Abstract
Background:
The junction fragment after the DMD gene deletion has been identified as a new specific DNA sequence formed by the reconnection of the ends. Our study aims to report a novel method for prenatal diagnosis of BMD by using PCR to detect junction fragments and deleted exons.
Methods:
We performed the prenatal diagnosis of a fetus with deletional BMD in this study. The proband of this family was the deletion of exons 3 to 5 of the DMD gene. The junction fragment primer designed after locating the breakpoint was used to PCR-amplify the junction fragments of the villus sample and the amniotic fluid genomic DNA. The exon 3 primer was used to amplify the deletion exons of the DMD gene from the villus sample and the amniotic fluid genomic DNA, respectively. At the same time, sex identification was carried out. Finally, the diagnosis results were analyzed.
Results:
The diagnosis of villus sampling was a contradictory result of obtaining the DMD gene deletion junction fragment and the absence of the exon deletion in the male fetus, suggesting that the villus sample was contaminated by maternal cells and the test was unsuccessful. The subsequent diagnosis of amniotic fluid was that the male fetus detected both the junction fragment and the corresponding exon deletion, and was diagnosed as a male fetus with BMD.
Conclusions:
Combining PCR to detect junction fragments and deleted exons in the prenatal diagnosis of BMD can effectively identify maternal cell contamination. The results were confirmed to be highly accurate and specific.
1 Introduction
Becker muscular dystrophy (BMD) is a common X-linked recessive genetic disease that primarily affects muscle tissue. BMD and Duchenne muscular dystrophy (DMD) are both considered allelic diseases that differ in severity and are caused by mutations in the DMD gene located in Xp21 (1). The large deletion mutation in the DMD gene has been determined to be responsible for the majority of DMD/BMD cases (~65%) (2). After deletion mutations are induced in the DMD gene, they are almost repaired by non-homologous end joining (3). In this process, the resulting junction fragment represents a new specific DNA sequence formed by the reconnection of the ends. Previously, under the condition that the full sequence of the DMD gene was basically clear, we have used a polymerase chain reaction (PCR)-based genome-walking method to accurately locate the missing breakpoint and we have directly amplified the junction fragment sequence by PCR (4).
The overall incidence of DMD/BMD is high, with ~1 of every 3,500 live-born boys affected (5). However, no effective treatment is currently available (6, 7). Approximately two-third of DMD/BMD cases are derived from familial inheritance (8, 9); thus, the prenatal diagnosis of pregnant women in DMD/BMD families is important to prevent the birth of a sick fetus and is currently the most effective way to reduce the incidence of DMD/BMD (10, 11). Commonly used detection technologies, such as multiplex PCR (12, 13), quantitative PCR (14, 15), fluorescence in situ hybridization (FISH) (16, 17), multiplex amplifiable probe hybridization (MAPH) (2, 18), and multiplex ligation-dependent probe amplification (MLPA) (19, 20), have been applied in the past decade to DMD/BMD pathogenic gene detection. However, prenatal diagnosis requires methods with greater detection ability. Prenatal diagnosis typically uses collected test specimens such as villus tissue, amniotic fluid, and umbilical cord blood; however, the small amount and possible maternal cell contamination presents difficulties in the detection and may interfere with the accuracy of the diagnostic results (21, 22). The aforementioned MLPA, FISH, quantitative PCR, etc. can be used to detect and analyse pathogenic genes in the prenatal diagnosis of DMD/BMD, but when the test specimen is contaminated with maternal cells, it cannot be additionally identified, which affects the accuracy of the test results. Therefore, laboratory testing of maternal cell contamination of each prenatal diagnostic sample has become a standard part of prenatal diagnosis (23). In this study, we diagnosed the fetus of a pregnant woman who is a carrier of BMD by detecting the missing exons, while simultaneously detecting the sequence of the junction fragment. We found that for male fetuses, maternal cell contamination can be effectively identified while achieving a prenatal diagnosis of DMD/BMD. However, for female fetuses, due to the fact that the mother is a female carrier, it is difficult to accurately identify whether the specimen being tested is contaminated by maternal cells using this method. Therefore, it is impossible to distinguish whether the fetus is a normal female or a carrier, which is the limitation of this method.
2 Materials and methods
2.1 Case information
Family members of a BMD family from Zhaoqing, Guangdong Province, China comprised the participants in this study. Proband III1 was a clinically diagnosed BMD patient and the deletion of exons 3 to 5 in the DMD gene were confirmed in this individual. After confirming that the 28 years old female III3 in the family was a carrier, she provided written informed consent to become a research participant in this study. Carrier III3 was provided genetic counseling and had three pregnancies, with the first two ending in abortion due to accidents. IV3 was the third fetus to be diagnosed at pregnancy. A villus sample was obtained (about 30 mg) at 11 weeks of pregnancy and an amniotic fluid sample (20 ml) was obtained at 20 weeks of pregnancy for prenatal genetic diagnosis (Figure 1).
Figure 1

Pedigree of a BMD family.
2.2 Identification of proband genotype and carrier detection
In this family, genomic DNA was extracted from proband III1 and possible carriers III3 by proteinase K and phenol extraction. For PCR detection, 18 exon primers (12, 24), in conjunction with primers from the second and fifth exons [refer to the primer design provided by Bakker and Kneppers on the Leiden muscular dystrophy web page (http://www.dmd.nl)], were used and the proband was identified as the DMD gene deletion of exons 3 to 5.
After determining the extent of the exon deletion, the breakpoint was located at the corresponding intron by the PCR walking method of primer design involved the design of 5 pairs of primers in the second intron with an average sequence of about 30 kb. After detecting the region of the breakpoint in the first PCR reaction, we continued to design 1 pair of primers per 3 kb sequence in this region. In the fifth intron, we continued to directly design 1 pair of primers per 3 kb sequence. Each primer was designed to limit the length of its PCR product to between 301 and 400 bp. Using the above primers, the PCR reaction was carried out to locate the breakpoint. The 5′ end of the breakpoint was located at a site about 153 kb away from the first base in intron 2, while the 3′ end of the breakpoint was located near exon 6 in intron 5. A pair of primers (D1-F/R) was subsequently designed near the breakpoints of the two introns to directly amplify the junction fragment. The product fragment of about 2 kb was obtained and sequenced after purification. This junction fragment has been accepted and published in GenBank with serial number EF434728. The sequences of introns 2 and 5 of the DMD gene were obtained from http://www.dmd.nl/seqs.
After confirming the detailed sequence of the junction fragment of proband III1, the pair of primers (D4-F/R) for amplifying the junction fragment was redesigned to reduce the length of the PCR product to 495 bp, which improved the success rate of the junction fragment amplification compared with conventional PCR. The peripheral blood genomic DNA of III3 was then amplified using PCR with the D4-F/R primer pair and the 495-bp positive product fragment result was confirmed by sequencing. III3 of this family was subsequently diagnosed as a BMD female carrier.
2.3 Prenatal diagnosis
Genomic DNA was prepared immediately after samples for the prenatal diagnosis were collected. The villus samples were collected at the 11th week of pregnancy and were repeatedly washed with normal saline. The washed small pieces of tissue were then ground and crushed and the genomic DNA was extracted by the proteinase K and phenol method. The amniotic fluid sample (20 ml) was collected at the 20th week of pregnancy and was immediately centrifuged to collect the precipitate. The genomic DNA was extracted by the protease K and phenol method.
The primer was used to PCR-amplify the junction fragments of the villus sample and the amniotic fluid genomic DNA, with the junction fragment of Proband III1 used as a control. The primer (12) was used to amplify exon 3 of the DMD gene from the villus sample and the amniotic fluid genomic DNA, respectively. The ex6-F/R primer (12) was used to amplify the non-deleted exon 6 as a normal control to determine if there was an exon deletion corresponding to the proband. The sex identification primers, SRY-109F and SRY-245R (25), were used to identify the genomic DNA of the villus samples and amniotic fluid cells.
If a positive result was obtained from the above samples by amplifying the junction fragments and the corresponding deletion of the associated exons was found, the PCR products were further sequenced for verification.
2.4 Sequencing of the junction fragments
To verify the accuracy of the diagnostic results of this method, sequencing analysis was performed on the junction fragments of the proband III1, carrier III3, and the confirmed specimen from prenatal diagnosis. The aforementioned PCR products were sequenced after purification and electrophoresis observation. The sequencing work for this study was completed by Shanghai Invitrogen Biotechnology Co., Ltd., China, and the sequences of the PCR products were fully determined in both directions. The sequencing primers are shown in Appendix.
3 Results
3.1 Villus sampling diagnosis
PCR results of the junction fragment from the genomic DNA of the villus tissue sample collected at the 11th week of pregnancy showed that a 495-bp product was amplified. This was the same length as the junction fragment product of the proband III1 of this family. However, testing of exon 3 showed a positive amplification without deletion. In addition, the gender was identified as male (Figure 2). The contradictory result of obtaining the DMD gene deletion junction fragment and the absence of the exon deletion in the male fetus indicated that the villus sample was contaminated by maternal cells and the test was unsuccessful.
Figure 2

PCR diagnosis results of chorionic villi sampling of tertigravida III3. (A) PCR amplification of the junction fragment and the corresponding exons; Marker: 100 bp ladder marker; lane CVS sample: PCR amplification result of the junction fragment of the chorionic villi sample; lane proband III1: PCR amplification result of the junction fragment of proband III1; lane exon 3: PCR amplification result of exon 3 of the chorionic villi sample; lane exon 6: PCR amplification result of exon 6 of the chorionic villi sample; (B) PCR amplification of the SRY gene. Marker: 100 bp ladder marker; lane CVS sample: PCR amplification result of the SRY gene of the chorionic villi sample; lane male: normal male control; lane female: normal female control.
3.2 Amniotic fluid diagnosis
PCR results of the junction fragment from the genomic DNA of the amniotic fluid cells taken at the 20th week of pregnancy showed that the 495-bp product was amplified, which was consistent with the length of the fragment product of the proband III1 of this family. The amplification of exon 3 was negative, demonstrating that the corresponding DMD gene region was deleted. The gender identification results were the same, indicating the fetus was male (Figure 3). Therefore, the fetal IV3 was diagnosed as a male fetus with BMD. Given this information, the pregnant woman III3 chose to terminate the pregnancy. The fetal tissue was subsequently analyzed and the diagnostic results were confirmed.
Figure 3

PCR diagnosis results of the amniotic fluid of tertigravida III3. (A) PCR amplification results of the junction fragment and the corresponding exons; Marker: 100 bp ladder marker; lane AFS sample: PCR amplification result of the junction fragment of the amniotic fluid sample; lane proband III1: PCR amplification result of the junction fragment of proband III1; lane exon 3: PCR amplification result of exon 3 of the amniotic fluid sample; lane exon 6: PCR amplification result of exon 6 of the amniotic fluid sample; (B) PCR amplifications of the SRY gene. Marker: 100 bp ladder marker; lane AFS sample: PCR amplification result of the SRY gene of the amniotic fluid sample; lane male: normal male control; lane female: normal female control.
3.3 Sequencing results
A total of 2,113 bp of valid sequences were obtained from the sequencing of the PCR products of proband III1, while 495 bp of valid sequences were obtained from the sequencing of the PCR products of carrier III3 and the amniotic fluid sample for prenatal diagnosis. These sequences were all confirmed to be the deletion junction fragments of the DMD gene through alignment by BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Meanwhile, the sequencing results of the junction fragment in the amniotic fluid sample showed no difference from those of the junction fragments in proband III1 and carrier III3 in this family (Figure 4), indicating that they have the same genotype. The pathogenic gene is a new junction sequence formed by the insertion of a 26-bp sequence (TATATTTAAAGGGGCTTATATTTAAT) after the large fragment deletion of exons 3-5 of the DMD gene, which further verified the PCR diagnosis result that fetus IV3 is a male fetus with BMD.
Figure 4
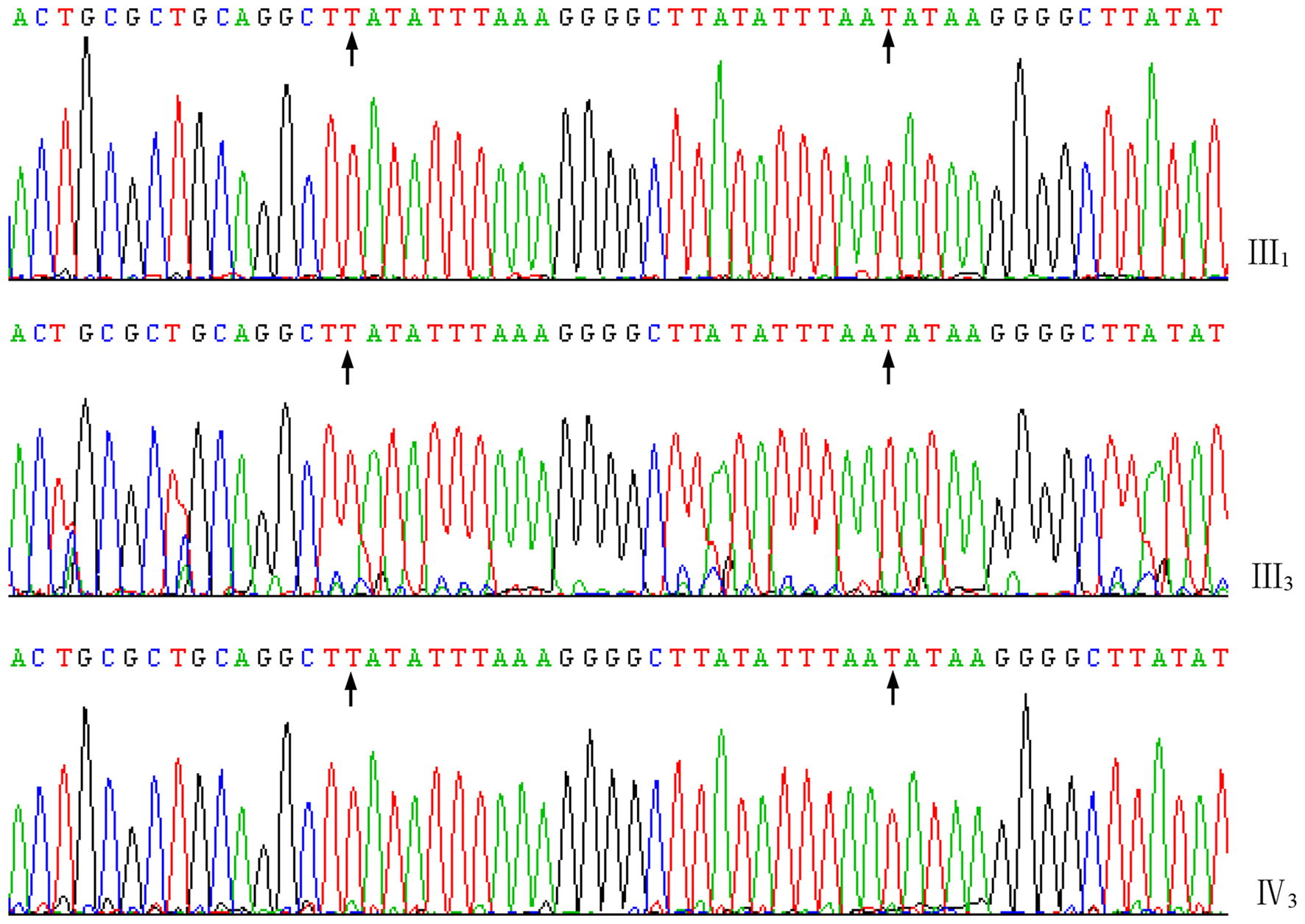
Forward sequences of the junction fragments of III1, III3, and IV3 of the family. The characters between the arrows indicate the 26 bp nucleotides inserted in the joint.
4 Discussion
Female carriers of DMD/BMD have a 50% risk of passing the disease-causing mutant gene onto their offspring. Among the potential offspring, boys who acquire the disease gene will become sick, while the girls will become healthy carriers. Therefore, the overall risk of giving birth to a sick infant is 25% (26). Therefore, the prenatal diagnosis of DMD/BMD is necessary to provide the family with accurate and timely information regarding the health of the unborn fetus. Accordingly, the prenatal diagnosis of DMD/BMD necessitates 100% accuracy, with a highly specific diagnosis method. In general, the prenatal diagnosis of DMD/BMD is based on invasive methods (27, 28) and uses test specimens such as villus tissue, amniotic fluid, or umbilical cord blood to directly analyze the fetus (29, 30). The diagnostic results are reliable and considered the gold standard for prenatal diagnosis. However, due to the risk of maternal cell contamination in these specimens, the diagnostic results may still be incorrect (21, 31, 32). Therefore, the identification and exclusion of maternal cells during the prenatal diagnosis process remains an obstacle that most laboratories have been unable to avoid. However, because it can lead to a false negative diagnosis, extraordinary measures must be taken to address this issue (21, 33). The identification of maternal cell contamination is generally performed by short tandem repeat polymorphism linkage analysis (34, 35). Based on the fact that the junction fragment is a specific DNA sequence belonging to a patient or carrier with deletion DMD/BMD, we believe that a diagnosis method using PCR to detect both the junction fragment and the corresponding deleted exons can effectively identify the maternal cell pollution while simultaneously detecting the pathogenic gene. In this regard, the accuracy and reliability of the prenatal diagnosis results can be improved.
Since DMD/BMD is a sex-linked inheritable disease, the method described in this study first performs sex identification of the embryo or fetuse. For female fetuses, the protocol is to record the experimental results and continue the pregnancy but inform the pregnant women that the fetus may be a carrier, something that can be verified after delivery. For male fetuses, if the junction fragment is amplified positively, suggesting that the same genotype as the proband, then theoretically the corresponding exons should be deleted (negative amplification) and the fetus would be considered to have DMB/BMD. If the junction fragment is positive and the corresponding exons are subsequently found to be deleted, it can be confirmed that the fetus has DMD/BMD. This information should be provided to the pregnant woman in a timely manner so that a decision can be made whether to terminate the pregnancy. If the woman chooses to terminate the pregnancy, the aborted fetal tissues should be collected and the DNA prepared for diagnostic verification.
Before the decision to terminate the pregnancy is made, if testing indicates that the junction fragment results are positive but the corresponding exons are detected without deletion (positive amplification), the test specimen can be considered to be contaminated with maternal cells and testing should be repeated. If the junction fragment results are negative and the corresponding exons are detected without deletion, the fetus can be considered to not have the junction fragment and there is no exon deletion corresponding to the proband. In the latter case, this represents a normal male fetus and the pregnancy can be continued without worry. In both cases, the results can be verified postpartum.
In this study, during the prenatal diagnosis of carrier III3, we performed villus sampling and amniotic fluid detection. The sex of the fetus was identified as male. A junction fragment consistent with Proband III1 was detected in the villus sample, but there was no detection of the associated deletion in the corresponding exons. This suggested that the specimen was contaminated with maternal cells and testing was repeated. In this process, the amniotic fluid sample was tested again and the junction fragment was still positive. However, the deletion of the corresponding exons was detected this time. These results ruled out the possibility of maternal cell contamination and confirmed the diagnosis of fetal IV3 with BMD. The probability of villus samples being contaminated with maternal cells during prenatal diagnostic sampling may be slightly higher than that of amniotic fluid samples (36). However, a disadvantage of amniotic fluid and umbilical cord blood analysis is that it can only be performed in the second trimester of pregnancy, making these types of samples not suitable for early diagnosis. Therefore, to achieve the best prenatal diagnosis results, laboratories need to complete the diagnosis as early as possible. At the same time, the diagnosis method should have procedures in place to effectively identify whether the specimen to be tested is contaminated with maternal cells (21, 37, 38).
While the method described in this study solves the problem of identifying whether the specimen to be tested is contaminated with maternal cells for male fetuses, it remains difficult to accurately identify whether the specimen to be tested is contaminated with maternal cells for female fetuses because the mother is a female carrier. For this reason, it is advised to wait for postpartum examination and verification when the fetus has been identified as female. In addition, the use of the junction fragment for DMD/BMD prenatal diagnosis requires a detailed analysis of the genotypes of the probands in the family, which has the disadvantage of requiring a number of complicated steps. Indeed, because the DMD gene sequence is extremely large and the mutation types are complex, each DMD gene detection method has their own advantages and disadvantages. For the actual application of these methods in the clinical setting, each laboratory is required to adapt their method to the specific circumstances of the case. For example, MAPH and MLPA technologies (18, 39), which are considered to be the most promising methods for detecting DMD gene deletions and repeated mutations, are not perfect techniques. In fact, studies have repeatedly identified these methods as time-consuming and cumbersome (40, 41), with a relatively high cost (42) and inability to apply to single cell analysis. In addition, many difficulties remain in terms of preimplantation genetic diagnosis (PGD) (26). In contrast, the method developed in this study only requires a small amount of test samples, making it feasible for application in PGD. Moreover, for male fetuses, this method has a distinctive advantage: it can effectively identify maternal cell contamination while performing prenatal diagnosis.
5 Conclusions
During the process of detecting the deleted exons and the junction fragments in two samples of villus and amniotic fluid via PCR, we found that maternal cell contamination can be effectively identified while conducting prenatal diagnosis for DMD/BMD in male fetuses. Our results have been confirmed to be highly accurate and specific. Overall, our findings indicate that this method for identifying maternal cell contamination during the diagnosis of male fetuses has a distinct advantage that can be implemented in the clinical setting. However, for female fetuses, it is still impossible to accurately distinguish between normal females and female carriers.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The studies involving humans were approved by the Medical Ethical Committee of Nanfang Hospital of Southern Medical University (Approval Number: NFEC-2024-437). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LC: Conceptualization, Validation, Writing – original draft. WL: Data curation, Methodology, Writing – original draft. MZ: Conceptualization, Funding acquisition, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the following funding sources: The Guangdong Science and Technology Plan Project(No. 2014A020212539), the President Foundation of Nanfang Hospital Baiyun Branch, Southern Medical University(No. BYYZ23010).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Patterson G Conner H Groneman M Blavo C Parmar MS . Duchenne muscular dystrophy: current treatment and emerging exon skipping and gene therapy approach. Eur J Pharmacol. (2023) 947:175675. 10.1016/j.ejphar.2023.175675
2.
White S Kalf M Liu Q Villerius M Engelsma D Kriek M et al . Comprehensive detection of genomic duplications and deletions in the DMD gene, by use of multiplex amplifiable probe hybridization. Am J Hum Genet. (2002) 71:365–74. 10.1086/341942
3.
Sironi M Pozzoli U Cagliani R Giorda R Comi GP Bardoni A et al . Relevance of sequence and structure elements for deletion events in the dystrophin gene major hot-spot. Hum Genet. (2003) 112:272–88. 10.1007/s00439-002-0881-5
4.
Zhong M Pan SY Lu BX Li W . Cloning and sequencing of junction fragment with exons 45-54 deletion of dystrophin gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2006) 23:138–41. 10.3760/j.issn:1003-9406.2006.02.004
5.
Eslahi A Alizadeh F Avan A Ferns GA Moghbeli M Reza Abbaszadegan M et al . New advancements in CRISPR based gene therapy of Duchenne muscular dystrophy. Gene. (2023) 867:147358. 10.1016/j.gene.2023.147358
6.
Manini A Abati E Nuredini A Corti S Comi GP . Adeno-Associated Virus (AAV)-Mediated Gene Therapy for Duchenne Muscular Dystrophy: the Issue of transgene persistence. Front Neurol. (2022) 12:814174. 10.3389/fneur.2021.814174
7.
Tang A Yokota T . Duchenne muscular dystrophy: promising early-stage clinical trials to watch. Expert OpinInvestig Drugs. (2024) 33:201–17. 10.1080/13543784.2024.2313105
8.
Cohen G Shtorch-Asor A Ben-Shachar S Goldfarb-Yaacobi R Kaiser M Rosenfeld R et al . Large scale population screening for Duchenne muscular dystrophy-Predictable and unpredictable challenges. PrenatDiagn. (2022) 42:1162–72. 10.1002/pd.6201
9.
Mukherjee M Chaturvedi LS Srivastava S Mittal RD Mittal B De . novo mutations in sporadic deletional Duchenne muscular dystrophy (DMD) cases. ExpMol Med. (2003) 35:113–7. 10.1038/emm.2003.16
10.
Massalska D Zimowski JG Roszkowski T Bijok J Pawelec M Bednarska-Makaruk M . Prenatal diagnosis of Duchenne and Becker muscular dystrophies: underestimated problem of the secondary prevention of monogenetic disorders. J ObstetGynaecol Res. (2017) 43:1111–21. 10.1111/jog.13344
11.
Zhao G Wang X Liu L Dai P Kong X . Noninvasive prenatal diagnosis of duchenne muscular dystrophy in five Chinese families based on relative mutation dosage approach. BMC Med Genomics. (2021) 14:275. 10.1186/s12920-021-01128-1
12.
Beggs AH Koenig M Boyce FM Kunkel LM . Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet. (1990) 86:45–8. 10.1007/BF00205170
13.
Oudet C Hanauer A Clemens P Caskey T Mandel JL . Two hot spots of recombination in the DMD gene correlate with the deletion prone regions. Hum Mol Genet. (1992) 1:599–603. 10.1093/hmg/1.8.599
14.
Pastore L Caporaso MG Frisso G Orsini A Santoro L Sacchetti L et al . A quantitative polymerase chain reaction (PCR) assay completely discriminates between Duchenne and Becker muscular dystrophy deletion carriers and normal females. Mol Cell Probes. (1996) 10:129–37. 10.1006/mcpr.1996.0018
15.
Hayat Nosaeid M Mahdian R Jamali S Maryami F Babashah S Maryami F et al . Validation and comparison of two quantitative real-time PCR assays for direct detection of DMD/BMD carriers. ClinBiochem. (2009) 42:1291–9. 10.1016/j.clinbiochem.2009.04.016
16.
Xiao Y Jiang X Wang R . Screening for DMD/BMD deletion carriers by fluorescence in situ hybridization. Genet Test. (2003) 7:195–201. 10.1089/109065703322537205
17.
Velázquez-Wong AC Hernández-Huerta C Márquez-Calixto A Hernández-Aguilar FO Rodríguez-Cruz M Salamanca-Gómez F et al . Identification of duchenne muscular dystrophy female carriers by fluorescence in situ hybridization and RT-PCR. Genet Test. (2008) 12:221–3. 10.1089/gte.2007.0081
18.
Sellner LN Taylor GR MLPA . and MAPH: new techniques for detection of gene deletions. Hum Mutat. (2004) 23:413–9. 10.1002/humu.20035
19.
Schouten JP McElgunn CJ Waaijer R Zwijnenburg D Diepvens F Pals G . Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. (2002) 30:e57. 10.1093/nar/gnf056
20.
Zhong J Xie Y Bhandari V Chen G Dang Y Liao H et al . Clinical and genetic characteristics of female dystrophinopathy carriers. Mol Med Rep. (2019) 19:3035–44. 10.3892/mmr.2019.9982
21.
Schrijver I Cherny SC Zehnder JL . Testing for maternal cell contamination in prenatal samples: a comprehensive survey of current diagnostic practices in 35 molecular diagnostic laboratories. J MolDiagn. (2007) 9:394–400. 10.2353/jmoldx.2007.070017
22.
Cariati F Savarese M D'Argenio V Salvatore F Tomaiuolo R . The SEeMORE strategy: single-tube electrophoresis analysis-based genotyping to detect monogenic diseases rapidly and effectively from conception until birth. ClinChem Lab Med. (2017) 56:40–50. 10.1515/cclm-2017-0147
23.
Buchovecky CM Nahum O Levy B . Assessment of Maternal Cell Contamination in Prenatal Samples by Quantitative Fluorescent PCR (QF-PCR). Methods Mol Biol. (2019) 1885:117–27. 10.1007/978-1-4939-8889-1_8
24.
Chamberlain JS Gibbs RA Ranier JE Nguyen PN Caskey CT . Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. (1988) 16:11141–56. 10.1093/nar/16.23.11141
25.
Lo YM Tein MS Lau TK Haines CJ Leung TN Poon PM et al . Quantitative analysis of fetal DNA in maternal plasma and serum: implications for noninvasive prenatal diagnosis. Am J Hum Genet. (1998) 62:768–75. 10.1086/301800
26.
Malmgren H White I Johansson S Levkov L Iwarsson E Fridström M et al . PGD for dystrophin gene deletions using fluorescence in situ hybridization. Mol Hum Reprod. (2006) 12:353–6. 10.1093/molehr/gal039
27.
Geifman-Holtzman O Ober Berman J . Prenatal diagnosis: update on invasive versus noninvasive fetal diagnostic testing from maternal blood. Expert Rev MolDiagn. (2008) 8:727–51. 10.1586/14737159.8.6.727
28.
Hui L Tabor A Walker SP Kilby MD . How to safeguard competency and training in invasive prenatal diagnosis: 'the elephant in the room'. Ultrasound Obstet Gynecol. (2016) 47:8–13. 10.1002/uog.15806
29.
Vaiopoulos AG Athanasoula KC Papantoniou N Kolialexi A . Review: advances in non-invasive prenatal diagnosis. In Vivo. (2013) 27:165–70.
30.
Sheth F Rahman M Liehr T Desai M Patel B Modi C et al . Prenatal screening of cytogenetic anomalies - a Western Indian experience. BMC Pregnancy Childbirth. (2015) 15:90. 10.1186/s12884-015-0519-y
31.
Antoniadi T Yapijakis C Kaminopetros P Makatsoris C Velissariou V Vassilopoulos D et al . A simple and effective approach for detecting maternal cell contamination in molecular prenatal diagnosis. PrenatDiagn. (2002) 22:425–9. 10.1002/pd.325
32.
Mann K Donaghue C Fox SP Docherty Z Ogilvie CM . Strategies for the rapid prenatal diagnosis of chromosome aneuploidy. Eur J Hum Genet. (2004) 12:907–15. 10.1038/sj.ejhg.5201224
33.
HadjFredj S Ouali F Siala H Bibi A Othmani R Dakhlaoui B et al . Prenatal diagnosis of cystic fibrosis: 10-years experience. PatholBiol (Paris). (2015) 63:126–9. 10.1016/j.patbio.2015.04.002
34.
Saadi AV Girisha KM Gopinath PM Satyamoorthy K . Analysis of cosegregation of intragenic DNA sequence variations as markers of maternal cell contamination in prenatal diagnosis of β-thalassemia. Transl Res. (2011) 157:150–5. 10.1016/j.trsl.2010.12.005
35.
Jorge P Mota-Freitas MM Santos R Silva ML Soares G Fortuna AM et al . 26-Year Experience in Chorionic Villus Sampling Prenatal Genetic Diagnosis. J Clin Med. (2014) 3:838–48. 10.3390/jcm3030838
36.
Rueangchainikhom W Sarapak S Orungrote N . Chorionic villus sampling for early prenatal diagnosis at BhumibolAdulyadej Hospital. J Med Assoc Thai. (2008) 91:1–6.
37.
Messaoud O Ben Rekaya M Jerbi M Ouertani I Kefi R Laroussi N et al . The experience of a Tunisian referral centre in prenatal diagnosis of Xerodermapigmentosum. Public Health Genomics. (2013) 16:251–4. 10.1159/000354584
38.
Kessler L Adams R Mighion L Walther S Ganguly A . Prenatal diagnosis in haemophilia A: experience of the genetic diagnostic laboratory. Haemophilia. (2014) 20:e384–91. 10.1111/hae.12517
39.
El Kadiri Y Selouani Y Ratbi I Lyahyai J Zrhidri A Sahli M et al . Molecular diagnosis of dystrophinopathies in Morocco and report of six novel mutations. Clin Chim Acta. (2020) 506:28–32. 10.1016/j.cca.2020.03.018
40.
Traverso M Malnati M Minetti C Regis S Tedeschi S Pedemonte M et al . Multiplex real-time PCR for detection of deletions and duplications in dystrophin gene. BiochemBiophys Res Commun. (2006) 339:145–50. 10.1016/j.bbrc.2005.11.006
41.
Borun P Kubaszewski L Banasiewicz T Walkowiak J Skrzypczak-Zielinska M Kaczmarek-Rys M et al . Comparative-high resolution melting: a novel method of simultaneous screening for small mutations and copy number variations. Hum Genet. (2014) 133:535–45. 10.1007/s00439-013-1393-1
42.
Atehortúa SC Lugo LH Ceballos M Orozco E Castro PA Arango JC et al . Cost-Effectiveness Analysis of Diagnosis of Duchenne/Becker Muscular Dystrophy in Colombia. Value Health Reg Issues. (2018) 17:1–6. 10.1016/j.vhri.2017.10.003
Appendix
PCR primer sequences in introns 2 and 5:
2-B1F 5′- GTTTGCTGAATCTGCCTCTGT -3′
2-B1R 5′- GGCCTTACGTGACCTGACAT -3′
2-C1F 5′- CAGGCATGAAGGTTGCCTAT -3′
2-C1R 5′- TCTCACCCATTTTGCCACTC -3′
2-D1F 5′- CGGAAGGGAGCTTAGTAGCA -3′
2-D1R 5′- ACTGAATGGATTTCGGGATG -3′
2-E1F 5′- AGCAGCCCTCATTGCATATT -3′
2-E1R 5′- GAGCCAGTGGCGTTAAACAG -3′
2-F1F 5′- AGGCAGGGTAGGACAATCAC -3′
2-F1R 5′- AAATAATGGCTCGGTCCACA -3′
2-F2F 5′- GCCAACCCAGATTTAAGAAGC -3′
2-F2R 5′- CCATCCGAGTGTACCGACTT -3′
2-F3F 5′- TCGAATGCACATCTGTTTGA -3′
2-F3R 5′- GGAACACCGTCGTCTTCTTT -3′
2-F4F 5′- TGTGTGTGAGATCTTGCTTCG -3′
2-F4R 5′- GGCAAATCGTATGCACTCAA -3′
2-F5F 5′- CAGCATTGATGTTGCCTGTC -3′
2-F5R 5′- GCAATACCCTACCAAGCACAA -3′
2-F6F 5′- TTGATGCTTGCCATCTAGGA -3′
2-F6R 5′- ATGGCATTTCTGGACTCACC -3′
2-F7F 5′- GACCCATCACCTGACGTTTT -3′
2-F7R 5′- TTCACCAAATCGTTTTCTGC -3′
5-1F 5′- CCATGGTCCCTTCACTATCAA -3′
5-1R 5′- TTGAGAGGCTACCAAGAACCA -3′
5-2F 5′- TGCCCCTTCATCAGTCAATAG -3′
5-2R 5′- CCCAAAGGTGAGCCTGTAAA -3′
PCR primer sequences for the junction fragment:
D1-F 5′- AAATCAAGAGAAGGTTAATGTGGAC -3′
D1-R 5′- CTTACCTATGACTATGGATGAGAGCA -3′
D1-F2 5′- TGGGCAAGGAATACAACCTTA -3′
D1-F3 5′- ACACCTCTTAGGGATAAATA -3′
D1-R2 5′- AGGAGGAATACTATAGATAA -3′
D1-R3 5′- GCAACCTCCGTCTCCCAGTG -3′
D4-F 5′- ATTTGAAAGTTGGCCGGGTA -3′
D4-R 5′- TCATAGGCTTCGTGCATGTG -3′
Summary
Keywords
Becker muscular dystrophy, DMD gene, junction fragments, maternal cell contamination, prenatal diagnosis
Citation
Cai L, Li W and Zhong M (2025) Combining PCR to detect junction fragments and deleted exons in the prenatal diagnosis of BMD can effectively identify maternal cell contamination. Front. Med. 12:1599498. doi: 10.3389/fmed.2025.1599498
Received
25 March 2025
Accepted
18 August 2025
Published
03 September 2025
Volume
12 - 2025
Edited by
Santasree Banerjee, Jilin University, China
Reviewed by
Hitham Aldharee, Qassim University, Saudi Arabia
Adhi Pribadi, Padjadjaran University, Indonesia
Updates
Copyright
© 2025 Cai, Li and Zhong.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Zhong pomelomin@aliyun.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.