Abstract
Birt–Hogg–Dubé syndrome (BHDS), an autosomal dominant disease, is caused by germline mutations in the folliculin (FLCN, NM_144997) gene. This rare disorder is characterized by a clinical triad, which includes fibrofolliculomas (FFs), renal cell carcinoma (RCC), and pulmonary manifestations such as multiple pulmonary cysts (PCs) and pneumothorax. To investigate the clinical features and genetic mutations of five unrelated BHDS families in a long-term follow-up study at the Second Xiangya Hospital of Central South University, five families and their affected patients, who met the clinical and histological criteria for BHDS and were confirmed to have FLCN germline mutations, were evaluated. All participants underwent a comprehensive physical examination along with other relevant tests. Three novel mutations (c.246C > A, c.625_626insAGGCAGAGCAGTTTGGAT, and c.1542_1542delA) and one previously reported mutation (c.1429C > T) in the FLCN gene were identified. These mutations are predicted to cause truncation of the folliculin protein, likely resulting in decreased folliculin expression. Our study expands the genetic landscape associated with BHDS and provides valuable insights for future genetic counseling and the clinical management of individuals with BHDS.
1 Introduction
Birt–Hogg–Dubé syndrome (BHDS) is an uncommon autosomal dominant hereditary disorder caused by germline mutations in the folliculin (FLCN) gene, which is located on chromosome 17p11.2 (1, 2). First described by Birt, Hogg, and Dubé in 1977, and later associated with the FLCN gene by Nickerson et al. in 2002, BHDS is characterized by phenotypic heterogeneity, with major clinical manifestations involving the skin (benign cutaneous fibrofolliculomas (FFs)), kidney (renal tumors), and lung (multiple pulmonary cysts (PCs), with a risk of spontaneous pneumothorax (PTX)) (3, 4). The FLCN gene, a known tumor suppressor, encodes the folliculin protein, comprising 14 exons. Pathogenic variants in FLCN include frameshift, nonsense, splice-site, and missense mutations, most of which result in the loss of function through the generation of premature termination codons (PTCs) and subsequent nonsense-mediated mRNA decay (NMD) (4, 5). While the precise molecular functions of folliculin remain incompletely understood, emerging evidence has significantly advanced our understanding of its role in cellular signaling. Folliculin interacts with key regulatory pathways, including AMP-activated protein kinase (AMPK) and the mechanistic target of rapamycin (mTOR) signaling cascades (6). Moreover, in complex with folliculin-interacting proteins 1 and 2 (FNIP1/2), folliculin functions as a GTPase-activating protein (GAP) for the RagC and RagD GTPases, promoting the mechanistic target of rapamycin complex 1 (mTORC1) activation in response to amino acid availability (7). Additionally, folliculin regulates the transcription factors TFEB and TFE3 by promoting their phosphorylation by mTORC1: mechanistic target of rapamycin complex 1, thereby preventing their nuclear translocation and activation. Loss of FLCN function leads to constitutive activation and nuclear localization of transcription factor EB (TFEB) and TFE3, which drives renal cystogenesis and tumorigenesis (8).
In our study, we conducted a long-term follow-up study of five unrelated Chinese families with genetically confirmed BHDS. All probands and selected family members were recruited from central southern China (Hunan Province) and underwent germline FLCN mutational analysis via direct Sanger sequencing. Comprehensive clinical data were collected, and genotype–phenotype correlations were evaluated to explore the relationship between FLCN variants and the diverse clinical presentations observed.
2 Materials and methods
Five families with BHDS were recruited from the Second Xiangya Hospital of Central South University. This study was approved by the Institutional Review Board of the Second Xiangya Hospital, and written informed consent was obtained from all family members. All participants underwent a comprehensive physical examination and other relevant examinations. Genomic DNA was extracted from a 6-ml peripheral blood sample using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA). The polymorphism phenotyping-2 (PolyPhen-2), Sorting Intolerant From Tolerant (SIFT), and Mutation Taster programs were used to analyze the effects of mutations on the function of the proteins. Genetic analysis was performed by Sanger sequencing, amplifying all coding exons (exons 4–14) and flanking intronic sequences of the FLCN gene using the previously described primers (9). The SWISS-MODEL tool1 was used for structural modeling of mutations due to its accuracy in predicting protein conformation changes (10).
3 Results
3.1 Clinical data
3.1.1 Family 1 (F1)
The proband (F1: I-1, Figure 1A), a 57-year-old woman, was admitted to our hospital with a history of chest pain lasting for more than 1 month. High-resolution computed tomography (HRCT) revealed multiple thin-walled PCs of varying sizes, predominantly distributed in the subpleural regions of the lungs (Figure 1C). Despite the absence of typical cutaneous or renal manifestations commonly associated with BHDS (Figure 1D), genetic testing confirmed the presence of a pathogenic variant in FLCN (Figure 1B). Her 32-year-old eldest son and 30-year-old second son both underwent comprehensive physical examinations at other hospitals, which revealed no findings suggestive of BHDS. The proband’s 28-year-old youngest daughter reported no symptoms but declined to participate in diagnostic assessments. Genetic testing consent from other family members was not obtained.
Figure 1

(A) Pedigree of the patient’s Family 1. Generations are identified by Roman numerals and individuals with Arabic numbers. Squares indicate male family members; circles indicate female members; and arrow indicates the proband. (B) Sanger sequencing of FLCN confirmed a mutation (c.246C > A). (C) Lung CT testing results of proband in Family 1. (D) Kidney CT testing result for proband in Family 1.
3.1.2 Family 2 (F2)
The proband (F2: II-1, Figure 2A) was a 53-year-old woman who was admitted for chest pain. HRCT revealed multiple PCs of varying sizes, predominantly distributed in the subpleural regions (Figures 3A,B). Abdominal ultrasonography showed no cystic lesions in the kidneys or liver. FFs were observed on her face and neck (Figure 2C). CT imaging of her older brother revealed a left renal mass (45 × 35 × 30 mm) suspicious of malignancy, with no evidence of metastasis. Histopathologic analysis confirmed papillary renal cell carcinoma (Figure 2D). Her youngest son had an episode of primary spontaneous pneumothorax at the age of 27 years, whereas the other two children remained asymptomatic. Given the patient’s multiple skin papules, cystic lesions, and family history of pneumothorax and renal tumors, BHDS was suspected. Genetic testing confirmed the presence of a pathogenic variant in the FLCN gene (Figure 2B).
Figure 2
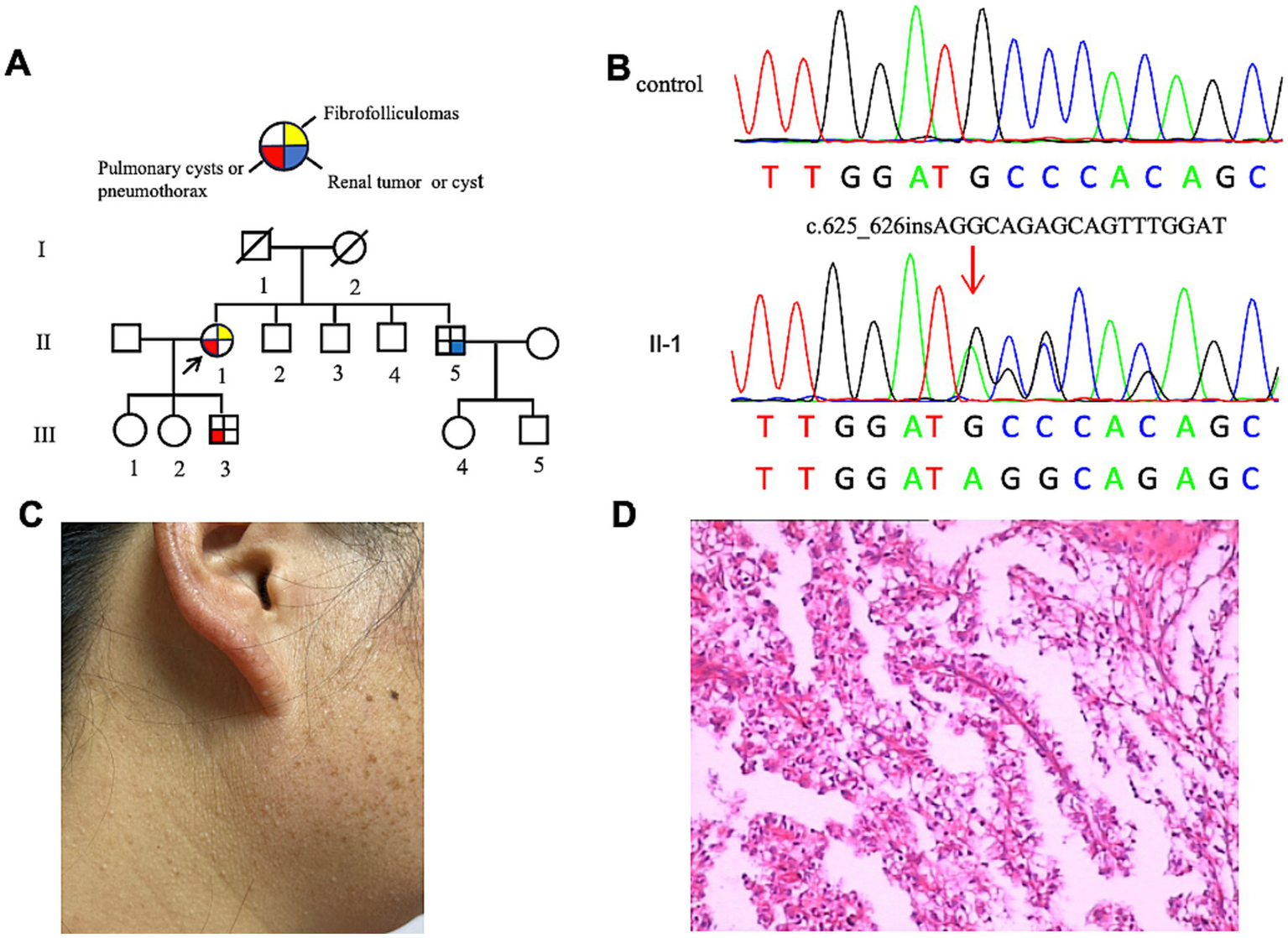
(A) Pedigree of the patient’s Family 2. Generations are identified by Roman numerals and individuals with Arabic numbers. Squares indicate male family members; circles indicate female members; and arrow indicates the proband. (B) Sanger sequencing of FLCN confirmed a mutation (c.625_626insAGGCAGAGCAGTTTGGAT). (C) Fibrofolliculomas in the face and neck of the proband in Family 2. (D) Pathological image of the renal tumor tissue in Family 2.
Figure 3

(A,B) represent the lung CT results of the proband in Family 2 in May 2019 and January 2024, respectively. (C,D) represent the lung CT results of the proband in Family 4 in August 2017 and May 2024, respectively. (E,F) represent the lung CT results of the proband in Family 5 in November 2023 and August 2024, respectively.
3.1.3 Family 3 (F3)
The proband, a 61-year-old woman (F3: II-5, Figure 4A), presented with dyspnea and was diagnosed with a left-sided pneumothorax based on HRCT (Figure 4C). Ultrasound revealed no renal cysts or tumors, and no skin papules were observed on the face, scalp, neck, or upper chest. Her father (F3: I-1) and older son (F3: III-1) both had a history of pulmonary bullae and spontaneous pneumothorax, which occurred at the ages of 45 and 28, respectively (Figure 4A). According to the proband, her brother (F3: II-1) died of kidney cancer at the age of 60, although medical records were unavailable. Genetic testing was performed only on the proband and her son, both of whom were confirmed to carry a pathogenic variant in the FLCN gene (Figure 4B).
Figure 4
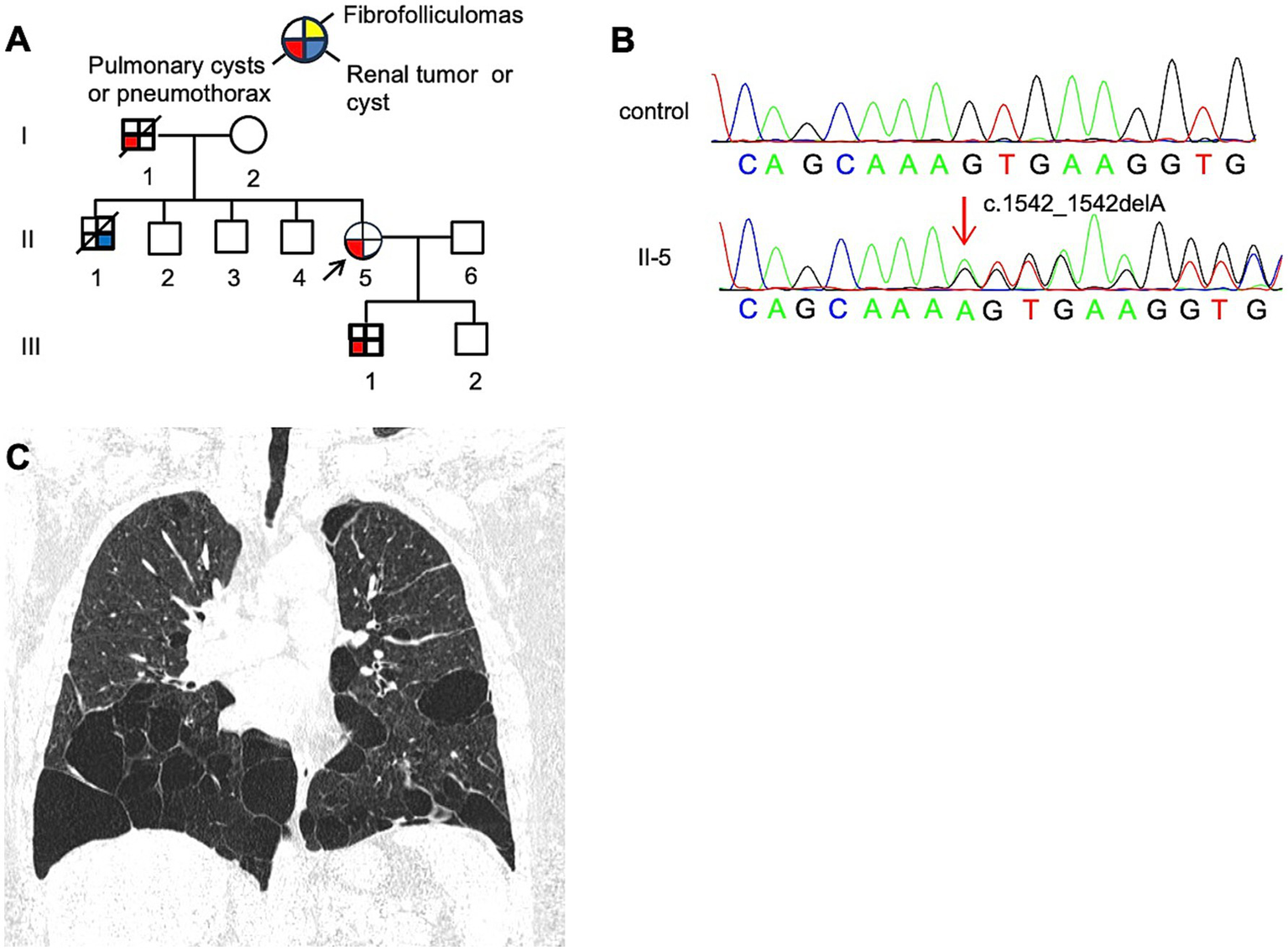
(A) Pedigree of the patient’s Family 3. Generations are identified by Roman numerals and individuals with Arabic numbers. Squares indicate male family members; circles indicate female members; and arrow indicates the proband. (B) Sanger sequencing of FLCN confirmed a mutation (c.1542_1542delA). (C) Lung CT testing result of proband in Family 3.
3.1.4 Family 4 (F4)
The proband (F4: II-5, Figure 5A), a 53-year-old woman, was admitted to the hospital in August 2017 with a 28-year history of recurrent chest tightness and shortness of breath, as well as a recent 20-day episode of right-sided chest pain. HRCT revealed multiple bilateral thin-walled PCs (Figures 3C,D). FFs were observed on the face and neck. During follow-up in May 2024, a left thyroid nodule was detected on HRCT (Figure 5C), while abdominal ultrasonography showed no significant abnormalities. Other family members reported no BHD-related symptoms and declined further clinical evaluation. Genetic testing was performed only on the proband, which confirmed the presence of a pathogenic variant (Figure 5B).
Figure 5
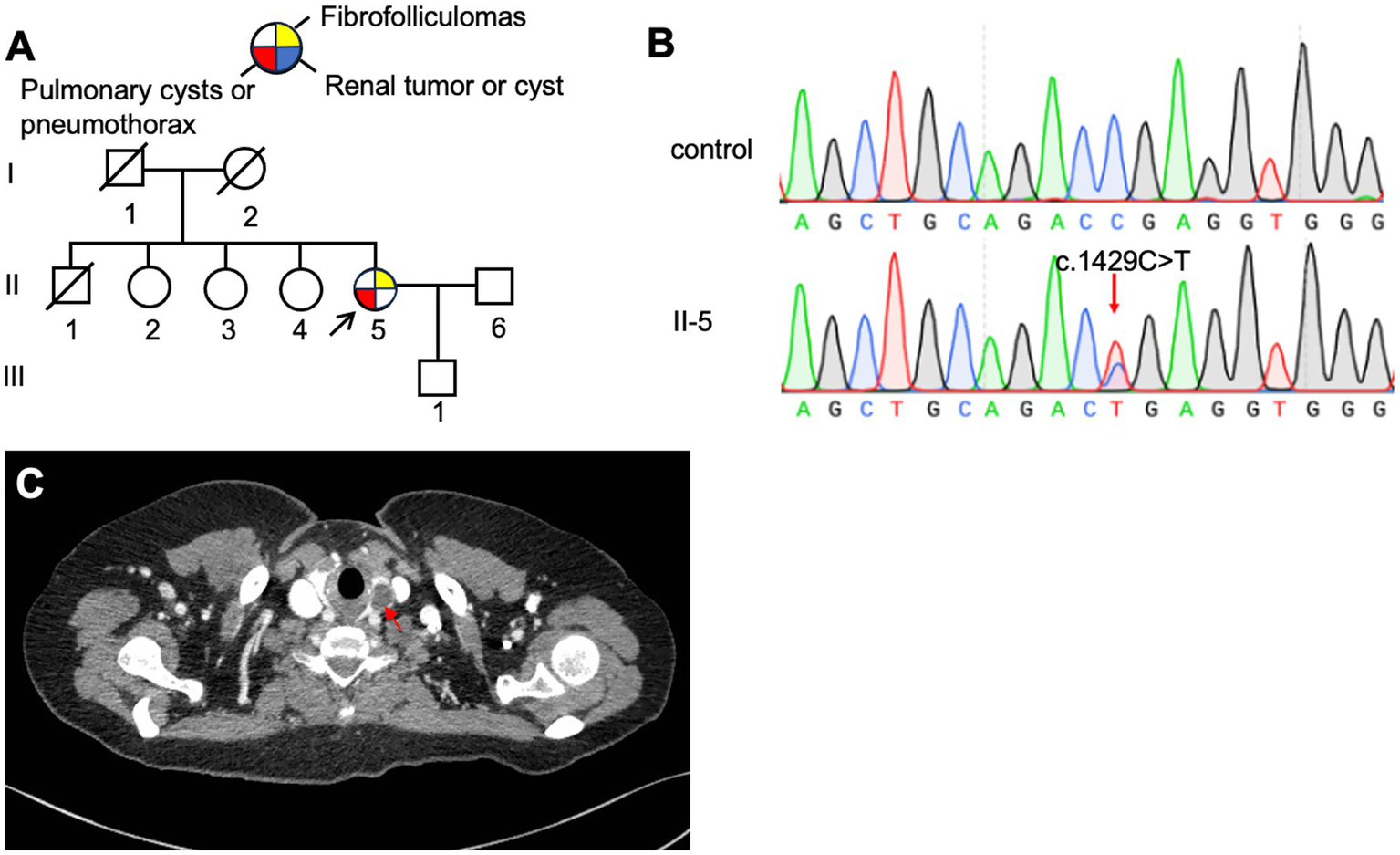
(A) Pedigree of the patient’s Family 4. Generations are identified by Roman numerals and individuals with Arabic numbers. Squares indicate male family members; circles indicate female members; and arrow indicates the proband. (B) Sanger sequencing of FLCN confirmed a mutation (c.1429C > T). (C) Thyroid CT testing result of proband in Family 4, red arrow, thyroid nodule.
3.1.5 Family 5 (F5)
The proband (F5: II-1, Figure 6A), a 65-year-old man, was admitted to our hospital in October 2023 with sudden dyspnea and right-sided chest pain. HRCT revealed a right-sided pneumothorax and multiple PCs (Figure 3E). He was managed conservatively with tube thoracostomy. Abdominal contrast-enhanced CT revealed bilateral renal cysts (Figure 6C). No cutaneous lesions suggestive of BHDS were observed in any family members. Genetic testing was performed on the proband and four relatives (II-2, II-3, III-1, and III-2), among whom only the proband was found to carry a pathogenic FLCN variant (Figure 6B).
Figure 6
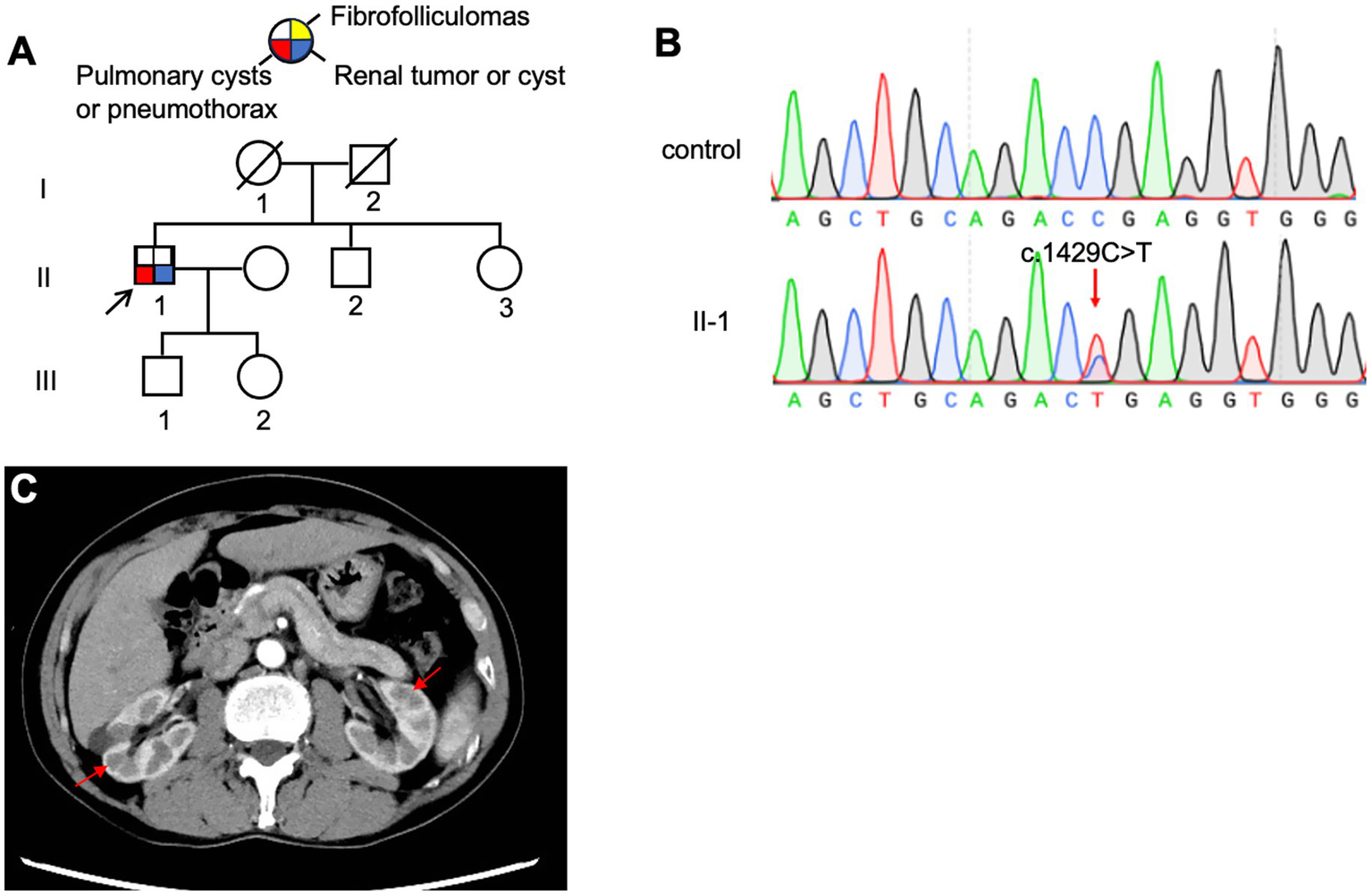
(A) Pedigree of the patient’s Family 5. Generations are identified by Roman numerals and individuals with Arabic numbers. Squares indicate male family members; circles indicate female members; and arrow indicates the proband. (B) Sanger sequencing of FLCN confirmed a mutation (c.1429C > T). (C) Abdominal CT testing result of proband in Family 5.
3.2 Mutation analysis of the FLCN gene
Genetic testing confirmed four distinct mutations of the FLCN gene in five families (Table 1). All mutations detected were unique, with three being novel. The mutation in each family cosegregated with the disease phenotype and was absent in at least 200 unaffected individuals. A novel (c.246C > A/p.C82*) variant was identified in F1, located in exon 4. This nonsense mutation results in a premature stop codon at position 82. In F2, a novel mutation (c.625_626insAGGCAGAGCAGTTTGGAT/p.C215*) in exon 7 of the FLCN gene was detected. This mutation introduces a premature stop codon at codon 215. In F3, a novel nonsense mutation (c.1542_1542delA/p.V515*) in exon 14 of the FLCN gene was confirmed. This nonsense mutation led to a premature stop codon at position 515. A previously reported nonsense mutation (c.1429C > T/p.R477*), located in exon 12 of the FLCN gene, was identified in both F4 and F5. This nonsense mutation encodes an early termination codon.
Table 1
| Family | Patient No. | Age/Sex | Skin | Lung | Kidney | Others | Site of FLCN gene | Nucleotide change | Protein change |
|---|---|---|---|---|---|---|---|---|---|
| F1 | I-1 | 57/F | – | Pulmonary cysts | – | – | exon 4 | c.246C > A | p. C82* |
| F2 | II-1 | 53/F | FFs | Pulmonary cysts | – | – | exon 7 | c.625_626insAGGCAGAGCAGTTTGGAT | p. C215* |
| F2 | II-5 | 43/M | – | – | renal tumors | – | exon 7 | c.625_626insAGGCAGAGCAGTTTGGAT | p. C215* |
| F2 | III-1 | 32/F | – | Pulmonary cysts/PTX | – | – | exon 7 | c.625_626insAGGCAGAGCAGTTTGGAT | p. C215* |
| F3 | II-5 | 61/F | – | Pulmonary cysts/PTX | – | – | exon 14 | c.1542_1542delA | p. V515* |
| F3 | III-1 | 28/M | – | Pulmonary cysts/PTX | – | – | exon 14 | c.1542_1542delA | p. V515* |
| F4 | II-5 | 53/F | FFs | Pulmonary cysts/PTX | – | thyroid nodule | exon 12 | c.1429C > T | p. R477* |
| F5 | II-1 | 65/M | – | Pulmonary cysts/PTX | renal cysts | liver cysts | exon 12 | c.1429C > T | p. R477* |
Clinical data and mutation analysis.
FFs, fibrofolliculomas; PTX, pneumothorax.
Mutation Taster, PolyPhen2, FATHMM, and PROVEAN all predicted the four mutations to be “disease-causing,” “probably damaging,” “damaging,” or “deleterious.” Moreover, the newly identified mutation was absent from the dbSNP and Exome Variant Server database.2
3.3 Disease course and clinical outcomes
Throughout the follow-up period, recurrent pneumothorax was observed in two patients (F4: II-5: 81 months and F5: II-1: 10 months), whereas the three other patients (F1: I-1 at 55 months; F2: II-1 at 56 months; and F3: II-5 at 75 months) remained clinically stable. Follow-up HRCT showed a left thyroid nodule in patient F4: II-5 (Figure 5C), with no significant abnormalities detected on abdominal ultrasonography. Thyroid function tests were within normal limits. HRCT imaging revealed multiple PCs of varying sizes and quantities in three patients (Figure 3), demonstrating a progression in both size and number. No renal tumors were detected in any affected individuals at final follow-up, and the cohort exhibited no mortality throughout the study duration.
3.4 Prediction of FLCN-mutated protein structure
To investigate the spatial configuration of the FLCN mutations, we utilized the SWISS-MODEL online software for structural modeling. Compared to the wild-type FLCN protein, the four mutations exhibited significant alterations in their three-dimensional structures, as shown in Figure 7. The four mutations (p.C82*, p.C215*, p.R477*, and p.V515*) introduce premature stop codons in exons 4, 7, 12, and 14, respectively, of the FLCN gene. These structural alterations suggest that the mutations produce premature stop codons, leading to the synthesis of truncated proteins that may be intrinsically unstable.
Figure 7
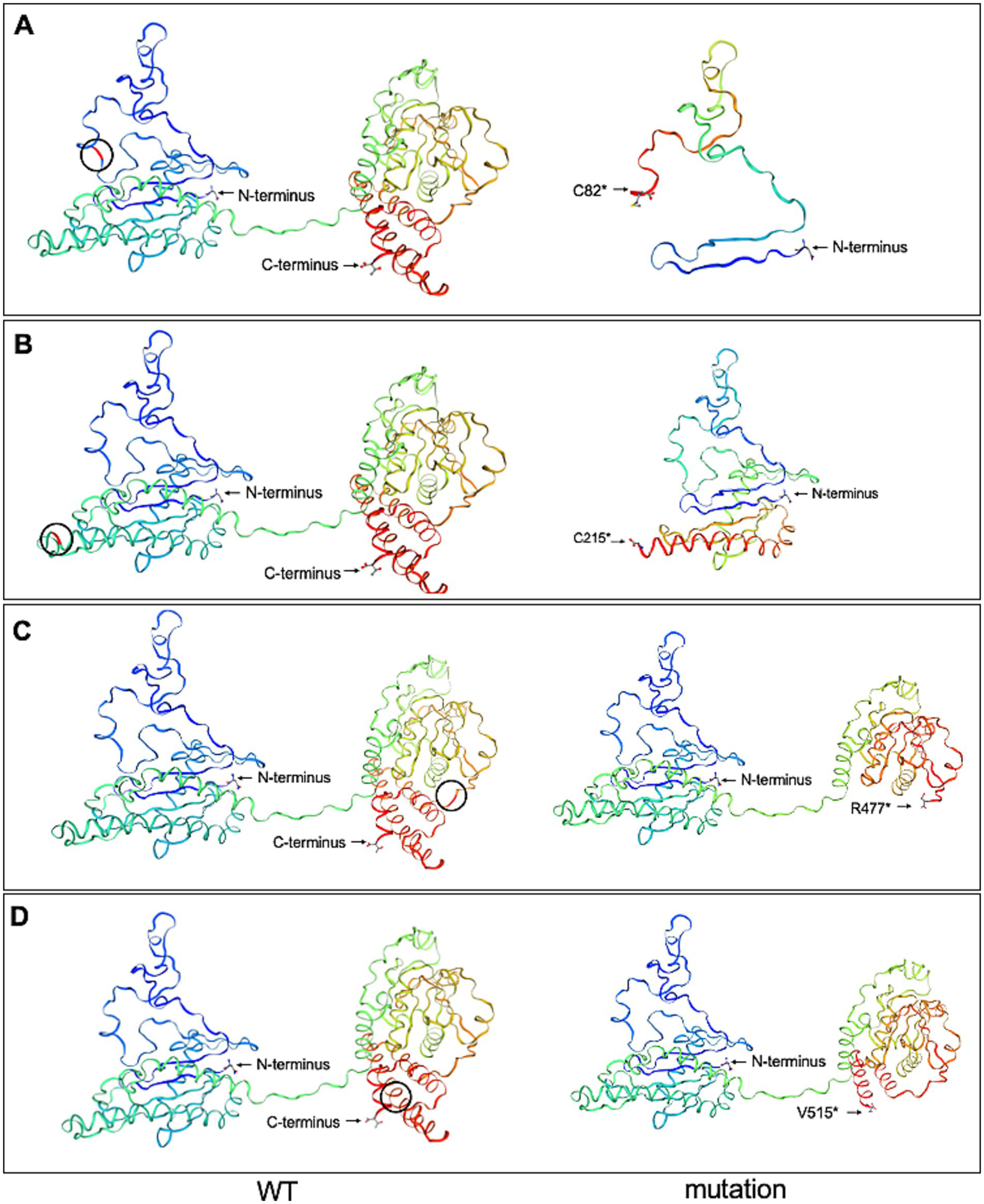
Wild-type FLCN (WT) protein structure and the four mutant FLCN protein structures were predicted using the SWISS-MODEL online software. (A) The mutant FLCN (c.246C > A/p. C82*) results in the truncation of the protein at codon 82. (B) The mutant FLCN (c.625_626insAGGCAGAGCAGTTTGGAT/p. C215*) results in the truncation of the protein at codon 215. (C) The mutant FLCN (c.1429C > T/p. R477*) results in the truncation of the protein at codon 477. (D) The mutant FLCN (c.1542_1542delA/p. V515*) results in the truncation of the protein at codon 515. Blue marks the N-terminus (start) and red marks the C-terminus (end) of the protein. Black circles indicate the positions of the changes between WT and mutated proteins.
4 Discussion
In this study, we identified four pathogenic FLCN mutations across five unrelated Chinese families with BHDS, including three novel variants—p.C82*, p.C215*, and p.V515*—and one previously reported mutation, p.R477*. These variants are located in exons 4, 7, 14, and 12, respectively.
Functionally, the loss of FLCN impairs its role in regulating autophagy and signaling pathways. The loss of FLCN function upregulates mTORC1 activity, leading to impaired autophagy and elevated levels of mTOR pathway downstream effectors, including vascular endothelial growth factor. These molecular changes may contribute to cyst development (11–13). Additionally, Khabibullin et al. demonstrated that FLCN-deficient cells exhibit increased cell–cell adhesion forces, which may predispose the lungs to stretch-induced alveolar injury and cyst formation (14–16). All affected individuals in our study consistently exhibited multiple PCs, most of which were irregularly shaped and predominantly located in the basal and peripheral lung regions. Recent structural studies revealed that the N-terminal Longin domain of FLCN binds to the Longin domain of FNIP1, while the C-terminal differentially expressed in normal and neoplastic cells (DENN) domain interacts with the DENNc domain of FNIP1. Through FNIP1, FLCN associates with RagA, RagC, and the Ragulator complex to regulate mTORC1 activation (17, 18). The four mutations identified in our study introduce PTCs, resulting in truncated folliculin proteins that may lack one or both of the functional domains. Such truncations are predicted to destabilize the protein and disrupt critical interactions with FNIP1 and the Rag GTPases, thereby impairing mTORC1 activation and lysosomal signaling. These molecular disruptions may explain the observed phenotypic variability among patients, including differences in pulmonary, renal, and cutaneous involvement.
Notably, the p.R477* mutation, located near the 3′ end of exon 12, was detected in two families (F4 and F5). Previous reports of p.R477* have described cases with cutaneous FFs (19), lung cysts and pneumothorax without renal involvement (20), isolated renal cell carcinoma (21), and mixed renal/pulmonary phenotypes (22). Intriguingly, bioinformatics analysis suggests that c.1429C > T may not only introduce a stop codon but also disrupt FLCN splicing, which could further contribute to functional loss (23). In our study, two individuals (F5: II-5 and F6: II-1) carrying this variant presented with bilateral lung cysts, with one of them (F5: II-5) later developed a thyroid nodule during follow-up. Although thyroid lesions are not considered hallmark features of BHDS, a cohort study has suggested a higher prevalence of thyroid nodules in BHDS patients (24). However, given the lack of a control group, causality remains unconfirmed, and age-related factors must be considered.
In our study, renal involvement was relatively uncommon. Only one patient (F2: II-5) was diagnosed with renal cell carcinoma, and another (F5: II-1) had renal and liver cysts. This low frequency may be attributed to referral bias, as most participants were recruited from a respiratory clinic, and some relatives declined abdominal imaging. Nonetheless, all probands were advised to undergo annual renal surveillance with ultrasound or CT scans due to the known risk of renal malignancy in BHDS.
Interestingly, only two cases of typical FFs were identified (F2: II-1, F4: II-5), further supporting previous observations that Asian BHDS patients may have a lower prevalence of cutaneous manifestations compared to Caucasian cohorts. For example, while Toro et al. (25) reported FFs in 90% of patients and renal tumors in 34%, studies from China, Japan, and the Republic of Korea have consistently shown higher rates of pulmonary involvement and lower rates of skin and renal lesions (26–29). This geographical and ethnic variability underscores the need for region-specific genotype–phenotype studies.
Longitudinal data from one patient (F2: II-1) showed progressive enlargement and an increased number of pulmonary cysts over 56 months, suggesting a possible correlation between novel truncating variants and disease progression. Mutations in exon 12 (25), such as p.R477*, have previously been associated with more severe pulmonary phenotypes, including increased cyst burden and pneumothorax risk, which is consistent with our findings.
Overall, our study expands the mutational spectrum of FLCN in the Chinese population by identifying three novel truncating mutations in exons 4, 7, and 14. These findings reinforce the pathogenic significance of truncating variants in BHDS and highlight the clinical and genetic heterogeneity of the disease. Molecular characterization of FLCN mutations not only facilitates early diagnosis and risk stratification but also provides valuable information for genetic counseling and family screening. Future large-scale studies integrating genotype, expression profiling, and longitudinal clinical outcomes are warranted to deepen our understanding of BHDS pathogenesis and its diverse phenotypic spectrum.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving humans were approved by institutional review board of the Second Xiangya Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
XK: Project administration, Validation, Supervision, Writing – review & editing, Methodology, Data curation, Conceptualization, Investigation, Writing – original draft, Funding acquisition, Resources, Software, Visualization, Formal analysis. TG: Writing – review & editing, Validation, Data curation, Investigation, Software, Methodology, Resources, Visualization, Conceptualization, Supervision, Project administration, Formal analysis, Funding acquisition. AB: Software, Writing – review & editing, Funding acquisition, Resources, Methodology, Formal analysis, Validation, Investigation, Data curation, Visualization, Supervision, Conceptualization, Project administration. LL: Supervision, Methodology, Validation, Conceptualization, Investigation, Resources, Data curation, Software, Funding acquisition, Writing – review & editing, Formal analysis, Project administration, Visualization. HL: Data curation, Supervision, Investigation, Conceptualization, Software, Methodology, Writing – review & editing, Resources, Visualization, Funding acquisition, Project administration, Validation, Formal Analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by grants from the National Natural Science Foundation of China (81900002 to TG, 82070003 to HL) and the Natural Science Foundation of Hunan Province, China (2020JJ5805 to TG, 2021JJ30943 to HL).
Acknowledgments
We would like to acknowledge all participants for their involvement in this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/fmed.2025.1613154/full#supplementary-material
References
1.
Schmidt LS Warren MB Nickerson ML Weirich G Matrosova V Toro JR et al . Birt-hogg-dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11.2. Am J Hum Genet. (2001) 69:876–82. doi: 10.1086/323744
2.
Khoo SK Bradley M Wong FK Hedblad MA Nordenskjold M Teh BT . Birt-Hogg-Dubé syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene. (2001) 20:5239–42. doi: 10.1038/sj.onc.1204703
3.
Birt AR Hogg GR Dube WJ . Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. (1977) 113:1674–7. doi: 10.1001/archderm.1977.01640120042005
4.
Nickerson ML Warren MB Toro JR Matrosova V Glenn G Turner ML et al . Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. (2002) 2:157–64. doi: 10.1016/s1535-6108(02)00104-6
5.
Zheng CM Hu XX Gao YL Miao JB Li H . Recurrent primary spontaneous pneumothorax in a large Chinese family: a clinical and genetic investigation. Chin Med J. (2019) 132:2402–7. doi: 10.1097/cm9.0000000000000442
6.
Baba M Hong SB Sharma N Warren MB Nickerson ML Iwamatsu A et al . Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci USA. (2006) 103:15552–7. doi: 10.1073/pnas.0603781103
7.
Tsun ZY Bar-Peled L Chantranupong L Zoncu R Wang T Kim C et al . The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. (2013) 52:495–505. doi: 10.1016/j.molcel.2013.09.016
8.
Di Malta C Zampelli A Granieri L Vilardo C De Cegli R Cinque L et al . TFEB and TFE3 drive kidney cystogenesis and tumorigenesis. EMBO Mol Med. (2023) 15:e16877. doi: 10.15252/emmm.202216877
9.
Liu L Yang K Wang X Shi Z Yang Y Yuan Y et al . Detection of Folliculin gene mutations in two Chinese families with Birt-Hogg-Dube syndrome. Biomed Res Int. (2017) 2017:8751384. doi: 10.1155/2017/8751384
10.
Biasini M Bienert S Waterhouse A Arnold K Studer G Schmidt T et al . SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. (2014) 42:W252–8. doi: 10.1093/nar/gku340
11.
Hasumi Y Baba M Ajima R Hasumi H Valera VA Klein ME et al . Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci USA. (2009) 106:18722–7. doi: 10.1073/pnas.0908853106
12.
Furuya M Nakatani Y . Birt-hogg-dube syndrome: clinicopathological features of the lung. J Clin Pathol. (2013) 66:178–86. doi: 10.1136/jclinpath-2012-201200
13.
Nishii T Tanabe M Tanaka R Matsuzawa T Okudela K Nozawa A et al . Unique mutation, accelerated mTOR signaling and angiogenesis in the pulmonary cysts of Birt-Hogg-Dubé syndrome. Pathol Int. (2013) 63:45–55. doi: 10.1111/pin.12028
14.
Medvetz DA Khabibullin D Hariharan V Ongusaha PP Goncharova EA Schlechter T et al . Folliculin, the product of the Birt-Hogg-Dube tumor suppressor gene, interacts with the adherens junction protein p0071 to regulate cell-cell adhesion. PLoS One. (2012) 7:e47842. doi: 10.1371/journal.pone.0047842
15.
Khabibullin D Medvetz DA Pinilla M Hariharan V Li C Hergrueter A et al . Folliculin regulates cell-cell adhesion, AMPK, and mTORC1 in a cell-type-specific manner in lung-derived cells. Physiol Rep. (2014) 2:e12107. doi: 10.14814/phy2.12107
16.
Kennedy JC Khabibullin D Henske EP . Mechanisms of pulmonary cyst pathogenesis in Birt-Hogg-Dube syndrome: the stretch hypothesis. Semin Cell Dev Biol. (2016) 52:47–52. doi: 10.1016/j.semcdb.2016.02.014
17.
Shen K Rogala KB Chou HT Huang RK Yu Z Sabatini DM . Cryo-EM structure of the human FLCN-FNIP2-rag-Ragulator complex. Cell. (2019) 179:1319–29.e8. doi: 10.1016/j.cell.2019.10.036
18.
Lawrence RE Fromm SA Fu Y Yokom AL Kim DJ Thelen AM et al . Structural mechanism of a rag GTPase activation checkpoint by the lysosomal folliculin complex. Science. (2019) 366:971–7. doi: 10.1126/science.aax0364
19.
Fuertes I Mascaró-Galy JM Ferrando J . Birt-Hogg-Dubé syndrome in a patient with cutaneous symptoms and a c.1429 C > T;p.R477X mutation in exon 12 of the folliculin gene. Actas Dermosifiliogr. (2009) 100:227–30. doi: 10.1016/S1578-2190(09)70049-6
20.
Lee JH Jeon MJ Song JS Chae EJ Choi JH Kim GH et al . Birt-Hogg-Dubé syndrome in Korean: clinicoradiologic features and long term follow-up. Korean J Intern Med. (2019) 34:830–40. doi: 10.3904/kjim.2018.119
21.
Mikhaylenko DS Matveev VB Filippova MG Anoshkin KI Kozlov NA Khachaturyan AV et al . Comprehensive molecular genetic diagnostics of Birt-Hogg-Dube syndrome in a Russian patient with renal Cancer and lung cysts: a case report. Case Rep Oncol. (2021) 14:963–71. doi: 10.1159/000516763
22.
Li J Liu F Liu X Hu Y Liu Z Shen Y et al . Heterozygous germline FLCN mutation in Birt-Hogg-Dubé syndrome with bilateral renal hybrid oncocytic/chromophobe tumor and unilateral renal chromophobe cell carcinoma: a case report. J Cancer Res Clin Oncol. (2023) 149:2319–25. doi: 10.1007/s00432-022-04417-z
23.
Soukarieh O Gaildrat P Hamieh M Drouet A Baert-Desurmont S Frébourg T et al . Exonic splicing mutations are more prevalent than currently estimated and can be predicted by using in silico tools. PLoS Genet. (2016) 12:e1005756. doi: 10.1371/journal.pgen.1005756
24.
Kluger N Giraud S Coupier I Avril MF Dereure O Guillot B et al . Birt-Hogg-Dubé syndrome: clinical and genetic studies of 10 French families. Br J Dermatol. (2010) 162:527–37. doi: 10.1111/j.1365-2133.2009.09517.x
25.
Toro JR Pautler SE Stewart L Glenn GM Weinreich M Toure O et al . Lung cysts, spontaneous pneumothorax, and genetic associations in 89 families with Birt-Hogg-Dubé syndrome. Am J Respir Crit Care Med. (2007) 175:1044–53. doi: 10.1164/rccm.200610-1483OC
26.
Furuya M Yao M Tanaka R Nagashima Y Kuroda N Hasumi H et al . Genetic, epidemiologic and clinicopathologic studies of Japanese Asian patients with Birt-Hogg-Dubé syndrome. Clin Genet. (2016) 90:403–12. doi: 10.1111/cge.12807
27.
Liu Y Xu Z Feng R Zhan Y Wang J Li G et al . Clinical and genetic characteristics of chinese patients with Birt-Hogg-Dubé syndrome. Orphanet J Rare Dis. (2017) 12:104. doi: 10.1186/s13023-017-0656-7
28.
Park HJ Kim YJ Kim MJ Kim HC . Epidemiology and clinical features of Birt-Hogg-Dubé syndrome: a nationwide population-based study in South Korea. PLoS One. (2022) 17:e0269358. doi: 10.1371/journal.pone.0269358
29.
Zhou W Liu K Xu KF Liu Y Tian X . Clinical and genetic comparison of Birt-Hogg-Dubé syndrome (Hornstein-Knickenberg syndrome) in Chinese: a systemic review of reported cases. Int J Gen Med. (2022) 15:5111–21. doi: 10.2147/ijgm.S359660
Summary
Keywords
Birt–Hogg–Dubé syndrome, pulmonary cysts, pneumothorax, fibrofolliculomas, renal cell carcinoma
Citation
Kang X, Guo T, Basit A, Liu L and Luo H (2025) Clinical and genetic investigations of five Chinese families with Birt–Hogg–Dubé syndrome: a long-term follow-up study. Front. Med. 12:1613154. doi: 10.3389/fmed.2025.1613154
Received
16 April 2025
Accepted
21 July 2025
Published
08 September 2025
Volume
12 - 2025
Edited by
Stephen C. Land, University of Dundee, United Kingdom
Reviewed by
Laura Schmidt, National Cancer Institute at Frederick (NIH), United States
Abhishek Singla, University of Cincinnati, United States
Updates
Copyright
© 2025 Kang, Guo, Basit, Liu and Luo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lv Liu, docliulv@csu.edu.cnHong Luo, luohonghuxi@csu.edu.cn
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.