collard E Byrne C Georgiou M Michaelides M Dixit A . Joubert syndrome diagnosed renally late. Clin Kidney J. (2021) 14:1017–9. doi: 10.1093/ckj/sfaa007
33.
Srivastava S Molinari E Raman S Sayer JA . Many genes-one disease? Genetics of Nephronophthisis (NPHP) and NPHP-associated disorders. Front Pediatr. (2017) 5:287. doi: 10.3389/fped.2017.00287
34.
Takagi Y Miura K Yabuuchi T Kaneko N Ishizuka K Takei M et al . Any modality of renal replacement therapy can be a treatment option for Joubert syndrome. Sci Rep. (2021) 11:462. doi: 10.1038/s41598-020-80712-4
35.
Ramsbottom SA Molinari E Srivastava S Silberman F Henry C Alkanderi S et al . Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model. Proc Natl Acad Sci USA. (2018) 115:12489–94. doi: 10.1073/pnas.1809432115
36.
Maria BL Hoang KB Tusa RJ Mancuso AA Hamed LM Quisling RG et al . "Joubert syndrome" revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. (1997) 12:423–30. doi: 10.1177/088307389701200703
Summary
Keywords
chronic kidney disease (CKD), hereditary kidney disease, TMEM216 gene, ciliopathy, gene mutation, case report
Citation
Sun L, Xu M, Deng X and Liu X (2025) Renal insufficiency caused by TMEM216 gene mutation: Case Report. Front. Med. 12:1579732. doi: 10.3389/fmed.2025.1579732
Received
19 February 2025
Accepted
09 April 2025
Published
29 April 2025
Volume
12 - 2025
Edited by
Gregory Braden, University of Massachusetts Medical School, United States
Reviewed by
Margareta Fistrek Prlic, University Hospital Centre Zagreb, Croatia
Taylor Richards, University of Wolverhampton, United Kingdom
Updates
Copyright
© 2025 Sun, Xu, Deng and Liu.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Liu, xiaoyanliu21@163.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.