Abstract
Background:
Abernethy malformation is a rare condition in which the portomesenteric blood drains into systemic circulation, bypassing the liver. With advancements in imaging techniques and increased awareness of this malformation, there has been a growing number of reported cases in recent years. We present a case report and literature review in an effort to further the understanding of Abernethy malformation.
Case presentation:
We report a 21-year-old male presenting with pulmonary hypertension (PH) and right heart enlargement for 7 days. Portal CT angiography (CTA) revealed a vessel communication between the portal vein (PV) and the IVC, located on the left side of the abdominal aorta below the renal vein, and multiple nodular liver lesions (NLL). Finally, Abernethy malformation type II was diagnosed, which is extremely rare due to the absence of polysplenia while co-existing with anomalies of inferior vena cava (AIVC). He was discharged with stable vital signs on symptomatic therapy.
Conclusion:
Abernethy malformation presents with a range of clinical manifestations. It should be considered in patients with unexplained PH.
Introduction
Abernethy malformation, or congenital extrahepatic portosystemic shunt (CEPS), is a rare condition in which the portomesenteric blood bypasses the liver and drains directly into systemic circulation. John Abernethy first reported it in 1793 (1). Morgan and Superina (2) classified Abernethy malformation into two types. Type I refers to the total aplasia of intrahepatic portal branches with complete extrahepatic shunting of portal blood into the systemic circulation. Type I is further divided into type Ia (the splenic vein and superior mesenteric vein drain separately into a systemic vein) and type Ib (the splenic vein and superior mesenteric vein drain together after joining to form a common trunk). Type II refers to hypoplastic intrahepatic portal branches with partial extrahepatic shunting of portal blood into the systemic circulation.
Abernethy malformation presents with diverse clinical manifestations and is associated with various congenital anomalies, including AIVC (3). Notably, polysplenia demonstrates a particularly high co-occurrence rate with AIVC in these cases (4). A previous report by Samir Shakya et al. (5) described a case of Abernethy malformation without polysplenia, accompanied by AIVC, in a pediatric patient. Our patient is an adult male with such a condition, along with PH and NLL.
Case presentation
A 21-year-old male presenting with PH and right heart enlargement for 7 days. His symptoms began over 5 years ago with breathlessness and anterior chest discomfort occurring primarily before bedtime. These symptoms gradually resolved after lying in a lateral position, and no specialized treatment was administered. PH and right heart enlargement were incidentally identified on an echocardiogram performed as part of a pre-employment medical examination 7 days ago. The physical examination showed that his sclera appeared slightly jaundiced. The timeline of the history is shown in Figure 1.
FIGURE 1

The timeline of the history.
Laboratory tests showed decreases in white blood cell (3.23 × 109/L; reference range, 3.5–9.5 × 109/L), platelet counts (93 × 109/L; reference range, 125–350 × 109/L) and serum albumin levels (36.7 g/L; reference range, 40–55 g/L), and an increase in glutamate dehydrogenase (7.9 μmol/L; reference range, < 7.4 μmol/L), total bilirubin (35.1 μmol/L; reference range, 5.0–21.0 μmol/L), direct bilirubin (12.3 μmol/L; reference range, < 6.0 μmol/L), indirect bilirubin (22.8 μmol/L; reference range, 2.0–15.0 μmol/L), cholyglycine (42.34 μmol/L; reference range, < 2.7 μmol/L) and homocysteine (45.4 μmol/L; reference range, < 15.0 μmol/L). Activated partial thromboplastin time ratio was 1.33 (reference range 0.80–1.20) and fibrinogen was 1.81 (reference range 2.00–4.00). Urine protein was + – (reference –) and urobilinogen was 4 + (reference – or + –).
Echocardiography revealed the dilated right atrium (area: 23 cm2) and right ventricle (mid-cavity diameter: 34 mm, basal diameter: 44 mm), widening of the pulmonary trunk (31 mm), and severe PH. Pulmonary artery systolic pressure of about 82 mmHg estimated by the tricuspid regurgitation pressure gradient method (see Supplementary Data Sheet 1–S1). CT pulmonary angiogram and portal CTA demonstrated that the pulmonary trunk was dilated with a diameter of approximately 4.0 cm. The portal vein was dilated, with a diameter of approximately 1.6 cm. The left and right branches of the portal vein were poorly visualized. Additionally, splenomegaly was noted. An arcuate bridging vessel was observed communicating between the PV and the IVC, with a diameter of approximately 2.3 cm (Figure 2). The IVC was on the left side of the abdominal aorta below the renal vein (Figure 2). The liver displayed multiple enhanced nodules in the arterial phase (Figure 2). Rheumatologic serology, echocardiography, virological testing and CT pulmonary angiogram excluded connective tissue diseases, congenital heart disease, HIV infection, left heart disease, chronic lung disease, and chronic thromboembolic disease as potential etiologies of PH. Abernethy malformation type II with left IVC, PH and NLL was considered. A right heart catheterization confirmed precapillary PH with a mean pulmonary arterial pressure of 42 mmHg, pulmonary arterial wedge pressure of 3 mmHg, pulmonary vascular resistance of 4.88 Wood units, right atrial pressure of 4 mmHg, cardiac output of 8.0 L/min, and a cardiac index of 4.57 L/min/m2 (see Supplementary Data Sheet 1–S2).
FIGURE 2
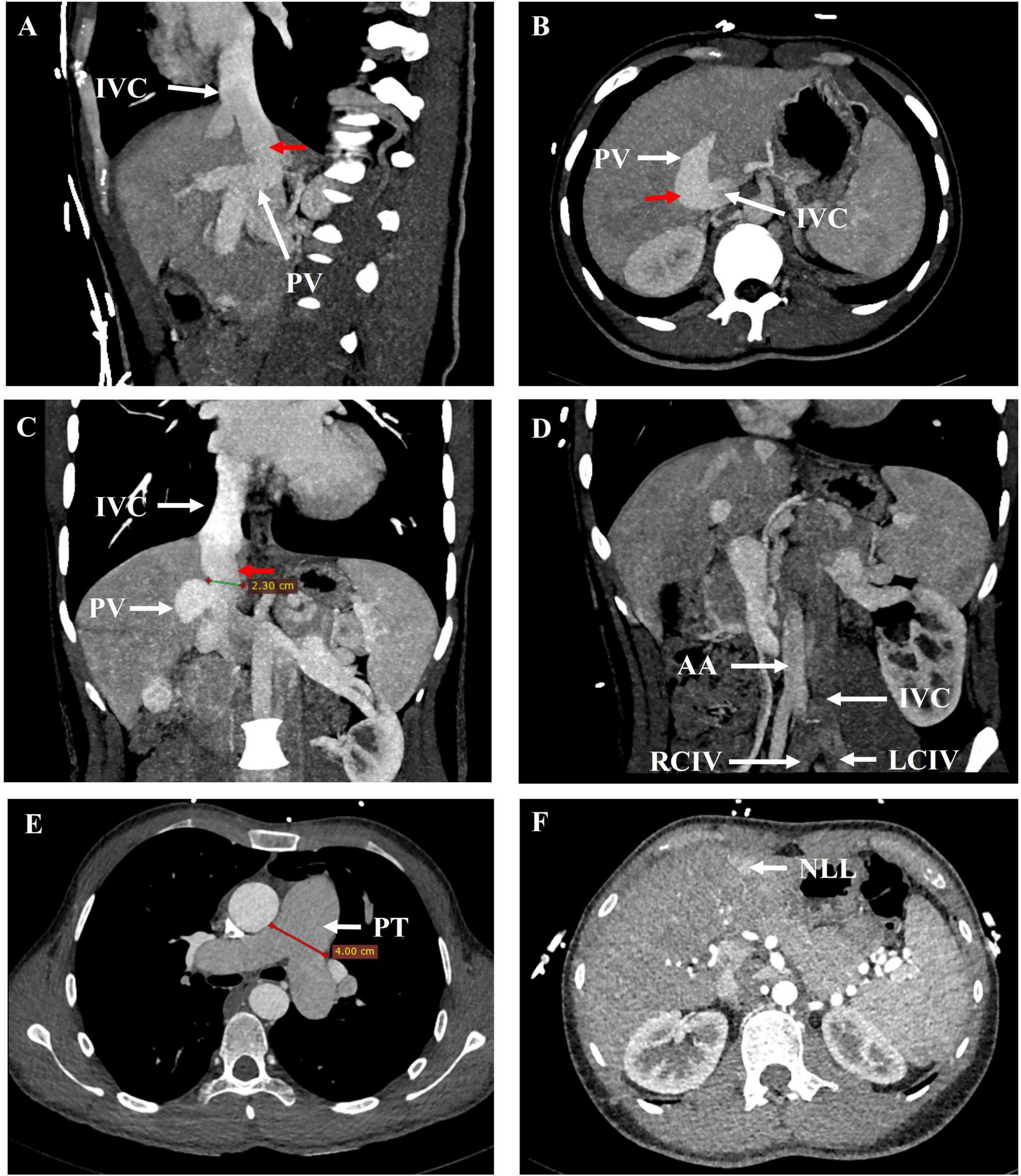
21 years-old male with Abernethy malformation type II, left ICV, and NLL. (A) Sagittal, (B) axial and (C) coronal maximum intensity projection image from contrast-enhanced CT revealed a vessel communication (red arrow) between the PV and the IVC. (D) Oblique coronal maximum intensity projection image demonstrated the IVC was on the left side of the abdominal aorta. (E, F) Arterial phase axial images from contrast-enhanced CT showed the dilated pulmonary trunk and the nodular liver lesions (NLL). AA, abdominal aorta; IVC, inferior vena cava; LCIV, left common iliac vein; NLL, nodular liver lesions; PV, portal vein; PT, pulmonary trunk; RCIV, right common iliac vein.
The patient was administered ambrisentan and tadalafil targeted therapy for PH and was subsequently discharged with stable vital signs. Following discharge, oral medication was continued.
Discussion
Abernethy malformation is a rare condition in which the portomesenteric blood drains into systemic circulation, bypassing the liver. Portal CTA showed that the left and right branches of the portal vein were poorly visualized and an arcuate bridging vessel was observed communicating between the PV and the IVC. According to the classification proposed by Morgan and Superina (2), these features are classified as Abernethy malformation type II. Of note, this case represents an exceptionally rare clinical occurrence of Abernethy malformation in an adult male with absence of polysplenia and coexistence of AIVC.
Abernethy malformation is a rare condition, with an increasing number of case reports in recent years due to advances in imaging techniques and greater awareness of this malformation. The literatures about Abernethy malformation published before September, 2024 were systematically collected. We searched PubMed, Web of Science, Embase and Cochrane databases using the search terms “Abernethy malformation*,” “congenital absence of portal vein,” “congenital absence of the portal vein,” “congenital extrahepatic portosystemic shunt*,” “congenital extrahepatic portocaval shunt*,” “congenital extrahepatic shunt*.” The following studies were excluded: those involving patients with clinical problems outside the target group (e.g., surgical shunts), fetal cases, autopsy cases, duplicate cases, literature such as conference abstracts and reviews, and studies on animals. A total of 295 studies (Supplementary Data Sheet 1–S3) involving 341 Abernethy malformation patients were included. The classification, gender, age, reported region, associated congenital diseases and complications, and mortality cases of the included patients were reported as case numbers. Intergroup differences were assessed by chi-squared (χ2) test and Fisher’s exact test, with P < 0.05 considered statistically significant. SPSS 27.0 software was used for statistical analyses.
There were 170 cases of type I, 169 cases of type II, and 2 cases of unknown type; 169 males, 169 females, and 3 patients with unknown gender or transgender; 221 children (≤ 18 years old) and 120 adults (> 18 years old) (Table 1); 181 cases in Asia, 150 cases in Europe and North America, and 10 cases in other regions. Aligning with the review by Kumar P et al. (6), there is no clear sex and age preponderance. Furthermore, among the Type I patients, there were 19 cases of Type Ia, 122 cases of Type Ib, and 29 cases of an unknown type.14 patients initially diagnosed as Type I were subsequently reclassified as Type II following further diagnostic procedures, including balloon occlusion angiography (7 cases), angiography (3 cases), liver biopsy (3 cases), and intraoperative findings (1 case).
TABLE 1
| Type | Gender | P value | Age | P value | |||
| Male (n = 169) |
Female (n = 169) |
Unknow or transgender (n = 3) |
Child (n = 221) |
Adult (n = 120) |
|||
| I (n = 170) | 77 | 90 | 3 | 0.190a | 116 | 54 | 0.238b |
| II (n = 169) | 90 | 79 | 0 | 105 | 64 | ||
| Unknow (n = 2) | 2 | 0 | 0 | 0 | 2 | ||
Comparison of gender and age distributions by Abernethy Malformation type.
a Chi-square test comparing type (Type I, Type II) and gender (male, female);
b Chi-square test comparing type (Type I, Type II) and age (children, adult).
Abernethy malformation can cause a broad spectrum of clinical manifestations, which can be divided into three classes (7): (1) symptoms associated with coexisting congenital abnormalities; (2) those related to abnormal hepatic development, ranging from fatty liver to nodular liver lesions; (3) those resulting from the shunt, including hyperammonemia, PH, hepatopulmonary syndrome.
Abernethy malformation is often observed in association with other congenital anomalies (7). Of the 341 patients, 151 (44.3%) were diagnosed with one or more other congenital anomalies, with congenital heart disease (78/151, 51.7%) being the most common. 29 patients (8.5%) exhibited comorbid AIVC, predominantly in pediatric patients (P < 0.05, Table 2). It is hypothesized that this may be associated with the increased likelihood of earlier detection of combinations of these congenital diseases. Our patient is an adult male with a left IVC. While congenital anomalies are more commonly seen in type I as compared to type II (6), our analysis suggests that no significant difference in the incidence of IVC abnormalities between the two types. This could be partly attributable to the inherent relative rarity of IVC anomalies limiting statistical power. A more plausible explanation involves their shared embryological origin. The association of AIVC with Abernethy malformation may be attributed to their close embryological development. Right-sided venous hypoplasia or agenesis affects both the vitelline (portal) and subcardinal (infrahepatic caval vein) venous systems (3). The coincidence of associations in both types also suggests that the two variants are closely related (7). Future studies with detailed phenotyping and larger cohorts are warranted to further elucidate these relationships.
TABLE 2
| Clinical features | Number | Type | P value | Gender | P value | Age | P value | Regional distribution | P value | ||||
| I | II | Male | Female | Child | Adult | Asia | Europe and North America | ||||||
| Inferior vena cava anomalies | 29 | 14 | 15 | 0.833 | 17 | 12 | 0.332 | 24 | 5 | 0.034 | 14 | 13 | 0.758 |
| Nodular liver lesions | 124 | 83 | 40 | <0.001 | 59 | 65 | 0.498 | 77 | 47 | 0.498 | 54 | 66 | 0.027 |
| FNH | 58 | 40 | 18 | 0.002 | 18 | 40 | 0.002 | 43 | 15 | 0.086 | 33 | 24 | 0.403 |
| NRH | 37 | 26 | 10 | 0.005 | 23 | 14 | 0.117 | 23 | 14 | 0.765 | 16 | 18 | 0.469 |
| HCC | 26 | 20 | 6 | 0.004 | 9 | 17 | 0.102 | 6 | 20 | <0.001 | 4 | 22 | <0.001 |
| Adenoma | 21 | 15 | 6 | 0.044 | 7 | 14 | 0.115 | 12 | 9 | 0.474 | 5 | 16 | 0.006 |
| Pulmonary hypertension | 66 | 31 | 34 | 0.660 | 30 | 35 | 0.490 | 43 | 23 | 0.948 | 35 | 29 | 0.999 |
| Death case | 20 | 10 | 9 | 0.824 | 10 | 10 | 1.000 | 15 | 5 | 0.325 | 6 | 12 | 0.061 |
Inferior vena cava anomalies, nodular liver lesions, pulmonary hypertension, and death case in patients with Abernethy malformation.
Additionally, Abernethy malformation is often complicated by NLL, which can be divided into benign and malignant according to their pathology. Benign nodules include focal nodular hyperplasia (FNH), nodular regenerative hyperplasia (NRH), adenoma and hemangioma, while malignant nodules include HCC and hepatoblastoma. Of the 341 patients, 124 (36.4%) had combined liver lesions, mainly including FNH (58/124,46.8%), NRH (37/124,29.8%), HCC (26/124,21.0%) and adenoma (21/124,16.9%). Statistically significant differences were observed between Type I and Type II patients in the overall incidence of NLL, as well as in the incidence of FNH, NRH, HCC and adenoma (all P < 0.05, Table 2), with these lesions being more commonly seen in Type I. The incidence of FNH also differed significantly between male and female patients (P < 0.05, Table 2), with a higher prevalence in females. The incidence of HCC was significantly higher in adult patients compared to pediatric patients (P < 0.05, Table 2). Additionally, significant regional differences were observed in the incidence of NLL, HCC, and adenomas among reported cases from Asia and Europe and North America (all P < 0.05, Table 2), with a higher prevalence in Europe and North America. Other less common NLL included hepatoblastoma (9 cases), hemangioma (4 cases), and combined hepatocellular carcinoma and cholangiocarcinoma (cHCC-CCA) (1 case). Patients could presented with multiple NLL concurrently. In our patient, MRI revealed multiple NLL, and FNH and solitary necrotic nodule of the liver were considered the most probable diagnoses. Contrast-enhanced CT also showed the multiple enhanced NLL in the arterial phase.
Abnormal oxygen supply appears to be a key driver in causing NLL to form (8). Insufficient portal venous supply results in dilation of the hepatic arteries and increased arterial blood flow, leading to increased oxygen supply. Patients with Abernethy malformation may experience increased exposure of hepatocytes to oxygen free-radicals (9). This may elucidate the observation that NLL are more prevalent in Type I, where the portal vein is entirely absent. Concurrently, shunting results in aberrant hepatotrophic substances composition, including estrogen. Estrogen is clearly associated with the development of FNH (10). Up to three quarters of females with FNH were found to be long-term users of oral contraceptives, suggesting a potential link to its pathogenesis (11). Estrogen receptors have been detected in FNH lesions and surrounding liver parenchyma (12). This may be attributed to the fact that focal nodular hyperplasia is more prevalent in females. The progression of benign NLL complicated by Abernethy malformation to malignancy is a potential concern (13–16). Therefore, such patients should be followed up, and the imaging alterations or elevated tumor markers should prompt consideration of malignant progression, and liver biopsy should be performed if necessary. Following the closure of the shunt, the NLL may resolve completely or partially (10).
Common non-hepatic complications of Abernethy malformation include hyperammonemia (108/341, 31.7%), PH (66/341, 19.4%), hepatopulmonary syndrome (58/341, 17.0%), hepatic encephalopathy (HE) (51/341, 15.0%), gastrointestinal hemorrhage (34/341, 10.0%), developmental delay (33/341, 9.7%) and intellectual disability (23/341, 6.7%). Less common complications include hypoglycemia, glomerular disease, hyperandrogenemia, osteoporosis and hypothyroxinemia. PH is a common pulmonary complication. The pathophysiology of PH in the setting of Abernethy malformation is considered a portopulmonary hypertension, although portal hypertension is not a feature of Abernethy malformation (17). The portosystemic shunt diverts portal blood flow into the systemic circulation, enabling the bypass of the liver and the direct entry of vascular mediators, proinflammatory cytokines, proangiogenic factors, and bacterial endotoxins into the pulmonary circulation from the mesenteric circulation. They can inflict damage upon the vascular pulmonary endothelium and stimulate endothelial cell proliferation, smooth muscle hypertrophy, and in situ thrombosis. Furthermore, blood clots from the mesenteric circulation can enter the pulmonary circulation via the portosystemic shunt. High cardiac output also promotes hypertrophy, proliferation and vasoconstriction of pulmonary endothelial cells. The aforementioned factors all contribute to the development of PH (18).
The patient presented with dilated portal vein and splenomegaly. We speculate that this patient may have a congenital hypoplastic portal vein system. Aligning with the report by Yao X et al. (19), because of the thin portal vein branch, the blood flow returning to the liver was blocked, forming regional portal hypertension and splenomegaly. The dilatation main portal vein was considered compensatory dilatation. It is a rare case of Abernethy malformation manifesting both shunt-mediated pulmonary hypertension and regional portal hypertension.
In addition, cardiopulmonary causes are the main causes of death in patients with Abernethy malformation. A total of 20 deaths were reported in 341 cases. Of these, 8 were due to cardiopulmonary causes, 6 of which were combined with PH (20–24). 2 patients died as a result of coronary artery disease caused by compression of the coronary arteries following pulmonary vasodilatation in PH (21, 24). 1 patient died as a result of pulmonary heart disease caused by PH (20). 1 patient, due to prolonged absence of intervention for PH, surgical treatment for CEPS had to be performed when treatment for PH was initiated but the target hemodynamic profile was not achieved, resulting in the patient’s death in the early postoperative period (25). It is therefore recommended that active intervention be performed in cases of Abernethy malformation with PH, in order to avoid further progression and to be alert to the poor prognosis that may result. Optimal management of patients with PH requires a multidisciplinary approach to determine the timing of shunt closure, medical PAH therapy and the indication for lung, liver, or combined transplantation in the most severe cases (26). Life-threatening visceral aneurysms may also occur, and the awareness of this unusual entity is crucial for the prevention and close monitoring of possible complications, such as abdominal hemorrhage (27). For patients with aneurysms, attention should be paid to the treatment strategy to prevent a potentially fatal aneurysm rupture (28).
Conclusion
We present the case of a 21-year-old male patient with Abernethy malformation type II with a left IVC, PH and NLL. Abernethy malformation presents with a range of clinical manifestations. It should be considered in patients with unexplained PH in clinical practice. Although rare, it may cause portal hypertension. Imaging examinations such as CTA can assist in confirming the diagnosis and detecting concomitant diseases, including nodular hepatic lesions and AIVC. It is crucial to diagnose and treat Abernethy malformation promptly, as well as to screen and prevent related diseases for a better prognosis.
Statements
Data availability statement
The original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Qilu Hospital of Shandong University Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XZ: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. MP: Investigation, Supervision, Validation, Writing – review & editing. RR: Software, Validation, Writing – review & editing. WL: Software, Supervision, Writing – review & editing. QZ: Supervision, Validation, Visualization, Writing – review & editing. CQ: Software, Supervision, Validation, Writing – review & editing. YZ: Funding acquisition, Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study has received funding by the Natural Science Foundation of Shandong Province (No. ZR2024ZD23).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1604853/full#supplementary-material
Abbreviations
AIVC, anomalies of inferior vena cava; CEPS, congenital extrahepatic portosystemic shunt; CTA, CT angiography; FNH, focal nodular hyperplasia; NLL, nodular liver lesions; NRH, nodular regenerative hyperplasia; PH, pulmonary hypertension; PV, portal vein.
References
1.
Abernethy J . Account of two instances of uncommon formation in the viscera of the human body: from the philosophical transactions of the royal society of London.Med Facts Obs. (1797) 7:100–8.
2.
Morgan G Superina R . Congenital absence of the portal vein: two cases and a proposed classification system for portasystemic vascular anomalies.J Pediatr Surg. (1994) 29:1239–41. 10.1016/0022-3468(94)90812-5
3.
Sheth R Sivakumar K . The Abernethy malformation with inferior caval vein hypoplasia: a tailored technique for transcatheter closure and an insight into embryological perspective.Cardiol Young. (2018) 28:1169–71. 10.1017/S1047951118000884
4.
Papamichail M Pizanias M Heaton N . Congenital portosystemic venous shunt.Eur J Pediatr. (2018) 177:285–94. 10.1007/s00431-017-3058-x
5.
Shakya S Saxena A Ramakrishnan S . Transcatheter closure of Abernethy malformation associated with interrupted inferior caval vein and other systemic venous anomalies.Cardiol Young. (2022) 32:337–9. 10.1017/S1047951121002900
6.
Kumar P Bhatia M Garg A Jain S Kumar K . Abernethy malformation: a comprehensive review.Diagn Interv Radiol. (2022) 28:21–8. 10.5152/dir.2021.20474
7.
Alonso-Gamarra E Parrón M Pérez A Prieto C Hierro L López-Santamaría M et al Clinical and radiologic manifestations of congenital extrahepatic portosystemic shunts: a comprehensive review. RadioGraphics. (2011) 31:707–22. 10.1148/rg.313105070
8.
Jungermann K Kietzmann T . Oxygen: modulator of metabolic zonation and disease of the liver.Hepatology. (2000) 31:255–60. 10.1002/hep.510310201
9.
Tyraskis A Deganello A Sellars M De Vito C Thompson R Quaglia A et al Portal venous deprivation in patients with portosystemic shunts and its effect on liver tumors. J Pediatr Surg. (2020) 55:651–4. 10.1016/j.jpedsurg.2019.05.027
10.
Glonnegger H Schulze M Kathemann S Berg S Füllgraf H Tannapfel A et al Case report: hepatic adenoma in a child with a congenital extrahepatic portosystemic shunt. Front Pediatr. (2020) 8:501. 10.3389/fped.2020.00501
11.
Zimmermann A . Focal nodular hyperplasia (FNH) of the Liver. In Tumors and Tumor-Like Lesions of the Hepatobiliary Tract.Cham: Springer (2017). 10.1007/978-3-319-26956-6_117
12.
Chandrasegaram M Shah A Chen J Ruszkiewicz A Astill D England G et al Oestrogen hormone receptors in focal nodular hyperplasia. HPB. (2015) 17:502–7. 10.1111/hpb.12387
13.
Barton Iii J Keller M . Liver transplantation for hepatoblastoma in a child with congenital absence of the portal vein.Pediatr Radiol. (1989) 20:113–4. 10.1007/BF02010653
14.
Morse S Taylor K Strauss E Ramirez E Seashore J . Congenital absence of the portal vein in oculoauriculovertebral dysplasia (Goldenhar syndrome).Pediatr Radiol. (1986) 16:437–9. 10.1007/bf02386831
15.
Chiang J Chiu H Moriarty J McWillliams J . Hyperandrogenism and malignant degeneration of hepatic adenomas in the setting of Abernethy malformation.Radiol Case Rep. (2020) 15:2701–5. 10.1016/j.radcr.2020.10.026
16.
Scheuermann U Foltys D Otto G . Focal nodular hyperplasia proceeds hepatocellular carcinoma in an adult with congenital absence of the portal vein.Transpl Int. (2012) 25:e67–8. 10.1111/j.1432-2277.2012.01454.x
17.
Bahadori A Kuhlmann B Debray D Franchi-Abella S Wacker J Beghetti M et al Presentation of congenital portosystemic shunts in children. Children. (2022) 9:243. 10.3390/children9020243
18.
Tanaka H Saijo Y Tomonari T Tanaka T Taniguchi T Yagi S et al An Adult case of congenital extrahepatic portosystemic shunt successfully treated with balloon-occluded retrograde transvenous obliteration. Intern Med. (2021) 60:1839–45. 10.2169/internalmedicine.5914-20
19.
Yao X Liu Y Yu L Qin J . Rare portal hypertension caused by Abernethy malformation (Type IIC): a case report.World J Radiol. (2023) 15:250–5. 10.4329/wjr.v15.i8.250
20.
Schaeffer D Laiq S Jang H John R Adeyi O . Abernethy malformation type II with nephrotic syndrome and other multisystemic presentation: an illustrative case for understanding pathogenesis of extrahepatic complication of congenital portosystemic shunt.Hum Pathol. (2013) 44:432–7. 10.1016/j.humpath.2012.08.018
21.
Rout A Chongthammakun V Siegel Y Mendoza D Damluji A . Abernethy malformation with massively dilated main pulmonary artery manifesting as acute myocardial infarction.J Cardiovasc Imaging. (2021) 29:379–81. 10.4250/JCVI.2021.0011
22.
Kobayashi D Edwards H Singh J Nadkarni M Lantz P Cook A et al Portopulmonary hypertension secondary to congenital extrahepatic portosystemic shunt with heterotaxy and polysplenia: a cause of sudden death in an infant. Pediatr Pulmonol. (2011) 46:1041–4. 10.1002/ppul.21463
23.
Jun S Lee W Cheon J et al Congenital absence of the portal vein presenting as pulmonary hypertension. J Korean Radiol Soc. (2007) 57:423–8. 10.3348/jkrs.2007.57.5.423
24.
Hlavata T Kaldararova M Klauco F Drangova E Reptova A Simkova I . Congenital absence of the portal vein as a rare cause of portopulmonary Hypertension-A case study series.Med Kaunas Lith. (2022) 58:1484. 10.3390/medicina58101484
25.
Miklashevich IM Potrokhova EA Morozov DA Isaeva Y . Pulmonary arterial hypertension associated with type II Abernethy malformation in an adolescent: a case report.Cardiovasc. Therapy Prevent. (2024) 23:3754. 10.15829/1728-8800-2024-3754
26.
McLin V Franchi-Abella S Brütsch T Bahadori A Casotti V de Ville de Goyet J et al Expert management of congenital portosystemic shunts and their complications. JHEP Rep. (2023) 6:100933. 10.1016/j.jhepr.2023.100933
27.
Rüksan Ütebey A Ufuk F Aykota MR . Less known but important complications and associated anomalies of Abernethy malformation.Diagn Interv Radiol. (2023) 29:410–1. 10.5152/dir.2022.211146
28.
Subramanian A Bharath A Barthur A Jayranganath M . Congenital portosystemic shunt with multiple splenic artery aneurysms: reversing pulmonary hypertension and preventing aneurysm rupture.Ann Pediatr Cardiol. (2022) 15:300–3. 10.4103/apc.apc_142_21
Summary
Keywords
Abernethy malformation, congenital extrahepatic portosystemic shunt, pulmonary hypertension, anomalies of inferior vena cava, nodular liver lesions
Citation
Zhang X, Peng M, Ren R, Li W, Zhao Q, Qi C and Zhang Y (2025) Pulmonary hypertension secondary to Abernethy malformation with left inferior vena cava: a case report and literature review. Front. Med. 12:1604853. doi: 10.3389/fmed.2025.1604853
Received
02 April 2025
Accepted
06 August 2025
Published
22 August 2025
Volume
12 - 2025
Edited by
Suellen Darc Oliveira, University of Illinois Chicago, United States
Reviewed by
Ayse Ruksan Utebey, TC Saglik Bakanligi Sungurlu Devlet Hastanesi, Türkiye
Thenappan Thenappan, University of Minnesota Twin Cities, United States
Updates
Copyright
© 2025 Zhang, Peng, Ren, Li, Zhao, Qi and Zhang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Zhang, drzhy001@163.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.