Abstract
We present the case of a 68-year-old man with acute kidney injury that occurred following radiofrequency cardiac ablation. Laboratory tests revealed elevated serum creatinine (SCr), positivity for antinuclear and anti-double-stranded DNA (anti-dsDNA) antibodies, elevated IgG4 levels, and hypocomplementemia. Renal biopsy identified coexisting mesangial proliferative lupus nephritis (LN) (Class II) and IgG4-related tubulointerstitial nephritis (IgG4-TIN). The patient responded well to glucocorticoid-based immunosuppressive therapy, with significant improvement in renal function. This case illustrates the rare coexistence of systemic lupus erythematosus (SLE) and IgG4-related kidney disease (IgG4-RKD), highlighting the importance of renal biopsy in distinguishing overlapping autoimmune nephropathies.
Introduction
Chronic kidney disease is a major cause of morbidity and mortality globally, affecting more than 12% of the adult population (1, 2), which underscores the need to uncover molecular pathogenic mechanisms and treatment targets (3–5). Immunoglobulin (Ig)G4-related disease (IgG4-RD) involves inflammation and fibrosis of one or more organs and is characterized by three features: (1) IgG4-positive plasma cell infiltration; (2) varying degrees of fibrosis, typically displaying mat pattern fibers; and (3) increased blood IgG4 levels (6). IgG4-related kidney disease (IgG4-RKD), a subset of IgG4-RD, is divided into three categories: tubulointerstitial inflammation (IgG4-TIN), retroperitoneal fibrosis, and membranous nephropathy. IgG4-TIN is the most common of these categories.
Systemic lupus erythematosus (SLE) is an autoimmune disease characterized by a range of clinical manifestations. Immune disorders lead to the production of autoantibodies, affecting the normal functioning of many organs and systems. Abnormal antinuclear antibodies (ANAs), complement, and immunoglobulins form immune complexes and deposits in the glomeruli, leading to lupus nephritis (LN). LN, one of the most serious clinical manifestations of SLE, is characterized by proteinuria and progressive renal dysfunction (7).
Both IgG4-TIN and LN are autoimmune inflammatory diseases, but their coexistence is rare. Here, we report a case with simultaneous IgG4-TIN and LN.
Case presentation
One month before admission to our hospital, a routine physical examination of a 68-year-old male patient revealed an abnormally elevated serum creatinine (SCr) level of 2.09 mg/dL (reference range: 0.67–1.18 mg/dL), despite the absence of other symptoms or signs of renal dysfunction. No further tests were performed at that time to determine the cause of the elevated SCr. Two weeks prior to admission, the patient experienced frequent premature ventricular contractions (PVCs) and palpitations, for which he underwent radiofrequency cardiac ablation using a small amount of contrast medium. Although SCr was 2.5 mg/dL during the post-procedural evaluation, no specific intervention was initiated, and the patient was advised to undergo regular follow-up. Five days before admission, the patient developed new-onset periorbital and bilateral lower extremity edema, accompanied by an increase in SCr to 5.74 mg/dL. Therefore, he was admitted to our hospital for treatment.
The patient had a history of PVCs, hypertension, benign prostatic hyperplasia, and gallstones. He was taking tamsulosin hydrochloride (0.2 mg, sustained-release tablet) once nightly, atorvastatin calcium (20 mg) once nightly, metoprolol succinate (47.5 mg, sustained-release) once daily, and nifedipine (30 mg, sustained-release) twice daily. Physical examination showed pallid conjunctiva, facial pallor, and edema of the eyelids and both lower limbs, and no other abnormal findings.
Laboratory tests revealed positivity for ANAs at a titer of 1:1000, elevated anti-double-stranded DNA (anti-dsDNA) antibodies at 197 IU/mL (reference: 0–120 IU/mL), and hypocomplementemia with reduced C3 (0.23 g/L; reference: 0.9–1.8 g/L) and C4 (0.02 g/L; reference: 0.1–0.4 g/L). Proteinuria was noted at 0.6 g/day (reference: 0–0.15 g/24 h), while antiphospholipid antibodies tested negative. Additionally, SCr was markedly elevated at 5.74 mg/dL (reference: 0.7–1.3 mg/dL), with serum IgG4 elevated to 1,180 mg/dL (reference: 3–201 mg/dL) and serum IgG to 2,030 mg/dL (reference: 700–1,600 mg/dL).
Chest computed tomography, cardiac ultrasound, and abdominal sonography showed liver cysts and gallstones, but no other abnormalities. Urological ultrasound showed preserved corticomedullary differentiation and mild separation of the right renal pelvis.
Renal biopsy findings under light microscopy showed mild mesangial hypercellularity (3–5 cells per mesangial area) and matrix expansion, with patent capillary loops and no evidence of basement membrane thickening, necrosis, or leukocyte infiltration (Figure 1A). Endothelial cells appeared unremarkable. Electron microscopy revealed prominent vacuolar changes in glomerular endothelial cells and mesangial matrix expansion with scattered electron-dense deposits in the mesangial areas (Figure 1B). Capillary lumina remained open. Immunofluorescence demonstrated granular mesangial deposition of immune complexes, including IgG (++), IgA (++), IgM (++), complement component C1q (+), Ig light chain Kappa (κ) (++), and Ig light chain Lambda (λ) (++) (Figure 2). These findings supported an immune complex-mediated injury pattern, consistent with the International Society of Nephrology (ISN)/Renal Pathology Society (RPS) Class II LN.
Figure 1

Renal biopsy findings. (A) Periodic Acid-Schiff staining shows mild mesangial cell and matrix proliferation (×400); arrow: mesangial hypercellularity. (B) Electron microscopy staining: electron-dense deposits (arrow) in the mesangial area (×1,900).
Figure 2
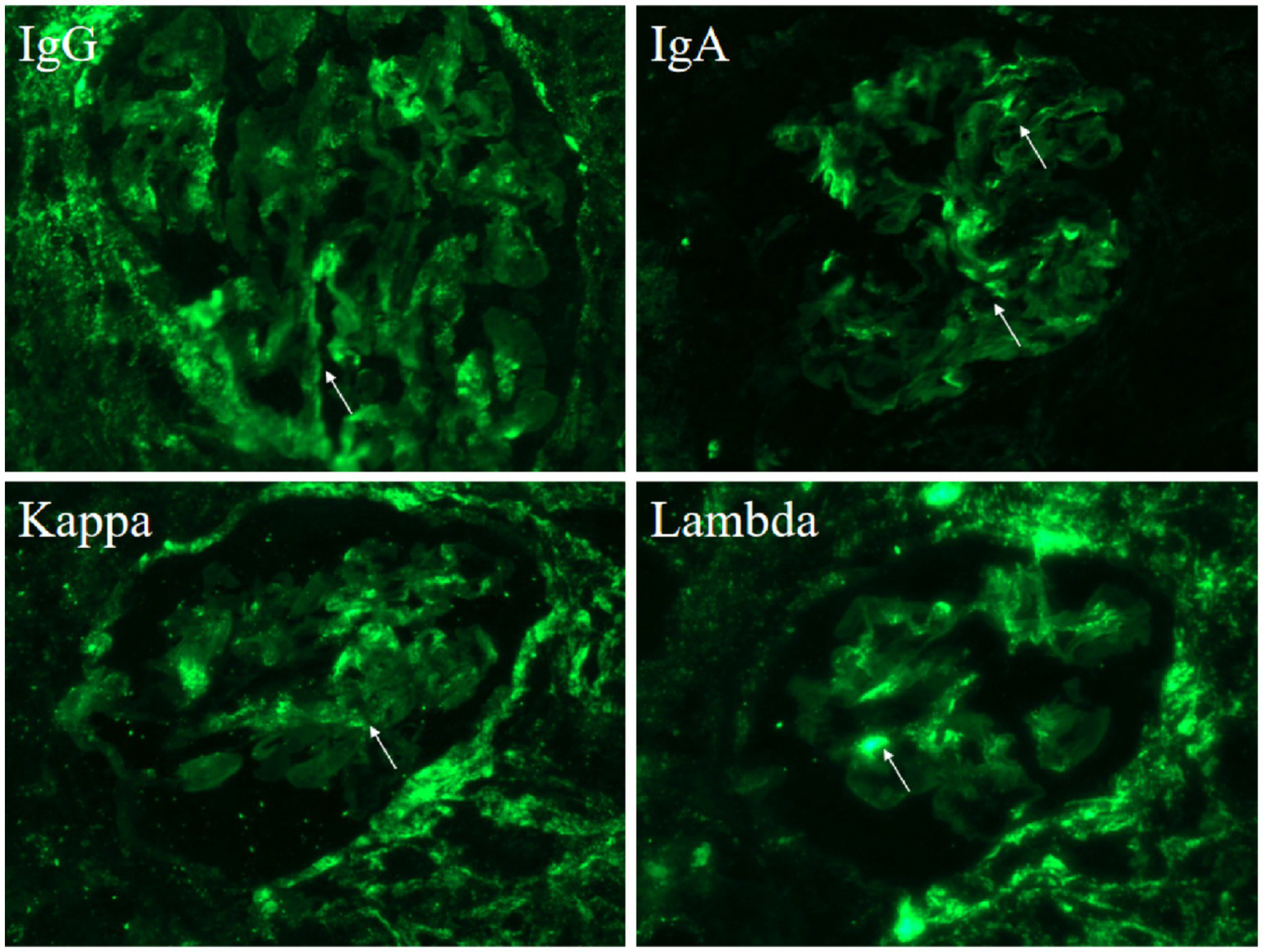
Immunofluorescence shows deposition of various immune complexes (arrow): IgG (++), IgA (++), Kappa (++), Lambda (++).
Hematoxylin–eosin staining revealed diffuse interstitial inflammation rich in plasma cells and associated tubular injury (Figure 3A), while Masson’s trichrome staining showed prominent interstitial fibrosis with a storiform pattern (Figure 3B). Immunohistochemistry revealed an abundance of IgG4-positive plasma cells (greater than 30 per high-power field (/HPF)), with an IgG4/IgG-positive cell ratio greater than 40%, along with CD38 (Figure 4). These features indicated IgG4-RKD.
Figure 3
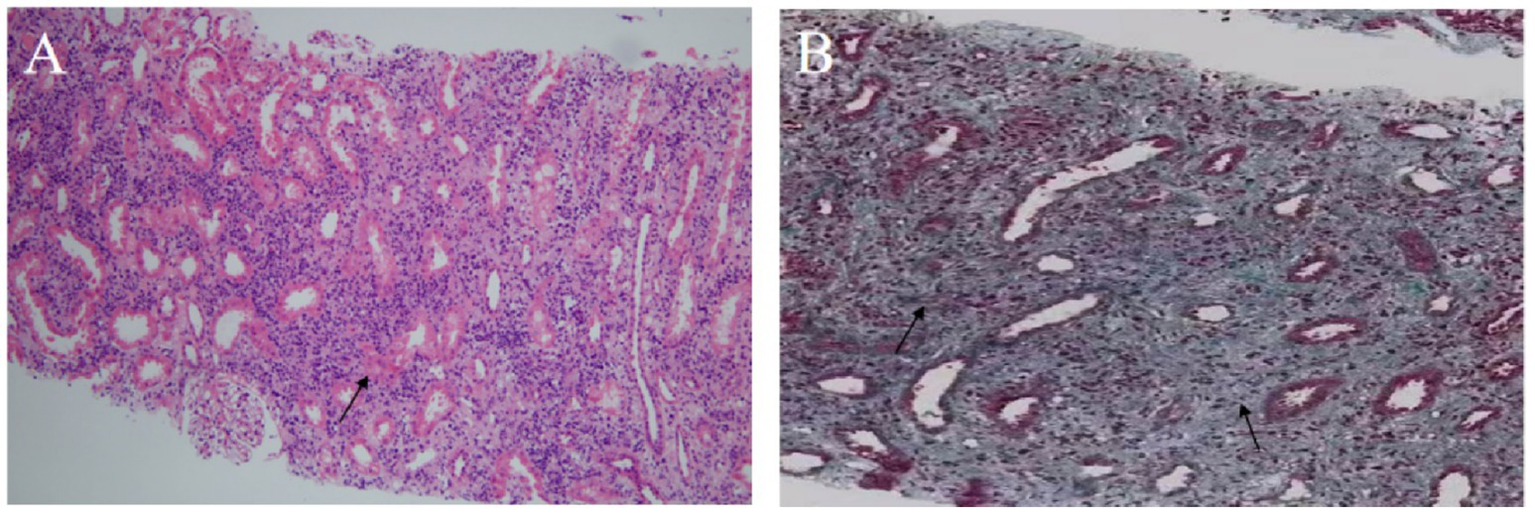
Renal biopsy findings. (A) Hematoxylin–eosin staining shows diffuse interstitial inflammatory infiltration with numerous plasma cells and tubular atrophy (×100); arrow: plasma cell infiltration. (B) Masson’s staining demonstrates storiform interstitial fibrosis (×200); arrow: area of storiform fibrosis.
Figure 4
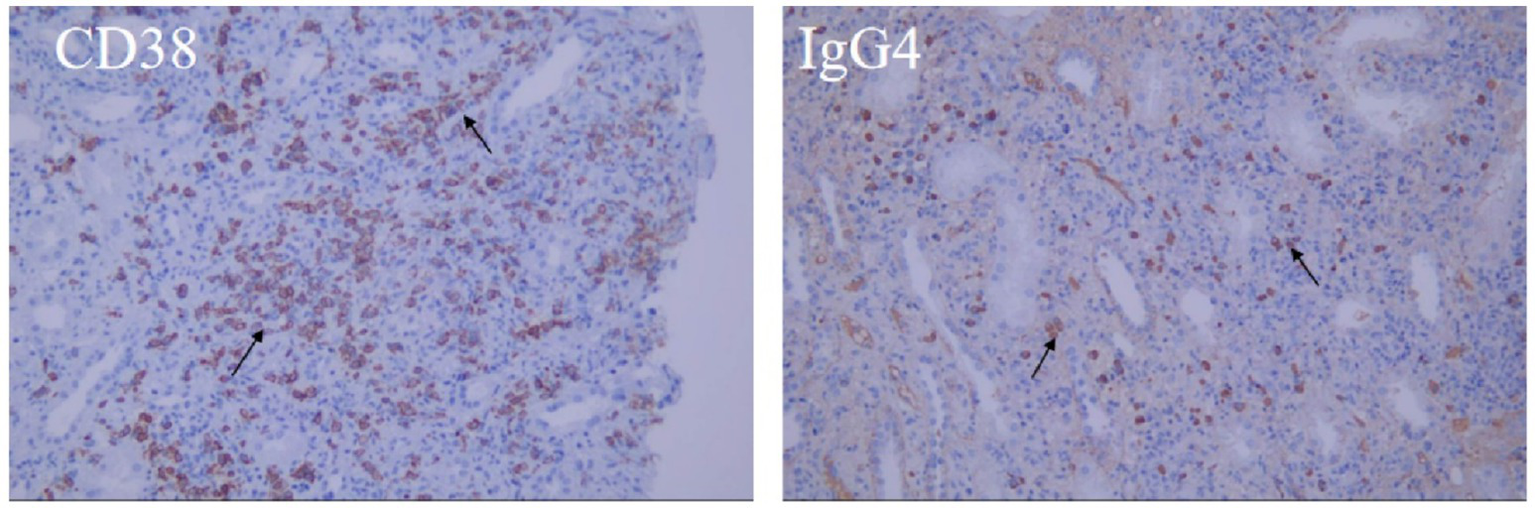
Immunohistochemistry: Multifocal cells of renal interstitium were positive for CD38, IgG4 (arrow).
In summary, our patient presented with elevated ANA titers, increased anti-dsDNA antibodies, and hypocomplementemia, consistent with ISN/RPS Class II LN, as confirmed by renal biopsy. The 2019 European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria yielded a total score of 18, fulfilling the diagnostic threshold for SLE (see Table 1) (8). Additionally, the patient exhibited elevated SCr and markedly increased IgG4 levels, with renal histopathology revealing storiform fibrosis, which was greater than 30 IgG4-positive plasma cells per high-power field, and the IgG4/IgG-positive cell ratio was greater than 40%. These findings met the 2011 diagnostic criteria for IgG4-RKD (items 1 + 3 + 4a + 4b), as defined by the Japanese Society of Nephrology (see Table 2) (9).
Table 1
| Domain | Item | In this case | Score |
|---|---|---|---|
| Entry criterion | ANA | Present | positive |
| Clinical domains and criteria | |||
| Constitutional | Absent | - | |
| Fever | - | ||
| Hematologic | Absent | ||
| Leukopenia | - | ||
| Thrombocytopenia | - | ||
| Autoimmune hemolysis | - | ||
| Neuropsychiatric | Absent | - | |
| Delirium | - | ||
| Psychosis | - | ||
| Seizure | - | ||
| Mucocutaneous | Absent | - | |
| Non-scarring alopecia | - | ||
| Oral ulcers | - | ||
| Subacute cutaneous or discoid lupus | - | ||
| Acute cutaneous lupus | - | ||
| Serosal | Absent | - | |
| Pleural or pericardial effusion | - | ||
| Acute pericarditis | - | ||
| Musculoskeletal | Absent | - | |
| Joint involvement | - | ||
| Renal | - | ||
| Proteinuria >0.5 g/24 h | Present | - | |
| Renal biopsy Class II or V lupus nephritis | Present | 8 | |
| Renal biopsy Class III or IV lupus nephritis | Absent | - | |
| Immunology domains and criteria | |||
| Antiphospholipid antibodies | - | ||
| Anti-cardiolipin antibodies OR anti-β2GP1 antibodies OR Lupus anticoagulant | Absent | - | |
| Complement proteins | - | ||
| Low C3 OR low C4 | - | ||
| Low C3 AND low C4 | Present | 4 | |
| SLE-specific antibodies | - | ||
| Anti-dsDNA antibodies OR anti-Smith antibodies | Present | 6 | |
| Total score | ≥10: SLE classified | 18 | |
Diagnostic criteria fulfilled by the patient: 2019 EULAR/ACR criteria for SLE.
SLE diagnostic criteria: ANA >1: 80; total score ≥10 (2019 EULAR/ACR). SLE, systemic lupus erythematosus; ANA, antinuclear antibody; Anti-dsDNA, anti-double-stranded DNA antibody.
Table 2
| Diagnostic criteria for IgG4-related kidney disease (IgG4-RKD) 2011 | Meets criteria |
|---|---|
| 1. Presence of some kidney damage, as manifested by abnormal urinalysis or urine marker(s) or decreased kidney function with either elevated serum IgG level, hypocomplementemia, or elevated serum IgE level | Present |
| 2. Abnormal renal radiologic findings: | Absent |
|
|
|
|
|
|
|
|
| 3. Elevated serum IgG4 level (IgG4 ≥ 135 mg/dL) | Present |
| 4. Histologic findings in the kidney: | |
|
Present |
|
Present |
5. Histologic findings in extra-renal organ(s):
|
Absent |
| This case fulfills items | 1 + 3 + 4a + 4b |
Diagnostic criteria fulfilled by the patient: 2011 JSN criteria for IgG4-related kidney disease.
IgG4-RKD definite diagnostic criteria: 1 + 3 + 4a + 4b, or 2 + 3 + 4a + 4b, or 2 + 3 + 5a, or 1 + 3 + 4a + 5a or 5b, or 2 + 3 + 4a + 5b.
Immunosuppressive therapy was initiated based on the patient’s SLE Disease Activity Index (SLEDAI) score of 12 points, in accordance with the 2023 EULAR recommendations for LN and the international consensus on IgG4-RKD (10–12). A personalized treatment strategy was adopted, starting with intravenous methylprednisolone (40 mg/day) combined with low-dose cyclophosphamide (CTX, 0.2 g per dose × 3) to induce remission. In October 2022, the patient’s SCr declined to 1.69 mg/dL, anti-dsDNA antibody titer decreased to 53 IU/mL, ANAs became negative, and the SLEDAI score dropped to 8. Prednisone was reduced to 20 mg/day, and the cumulative CTX dose reached 1.8 g. By January 2023, the patient’s renal function normalized (SCr: 1.18 mg/dL), and mycophenolate mofetil (MMF; 0.75 g twice daily) was introduced while prednisone was tapered to 5 mg/day. At the recent follow-up in January 2025, SCr had further improved to 0.85 mg/dL, with persistently negative ANAs, normalized complement levels (C3: 0.74 g/L; C4: 0.20 g/L), a low anti-dsDNA antibody titer (49 IU/mL), and a SLEDAI score of 0, indicating sustained disease remission. The key follow-up indicators and treatment course of the patient are summarized in Table 3.
Table 3
| Date | SCr (mg/dL) | eGFR* (ml/min × 1.73 m2) | ANA | A-dsDNA (IU/mL) | C3 (g/L) | C4 (g/L) | SLE DAI | Treatment |
|---|---|---|---|---|---|---|---|---|
| July 2022 | 5.74 | 13.11 | Positive | 197 | 0.23 | 0.02 | 12 | Methylprednisolone 40 mg/day (IV) + CTX (IV) 0.2 g per dose × 3 |
| Sep 2022 | 2.37 | 26.01 | Positive | 88 | 0.69 | 0.19 | 10 | Oral prednisone 40 mg/day tapering; CTX (IV) 0.2 g per dose × 3 |
| Oct 2022 | 1.69 | 37.42 | Negative | 53 | 0.63 | 0.19 | 8 | Prednisone 20 mg/day; CTX (cumulative 1.8 g) |
| Jan 2023 | 1.18 | 49.65 | - | - | - | - | - | Prednisone 5 mg/day +MMF started (0.75 g BID) |
| Apr 2023 | 1.1 | 56.96 | - | - | - | - | - | MMF maintenance |
| July 2023 | 0.88 | 79.57 | Negative | 91 | 0.73 | 0.17 | 4 | MMF tapering phase |
| Mar 2024 | 0.99 | 76.75 | Negative | 69.1 | 0.82 | 0.19 | 0 | MMF withdrawal |
| Jan 2025 | 0.85 | 87.65 | Negative | 49 | 0.74 | 0.20 | 0 | Prednisone 5 mg/day only |
Key follow-up indicators and treatment timeline of the patient.
*eGFR was calculated using the CKD-EPI equation (Chronic Kidney Disease Epidemiology Collaboration) (32); ANA, antinuclear antibody; A-dsDNA, anti-double-stranded DNA antibody; SCr, serum creatinine; SLE DAI, Systemic Lupus Erythematosus Disease Activity Index; MMF, mycophenolate mofetil; CTX, cyclophosphamide; IV, intravenous injection; −, not checked.
Discussion
Previous kidney biopsy analyses showed IgG4-TIN with concurrent glomerular injury-associated kidney disease (13, 14). Here, we present a rare case of the simultaneous diagnosis of SLE with IgG4-RKD, highlighting two distinct autoimmune diseases with overlapping renal manifestations. Although both conditions can involve the kidneys, their coexistence within the same individual is exceedingly rare, posing unique diagnostic and therapeutic challenges.
Our case patient experienced frequent PVCs. Although his antiphospholipid antibodies were negative, PVCs were reported during the active phases of SLE, which was likely attributed to autoimmune-mediated myocardial or conduction system involvement (15). Thus, an SLE-related arrhythmia could not be excluded. Additionally, there are currently no documented cases of IgG4-RD presenting with cardiac arrhythmias, making IgG4-RD an unlikely contributor in this context. During arrhythmia management, the patient underwent radiofrequency catheter ablation, requiring the administration of iodinated contrast media. Given the elevated SCr, contrast-induced nephropathy (CIN) was initially suspected. However, renal biopsy showed predominant plasma cell infiltration and storiform interstitial fibrosis, without eosinophilic infiltration or interstitial edema typical of CIN. These histopathologic features ruled out CIN in this patient.
Systemic lupus erythematosus is a multisystem autoimmune disease with strong female predominance, typically affecting women aged 20–40 years. LN is a common and serious complication of SLE, affecting approximately 14–55% of patients, with prevalence varying based on ethnicity and geographic region (16). The 2018 revised ISN/RPS classification stratifies LN based on immune complex deposition patterns, with Class II characterized by mesangial hypercellularity and immune deposits without significant glomerular basement membrane involvement (17). Serologically, positive anti-dsDNA antibodies and low complement (C3, C4) levels reflect disease activity.
IgG4-RD is a systemic, immune-mediated fibroinflammatory condition characterized by organ enlargement, elevated serum IgG4, and tissue infiltration by IgG4-positive plasma cells. In the kidney, the predominant manifestation is IgG4-TIN, typically observed in middle-aged and elderly men (9).
Despite overlapping renal manifestations, SLE and IgG4-RKD represent distinct immunologic entities. Histologically, LN typically affects glomeruli, whereas IgG4-RKD predominantly involves the tubulointerstitial compartment. Our patient simultaneously met the diagnostic criteria for both SLE (EULAR/ACR 2019) and IgG4-RKD (Japanese Society of Nephrology 2011) (8, 9). The clinical significance of differentiating SLE from IgG4-RKD lies in their distinct therapeutic strategies and prognostic implications. Although both conditions can affect the kidneys, misclassification could lead to overtreatment or undertreatment. For instance, unnecessary exposure to cytotoxic immunosuppressants can be avoided in patients correctly diagnosed with IgG4-TIN, which often responds well to corticosteroids alone. Conversely, failure to recognize LN can delay the timely initiation of disease-modifying therapies, increasing the risk of irreversible renal damage. Therefore, establishing an accurate diagnosis is essential for tailoring appropriate treatment and optimizing patient outcomes.
To better contextualize our case, we reviewed five published reports of patients diagnosed with both SLE and IgG4-RD (18–22), which are summarized in Table 4. This revealed a consistent pattern of elderly male predominance, mixed systemic and renal symptoms, and dual serologic and histopathologic features. All patients exhibited positivity for ANAs and anti-dsDNA antibodies, while complement levels varied. Renal biopsy findings ranged from lupus-associated glomerulonephritis to IgG4-TIN, often coexisting within the same specimen. Despite pathologic heterogeneity, the presence of abundant IgG4+ plasma cells (> 10–40/high-power field) (18, 20–22), storiform fibrosis (21, 22), and an IgG4/IgG-positive cell ratio > 40% (19, 20) in several of these cases supports true IgG4-RKD rather than incidental IgG4 elevation. All patients received glucocorticoid-based immunosuppressive therapy, with or without additional agents (MMF, HCQ, belimumab), and experienced clinical improvement. These cases, along with our case patient, highlight the diagnostic value of IgG subclass staining in lupus patients with atypical interstitial pathology. Standard SLE-directed therapy appears effective in such overlap syndromes, but the pathogenic role and prognostic relevance of IgG4 components warrant further investigation. Increasing evidence suggests that the molecular mechanisms underlying IgG4-TIN may be associated with retroperitoneal fibrosis, activation of the renin–angiotensin system, and microbial dysbiosis (23–25).
Table 4
| Author (Year) | Age/Sex | IgG4 (mg/dl) | Main clinical symptoms | Test Results | Pathology | Treatment plan | Therapeutic effect |
|---|---|---|---|---|---|---|---|
| Zaarour et al. (18) | 71/ female | 37.10 | Abdominal pain, vomiting, and diarrhea; AKI with hematuria and proteinuria | ANA positive; hypoalbuminemia; creatinine 9.56 mg/dL, BUN 88 mg/dL | Kidney: Acute tubular necrosis, with a maximum of 13 IgG4 + cells /HPF. “full-house” of immune reactants. | GC + MMF | Showed improvement |
| Arai et al. (19) | 74/male | 1,210 | Raynaud’s, arthritis, parotid swelling, and lymphadenopathy; later fever, dyspnea, and pleural effusion | ANA positive, low complement, elevated globulin, Anti-dsDNA positive | Kidney: Significant IgG4-positive infiltration in tubulointerstitium, IgG4+/ IgG + >40% | GC | Showed improvement |
| Yamamoto et al. (20) | 58/ female | 1,240 | Photosensitivity and extremity rash; 2016: fever and fatigue. | ANA and anti-dSDNA are positive, normal complement, elevated IgG, and massive proteinuria | Kidney: Abundant IgG4-positive cells in the renal tubulointerstitium | GC + MMF; GC + Belimumab | Showed improvement |
| Ying et al. (22) | 64/male | 2,960 | Recurrent lower limb erythema for 3 months | Elevated SCr, ANA and Anti-dsDNA positive, hemolytic anemia, 0.5 g/d proteinuria; CT: multiple lymphadenopathies | IgG4-related tubulointerstitial nephritis; mesangial proliferative glomerulonephritis with immune complex deposits | GC | Showed improvement |
| Xie et al. (21) | 67/male | 1700 | Loss of appetite and general weakness | Leukopenia, thrombocytopenia; ANA and Anti-dsDNA positive; low C3/C4; elevated IgG | Multifocal lymphoid hyperplasia and interstitial fibrosis; >10 IgG4 + cells/HPF | GC + HCQ + MMF | Showed improvement |
| This case | 68/male | 1,180 | periorbital and bilateral lower extremity edema | Elevated SCr, ANA and Anti-dsDNA positive; low C3/C4; and elevated IgG4 | “full-house” of immune reactants, >30 IgG4 + cells /HPF; IgG4+/IgG + >40% | GC + CTX; GC + MMF | Showed improvement |
Clinical data of 5 SLE patients complicated with IgG4-RD.
GC, Glucocorticoid; HCQ, Hydroxychloroquine; MMF, Mycophenolate mofetil; HPF, high-power visual field; CTX, Cyclophosphamide.
Although the overlapping pathophysiology of IgG4-RD and SLE remains unclear, dysregulation of the humoral immune system is a common feature of the two diseases. In IgG4-RD, the expansion of CD20+ B cells and plasmablasts, as well as antigen presentation to CD4+ cytotoxic T cells, have been implicated in disease pathogenesis (26–28). Notably, T follicular helper (Tfh) cells facilitate B-cell differentiation into plasmablasts and contribute to IgG4 class-switching, suggesting a pivotal role in IgG4-RD progression (29). In SLE, which is also an autoimmune disease, impaired humoral regulation and hyperactivation of CD4+ T cells are prominent features. Tfh cells, which are significantly increased in patients with SLE, are known to promote autoreactive B-cell responses and autoantibody production, thereby driving disease progression (30, 31). These findings suggest that Tfh cells may act as a shared immunological hub linking IgG4-RD and SLE. Whether the dysregulation of the Tfh cell axis constitutes a common pathogenic mechanism in the two diseases remains to be further elucidated (32). Investigating this potential convergence may provide deeper insights into their overlapping features and guide targeted immunomodulatory therapies.
Our case patient was diagnosed with overlapping Class II LN and IgG4-RKD, presenting with acute kidney injury, hypocomplementemia, and elevated disease activity. Although Class II LN typically requires limited immunosuppression, the presence of renal dysfunction (SCr 5.74 mg/dL; estimated glomerular filtration rate [eGFR] 13.1 mL/min) warranted an intensified approach. Following the 2023 EULAR recommendations for SLE and the 2015 international consensus on IgG4-RD management, we induced remission by initiating intravenous methylprednisolone and low-dose CTX (11, 12). As renal function improved, glucocorticoids were tapered, and MMF was introduced as a steroid-sparing agent for long-term maintenance. The patient achieved sustained clinical remission, allowing MMF tapering and maintenance with low-dose prednisone alone. This stepwise immunosuppressive strategy of balancing early disease control with long-term safety proved effective in managing both the LN and the IgG4-RKD.
Conclusion
In this study, we report a rare case of overlapping LN and IgG4-TIN that highlights the importance of distinguishing CIN from immune-mediated renal dysfunction. This case shows that SLE and IgG4-TIN can coexist, though reports remain scarce. Our treatment strategy involves glucocorticoids combined with CTX proved effective, offering valuable therapeutic insights into managing complex presentations of SLE with IgG4-TIN.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of Weifang Medical College Affiliated Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XJ: Writing – original draft, Conceptualization, Data curation, Formal analysis, Investigation, Validation. GJ: Writing – original draft, Conceptualization, Data curation, Formal analysis, Investigation, Validation. HS: Formal analysis, Project administration, Validation, Writing – original draft. XL: Methodology, Project administration, Writing – review & editing. XC: Funding acquisition, Writing – review & editing, Investigation, Methodology, Project administration, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Weifang Key Laboratory of Integrated Traditional Chinese and Western Medicine for Chronic renal Failure; the TCM science and technology project of Shandong Province (M-2023105 to Xuexun Chen); Weifang Soft Science Research Plan (2021RKX047 to Xuan Li); the Medical and Health Science and Technology Project of Shandong Province (202403050819 to Xuan Li); and the Science and Technology Development Project of the Affiliated Hospital of Shandong Second Medical University (2024FYQ011 to Xuan Li).
Acknowledgments
The authors thank all the physicians and nurses who took care of the patient. We thank the patient who agreed to collect and publish his data. We thank Michelle Kahmeyer-Gabbe, PhD, from Liwen Bianji (Edanz) (www.liwenbianji.cn) for editing the English text of a draft of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Francis A Harhay MN Ong A Tummalapalli SL Ortiz A Fogo AB et al . Chronic kidney disease and the global public health agenda: an international consensus. Nat Rev Nephrol. (2024) 20:473–85. doi: 10.1038/s41581-024-00820-6
2.
Khandpur S Mishra P Mishra S Tiwari S . Challenges in predictive modelling of chronic kidney disease: a narrative review. World J Nephrol. (2024) 13:97214. doi: 10.5527/wjn.v13.i3.97214
3.
Amatruda M Carucci NS Chimenz R Conti G . Immunoglobulin a vasculitis nephritis: current understanding of pathogenesis and treatment. World J Nephrol. (2023) 12:82–92. doi: 10.5527/wjn.v12.i4.82
4.
Salvadori M Rosso G . What is new in the pathogenesis and treatment of IgA glomerulonephritis. World J Nephrol. (2024) 13:98709. doi: 10.5527/wjn.v13.i4.98709
5.
Yamamoto T Isaka Y . Pathological mechanisms of kidney disease in ageing. Nat Rev Nephrol. (2024) 20:603–15. doi: 10.1038/s41581-024-00868-4
6.
Lanzillotta M Mancuso G Della-Torre E . Advances in the diagnosis and management of IgG4 related disease. BMJ. (2020) 369:m1067. doi: 10.1136/bmj.m1067
7.
Owen KA Grammer AC Lipsky PE . Deconvoluting the heterogeneity of SLE: the contribution of ancestry. J Allergy Clin Immunol. (2022) 149:12–23. doi: 10.1016/j.jaci.2021.11.005
8.
Aringer M Costenbader K Daikh D Brinks R Mosca M Ramsey-Goldman R et al . 2019 European league against rheumatism/American College of Rheumatology Classification Criteria for systemic lupus erythematosus. Arthritis Rheumatol. (2019) 71:1400–12. doi: 10.1002/art.40930
9.
Saeki T Kawano M Nagasawa T Ubara Y Taniguchi Y Yanagita M et al . Validation of the diagnostic criteria for IgG4-related kidney disease (IgG4-RKD) 2011, and proposal of a new 2020 version. Clin Exp Nephrol. (2021) 25:99–109. doi: 10.1007/s10157-020-01993-7
10.
Jesus D Matos A Henriques C Zen M Larosa M Iaccarino L et al . Derivation and validation of the SLE disease activity score (SLE-DAS): a new SLE continuous measure with high sensitivity for changes in disease activity. Ann Rheum Dis. (2019) 78:365–71. doi: 10.1136/annrheumdis-2018-214502
11.
Khosroshahi A Wallace ZS Crowe JL Akamizu T Azumi A Carruthers MN et al . International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. (2015) 67:1688–99. doi: 10.1002/art.39132
12.
Fanouriakis A Kostopoulou M Andersen J Aringer M Arnaud L Bae SC et al . EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann Rheum Dis. (2024) 83:15–29. doi: 10.1136/ard-2023-224762
13.
Hârza M Ismail G Mitroi G Gherghiceanu M Preda A Sinescu I . Inflammatory pseudotumors of the kidney due to IgG4-related tubulointerstitial nephritis. Romanian J Morphol Embryol. (2014) 55:419–23. PMID:
14.
Su T Wang H Wang S Yang L . Clinicopathological patterns and predictors of the functional restoration of immunoglobulin G4-related kidney disease: a Chinese single-center cohort study. Front Med. (2021) 8:736098. doi: 10.3389/fmed.2021.736098
15.
Frostegård J . Systemic lupus erythematosus and cardiovascular disease. J Intern Med. (2023) 293:48–62. doi: 10.1111/joim.13557
16.
Parikh SV Almaani S Brodsky S Rovin BH . Update on lupus nephritis: Core curriculum 2020. Am J Kidney Dis. (2020) 76:265–81. doi: 10.1053/j.ajkd.2019.10.017
17.
Bajema IM Wilhelmus S Alpers CE Bruijn JA Colvin RB Cook HT et al . Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. (2018) 93:789–96. doi: 10.1016/j.kint.2017.11.023
18.
Zaarour M Weerasinghe C Eter A El-Sayegh S El-Charabaty E . An overlapping case of lupus nephritis and IgG4-related kidney disease. J Clin Med Res. (2015) 7:575–81. doi: 10.14740/jocmr2189w
19.
Arai H Hayashi H Ogata S Uto K Saegusa J Takahashi K et al . Progression of immunoglobulin G4-related disease to systematic lupus erythematosus after gastric cancer surgery: a case report. Medicine (Baltimore). (2018) 97:e13545. doi: 10.1097/MD.0000000000013545
20.
Yamamoto M Aochi S Suzuki C Nakamura S Murakami R Ogawa Y et al . A case with good response to belimumab for lupus nephritis complicated by IgG4-related disease. Lupus. (2019) 28:786–9. doi: 10.1177/0961203319840430
21.
Xie R Li H Wang X Li X . IgG4-associated disease with systemic lupus erythematosus: a case report and review. Clin Nephrol. (2024) 102:166–73. doi: 10.5414/CN111343
22.
Ying J Lili Z Chenfeng J Shaoshan L Zhen C . A case of IgG4 related disease with SLE like manifestation. Chin J Nephrol Dial Trans. (2022) 31:395–400. doi: 10.3969/j.issn.1006-298X.2022.04.020
23.
Miao H Wang YN Su W Zou L Zhuang SG Yu XY et al . Sirtuin 6 protects against podocyte injury by blocking the renin-angiotensin system by inhibiting the Wnt1/β-catenin pathway. Acta Pharmacol Sin. (2024) 45:137–49. doi: 10.1038/s41401-023-01148-w
24.
Miao H Wang YN Yu XY Zou L Guo Y Su W et al . Lactobacillus species ameliorate membranous nephropathy through inhibiting the aryl hydrocarbon receptor pathway via tryptophan-produced indole metabolites. Br J Pharmacol. (2024) 181:162–79. doi: 10.1111/bph.16219
25.
Kawano M Saeki T Nakashima H . IgG4-related kidney disease and retroperitoneal fibrosis: an update. Mod Rheumatol. (2019) 29:231–9. doi: 10.1080/14397595.2018.1554321
26.
Kersten R Trampert DC Herta T Hubers LM de Buy M Wenniger LJ et al . IgG4-related cholangitis - a mimicker of fibrosing and malignant cholangiopathies. J Hepatol. (2023) 79:1502–23. doi: 10.1016/j.jhep.2023.08.005
27.
Lu C Li S Qing P Zhang Q Ji X Tang Z et al . Single-cell transcriptome analysis and protein profiling reveal broad immune system activation in IgG4-related disease. JCI Insight. (2023) 8:e167602. doi: 10.1172/jci.insight.167602
28.
Koga R Maehara T Aoyagi R Munemura R Murakami Y Doi A et al . Granzyme K-and amphiregulin-expressing cytotoxic T cells and activated extrafollicular B cells are potential drivers of IgG4-related disease. J Allergy Clin Immunol. (2024) 153:1095-112. doi: 10.1016/j.jaci.2023.11.916
29.
Xu J Zhai J Zhao J . Pathogenic roles of follicular helper T cells in IgG4-related disease and implications for potential therapy. Front Immunol. (2024) 15:1413860. doi: 10.3389/fimmu.2024.1413860
30.
Li X Sun W Huang M Gong L Zhang X Zhong L et al . Deficiency of CBL and CBLB ubiquitin ligases leads to hyper T follicular helper cell responses and lupus by reducing BCL6 degradation. Immunity. (2024) 57:1603–17.e7. doi: 10.1016/j.immuni.2024.04.023
31.
Shi X Liao T Chen Y Chen J Liu Y Zhao J et al . Dihydroartemisinin inhibits follicular helper T and B cells: implications for systemic lupus erythematosus treatment. Arch Pharm Res. (2024) 47:632–44. doi: 10.1007/s12272-024-01505-1
32.
Li H Boulougoura A Endo Y Tsokos GC . Abnormalities of T cells in systemic lupus erythematosus: new insights in pathogenesis and therapeutic strategies. J Autoimmun. (2022) 132:102870. doi: 10.1016/j.jaut.2022.102870
Summary
Keywords
systemic lupus erythematosus, lupus nephritis, IgG-4 related diseases, IgG4-related tubulointerstitial nephritis, renal biopsy
Citation
Ji X, Jing G, Sun H, Li X and Chen X (2025) Case report and literature review: IgG4-related tubulointerstitial nephritis coexistent with systemic lupus erythematosus. Front. Med. 12:1585351. doi: 10.3389/fmed.2025.1585351
Received
28 February 2025
Accepted
16 June 2025
Published
11 July 2025
Volume
12 - 2025
Edited by
Wenjun Lin, Shanghai Sixth People’s Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, China
Reviewed by
Yu Jia, First Affiliated Hospital of Zhengzhou University, China
Zhe Li, Nanjing University, China
Wenbo Zhao, Third Affiliated Hospital of Sun Yat-sen University, China
Updates
Copyright
© 2025 Ji, Jing, Sun, Li and Chen.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuan Li, fylixuan1992@sdsmu.edu.cn Xuexun Chen, fyxuexun_chen@sdsmu.edu.cn
†These authors have contributed equally to this work and share first authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.