 Shuwen Jiang
Shuwen Jiang Binfeng Xia
Binfeng Xia Hongyu Wu1
Hongyu Wu1 Xiaohua Li
Xiaohua Li Kaiyu Zhang
Kaiyu Zhang- 1Department of Infectious Diseases and Center of Infectious Diseases and Pathogen Biology, The First Hospital of Jilin University, Changchun, China
- 2Department of Gastroenterology, Lequn Branch, The First Hospital of Jilin University, Changchun, China
Introduction: Hemophagocytic lymphohistiocytosis (HLH), a rare and deadly disease, is typically classified as either primary (familial) or secondary (acquired), depending on the etiology and underlying cause. Secondary HLH often develops in the presence of infectious, malignant, rheumatologic, or metabolic conditions, with infections, especially Epstein–Barr virus (EBV) infection, being among the leading causes. Brucella infection-induced HLH is relatively rare, with only eight cases reported in the past decade, all of which had a favorable prognosis following timely diagnosis and treatment.
Case description: A 53-year-old man with brucellosis who developed secondary HLH and multiple organ dysfunction presented to our hospital with a 2-month history of fever and abnormal liver enzymes. Initial blood culture following admission confirmed Brucella spp. in the aerobic bottle after ⁓87.85 h of incubation. However, after the initial discharge, the patient did not adhere to the prescribed antibiotic therapy and subsequently developed symptoms of fever and abdominal discomfort, and was readmitted to our hospital. Laboratory examination also revealed pancytopenia. An additional blood culture further revealed the growth of Brucella spp. in the aerobic bottle after ⁓113.67 h of incubation. Other findings included decreased fibrinogen, increased ferritin, increased soluble IL-2 receptor α chain (sCD25), decreased Natural Killer (NK) cell activity, presence of hemophagocytic cells in the bone marrow smear, splenomegaly, and abnormal liver and kidney functions. The HScore score was 230 points. A thorough assessment was made, which led to the exclusion of other possible diseases, culminating in the identification of Brucella infection as the most probable cause of HLH. Consequently, the patient was given anti-infection (doxycycline, levofloxacin, etimicin, and rifampin), glucocorticoids (GCs), human immunoglobulin (HIG), and other symptomatic supportive treatments, which ultimately improved his condition.
Conclusion: Despite the generally poor prognosis of HLH patients, those with Brucella-induced HLH may have a favorable outcome with prompt intervention. Conversely, a delayed treatment could increase the risk of HLH onset and progression, leading to death in severe cases.
1 Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening immune-mediated disease characterized by uncontrolled cytotoxic lymphocyte and macrophage activation, leading to cytokine-mediated tissue injury and multiorgan dysfunction (1). Clinical manifestations include fever, hepatosplenomegaly, pancytopenia, and the presence of activated macrophages in hematopoietic organs (2). According to reports, HLH has a high mortality rate ranging between 42% and 88% (1) and can occur at any age (3), with adult patients often having a poor prognosis. Furthermore, depending on the etiology and underlying cause, HLH could be categorized as primary (familial) or secondary (acquired). Secondary HLH could be attributed to infectious, malignant, rheumatologic, or metabolic conditions, with a high mortality rate ranging from 50% to 70% (4). In adults, viral infection has been identified as the most common trigger for acquired HLH, with herpes viruses, Epstein–Barr virus (EBV), and Cytomegalovirus (CMV) as the most commonly reported viral pathogens (1, 5). Chronic viral infections such as human immunodeficiency virus (HIV) and hepatitis B and hepatitis C may also trigger HLH during the acute or chronic infection phase (1). Furthermore, although rare, bacterial, parasitic, and fungal agents could cause HLH, with intracellular pathogens such as Mycobacterium tuberculosis (TB), Pneumocystis jirovecii, and Plasmodium species as the most widely reported (1, 5). Certain zoonotic diseases, including tick-borne illnesses and dengue fever, could also trigger secondary HLH. In a systematic review of 98 cases of tick-borne infection-associated HLH, Dorde Jevtic et al. (6) found that HLH secondary to tick-borne pathogens is relatively uncommon, with Ehrlichia spp. as the most frequently implicated tick-borne agent; however, the mortality rate in their study was 16.3%. Similarly, Leong Tung Ong and Roovam Balasubramaniam (7) reported that while HLH prevalence is low among dengue patients, dengue-associated HLH still carries a significant mortality risk, with a reported overall mortality rate of 20.2%. On the other hand, Brucella infection-induced HLH is quite rare, with only eight cases reported in the past decade, all exhibiting a good prognosis following timely diagnosis and treatment (8–15) (Table 1), thus highlighting the significance of early diagnosis and treatment for Brucella infection-associated HLH.
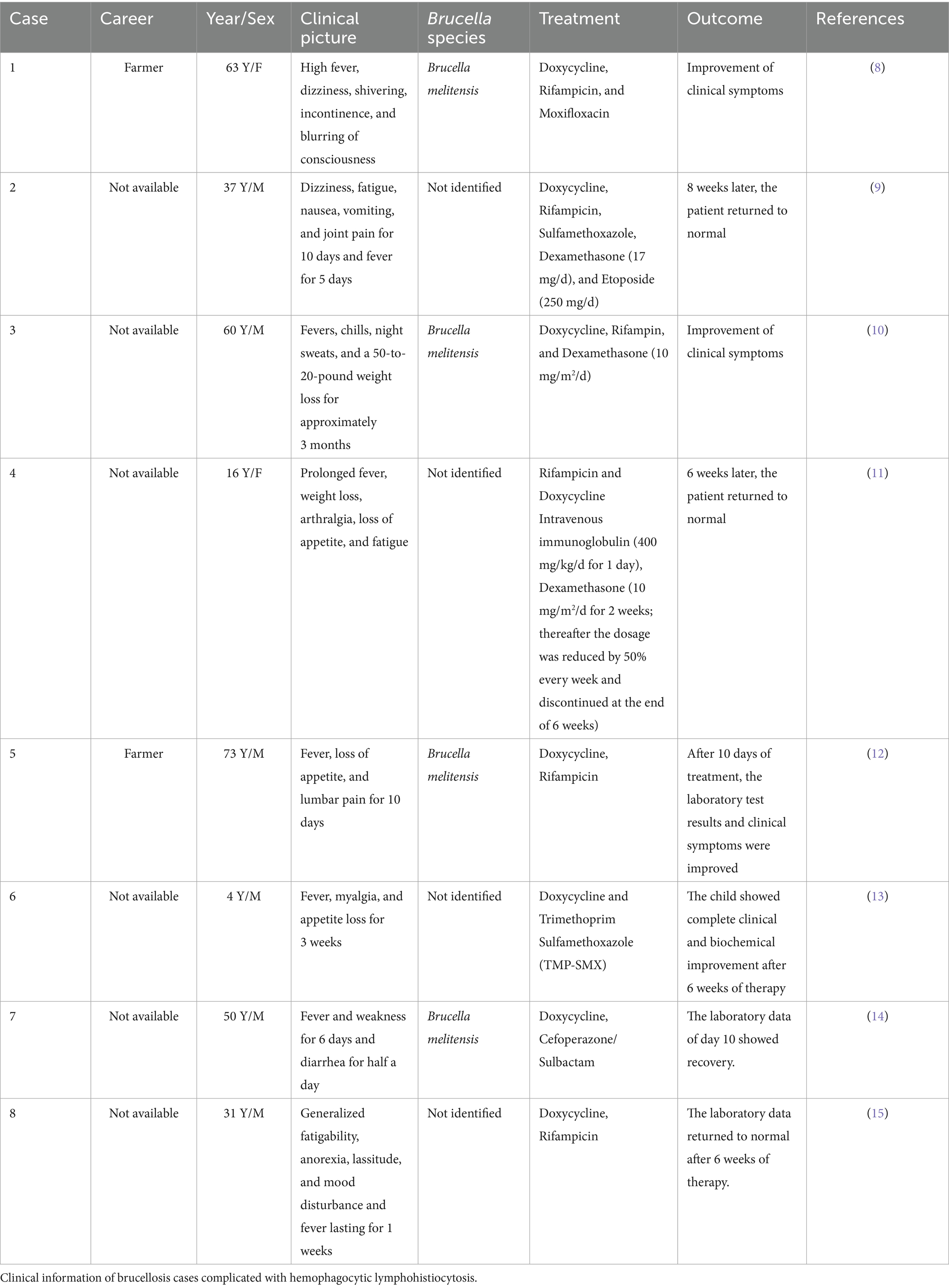
Table 1. Case reports of brucellosis complicated with hemophagocytic lymphohistiocytosis over the past 10 years.
Brucellosis is a pervasive zoonotic disease caused by various Brucella species (16). The Brucella genus comprises six species, each named after its principal host: B. melitensis (sheep and goats), B. abortus (cattle), B. suis (pigs), B. ovis (sheep), B. canis (dogs), and B. neotomae (wood desert rats). Notably, although the disease’s incubation period typically ranges from 2 to 4 weeks, it could last several months (16, 17). Additionally, according to a 2006 report, ⁓500,000 new cases of brucellosis are diagnosed globally each year (18) and contact with infected animals (e.g., cattle and sheep) and consumption of contaminated animal products have been established as the primary transmission pathways for the disease (19). Furthermore, its most common clinical symptoms include fever, fatigue, and liver and spleen enlargement. Moreover, the non-specific clinical symptoms of brucellosis could lead to a delayed diagnosis, allowing the disease to progress to HLH.
Herein, we systematically reviewed a brucellosis-induced HLH case and related HLH literature through a comprehensive search of the PubMed medical database (Search query: “Brucellosis” AND “Lymphohistiocytosis, Hemophagocytic”[Mesh] or “hemophagocytic lymphohistiocytosis”). We also cross-referenced existing review articles to obtain other relevant reports.
2 Case description
A 53-year-old man presented to our hospital with a 2-month history of fever and abnormal liver enzymes. Notably, the patient was a sheep breeder and had a prior history of contact with sheep. Blood samples were drawn from the patient into a Mérieux blood culture bottle and incubated at 35 °C for 5 days, and the initial blood culture (aerobic) revealed the presence of Brucella. Mass spectrometry also confirmed the presence of Brucella in the culture. Notably, due to inappropriate laboratory conditions, we could not identify the species of the Brucella genus. The patient was discharged after some time with Doxycycline and Levofloxacin prescriptions for oral antimicrobial therapy. However, the patient did not take the antibiotics as prescribed. Within 4 days, he presented to a local hospital again with complaints of fever and abdominal discomfort, as well as pancytopenia (the specific value was unknown). The patient was later re-admitted to our hospital following a platelet transfusion and bone marrow aspiration. Notably, the patient had a 2-month history of diabetes mellitus (DM) without diabetic complications. The glycosylated hemoglobin concentration 2 months before admission was 7.2% (NR:4.0%–6.0%) but did not receive systemic treatment. He also denied a history of other illnesses or a familial genetic history of specific diseases.
Upon admission, the preliminary physical examination revealed a temperature of 38.6 °C, a respiration rate (RR) of 26 breaths/min, a pulse of 88 beats/min, a blood pressure (BP) of 99/68 mmHg, a height of 168 cm, and a weight of 60 kg. The patient had no jaundice of the skin or sclera. The cardiopulmonary assessment revealed no abnormalities. The abdominal examination revealed mild tenderness in the right upper quadrant without rebound pain or rigidity. The liver and spleen were not palpable below the ribs. The lower limbs showed no edema.
Table 2 details the main laboratory test results during hospitalization and other related examinations. Routine coagulation tests revealed an activated Partial Thromboplastin Time (APTT) of 51.3 s (normal range [NR]: 21–33 s). Furthermore, the fasting triglyceride (Ftriglyceride), cholesterol, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) levels were 2 mmol/L(NR:0.28–1.8 mmol/L), 2.53 mmol/L (NR:2.6–6 mmol/L), 0.41 mmol/L(NR:0.76–2.1 mmol/L), and 1.62 mmol/L(NR:2.06–2.1 mmol/L), respectively. The glycosylated hemoglobin value at 2 months before admission was 7.2% (NR:4.0%–6.0%). A bone marrow smear identified histiocytes with occasional hemophagocytic cells (Figure 1A). On day 6 of hospitalization, a blood culture confirmed the growth of Brucella spp. in an aerobic bottle after ⁓113.67 h of incubation (Figure 1B). On day 8 of hospitalization, the blood culture revealed the growth of Brucella spp. in an aerobic bottle after ⁓94.87 h of incubation. Antinuclear antibody (ANA) profiles, antiplatelet antibodies, CMV, EPV, hepatitis B, hepatitis C, syphilis, and HIV were all negative.
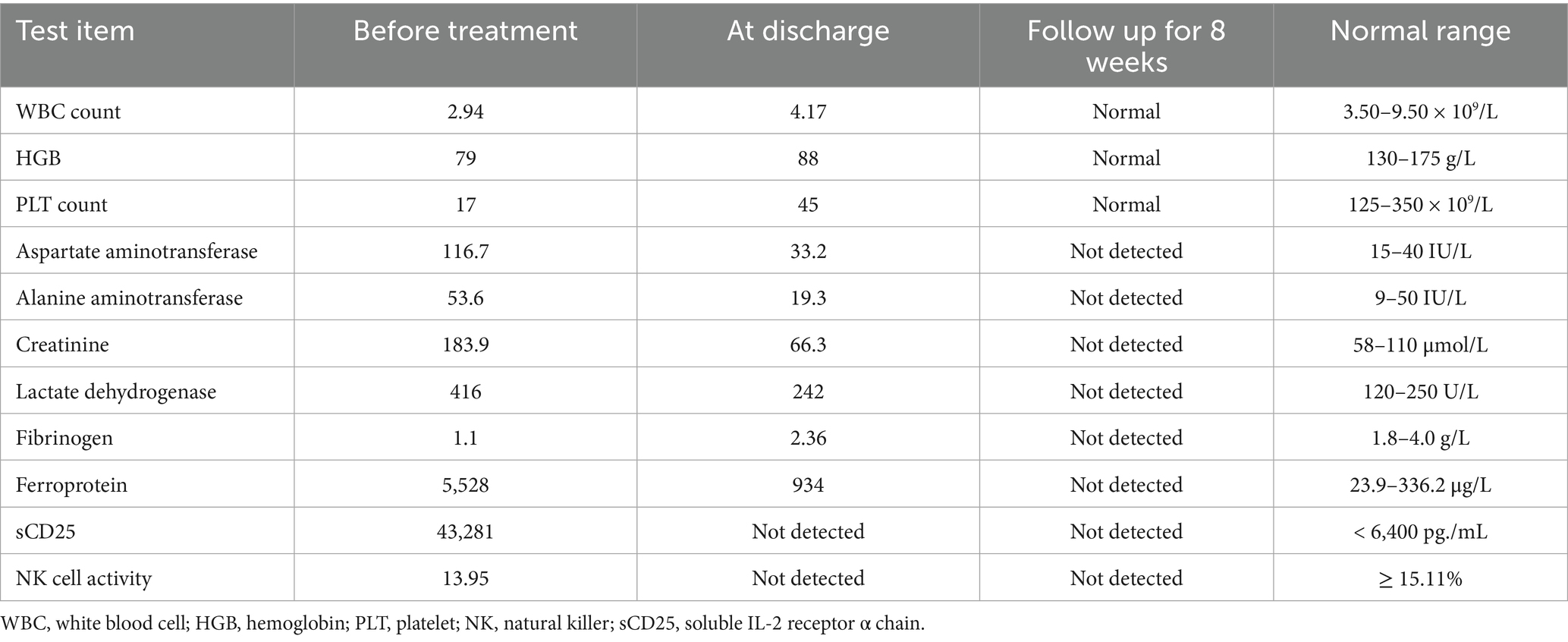
Table 2. Major laboratory test results of the patient during hospitalization.
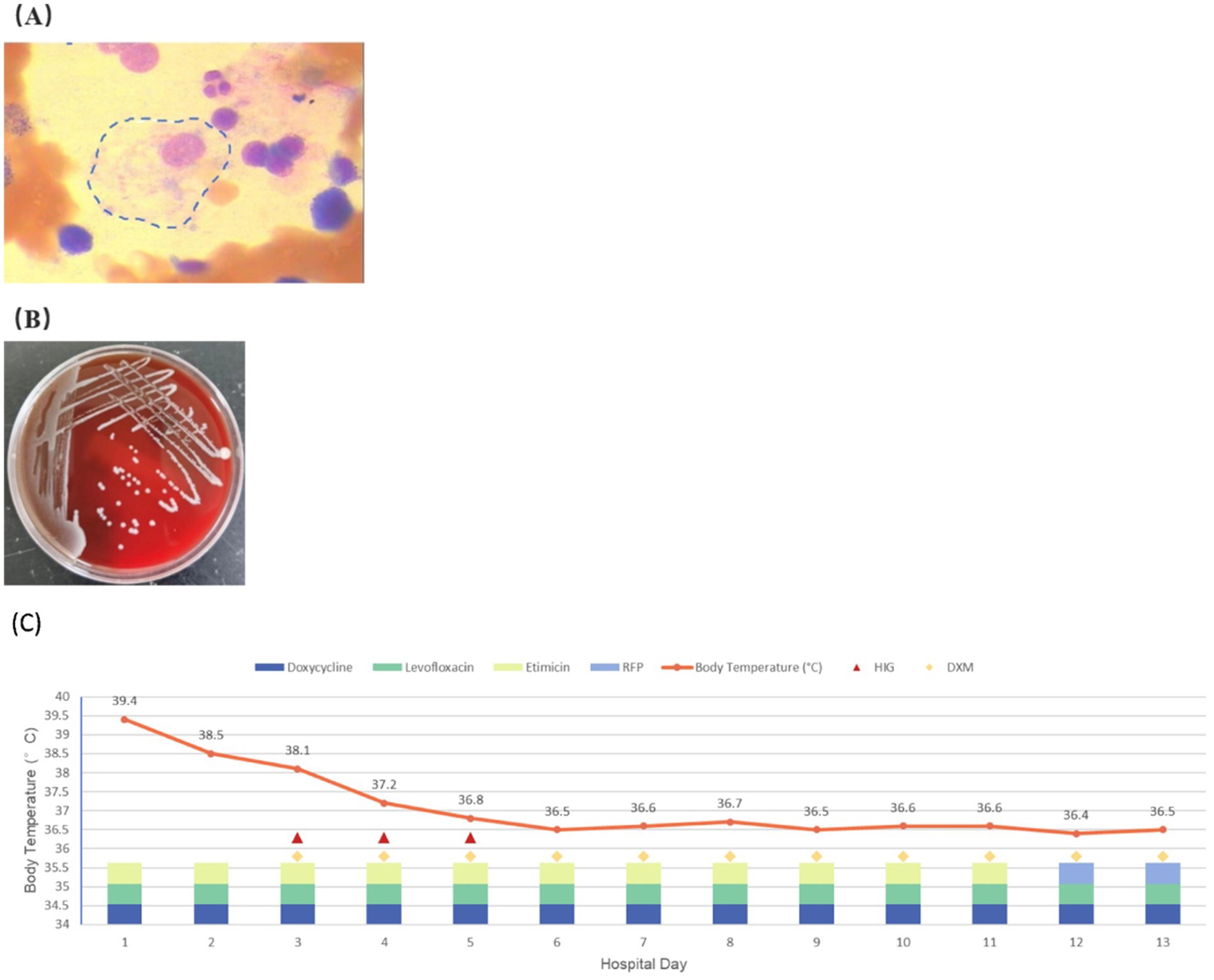
Figure 1. (A) Hemophagocytic cells are observed in the patient’s bone marrow smear. (B) Blood aerobic culture positive for Brucella. (C) Temperature changes and the treatment of the patient during hospitalization.
Regarding the imageological examination, aortic valve calcification was the key cardiac ultrasound finding. Meanwhile, the ultrasound findings of the digestive system included a fatty liver, hepatomegaly, gallbladder wall edema, splenomegaly, hypoechoic splenic lesions, and ascites.
The patient was diagnosed with brucellosis 2 months ago and put on doxycycline and levofloxacin for 1 week. However, he did not continue taking either prescription post-discharge. Fever was the main symptom during the patient’s second hospitalization. Based on the above laboratory findings, the patient met the HLH-2004 diagnostic criteria (20) (Table 3), with an HScore (21) of 230. Blood cultures detected Brucella spp. in aerobic bottles, leading to the diagnosis of secondary hemophagocytic lymphohistiocytosis (HLH) induced by Brucella infection. The patient received systemic anti-infective therapy, glucocorticoids, and intravenous immunoglobulin (IVIG). The specific dosages and treatment duration were as follows: The specific antibiotic treatment regimen was: doxycycline (100 mg every 12 h via Intravenous [IV] infusion during hospital days 1 to 5 and 100 mg administered orally every 12 h during hospital days 6 to 13); levofloxacin (200 mg every 24 h through IV infusions with the initial dose doubled during hospital days 1 to 4 and 200 mg every 12 h through IV during hospital days 5 to 13); etimicin (300 mg through IV every 24 h during hospital days 1 to 7, 200 mg through IV every 24 h during hospital days 8 to 10, and 300 mg through IV every 24 h in hospital day 11), and rifampin (600 mg administered orally every 24 h during hospital days 12 to13). Dexamethasone (15 mg/day from hospital days 3 to 11, then 10 mg/day from days 12 to 13). Intravenous immunoglobulin (IVIG) (250 mg/kg/day from hospital days 3 to 5).

Table 3. HLH-2004 diagnostic criteria.
During the early hospitalization period, the patient showed no significant improvement following initiation of antimicrobial therapy in clinical symptoms or laboratory parameters, including complete blood count, ferritin, and fibrinogen levels. Consequently, the treatment regimen was optimized by the inclusion of glucocorticoids and intravenous immunoglobulin (IVIG) in ongoing antimicrobial therapy. This combined therapeutic approach caused a significant clinical improvement relative to the status at admission, with significant normalization of hematological indices (complete blood count), ferritin, and lactate dehydrogenase (LDH) levels at the time of discharge. The patient achieved clinical stability by hospital day 13, and his fever curve and details of his therapeutic regimen during hospitalization are shown in Figure 1C. He was subsequently discharged. Post-discharge medication included rifampin, doxycycline, levofloxacin, and oral dexamethasone (10 mg/day). Complete recovery was achieved after 8 weeks of follow-up monitoring.
3 Discussion
Despite its significant clinical implications, the pathogenesis of HLH remains incompletely unclear. However, the disease has been largely associated with a significant reduction or obstruction of cytotoxic T cells and NK cytotoxic activity, resulting in ineffective clearance of viruses and other antigens. This immune dysfunction, combined with the persistent abnormal continuous activation of macrophages, can trigger a hyperinflammatory response in multiple organs, ultimately leading to immune-mediated tissue and organ damage (22). Under normal physiological conditions, CD8+ cytotoxic T cells and NK cells often release cytolytic granules containing perforin and granzyme when they come into contact with virus-infected or tumor cells. Herein, we reviewed a case of Brucella infection-induced secondary HLH. The resulting inability to clear antigen stimulation could, in turn, lead to immune response persistence and expansion. Activated immune cells have been established to release proinflammatory cytokines (IFN-γ, TNF-α, IL-1, IL-6, TNF-α, and so on), leading to high levels of macrophage activation, which, in turn, might cause hemophagocytosis, tissue damage, organ failure, and other inflammatory manifestations (23, 24). Relevant retrospective studies have shown that adult HLH is largely attributable to malignant tumors. Infections and autoimmune diseases are the other common causes of the disease, with viral infections, including EBV, HIV, CMV, or influenza, as the most common pathogens. Meanwhile, bacterial infection-induced HLH is relatively rare (25–27). Moreover, although Brucella—an intracellular bacterium that can manipulate host cell processes, disrupt phagocyte function, inhibit phagocytosis, and prevent host cell apoptosis—can cause secondary HLH, the underlying mechanisms remain underexplored.
Brucella subverts signaling pathways crucial for innate immunity, thus modulating the host immune response (16). Nonetheless, the mechanism underlying brucellosis-induced HLH remains unclear. Some researchers linked the hemophagocytic mechanism of non-viral infection to the excessive production of activating cytokines (such as TNF-α and interferon-γ), leading to macrophage activation. However, it could also be the result of poorly regulated or inappropriate T-helper lymphocyte responses to intracellular pathogens (28). Consequently, following Brucella infection, the host immune response produces numerous cytokines, leading to phagocytic cell activation and helper T lymphocyte dysfunction, ultimately inducing HLH. In this regard, it is noteworthy that the timely removal of bacteria, inflammation reduction, and immune response regulation would be particularly important interventions.
We compared and analyzed eight cases of hemophagocytic syndrome caused by Brucella infection (Table 1), which showed that the clinical manifestations and laboratory findings of brucellosis were comparable to those of HLH, especially in terms of hematologic abnormalities. Data show that Brucella species often replicate in host phagocytes, impairing immune responses (29). This phenomenon suggests that the patients’ blood system is often affected. Analysis of blood cells in 484 patients with brucellosis confirmed the presence of anemia (21.5%), thrombocytopenia (18.8%), leukopenia (14.6%), and pancytopenia (2%–14%) (30). Another study reported pancytopenia in 14.8% of 54 children with brucellosis (31). Furthermore, the pathogenesis of brucellosis-induced pancytopenia included the following: (1) Hemophagocytosis, (2) hypersplenism, (3) bone marrow dysgenesis, (4) bone marrow granuloma, and (5) immune destruction (32). Common histopathological manifestations of HLH include hemophagocytosis in the bone marrow and reticuloendothelial organs (5). Furthermore, complete blood count analyses in patients with HLH commonly reveal leukocytosis, anemia, thrombocytopenia, and elevated inflammatory markers such as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). Given these findings, the presence of Brucella-induced pancytopenia should prompt clinical consideration of HLH as a potential diagnosis.
Several measures have been developed for assessing HLH. Henter et al. developed the HLH2004 criteria (Table 3) whereby a diagnosis of HLH is confirmed if five out of the eight criteria are met (20). Additionally, Fardet et al. (21) developed the HScore1–a total sum of the score of nine variables that allow for the assessment of HLH risk (Table 4) (27). Unlike the HLH-2004 criteria, which comprise parameters derived from a pediatric population, the HScore was developed in an adult cohort, including patients aged ≥ 18 years (33). Furthermore, the pattern of inflammatory cytokines (IFN-γ and IL-10 upregulation, with only modestly elevated IL-6 levels) demonstrated a high diagnostic accuracy for secondary hemophagocytic lymphohistiocytosis secondary hemophagocytic lymphohistiocytosis (sHLH) and could be useful in differentiating infection-induced HLH and monitoring patients (27).
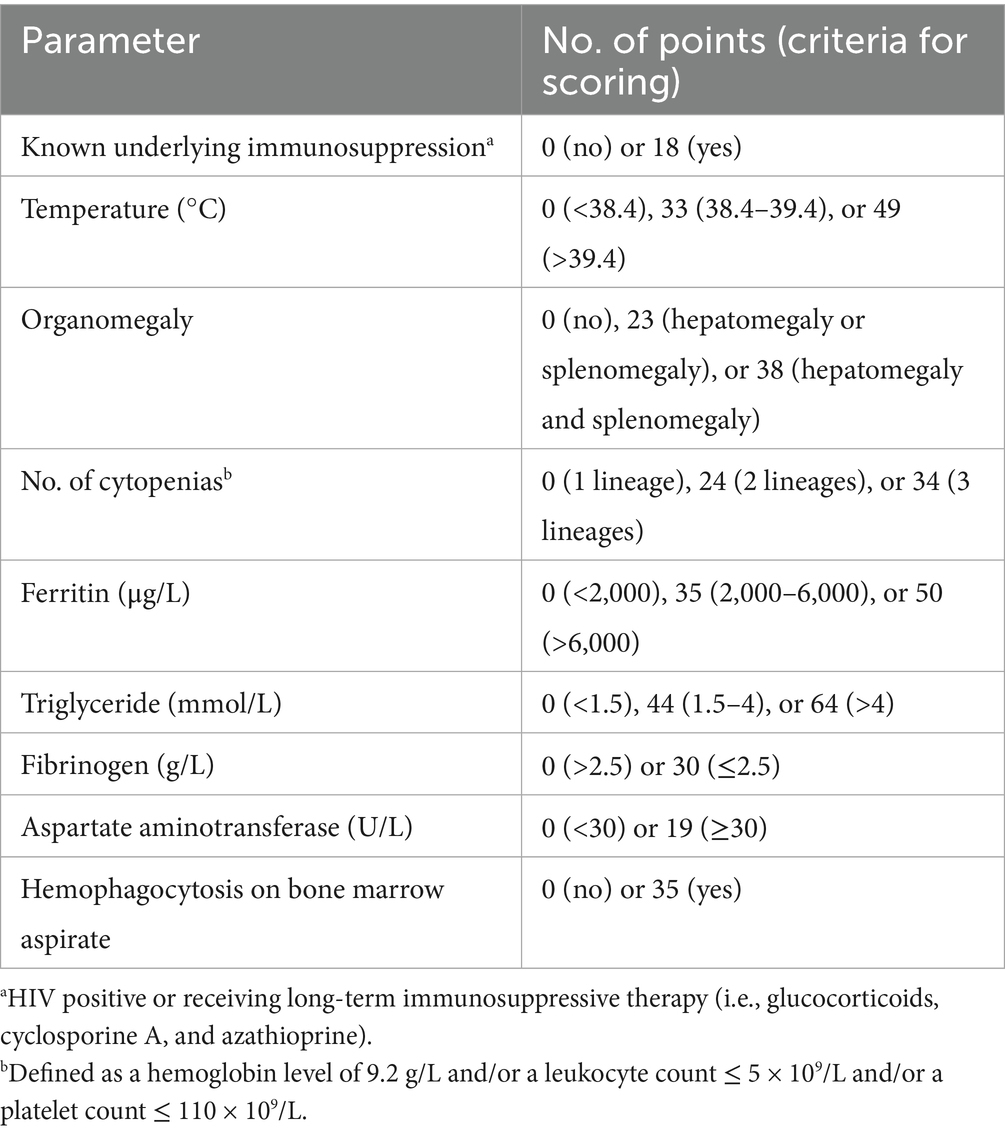
Table 4. Parameters and points in the HScore (27).
Our patient had positive blood and bone marrow cultures for Brucella. Meanwhile, the patient exhibited common clinical manifestations of brucellosis, including fever, splenomegaly, abnormal liver function, and elevated LDH levels, confirming the diagnosis of Brucella. Moreover, due to the presence of pancytopenia, especially thrombocytopenia, a bone marrow smear examination was performed, revealing hemophagocytic cells. Additionally, bone marrow aspiration was performed, revealing hemophagocytic cells. Based on these findings, along with laboratory ferritin, fibrinogen, soluble CD25, NK cell activity, and other test results, HLH diagnosis was confirmed.
Presently, the specific treatment for Brucella-induced HLH remains unknown, with most of the current interventions being empirical clinical treatments. The majority of HLH patients are currently being treated with antibiotic medications based on the HLH-2004/HLH-1994 protocol and La Rosée et al.’s recommendations for managing HLH in adults (hereinafter referred to as the recommendations) (20, 27, 34). The recommendations propose that the heterogeneity of adult HLH prohibits a “1-size-fits-all protocol” and that treatment should be tailored to control hyperinflammation and treat identified disease triggers, especially the disease-causing pathogen (Brucella). Qureshi et al. proposed doxycycline and RIF for treating brucellosis and minimizing the risk of relapse, with aminoglycosides frequently added during the initial 2–3 weeks of therapy (16, 35). While the WHO-recommended guidelines for brucellosis treatment have evolved, the optimal approach remains unclear (16). Moreover, important organs such as the liver and kidneys should always be protected during treatment. Therefore, appropriate antibiotics should be chosen for the whole course of treatment based on the patient’s organ function. Considering the aforementioned clinical guidelines and the severity of the patient’s condition, we initiated an antimicrobial therapy encompassing doxycycline, levofloxacin, and etimicin, with appropriate dosage adjustments based on the patient’s hepatic and renal function. Following a comprehensive treatment including the aforementioned antimicrobial agents, corticosteroids, and Intravenous Immunoglobulin (IVIG), the patient’s general condition and previously abnormal laboratory parameters improved significantly compared to admission status. Nonetheless, aerobic blood cultures obtained on hospital day 8 revealed the growth of Brucella species after ⁓94.87 h of incubation. Given the normalized liver function, we modified the antibiotic regimen to doxycycline, levofloxacin, and rifampicin to consolidate anti-infective therapy. Notably, some case reports indicated recovery from the disease with antibiotics alone (8, 12–15). On the other hand, the treatment drugs per the HLH-2004/HLH-1994 protocol and the recommendations mainly included GCs, immunosuppressants, biologic agents, and cytotoxic drugs. The main GCs were DEX (10 mg/m2/d for 2 weeks; thereafter reduced by 50% every 2 weeks and continued to the end of 8 weeks). In the majority of secondary HLH cases, the dosage and course of DEX should be stopped after controlling the primary disease per the clinical situation. Moreover, immunosuppressive agents and cytotoxic drugs may not be required when GCs are effective. The treatment of Brucella-associated HLH currently follows the principle of individualization. In this regard, it is noteworthy that intravenous immunoglobulin therapy has been successfully used in adults with multifactorial HLH. Two small series of adult HLH patients who received intravenous immunoglobulins exhibited promising results—especially those with HLH attributed to infections and autoimmune diseases—with an estimated survival rate (SR) of 59%–75% (36, 37). Furthermore, among the eight case reports from the past decade (Table 1), antibiotic therapy alone was effective in five patients, with the remaining three patients treated with the corticosteroid DEX in combination with anti-brucellosis therapy (HIG or etoposide in two cases). According to the recommendations, although patients infected by pathogens targeting the monocyte–macrophage system may develop HLH, immunosuppression as recommended in the HLH-94 protocol should be avoided, as they usually respond well to specific antimicrobial treatments. However, our patient’s cell count was not restored to normal after antimicrobial therapy with doxycycline, levofloxacin, and etimicin. Moreover, the platelet count and hemoglobin levels did not increase significantly following platelet transfusion. La Rosée et al. (27) proposed that IVIG could inhibit complement activation, antibody Fc fragments, and macrophage Fc receptors, as well as neutralize pro-inflammatory cytokines, thus exerting an anti-inflammatory effect. Considering the HLH-1994/HLH-2004 protocol and the recommendations, as well as the patient’s clinical status, we initiated treatment with corticosteroids and IVIG for the patient. Notably, the use of IVIG is quite debatable. The HLH-2004 protocol recommends IVIG at 0.5 g/kg once every 4 weeks (during initial and continuation therapy). On the other hand, La Rosée et al. (27) reported that IVIG should be administered over 2–3 days to a total dose of 1.6 g/kg. Furthermore, Georgia Griffin et al. (1) in their review of hemophagocytic syndrome-related literature, mentioned that Rong-Rong He et al. (38) reported comparable efficacy with regimens of 400 mg/kg/day for 5 days (IVIG5) and 1 g/kg/day for 2 days (IVIG2). Due to our patient’s financial constraints, the aforementioned doses could not be administered. However, the patient’s general condition and laboratory parameters improved after 3 days of alternative therapy. A dose of 250 mg/kg/day was given for 3 days, which represents a limitation as it is not the standard recommended dosage. Nonetheless, this treatment inhibited cellular inflammatory factor release and reduced phagocytic cell activation. The patient’s temperature was also restored to normal, while the white blood cell (WBC) count increased, and the hemoglobin and platelet levels remained stable without blood transfusion support. Based on these findings, we deduced that when treatment of Brucella-induced HLH with antibiotic therapy alone is not effective, timely application of GCs, such as DEX, along with HIG could shorten the treatment time.
Viral infection is the most common cause of adult HLH, with EBV being the most predominant pathogen. Although the timely initiation of the HLH-94 regimen could significantly improve the prognosis of EBV-HLH, the treatment intensity and duration should be stratified based on disease severity. For milder cases or patients with clinical improvements, a short-course corticosteroid regimen (with/without IVIG) could be a sufficient conservative approach. Nonetheless, etoposide must be administered immediately in cases of rapid clinical deterioration—particularly in untreated EBV-infected patients. Furthermore, while rituximab could eliminate EBV reservoirs in EBV-triggered HLH, it should not replace GC-based therapy (with/without etoposide) (27). Trym Fauchald et al. (39), in their study of TB-associated HLH, concluded that anti-mycobacterial therapy (with or without etoposide) could be a critical determinant of survival, with an overall survival (OS) rate marginally above 50%.
A summary of eight reports on cases of brucellosis-associated HLH (Table 1) was provided, which showed that the patients presented with diverse non-specific symptoms. Moreover, some patients lacked a clear epidemiological history associated with brucellosis. Fever was the main clinical manifestation (which lasted from a few days to several months), and was sometimes accompanied by splenomegaly. The common laboratory findings included cytopenia and increased ferritin levels, accompanied by abnormal liver function and LDH levels. In certain patients with prolonged fever, delayed identification of the underlying cause and the absence of timely intervention may contribute to the development of HLH. Notably, both the previously reported cases and the present case demonstrated marked improvement in clinical symptoms and laboratory findings following comprehensive and systematic treatment. Anti-infective therapy was the most effective treatment, and all patients received a standardized anti-infective regimen. Some patients improved with anti-infective therapy alone, while others required additional corticosteroids (with or without etoposide/human immunoglobulin) on top of the anti-infective treatment. Therefore, patients with prolonged fever and laboratory tests showing cytopenia, splenomegaly, and elevated ferritin levels should be actively screened for Brucella infection despite the absence of an epidemiological history of brucellosis. Meanwhile, further examinations (triglycerides, fibrinogens, marrow aspiration, sCD25, and NK cell activity) should be conducted to confirm the presence of HLH. In conclusion, early diagnosis is recommended to improve the prognosis of HLH patients.
This study highlights the fact that Brucella-induced HLH is rare. Furthermore, when encountering a patient with clinical manifestations such as fever, splenomegaly, and liver injury, clinicians should carefully consider the patient’s medical history, as well as the possibility of Brucella infection. Additionally, more laboratory tests should be conducted in cases displaying pancytopenia to confirm HLH diagnosis. Overall, early diagnosis and timely treatment can reduce disease progression and mortality rates in HLH patients, and patients with a confirmed Brucella infection should undergo regular antimicrobial therapy to prevent disease progression.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of the First Hospital of Jilin University, China. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
SJ: Validation, Writing – original draft. BX: Validation, Writing – original draft. HW: Writing – review & editing. PZ: Writing – review & editing. XL: Writing – review & editing. KZ: Conceptualization, Project administration, Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Jilin Provincial Department of Science and Technology (20200201616JC) to KZ and the Jilin Province Health Science and Technology Capacity Improvement Project (2021JC001) to KZ.
Acknowledgments
We thank all authors for their contributions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
1. Griffin, G, Shenoi, S, and Hughes, GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. (2020) 34:101515. doi: 10.1016/j.berh.2020.101515
2. Cisło, M, Nowicka, J, Baran, E, and Wojtala, R. Histiocytosis Maligna (histiocytic medullary Reticulocytosis). Przegl Dermatol. (1979) 66:275–80.
3. Degar, B. Familial hemophagocytic Lymphohistiocytosis. Hematol Oncol Clin North Am. (2015) 29:903–13. doi: 10.1016/j.hoc.2015.06.008
4. Schram, AM, and Berliner, N. How I treat hemophagocytic Lymphohistiocytosis in the adult patient. Blood. (2015) 125:2908–14. doi: 10.1182/blood-2015-01-551622
5. Ramos-Casals, M, Brito-Zerón, P, López-Guillermo, A, Khamashta, MA, and Bosch, X. Adult haemophagocytic syndrome. Lancet. (2014) 383:1503–16. doi: 10.1016/s0140-6736(13)61048-x
6. Jevtic, D, da Silva, MD, Haylock, AB, Nordstrom, CW, Oluic, S, Pantic, N, et al. Hemophagocytic lymphohistiocytosis (Hlh) in patients with tick-borne illness: a scoping review of 98 cases. Infect Dis Rep. (2024) 16:154–69. doi: 10.3390/idr16020012
7. Ong, LT, and Balasubramaniam, R. Prevalence and mortality of haemophagocytic lymphohistiocytosis in dengue fever: a systematic review and meta-analysis. Trans R Soc Trop Med Hyg. (2024) 118:711–9. doi: 10.1093/trstmh/trae032
8. Guo, M, Zhang, L, Chen, K, Huang, D, Feng, C, Liu, H, et al. Hemophagocytic lymphohistiocytosis associated with brucellosis. Diagn Microbiol Infect Dis. (2024) 109:116289. doi: 10.1016/j.diagmicrobio.2024.116289
9. Xu, M, Mo, S, and Fu, X. Brucella infection-induced hemophagocytic syndrome with subsequent development of the probable vanishing bile duct syndrome: a case report and literature review. SAGE Open Med Case Rep. (2023) 11:2050313x231207562. doi: 10.1177/2050313x231207562
10. Park, D, Yoon, K, Lo, A, and Bolos, D. Hemophagocytic Lymphohistiocytosis induced by brucellosis: a case report. Cureus. (2024) 16:e64287. doi: 10.7759/cureus.64287
11. Pekpak, E, and Sirvan, CB. Secondary hemophagocytic lymphohistocytosis in a child with brucellosis. J Pediatr Hematol Oncol. (2017) 39:e501–3. doi: 10.1097/mph.0000000000000849
12. Aydın, S, Günal, Ö, Taşkın, MH, Atilla, A, and Kılıç, SS. Brucellosis as a cause of hemophagocytic syndrome. Mikrobiyol Bul. (2015) 49:292–4. doi: 10.5578/mb.9332
13. Mittal, J, Kumar, P, Goyal, JP, and Purohit, A. Haemophagocytic lymphohistiocytosis secondary to brucellosis in a young child. BMJ Case Rep. (2021) 14. doi: 10.1136/bcr-2020-240759
14. Tian, LH, Dong, ZG, Chen, XY, Huang, LJ, and Xiao, PP. Brucellosis of unknown origin with Haemophagocytic syndrome: a case report. World J Clin Cases. (2021) 9:3649–54. doi: 10.12998/wjcc.v9.i15.3649
15. El Euch, M, Kaabar, MY, Bouaziz, R, Mahfoudhi, M, Jaziri, F, Kefi, A, et al. Successful resolution of hemophagocytic lymphohistiocytosis associated to brucellosis in the adult. Tunis Med. (2018) 96:458–61.
16. Qureshi, KA, Parvez, A, Fahmy, NA, Abdel Hady, BH, Kumar, S, Ganguly, A, et al. Brucellosis: epidemiology, pathogenesis, diagnosis and treatment-a comprehensive review. Ann Med. (2023) 55:2295398. doi: 10.1080/07853890.2023.2295398
17. Pappas, G, Akritidis, N, Bosilkovski, M, and Tsianos, E. Brucellosis. N Engl J Med. (2005) 352:2325–36. doi: 10.1056/NEJMra050570
18. Pappas, G, Papadimitriou, P, Akritidis, N, Christou, L, and Tsianos, EV. The new global map of human brucellosis. Lancet Infect Dis. (2006) 6:91–9. doi: 10.1016/s1473-3099(06)70382-6
19. Zheng, R, Xie, S, Lu, X, Sun, L, Zhou, Y, Zhang, Y, et al. A systematic review and meta-analysis of epidemiology and clinical manifestations of human brucellosis in China. Biomed Res Int. (2018) 2018:1–10. doi: 10.1155/2018/5712920
20. Henter, JI, Horne, A, Aricó, M, Egeler, RM, Filipovich, AH, Imashuku, S, et al. Hlh-2004: diagnostic and therapeutic guidelines for hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer. (2007) 48:124–31. doi: 10.1002/pbc.21039
21. Fardet, L, Galicier, L, Lambotte, O, Marzac, C, Aumont, C, Chahwan, D, et al. Development and validation of the Hscore, a score for the diagnosis of reactive hemophagocytic syndrome. Arthritis Rheumatol. (2014) 66:2613–20. doi: 10.1002/art.38690
22. Al-Samkari, H, and Berliner, N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol. (2018) 13:27–49. doi: 10.1146/annurev-pathol-020117-043625
23. Morimoto, A, Nakazawa, Y, and Ishii, E. Hemophagocytic lymphohistiocytosis: pathogenesis, diagnosis, and management. Pediatr Int. (2016) 58:817–25. doi: 10.1111/ped.13064
24. Kaçar, AG, and Celkan, TT. Hemophagocytic lymphohistiocytosis. Balkan Med J. (2022) 39:309–17. doi: 10.4274/balkanmedj.galenos.2022.2022-4-83
25. Rivière, S, Galicier, L, Coppo, P, Marzac, C, Aumont, C, Lambotte, O, et al. Reactive hemophagocytic syndrome in adults: a retrospective analysis of 162 patients. Am J Med. (2014) 127:1118–25. doi: 10.1016/j.amjmed.2014.04.034
26. Parikh, SA, Kapoor, P, Letendre, L, Kumar, S, and Wolanskyj, AP. Prognostic factors and outcomes of adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc. (2014) 89:484–92. doi: 10.1016/j.mayocp.2013.12.012
27. La Rosée, P, Horne, A, Hines, M, von Bahr, GT, Machowicz, R, Berliner, N, et al. Recommendations for the management of hemophagocytic lymphohistiocytosis in adults. Blood. (2019) 133:2465–77. doi: 10.1182/blood.2018894618
28. Filipovich, A, McClain, K, and Grom, A. Histiocytic disorders: recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. (2010) 16:S82–9. doi: 10.1016/j.bbmt.2009.11.014
29. Zhang, Y, Li, K, Yang, L, and Li, T. Bone marrow histiocytic hyperplasia and hemophagocytosis revealing brucellosis. Int J Lab Hematol. (2022) 44:49–50. doi: 10.1111/ijlh.13589
30. Uluğ, M, Yaman, Y, Yapici, F, and Can-Uluğ, N. Clinical and laboratory features, complications and treatment outcome of brucellosis in childhood and review of the literature. Turk J Pediatr. (2011) 53:413–24. doi: 10.24953/turkjped.2011.1793
31. Karakukcu, M, Patiroglu, T, Ozdemir, MA, Gunes, T, Gumus, H, and Karakukcu, C. Pancytopenia, a rare hematologic manifestation of brucellosis in children. J Pediatr Hematol Oncol. (2004) 26:803–6.
32. Yaman, Y, Gözmen, S, Özkaya, AK, Oymak, Y, Apa, H, Vergin, C, et al. Secondary hemophagocytic lymphohistiocytosis in children with brucellosis: report of three cases. J Infect Dev Ctries. (2015) 9:1172–6. doi: 10.3855/jidc.6090
33. Knaak, C, Nyvlt, P, Schuster, FS, Spies, C, Heeren, P, Schenk, T, et al. Hemophagocytic lymphohistiocytosis in critically ill patients: diagnostic reliability of Hlh-2004 criteria and Hscore. Crit Care. (2020) 24:244. doi: 10.1186/s13054-020-02941-3
34. Henter, JI, Samuelsson-Horne, A, Aricò, M, Egeler, RM, Elinder, G, Filipovich, AH, et al. Treatment of hemophagocytic lymphohistiocytosis with Hlh-94 immunochemotherapy and bone marrow transplantation. Blood. (2002) 100:2367–73. doi: 10.1182/blood-2002-01-0172
35. Bosilkovski, M, Keramat, F, and Arapović, J. The current therapeutical strategies in human brucellosis. Infection. (2021) 49:823–32. doi: 10.1007/s15010-021-01586-w
36. Emmenegger, U, Spaeth, PJ, and Neftel, KA. Intravenous immunoglobulin for hemophagocytic lymphohistiocytosis? J Clin Oncol. (2002) 20:599–601. doi: 10.1200/jco.2002.20.2.599
37. Larroche, C, Bruneel, F, André, MH, Bader-Meunier, B, Baruchel, A, Tribout, B, et al. Intravenously administered gamma-globulins in reactive Hemaphagocytic syndrome. Multicenter study to assess their importance, by the immunoglobulins Group of Experts of Cedit of the ap-hp. Ann Med Interne (Paris). (2000) 151:533–9.
38. Chen, RL, Lin, KH, Lin, DT, Su, IJ, Huang, LM, Lee, PI, et al. Immunomodulation treatment for childhood virus-associated haemophagocytic lymphohistiocytosis. Br J Haematol. (1995) 89:282–90. doi: 10.1111/j.1365-2141.1995.tb03302.x
Keywords: brucellosis, hemophagocytic lymphohistiocytosis, pathogenesis, diagnosis, treatment
Citation: Jiang S, Xia B, Wu H, Zhang P, Li X and Zhang K (2025) Brucellosis-associated hemophagocytic lymphohistiocytosis: a case report and literature review. Front. Med. 12:1609644. doi: 10.3389/fmed.2025.1609644
Edited by:
Julien Carvelli, Assistance Publique Hôpitaux de Marseille, FranceReviewed by:
Angel Robles-Marhuenda, La Paz Hospital, SpainAmandine Bichon, Assistance Publique Hôpitaux de Marseille, France
Copyright © 2025 Jiang, Xia, Wu, Zhang, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaiyu Zhang, a2FpeXVAamx1LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship