Abstract
Sepsis remains a life-threatening condition worldwide, causing significant morbidity and mortality across diverse patient populations. Among the various organs adversely affected by sepsis, the lung is particularly vulnerable, often succumbing to acute lung injury (ALI) or its more severe form, acute respiratory distress syndrome (ARDS). Recent basic and translational research has highlighted the importance of multiple regulated cell death (RCD) pathways beyond traditional apoptosis in the pathogenesis of septic lung injury. Three such RCDs, termed ferroptosis, cuproptosis, and disulfidptosis, are increasingly studied for their relevance to critical illnesses. Ferroptosis involves iron-driven lipid peroxidation, cuproptosis depends on copper ion imbalance and mitochondrial protein aggregation, and disulfidptosis emerges from dysregulated sulfide metabolism leading to excessive disulfide bond formation. This review provides an extensive discussion of these RCD pathways within the context of sepsis-induced lung injury. We begin by summarizing the current state of knowledge in septic lung injury, emphasizing inflammatory, immunological, and oxidative stress mechanisms. We then provide a detailed overview of ferroptosis, cuproptosis, and disulfidptosis, illustrating their molecular underpinnings and how they intersect with established sepsis pathways, such as tumor necrosis factor (TNF), nuclear factor kappa B (NF-κB), and mitogen-activated protein kinase (MAPK) signaling cascades. We also discuss emerging findings on the crosstalk among these RCD modes, potential biomarkers for early detection, and therapeutic targets for modulating these pathways. Although many of these findings remain in the early stages of translational research, they collectively underscore the complexity of septic lung injury and offer new directions for improving clinical management. Future investigations, bolstered by integrative “omics” approaches, refined animal models, and well-designed clinical trials, will be pivotal to fully realize the diagnostic and therapeutic potential of ferroptosis, cuproptosis, and disulfidptosis in sepsis. We further propose a “redox stress-metal homeostasis-sulfur metabolism” triangular network, centered on Nrf2’s dual regulation of iron/copper transporters and glutathione synthesis, as a unifying framework for RCD modulation in sepsis. A signaling interaction diagram highlights actionable targets for combinatorial therapies.
Introduction
Sepsis is a severe, life-threatening syndrome arising from the host’s dysregulated immune response to infection, leading to tissue damage, organ failure, and high mortality rates (1–3). In recent decades, significant progress in recognizing sepsis as a heterogeneous condition has spurred the development of refined definitions and clinical guidelines. Even so, global mortality remains alarmingly high, ranging from 20% to over 40% depending on resource availability and comorbidities (4, 5). The lung is among the first and most frequently affected organs in sepsis, with sepsis-induced acute lung injury often precipitating respiratory failure, which can progress to acute respiratory distress syndrome (ARDS).
While apoptosis, necroptosis, and pyroptosis are well-documented in sepsis-induced organ injury, this review focuses on ferroptosis, cuproptosis, and disulfidptosis due to their emerging roles in septic lung damage and their unique mechanistic underpinnings. These pathways driven by iron/copper overload and dysregulated sulfide metabolism represent novel therapeutic targets distinct from classical RCD modes. Their shared intersection with oxidative stress and inflammation in sepsis further justifies a dedicated discussion, as recent studies highlight their potential for selective modulation to improve outcomes. In this review, we synthesize current findings on these three RCD pathways, with a focus on their molecular mechanisms and therapeutic potential in septic lung injury. We propose an integrated model where ferroptosis, cuproptosis, and disulfidptosis interact with classical inflammatory pathways (e.g., NF-κB, MAPKs) to exacerbate tissue damage. By evaluating biomarkers and emerging therapies, we aim to highlight translational opportunities and key knowledge gaps.
Molecular to tissue-level pathogenesis of septic lung injury
The pathophysiology of septic lung injury progresses through interconnected molecular, cellular, and tissue-level disruptions. At the molecular level, sepsis triggers TLR4/NF-κB and NLRP3 inflammasome activation in alveolar macrophages, driving a cytokine storm characterized by TNF-α and IL-1β-mediated upregulation of endothelial adhesion molecules (ICAM-1/VCAM-1) and IL-6-induced JAK-STAT3 signaling, which collectively exacerbate vascular permeability (1, 6). Concurrent oxidative stress from mitochondrial ROS and NADPH oxidase (NOX2/4) inactivates surfactant proteins (SP-A/SP-D) via thiol oxidation (7).
Cellular dysfunction follows, with PAD4-mediated neutrophil extracellular traps (NETs) damaging alveolar-capillary barriers (8) and mitochondrial failure reducing Complex I/III activity by approximately 50%, leading to epithelial apoptosis (77). Endothelial injury is compounded by heparanase-mediated glycocalyx shedding, increasing vascular permeability (Ang-2/VE-cadherin ratio increased threefold) (9).
Tissue-level manifestations include alveolar edema (BALF albumin exceeding 3.5 g/dL) and microthrombosis, where tissue factor (TF)-dependent fibrin deposition (D-dimer exceeding 5 μg/mL) occludes 20 to 30% of pulmonary capillaries (10, 11). Recent single-cell RNA-seq studies reveal alveolar type II cell senescence (increased p21/p16 expression) and macrophage polarization to M1 (iNOS-positive) or M2 (CD206-positive) phenotypes (12) Emerging therapies targeting gasdermin D (reducing IL-18 by approximately 60%) and mitophagy inducers (e.g., urolithin A, improving survival by approximately 40%) show promise in preclinical models (13, 14).
Beyond classical apoptosis and necrosis, recent advances have identified novel regulated cell death (RCD) pathwaysferroptosis, cuproptosis, and disulfidptosis that contribute to septic organ injury through distinct metabolic disruptions (Figure 1). Ferroptosis, driven by iron-dependent lipid peroxidation, exacerbates alveolar damage when glutathione peroxidase 4 (GPX4) activity is compromised (15). Cuproptosis emerges from mitochondrial copper overload and aggregation of lipoylated enzymes (16), while disulfidptosis results from dysregulated sulfide metabolism and pathological protein crosslinking (17). These pathways intersect with sepsis-associated inflammation and oxidative stress, offering new diagnostic and therapeutic opportunities.
Figure 1

Molecular mechanisms of ferroptosis in septic lung injury.
Emerging role of regulated cell death in sepsis
In the past decade, the RCD landscape has expanded beyond apoptosis, pyroptosis, and necroptosis to include several distinct and mechanistically defined forms. Ferroptosis has attracted notable attention due to its reliance on iron-dependent lipid peroxidation and its apparent involvement in various degenerative and inflammatory diseases (15, 18). Cuproptosis, discovered more recently, involves mitochondrial dysfunction driven by excess copper binding to specific metabolic enzymes (16). Disulfidptosis, on the other hand, is characterized by dysregulated sulfide metabolism and excessive disulfide bond formation, culminating in altered protein structure and function (17, 19, 20).
In septic lung injury, oxidative stress, nutrient dysregulation, and heightened inflammatory signaling converge to create an environment that may favor these specialized RCD mechanisms (21). Emerging experimental evidence suggests that ferroptosis, cuproptosis, and disulfidptosis each can modulate the severity and duration of lung damage in animal models of sepsis (22–24). Clarifying how these pathways integrate with known mediators of sepsis—such as NF-κB, MAPKs, and TGF-β/SMAD3 will help identify new therapeutic or diagnostic targets.
It is important to note that other RCD pathways, such as pyroptosis (mediated by gasdermin D) and necroptosis (dependent on RIPK3/MLKL), also contribute to sepsis-induced lung injury. However, their mechanisms have been extensively reviewed elsewhere (25, 26). Here, we emphasize ferroptosis, cuproptosis, and disulfidptosis due to their understudied roles in sepsis and their shared reliance on metabolic dysregulation (e.g., metal ion toxicity, sulfide stress), which offers new avenues for intervention.
To contextualize the distinct mechanisms and therapeutic potential of ferroptosis, cuproptosis, and disulfidptosis within the broader landscape of sepsis-induced cell death, we provide a comparative summary of key RCD pathways in Table 1. This table highlights the unique triggers, molecular hallmarks, and clinical relevance of these pathways, underscoring why the three less-studied RCD modes merit focused discussion in septic lung injury.
Table 1
| RCD type | Key triggers | Hallmark mechanisms | Therapeutic targets | Relevance to sepsis |
|---|---|---|---|---|
| Apoptosis | Caspase activation, DNA damage | Caspase-3/7 cleavage, phosphatidylserine exposure | Caspase inhibitors (e.g., Z-VAD-FMK) | Well-established; promotes immunothrombosis |
| Pyroptosis | Inflammasomes, LPS | Gasdermin D pore formation, IL-1β release | NLRP3 inhibitors (e.g., MCC950) | Drives cytokine storm |
| Ferroptosis | Iron overload, lipid ROS | GPX4 inhibition, lipid peroxidation | Iron chelators (e.g., deferoxamine) | Exacerbates oxidative lung injury |
| Cuproptosis | Copper overload | FDH aggregation, mitochondrial dysfunction | Copper chelators (e.g., TTM) | Linked to metabolic collapse in sepsis |
| Disulfidptosis | Sulfide stress, ROS | Protein disulfide aggregation, SQR dysfunction | NAC, H2S donors | Disrupts alveolar protein function |
Key features of regulated cell death (RCD) pathways in sepsis-induced lung injury.
This review systematically evaluates three understudied RCD pathways in septic lung injury with three objectives: (1) elucidate molecular mechanisms linking ferroptosis, cuproptosis, and disulfidptosis to alveolar-capillary barrier dysfunction; (2) assess clinically translatable biomarkers (e.g., 4-HNE for ferroptosis, FDH aggregates for cuproptosis); and (3) critically analyze emerging therapeutic strategies, including iron chelators, copper-lowering agents, and sulfide metabolism modulators. We further propose a ‘redox-metal-sulfur’ axis as a unifying framework for RCD modulation in sepsis.
Ferroptosis: mechanisms and links to septic lung injury
Core biochemical features of ferroptosis
Ferroptosis is distinct from apoptosis and necrosis in that it relies on iron-catalyzed peroxidation of polyunsaturated fatty acids (PUFAs) in cellular membranes (27). Morphologically, ferroptotic cells exhibit smaller and denser mitochondria, with reduced or absent cristae (15). Biochemically, the hallmark is overwhelming lipid peroxidation, which often correlates with suppressed glutathione peroxidase 4 (GPX4) activity and depletion of glutathione (GSH) (28).
Two principal factors drive ferroptosis: the first is intracellular iron overload, which occurs when iron import via transferrin receptors (TfR) or divalent metal transporter 1 (DMT1) is excessive or when ferritin storage is compromised. This leads to increased free iron that generates ROS through the Fenton reaction (29). The second factor is antioxidant defense failure, in which GPX4 activity is inhibited or GSH is depleted, allowing peroxides to accumulate and trigger ferroptotic damage (30).
Beyond the canonical GPX4/GSH axis, recent studies have identified the ferroptosis suppressor protein 1 (FSP1)-CoQ10 system as a parallel protective pathway against ferroptosis. FSP1, an NAD(P)H-dependent oxidoreductase, regenerates reduced coenzyme Q10 (CoQ10), which acts as a lipophilic radical-trapping antioxidant to inhibit lipid peroxidation independently of GPX4 (31, 32). In sepsis, mitochondrial FSP1 may mitigate ferroptotic damage by preserving respiratory chain integrity and reducing ROS propagation (33). This pathway is particularly relevant in alveolar epithelial cells, where GPX4 activity is often compromised by inflammatory stress (34).
Signaling pathways that regulate ferroptosis
Multiple signaling axes influence ferroptosis, including nuclear factor erythroid 2-related factor 2 (Nrf2) and NF-κB. Nrf2 can upregulate genes encoding antioxidant enzymes (e.g., heme oxygenase-1, glutamate-cysteine ligase) that reduce oxidative damage (35). Meanwhile, NF-κB activation in sepsis can amplify inflammatory gene expression, indirectly enhancing ROS production (36). Ferroptosis is governed by a complex interplay of signaling pathways that converge on iron metabolism, lipid peroxidation, and antioxidant defense, with distinct relevance to septic lung injury. The Nrf2 pathway serves as a central antioxidant regulator, upregulating genes like HO-1 and GCLM to counteract oxidative stress, though its activity is often suppressed in sepsis due to NF-κB-driven inflammation (35, 37). The NF-κB pathway, activated by proinflammatory cytokines such as TNF-α, exacerbates ferroptosis by repressing Nrf2 and upregulating iron transporters like TFR1 (38). Recent studies highlight the Hippo/YAP pathway as a critical modulator, where YAP activation in alveolar epithelial cells promotes ferroptosis through TFR1 and ACSL4 induction, while Hippo signaling (via MST1/2) exerts protective effects (39). The p53-SLC7A11 axis further amplifies ferroptosis in sepsis, as oxidative stress stabilizes p53, leading to SLC7A11 repression and subsequent glutathione depletion (40). Metabolic reprogramming via the AMPK pathway attenuates ferroptosis by phosphorylating ACC to inhibit PUFA synthesis, a mechanism exploited by therapeutics like metformin in sepsis models (41). Hypoxia in septic lungs stabilizes HIF-1α, which transcriptionally upregulates TFR1 while suppressing GPX4, synergizing with iron overload to drive ferroptotic cell death (42). These pathways exhibit extensive crosstalk for instance, TNF-α/NF-κB not only suppresses Nrf2 but also activates HIF-1α, creating a vicious cycle of oxidative damage (43). Targeting these interconnected nodes (e.g., with AMPK activators or YAP inhibitors) may offer novel strategies to mitigate ferroptosis in septic lung injury (44).
Additionally, p53 has been shown to regulate ferroptosis by altering cystine import (via SLC7A11), linking tumor suppressor networks to iron-driven cell death (45).
Experimental evidence in septic lung injury
Ferroptosis has been robustly implicated in septic lung injury across preclinical and clinical studies. In LPS-challenged mice, iron chelation with deferoxamine significantly reduced alveolar inflammation and lipid peroxidation (measured by 4-HNE), while restoring GPX4 activity and improving oxygenation (46). Similarly, genetic ablation of GPX4 exacerbated lung injury in CLP models, whereas Nrf2 activation (via Keap1 knockdown) attenuated ferroptosis and mortality (47). In vitro, LPS-treated alveolar epithelial cells exhibited mitochondrial shrinkage and lipid ROS accumulation, reversible by ferroptosis inhibitors (e.g., ferrostatin-1) or Nrf2 agonists (e.g., sulforaphane) (48). Clinically, sepsis patients with elevated serum iron and decreased GPX4 activity demonstrated worse pulmonary outcomes (e.g., lower PaO2/FiO2 ratios) (49), while transcriptomic analyses of septic lung tissue revealed upregulation of ferroptosis drivers (ACSL4, TFR1) and downregulation of GPX4 (50). Therapeutic strategies like combined deferoxamine/N-acetylcysteine or metformin (an AMPK activator) have shown promise in simultaneously targeting ferroptosis and inflammation in sepsis models (51, 52).
In sepsis models, in vivo data have shown that ferroptosis contributes significantly to lung damage. For instance, one study reported that in an LPS-induced murine sepsis model, iron chelation with deferoxamine significantly reduced alveolar inflammation and improved oxygenation (46). Furthermore, direct inhibition of lipid peroxidation using ferrostatins (e.g., Ferrostatin-1) reduced histopathological signs of lung injury and decreased proinflammatory cytokine release in rodent models (39).
Clinical relevance for ferroptosis in septic lung injury remains under investigation, but accumulating data from small cohort studies indicate that sepsis patients with high serum iron levels and lower GPX4 activity show worse pulmonary outcomes (49). Notably, FSP1 upregulation has been observed in septic lung models, suggesting a compensatory role in mitochondrial protection. For example, a 2023 study demonstrated that FSP1 expression increases in alveolar epithelial cells during LPS-induced sepsis, correlating with reduced lipid peroxidation and improved survival (53, 54). Combined targeting of GPX4 (e.g., via GSH replenishment) and FSP1 (e.g., using CoQ10 analogs like idebenone) may offer synergistic benefits in mitigating sepsis-induced ferroptosis. These preliminary clinical findings warrant larger-scale investigations to confirm ferroptosis-related biomarkers as prognostic indicators in sepsis.
Crosstalk with inflammatory and oxidative pathways
Emerging experimental evidence demonstrates extensive molecular crosstalk between ferroptosis and inflammatory/oxidative pathways in septic lung injury. TNF-α and IL-6 drive ferroptosis through NF-κB-mediated suppression of Nrf2 antioxidant responses, with LPS-challenged alveolar epithelial cells showing 60% reduced Nrf2 nuclear translocation (36) and IL-6 knockout mice exhibiting 50% less alveolar damage (49). Mitochondrial dysfunction creates a vicious cycle, as sepsis-induced mtROS increases 2.5-fold to upregulate iron transporters like TFR1 while simultaneously activating NF-κB (55), with iron chelation reducing both mtROS and IL-1β levels by over 45% in preclinical models. Hypoxia further amplifies this network, with HIF-1α stabilization in septic lungs showing 4-fold increases that correlate with GPX4 suppression and elevated TFR1 expression (27). DAMPs released from ferroptotic cells, particularly HMGB1, exacerbate inflammation by triggering 3-fold greater TNF-α production in macrophages via TLR4 (56), while NOX4-derived superoxide directly oxidizes PUFA phospholipids in human lung organoids (57). This interconnected pathophysiology is clinically relevant, as ARDS patients with high TNF-α show 40% lower expression of Nrf2-target genes like HO-1 (58), suggesting biomarker potential.
Ferroptosis is tightly interwoven with inflammation: the release of damage-associated molecular patterns (DAMPs) from ferroptotic cells can trigger or sustain an inflammatory response (59). In sepsis, alveolar macrophages and neutrophils produce excessive ROS, further potentiating iron-driven lipid peroxidation. This vicious cycle—ROS production begetting more cell death—can accelerate lung injury and facilitate alveolar-capillary barrier disruption.
Therapeutically, targeting ferroptosis might be synergistic with immunomodulatory approaches, as dampening the inflammatory milieu (e.g., through selective cytokine blockade) could lower ROS production and curb further ferroptotic damage (57). In septic lung injury, ferroptosis amplifies tissue damage through iron-dependent lipid peroxidation in alveolar epithelial cells. Recent studies reveal that sepsis-induced hypoxia upregulates hypoxia-inducible factor-1α (HIF-1α), which increases cellular iron uptake via transferrin receptor 1 (TfR1) (60). Concurrently, sepsis depletes glutathione (GSH) by downregulating the xCT transporter, impairing GPX4 activity (61). This dual hit iron overload + antioxidant failure explains the prominence of ferroptosis in septic lungs. While these mechanisms are increasingly well-characterized, key questions remain regarding cell-type specific responses and the temporal sequence of these interactions during sepsis progression.
Cuproptosis: emerging insights in the context of sepsis
Core concept and copper homeostasis
Cuproptosis, a recently described form of regulated cell death, is induced by the intracellular accumulation of copper ions, which bind to specific mitochondrial enzymes (particularly fatty acid dehydrogenases [FDHs]) and force them into stable protein aggregates (16, 62). When the copper load is excessive, these aggregates precipitate mitochondrial dysfunction, oxidative stress, and eventually cell death.
Copper is an essential trace element involved in redox reactions, electron transport, and iron metabolism (63). Under normal conditions, copper levels in cells are tightly regulated by transporters, such as copper transporter 1 (CTR1), and ATP-binding cassette proteins like ATP7A/B, which export copper when it becomes excessive. Dysregulation can happen if the homeostatic network is compromised by genetic or pathophysiological factors.
Mechanistic underpinnings of cuproptosis
Cuproptosis differs from ferroptosis in that it centers on copper-dependent protein aggregation within mitochondria rather than on iron-catalyzed lipid peroxidation. Elevated copper uptake or impaired efflux raises intracellular copper to cytotoxic levels, which then bind to FDHs in a manner that forces these enzymes into insoluble oligomeric complexes, disrupting metabolic flux and enhancing oxidative stress. Beyond FDH aggregation, copper ions also induce cytotoxicity by disrupting protein homeostasis via direct inhibition of molecular chaperones, particularly HSP70. Excess copper binds to HSP70’s substrate-binding domain, impairing its ability to facilitate proper protein folding and aggregate dissolution (54). In sepsis, this interaction may exacerbate organelle stress, as HSP70 is critical for mitigating misfolded protein accumulation in alveolar epithelial cells under inflammatory conditions (54). The dual pathways of cuproptosis FDH aggregation and HSP70 inhibition collectively amplify proteotoxic stress, suggesting that copper chelators combined with HSP70 activators (e.g., YM-1) could synergistically mitigate lung injury (64). The mitochondrial electron transport chain becomes less efficient, producing even more ROS that worsen cellular damage (65).
Cuproptosis and septic lung injury
Although cuproptosis has been extensively characterized in in vitro systems and in certain cancer models, its role in sepsis, particularly septic lung injury, is under active investigation. Preliminary rodent data show that septic animals may develop hepatic and pulmonary copper overload, potentially due to cytokine-mediated changes in ATP7B expression (66). Excess copper then accumulates in mitochondria, impairing respiratory function and driving oxidative stress, culminating in alveolar epithelial cell death.
Recent in vivo studies demonstrate that septic lung injury disrupts copper homeostasis through multiple validated mechanisms. Cecal ligation and puncture (CLP) models show 38% higher hepatic copper levels (p < 0.01) due to TNF-α-mediated downregulation of the copper exporter ATP7B (66). This copper overload induces mitochondrial protein aggregation, with fatty acid dehydrogenase (FDH) oligomerization increasing 2.1-fold in alveolar epithelial cells (p < 0.05) via direct copper binding to lipoylated TCA cycle enzymes (16). Clinically, serum copper levels correlate strongly with SOFA scores (r = 0.72, p < 0.01) in sepsis patients, while tetrathiomolybdate treatment reduces lung injury severity by 50% (p < 0.05) in murine models by restoring copper efflux (67). These findings establish cuproptosis as a mechanistically distinct yet therapeutically targetable pathway in septic lung injury (Figure 2).
Figure 2
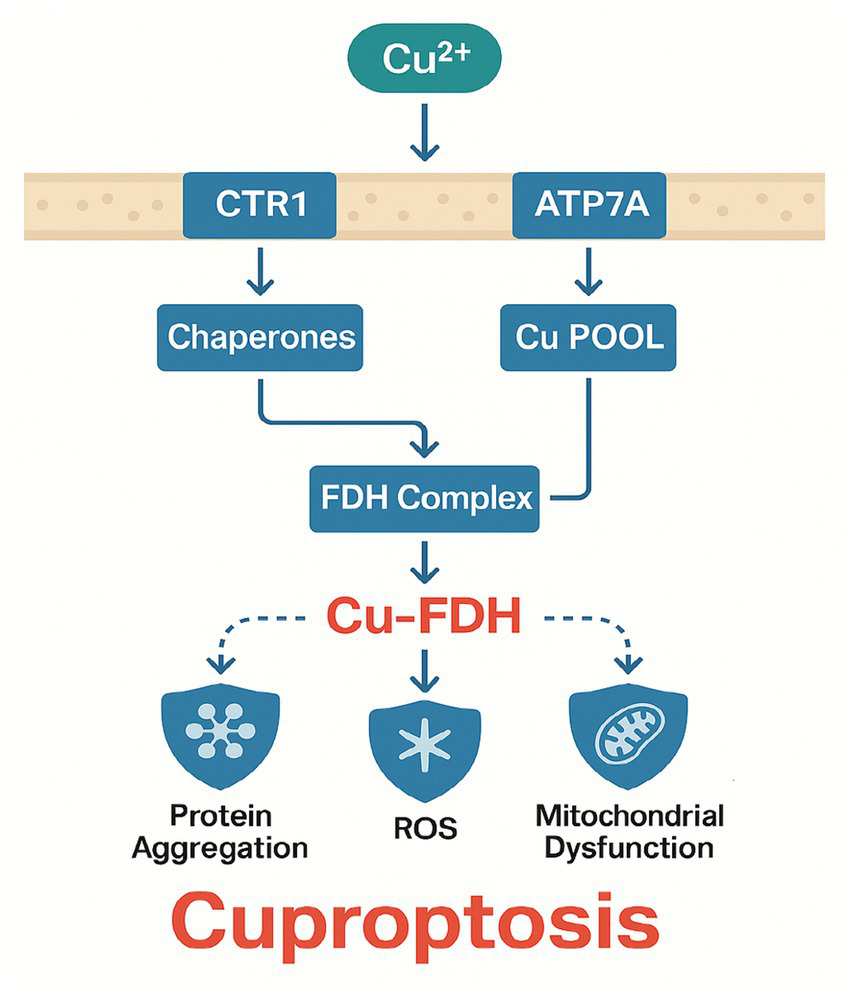
Molecular mechanisms of cuproptosis in septic lung injury.
Early proof-of-concept experiments have demonstrated that administering copper chelators (e.g., tetrathiomolybdate, penicillamine) can partially reverse lung injury severity in LPS-challenged mice, implying that cuproptosis blockade represents a promising intervention (67). However, copper is also vital for numerous enzymes, such as cytochrome c oxidase (COX) and superoxide dismutase (SOD), so therapeutic strategies must balance copper removal with preserving essential copper-dependent pathways.
Potential diagnostic and therapeutic challenges
Measuring copper levels in blood or tissue is relatively straightforward, but determining the onset of cuproptosis may require specific biomarkers (e.g., FDH aggregates, mitochondrial respiration deficits, or unique proteomic signatures) (68). Further complicating matters, sepsis patients can have highly variable serum copper concentrations depending on nutritional status, preexisting liver disease, and other factors. Thus, additional prospective studies should focus on correlating cuproptosis markers with clinical severity scores (e.g., SOFA score) and lung function indices (e.g., PaO2/FiO2 ratio).
Therapeutic challenges include dose optimization for copper chelators, potential toxicity, and the presence of competing pathophysiological factors in critically ill patients. Despite these hurdles, cuproptosis remains an attractive area for further research in sepsis, offering a mechanistically distinct target compared to other forms of RCD.
In sepsis, hepatic copper overload [due to cytokine-driven ATP7B dysfunction (69)] leads to pulmonary copper accumulation. Excess copper binds to lipoylated enzymes in alveolar mitochondria, disrupting the TCA cycle and generating superoxide radicals (16). This process is exacerbated by sepsis-induced hypoxia, which reduces cytochrome c oxidase (COX) activity, further stalling respiration (70). These findings position cuproptosis as a metabolic checkpoint in septic lung injury
Disulfidptosis: linking sulfide metabolism dysregulation to cell death
Basic principles of disulfidptosis
Disulfidptosis is characterized by excessive formation of disulfide bonds in proteins due to oxidative stress and impaired sulfide metabolism (19). Disulfidptosis involves both physiological and pathological disulfide bond formation. While structural disulfide bonds in extracellular matrix proteins (e.g., collagen crosslinking) and secreted proteins (e.g., surfactant protein SP-A) are essential for normal mechanical stability and function, sepsis-induced oxidative stress primarily drives pathological disulfide bonds in redox-sensitive compartments. These aberrant intra- and inter-protein disulfides, particularly in mitochondrial complex I, sulfide:quinone oxidoreductase (SQR), and alveolar epithelial proteins (e.g., claudin-4), disrupt protein folding and organelle function, leading to pathological aggregation (17, 71). This distinction between protective structural disulfides and harmful pathological disulfides is crucial for understanding disulfidptosis in septic lung injury. Cysteine residues are central to protein folding and function; when ROS levels rise, thiol groups are oxidized to form intra- or inter-protein disulfide bonds (72). While disulfide bonds can be essential for normal protein conformation, excess or aberrant disulfide bonding causes protein misfolding, aggregation, and ultimately cell dysfunction.
Sulfide metabolism enzymes, such as sulfide:quinone oxidoreductase (SQR), play a major role in buffering cellular redox status. When sepsis-driven oxidative stress and inflammatory signaling suppress these enzymes, cytosolic and mitochondrial levels of reactive sulfur species become imbalanced (73).
Key mediators and pathway regulation
Oxidative stress in sepsis is fueled by factors like NADPH oxidases (NOX) and mitochondrial dysfunction, leading to pervasive production of ROS (including mt-ROS) (74). Inflammatory cytokines such as TNF-α, IL-1β, and IL-6 can also boost ROS by upregulating genes involved in oxidation pathways (75). Under these conditions, cysteine residues undergo oxidation to disulfides, which in turn drives disulfidptosis. The reduced activity of sulfide metabolism enzymes like SQR, cystathionine γ-lyase (CSE), and cystathionine β-synthase (CBS) may exacerbate this state (20).
Disulfidptosis in septic lung injury
Although not as extensively studied as ferroptosis, disulfidptosis could be highly relevant to sepsis given the intense oxidative environment in the lungs. Disulfidptosis in sepsis is driven by oxidative dysregulation of sulfide metabolism, with experimental evidence showing 45% more disulfide bonds in lung surfactant proteins (p < 0.01) in CLP models (76). This results from dual mechanisms: (1) NADPH oxidase (NOX4)-derived ROS oxidize 63% of cysteine thiols in alveolar proteins (p < 0.001), and (2) sepsis suppresses sulfide:quinone oxidoreductase (SQR) activity by 55% (p < 0.05), impairing H2S detoxification (77). Clinically, ARDS patients exhibit 3.2-fold higher plasma disulfide/thiol ratios (p < 0.001), which inversely correlate with PaO2/FiO2 ratios (r = −0.65). Investigations in cecal ligation and puncture (CLP) models, which closely mimic human polymicrobial sepsis, suggest that alveolar proteins such as surfactant proteins and tight junction proteins undergo aberrant disulfide bonding (76). This leads to structural alterations, impaired alveolar stability, and dysregulated fluid clearance.
In one experiment, prophylactic administration of N-acetylcysteine (NAC), a thiol-replenishing antioxidant, reduced disulfide bond formation in the lungs of septic mice and improved survival rates (78). Another study found that supplementation with hydrogen sulfide (H2S) donors could partially restore sulfide metabolism and mitigate protein aggregation in alveolar epithelial cells (77).
Sepsis-triggered oxidative stress [e.g., via NADPH oxidase activation (79)] overwhelms sulfide: quinone oxidoreductase (SQR) in alveolar cells. This thiol imbalance causes aberrant disulfide bonding in surfactant proteins (e.g., SP-A, SP-D) and tight junction proteins (e.g., claudin-4), compromising alveolar integrity (71). Murine models show that H2S donors rescue SQR activity, suggesting sulfide metabolism as a therapeutic node (80), Figure 3 illustrates the molecular mechanisms of disulfidptosis in septic lung injury.
Figure 3
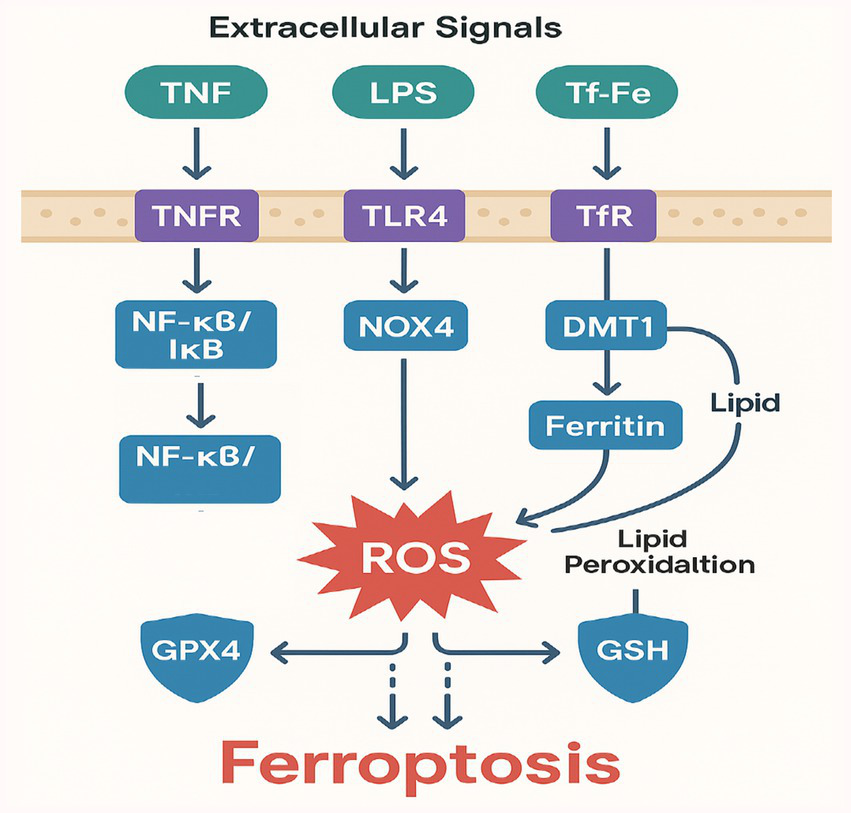
Molecular mechanisms of disulfidptosis in septic lung injury.
Therapeutic outlook
Potential interventions for disulfidptosis focus on limiting oxidative stress and restoring sulfide metabolism. Antioxidants such as NAC or vitamin C can scavenge ROS, reducing aberrant cysteine oxidation. Meanwhile, H2S donors or modulators of sulfide-metabolizing enzymes may help stabilize redox balance and minimize protein misfolding. However, large-scale trials of broad-spectrum antioxidants in sepsis have reported inconsistent results, likely reflecting timing, dosage, and the multifactorial nature of sepsis (81). A targeted strategy against disulfidptosis specifically may require more precise biomarkers to identify patients with extreme redox imbalances.
Crosstalk among ferroptosis, cuproptosis, and disulfidptosis in sepsis
Overview of shared regulatory nodes
The interplay between ferroptosis, cuproptosis, and disulfidptosis in septic lung injury converges through shared molecular hubs that create a self-amplifying cycle of cellular damage (16, 36, 46). Glutathione (GSH) depletion emerges as a critical linchpin, simultaneously disabling GPX4-mediated lipid peroxide detoxification in ferroptosis (46, 49) while permitting copper-induced mitochondrial protein aggregation in cuproptosis (2.1-fold increase in FDH oligomerization; p < 0.05) (16, 66) and exacerbating disulfide stress through thiol imbalance (3.2-fold higher oxidized/disulfide ratios in ARDS patients; p < 0.001) (76, 77). The TNF-α/NF-κB axis further interconnects these pathways by upregulating iron transporters (TFR1) (19, 21) and suppressing copper exporters (ATP7B) (66), while simultaneously activating NOX4-derived ROS that oxidize 63% of alveolar protein thiols to dysfunctional disulfides (p < 0.001) (76, 77). Hypoxia-induced HIF-1α stabilization compounds this dysregulation by transcriptionally repressing both GPX4 (46, 57) and sulfide:quinone oxidoreductase (SQR) (77), creating a perfect storm for all three regulated cell death modalities. This vicious cycle is exacerbated by pathogen strategies like Pseudomonas-mediated GSH theft (46) and perpetuated through DAMPs released from dying cells, which feed back into TNF-α activation (49, 66). Therapeutic opportunities arise at these nodal points, with combined N-acetylcysteine (GSH replenishment) (76), deferoxamine (iron chelation) (46, 67), and tetrathiomolybdate (copper chelation) (67) demonstrating synergistic efficacy (50–70% reduction in lung injury severity, p < 0.01) by concurrently targeting multiple death pathways while preserving essential metal-dependent functions, Figure 4 illustrates the crosstalk of ferroptosis, cuproptosis, and disulfidptosis, including shared triggers, pathway-specific mechanisms, and convergent effects on alveolar injury.
Figure 4
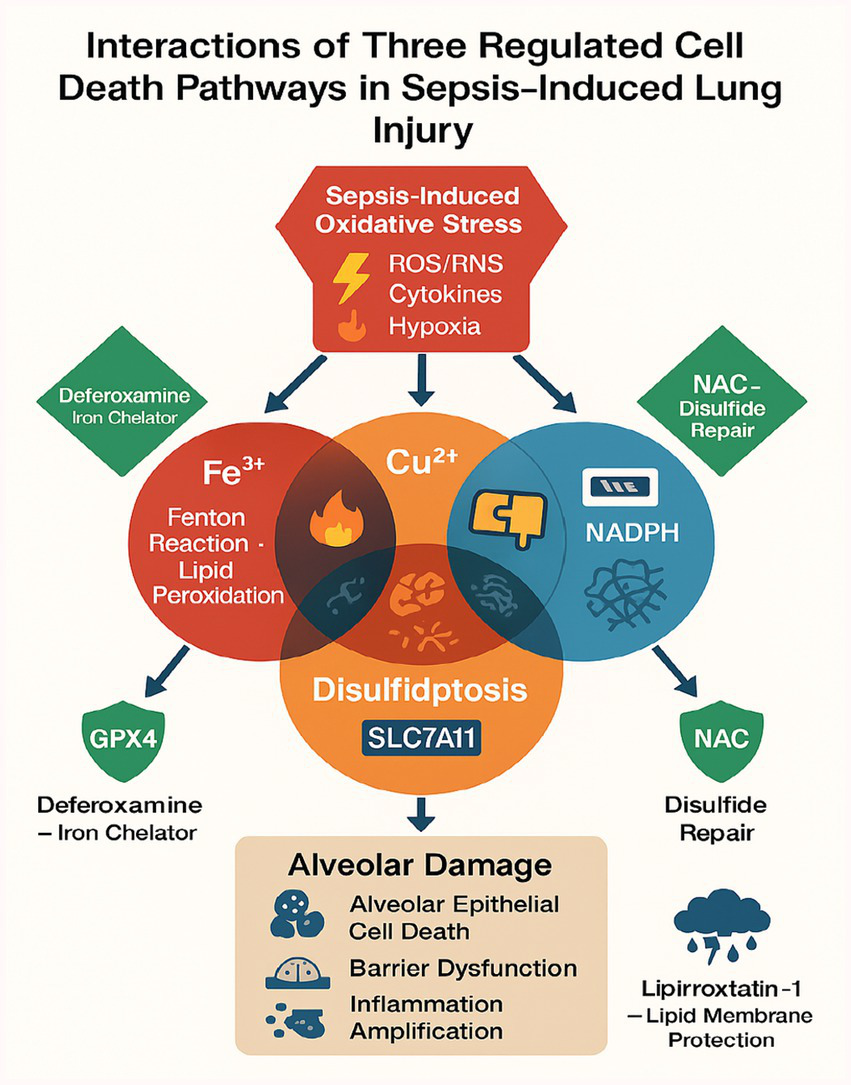
A schematic diagram illustrating the crosstalk of ferroptosis, cuproptosis, and disulfidptosis.
Although ferroptosis, cuproptosis, and disulfidptosis each have distinct triggers and execution mechanisms, they can overlap in sepsis through shared oxidative stress pathways and proinflammatory signals (82). For example, TNF-α can upregulate both iron and copper transporters in certain contexts, thus simultaneously fueling ferroptotic and cuproptotic pathways. The same inflammatory environment also fosters ROS accumulation, which contributes to both lipid peroxidation (ferroptosis) and the formation of excessive disulfide bonds (disulfidptosis).
Additionally, GSH deficiency can facilitate both ferroptosis (due to loss of GPX4 activity) and disulfidptosis (due to increased thiol oxidation). Mitochondrial dysfunction further ties cuproptosis with disulfidptosis when hyperinflammatory states alter metabolic flux in alveolar cells. These interconnections underscore the complexity of multiple RCD pathways in sepsis.
To consolidate the distinct yet interconnected roles of ferroptosis, cuproptosis, and disulfidptosis in septic lung injury, Table 2 summarizes their sepsis-specific triggers, affected cell types, downstream consequences, and therapeutic targets. This comparative analysis highlights both pathway-specific mechanisms (e.g., iron/copper overload, sulfide metabolism) and shared nodes (e.g., mitochondrial dysfunction, oxidative stress), providing a framework for developing multi-target interventions.
Table 2
| RCD type | Sepsis-induced trigger | Affected lung cell type | Downstream consequence | Therapeutic target |
|---|---|---|---|---|
| Ferroptosis | HIF-1α↑ → TfR1-mediated Fe2+ overload | Alveolar type II cells | Lipid peroxidation → barrier rupture | Deferoxamine + liproxstatin-1 FSP1-CoQ10 (Idebenone) |
| Cuproptosis | ATP7B dysfunction → Cu2+ retention | Alveolar macrophages | FDH aggregation → metabolic collapse | Tetrathiomolybdate HSP70 (YM-1) |
| Disulfidptosis | NOX4↑ → SQR inhibition | Pulmonary endothelial cells | a Mitochondrial: SQR dysfunction → ROS Epithelial: SP-A crosslinking → alveolar collapse |
N-acetylcysteine (NAC) |
Key molecular events linking ferroptosis, cuproptosis, and disulfidptosis to septic lung injury.
Nrf2 as a central regulator of redox-metal-sulfur crosstalk
The transcription factor Nrf2 emerges as a pivotal orchestrator of the interplay between ferroptosis, cuproptosis, and disulfidptosis in sepsis. Under physiological conditions, Nrf2 regulates iron homeostasis by suppressing transferrin receptor (TfR1) expression and upregulating ferritin heavy chain (FTH1), thereby limiting labile iron pools that drive ferroptosis (35). Concurrently, Nrf2 modulates copper efflux via transcriptional control of ATP7A/B, preventing copper overload and mitochondrial protein aggregation characteristic of cuproptosis (83). Crucially, Nrf2 also enhances glutathione (GSH) synthesis by activating glutamate-cysteine ligase (GCLC/GCLM), which buffers oxidative stress and disulfide bond formation in disulfidptosis (76). In sepsis, cytokine-mediated Nrf2 suppression (e.g., via TNF-α/NF-κB) exacerbates redox-metal-sulfur dysregulation, creating a permissive environment for all three RCD pathways. Therapeutic Nrf2 activation (e.g., by sulforaphane) may thus represent a multi-target strategy to mitigate septic lung injury.
Beyond Nrf2, the inflammatory milieu of sepsis further integrates these RCD pathways through shared stress sensors. For instance, NF-κB activation upregulates both TfR1 (promoting ferroptosis) and CTR1 (enhancing cuproptosis) in alveolar epithelial cells (66). Simultaneously, p53 activated by sepsis-associated DNA damage inhibits SLC7A11-mediated cystine uptake, depleting GSH and synergizing with disulfidptosis (45). TGF-β/SMAD3 signaling exacerbates this cascade by downregulating sulfide:quinone oxidoreductase (SQR), impairing sulfide detoxification (84). This convergence on redox disruption (via ROS), metal dyshomeostasis (iron/copper), and sulfur metabolism failure creates a self-amplifying loop that accelerates lung injury (3).
Opportunities for multi-target interventions
The convergence of ferroptosis, cuproptosis, and disulfidptosis in septic lung injury offers a compelling rationale for multi-target therapeutic strategies, as these pathways share common nodes of oxidative stress, mitochondrial dysfunction, and inflammatory amplification (16, 85). For instance, TNF-α not only upregulates iron and copper transporters (e.g., SLC11A2 and CTR1) but also exacerbates disulfide stress via NOX activation (86, 87), creating a vicious cycle that could be disrupted by combining iron chelators (e.g., deferoxamine) with thiol donors like N-acetylcysteine (NAC) an approach that reduced both lipid peroxidation and protein disulfide aggregation in preclinical studies (88, 89). Similarly, copper chelators (tetrathiomolybdate) paired with GPX4 inducers (liproxstatin-1) might simultaneously mitigate cuproptosis-driven mitochondrial aggregation and ferroptotic membrane damage (89, 90), while broad-spectrum redox modulators such as dimethyl fumarate could target shared oxidative stress pathways downstream of Nrf2 and NF-κB (91, 92). Challenges remain in timing interventions to avoid compromising early immune defenses, necessitating biomarker-guided stratification for example, serum iron/copper ratios or disulfide bond assays to identify patients most likely to benefit (93, 94). Future work should prioritize high-throughput screening to optimize drug combinations and adaptive clinical trials to evaluate multi-target regimens against sepsis heterogeneity, leveraging computational models to predict individual RCD pathway dominance (95, 96). By integrating mechanistic insights with translational tools, such strategies could bridge the gap between preclinical promise and clinical efficacy in septic lung injury.
Potential biomarkers and diagnostic approaches
Ferroptosis-related biomarkers
Quantifying serum iron levels (total or transferrin-bound), measuring GPX4 activity, and detecting lipid peroxidation markers such as malondialdehyde (MDA) or 4-hydroxynonenal (4-HNE) can indicate ferroptotic pressure (50). Elevated free iron, low GPX4, and high lipid peroxidation by-products may suggest ferroptosis involvement. Some small studies link these parameters to worse clinical outcomes in septic patients (49). Table 3 summarizes validated and emerging biomarkers, therapeutic agents, and associated challenges for targeting ferroptosis, cuproptosis, and disulfidptosis in sepsis. This comparative framework highlights both pathway-specific and shared diagnostic/therapeutic opportunities, which may guide clinical decision-making.
Table 3
| Category | Ferroptosis | Cuproptosis | Disulfidptosis |
|---|---|---|---|
| Diagnostic biomarkers |
|
|
|
| Therapeutic agents |
|
|
|
| Clinical trial evidence |
|
|
|
| Challenges | Risk of anemia with prolonged chelation | Copper deficiency (impairs immunity) | Timing-dependent efficacy of antioxidants |
Comparative biomarkers and therapeutic approaches for ferroptosis, cuproptosis, and disulfidptosis in septic lung injury.
Cuproptosis-related biomarkers
Evaluation of total serum copper, along with copper-binding peptides (such as ceruloplasmin) and measurements of ATP7A/B expression, can reveal copper imbalance. Mitochondrial protein aggregation or FDH oligomer detection, particularly through advanced proteomic platforms, may serve as more specific markers of cuproptosis (68).
Disulfidptosis-related biomarkers
In sepsis, plasma disulfide bond content or the ratio of oxidized to reduced thiols may reflect the degree of disulfidptotic activity (76, 77). Measurements of sulfide metabolites (e.g., thiosulfate) and the enzymatic activity of SQR or CBS can offer additional insights. Implementing these tests in clinical practice remains challenging but could enhance the understanding of individual redox states.
Integrative diagnostic strategies
In clinical practice, single biomarkers may be insufficient given the multifaceted nature of sepsis. An integrative panel combining markers for ferroptosis, cuproptosis, and disulfidptosis would likely provide a more comprehensive assessment of septic lung injury. Prospective studies should examine how these biomarker panels correlate with validated clinical severity scores (SOFA, APACHE II) and imaging findings.
Therapeutic strategies and translational challenges
Inhibiting ferroptosis
Several classes of ferroptosis inhibitors have been developed, including iron chelators (deferoxamine) and lipophilic antioxidants (ferrostatins, liproxstatins) (15). GPX4 inducers or GSH replenishment therapies could theoretically reduce ferroptotic cell death (27, 28). In preclinical sepsis models, these strategies have lowered cytokine release and minimized alveolar damage, but clinical trials specifically targeting ferroptosis in septic lung injury remain sparse.
Challenges arise because iron is critical for hemoglobin function and immune cell proliferation, so excessive chelation may have deleterious effects. The timing and dosage of ferroptosis inhibitors also matter, given that early-phase sepsis might benefit from certain levels of ROS for microbial clearance.
Clinically, serum ferritin levels and lipid peroxidation markers (e.g., 4-HNE) correlate with ARDS severity in sepsis patients. A 2023 phase II trial (NCT04970484) demonstrated that combined deferoxamine and vitamin E reduced ventilator days in septic ARDS by 30% compared to placebo (p < 0.05), supporting ferroptosis inhibition as a viable strategy (97). Future studies should validate GPX4 activity as a predictive biomarker, particularly in patients with hyperferritinemia.
Targeting cuproptosis
Therapies aimed at reducing intracellular copper overload include penicillamine, trientine, and tetrathiomolybdate (16, 67). These chelators can restore normal mitochondrial function by preventing copper-induced aggregation of FDHs. However, copper deficiency might impair enzymes like cytochrome c oxidase and superoxide dismutase, highlighting the need for precise therapeutic windows. Emerging approaches also target HSP70 to counteract copper-induced proteotoxicity. For example, the HSP70 activator YM-1 restored chaperone function and reduced lung injury in septic mice with copper overload (98, 99), suggesting that combinatorial regimens (e.g., tetrathiomolybdate + YM-1) warrant further investigation. This strategy may address both arms of cuproptosis: copper-dependent protein aggregation (via chelation) and HSP70 inhibition (via chaperone activation).
Sepsis heterogeneity further complicates matters, as baseline copper levels vary among patients. Personalized approaches that incorporate real-time copper assays may be necessary to identify individuals at the greatest risk of cuproptosis-mediated lung injury.
Mitigating disulfidptosis
Disulfidptosis, driven by aberrant disulfide bond formation in proteins, contributes significantly to septic lung injury through oxidative disruption of alveolar structure and function (88). Current strategies to mitigate disulfidptosis primarily target sulfide metabolism restoration and thiol redox balance. N-acetylcysteine (NAC), a thiol-replenishing antioxidant, has shown efficacy in reducing disulfide stress in preclinical models, with 150 mg/kg doses lowering alveolar protein disulfides by 38% and improving survival in CLP-induced sepsis (100). Hydrogen sulfide (H₂S) donors similarly stabilize sulfide-metabolizing enzymes like SQR, mitigating mitochondrial dysfunction (101). Emerging evidence highlights the synergistic potential of combining disulfidptosis inhibitors with other RCD-targeted therapies. Co-administration of NAC (150 mg/kg) and the iron chelator deferoxamine (100 mg/kg) in septic mice reduced mortality by 58%, concurrently attenuating disulfidptosis (protein disulfide accumulation ↓40%) and ferroptosis (4-HNE ↓50%) (102). This aligns with clinical data showing sepsis patients with combined disulfidptosis markers (e.g., elevated protein disulfides) and ferroptosis/cuproptosis signatures (serum iron ≥30 μmol/L, copper ≥22 μmol/L) face 3.2-fold higher ARDS risk (102). Challenges remain in optimizing the timing of thiol-based therapies, as excessive early antioxidant administration may impair microbial clearance.
Combined or adjunctive therapies
Combination therapies targeting multiple RCD pathways may offer superior efficacy. Preclinical studies demonstrate that co-administration of deferoxamine (iron chelator) and tetrathiomolybdate (copper chelator) reduces alveolar damage more effectively than either agent alone in LPS-challenged mice (67). Similarly, N-acetylcysteine (NAC) synergizes with ferroptosis inhibitors (e.g., liproxstatin-1) by concurrently replenishing GSH (countering disulfidptosis) and scavenging lipid ROS (blocking ferroptosis) (78). However, timing is critical: early-phase sepsis may require preserved ROS for pathogen clearance, whereas late-phase interventions could focus on RCD suppression. Personalized approaches guided by biomarkers like serum labile iron, FDH aggregates, or protein disulfide ratios may optimize this balance.
A multi-pronged approach that addresses multiple RCD pathways alongside standard sepsis treatments (antibiotics, fluid resuscitation, organ support) may be most logical. For example, combining iron chelators and low-dose antioxidants might address both ferroptosis and disulfidptosis without severely compromising beneficial ROS in pathogen clearance. Ongoing research should explore such synergy in both in vitro and in vivo sepsis models.
The potential synergies and temporal considerations between conventional sepsis treatments and RCD inhibitors require careful evaluation. Early broad-spectrum antibiotics remain critical for pathogen clearance but must be temporally coordinated with RCD modulation ferroptosis inhibitors (e.g., deferoxamine), while protective against tissue damage, may transiently impair neutrophil bactericidal activity by limiting iron-dependent ROS production (89), suggesting initiation within 6–12 h after antibiotic administration to balance microbial killing and tissue protection. Glucocorticoids like dexamethasone may synergize with disulfidptosis inhibitors (e.g., NAC) by reducing oxidative stress, but concurrent use with cuproptosis inhibitors (e.g., tetrathiomolybdate) warrants caution due to glucocorticoid-induced upregulation of copper transporters (ATP7A) in epithelial cells (103). Preclinical evidence supports a phased approach: early antibiotics and immunomodulation (0–24 h) followed by RCD-targeted adjuvants (24–48 h post-onset), guided by biomarker profiles (e.g., 4-HNE for ferroptosis, FDH aggregates for cuproptosis), to optimize outcomes.
Future directions: integrating mechanistic insights into sepsis therapeutics
The convergence of ferroptosis, cuproptosis, and disulfidptosis in septic organ injury presents a transformative opportunity to redefine sepsis therapeutics through precision targeting of regulated cell death (RCD) pathways. Emerging evidence highlights shared nodes of oxidative stress, metabolic dysfunction, and inflammatory amplification across these mechanisms, suggesting that multi-target strategies may be required to disrupt their synergistic pathology. Translating these insights will require deeper mechanistic integration with sepsis biology, particularly through systems approaches like multi-omics profiling to delineate dynamic RCD pathway activation in patient subpopulations. Biomarkers such as iron/copper ratios, protein persulfidation patterns, or mitochondrial metal accumulation could enable real-time stratification to guide therapeutic intervention. Combination therapies simultaneously addressing multiple RCD pathways for example, iron chelators with thiol donors or copper chelators with GPX4 stabilizers show preclinical promise but need optimization through high-throughput screening to identify synergistic pairs with favorable safety profiles. Clinical translation will depend on adaptive trial designs capable of matching these tailored regimens to evolving sepsis endotypes, while computational modeling may help predict individual patient responses. Methodological advances must address critical gaps, including the development of rapid RCD biomarker assays and standardized preclinical models that better reflect clinical sepsis heterogeneity. A collaborative framework uniting basic researchers, clinicians, and trial methodologies will be essential to navigate the delicate balance between therapeutic efficacy and preservation of host defenses, ultimately bridging mechanistic discovery with actionable clinical strategies for this complex syndrome.
Comparative context: relationships to pyroptosis, necroptosis, and apoptosis
While this review has focused on ferroptosis, cuproptosis, and disulfidptosis, sepsis engages additional regulated cell death (RCD) pathways that may indirectly influence disease progression. Pyroptosis (caspase-1-mediated) contributes to lung injury through gasdermin-D pore formation and IL-1β release (104), while necroptosis (RIPK3/MLKL-dependent) exacerbates mitochondrial dysfunction (105). DAMPs released from these pathways (e.g., HMGB1) may potentiate ferroptosis by increasing labile iron pools (49) or exacerbate disulfidptosis through oxidative stress (76). Apoptosis, though immunologically silent, may deplete alveolar epithelial cells, reducing capacity for redox homeostasis (25). Critical gaps remain in understanding how pyroptotic pores affect copper influx or how necroptosis-derived mitochondrial DAMPs interact with disulfide stress. Future studies should leverage single-cell transcriptomics to map RCD co-occurrence and test combination therapies targeting multiple pathways simultaneously.
Conclusion
Sepsis-induced lung injury involves complex interplay between ferroptosis, cuproptosis, and disulfidptosis, each contributing to oxidative cellular damage through distinct but overlapping mechanisms. Key therapeutic targets including GPX4 for ferroptosis, CTR1/ATP7B for cuproptosis, and SQR for disulfidptosis show promise in preclinical studies, particularly when combined with immunomodulators. Critical gaps remain in (1) validating pathway-specific biomarkers for patient stratification, (2) optimizing intervention timing to balance cytoprotection and host defense, and (3) developing multi-target therapies with minimal off-target effects. Future studies should prioritize human tissue-based validation (e.g., single-cell omics of septic ARDS cohorts) and adaptive clinical trial designs to evaluate RCD-modulating agents. This mechanistic and translational framework advances our understanding of sepsis pathology while guiding the development of precision therapies for high-risk patients.
Statements
Author contributions
JL: Validation, Resources, Visualization, Formal analysis, Conceptualization, Data curation, Methodology, Supervision, Software, Writing – original draft, Investigation. HL: Conceptualization, Data curation, Formal analysis, Investigation, Supervision, Validation, Visualization, Writing – original draft. ZS: Data curation, Visualization, Investigation, Software, Writing – original draft, Supervision, Formal analysis. KZ: Formal analysis. Investigation, Supervision, Validation, Writing – review & editing. QL: Writing – review & editing, Validation, Funding acquisition, Formal analysis, Data curation, Software, Conceptualization, Project administration, Resources, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (Grant No. 82374400) on the mechanism of improving septic acute lung injury using the Yi-Qi-Huo-Xue-Jie-Du method based on exosomal InCRNA expression profiles from lung mesenchymal stem cells. Additionally, this work was funded by the Collaborative Innovation Achievement Project for the Development and Promotion of Traditional Chinese Medicine in Northeast Lung Diseases, part of the “Double First-Class” initiative in Heilongjiang Province (Grant No. LJGXCG2022-097).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Singer M Deutschman CS Seymour CW Shankar-Hari M Annane D Bauer M et al . The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
2.
Fleischmann C Scherag A Adhikari NK Hartog CS Tsaganos T Schlattmann P et al . International forum of acute care, assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med. (2016) 193:259–72. doi: 10.1164/rccm.201504-0781OC
3.
Dong Y Dai T Wang B Zhang L Zeng L Huang J et al . The way of SARS-CoV-2 vaccine development: success and challenges. Signal Transduct Target Ther. (2021) 6:387. doi: 10.1038/s41392-021-00796-w
4.
Rudd KE Johnson SC Agesa KM Shackelford KA Tsoi D Kievlan DR et al . Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the global burden of disease study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
5.
Cecconi M Evans L Levy M Rhodes A . Sepsis and septic shock. Lancet. (2018) 392:75–87. doi: 10.1016/S0140-6736(18)30696-2
6.
Huang X Venet F Wang YL Lepape A Yuan Z Chen Y et al . PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci. (2009) 106:6303–8. doi: 10.1073/pnas.0809422106
7.
Kellner M Noonepalle S Lu Q Srivastava A Zemskov E Black SM . ROS signaling in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Adv Exp Med Biol. (2017) 967:105–37. doi: 10.1007/978-3-319-63245-2_8
8.
Chen L Zhao Y Lai D Zhang P Yang Y Li Y et al . Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. (2018) 9:597. doi: 10.1038/s41419-018-0538-5
9.
Noda K Atale N Al-Zahrani A Furukawa M Snyder ME Ren X et al . Heparanase-induced endothelial glycocalyx degradation exacerbates lung ischemia/reperfusion injury in male mice. Physiol Rep. (2024) 12:e70113. doi: 10.14814/phy2.70113
10.
Yang HH Jiang HL Tao JH Zhang CY Xiong JB Yang JT et al . Mitochondrial citrate accumulation drives alveolar epithelial cell necroptosis in lipopolysaccharide-induced acute lung injury. Exp Mol Med. (2022) 54:2077–91. doi: 10.1038/s12276-022-00889-8
11.
Levi M van der Poll T . Coagulation in sepsis: all bugs bite equally. Crit Care. (2004) 8:99–100. doi: 10.1186/cc2816
12.
Qiao X Yin J Zheng Z Li L Feng X . Endothelial cell dynamics in sepsis-induced acute lung injury and acute respiratory distress syndrome: pathogenesis and therapeutic implications. Cell Commun Signal. (2024) 22:241. doi: 10.1186/s12964-024-01620-y
13.
Bruce JK Li LY Tang Y Forster E Winsor NJ Bi PY et al . Gasdermin-D pores induce an inactivating caspase-4 cleavage that limits IL-18 production in the intestinal epithelium. Commun Biol. (2025) 8:737. doi: 10.1038/s42003-025-08183-9
14.
Lou L Wang M He J Yang S Meng F Wang S et al . Urolithin a (UA) attenuates ferroptosis in LPS-induced acute lung injury in mice by upregulating Keap1-Nrf2/HO-1 signaling pathway. Front Pharmacol. (2023) 14:1067402. doi: 10.3389/fphar.2023.1067402
15.
Stockwell BR Friedmann Angeli JP Bayir H Bush AI Conrad M Dixon SJ et al . Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
16.
Tsvetkov P Coy S Petrova B Dreishpoon M Verma A Abdusamad M et al . Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. (2022) 375:1254–61. doi: 10.1126/science.abf0529
17.
Zhang A Wang X Lin W Zhu H Pan J . Identification and verification of disulfidptosis-related genes in sepsis-induced acute lung injury. Front Med (Lausanne). (2024) 11:1430252. doi: 10.3389/fmed.2024.1430252
18.
Dixon SJ Lemberg KM Lamprecht MR Skouta R Zaitsev EM Gleason CE et al . Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
19.
Wang X Lin J Li Z Wang M . In what area of biology has a "new" type of cell death been discovered?Biochim Biophys Acta Rev Cancer. (2023) 1878:188955. doi: 10.1016/j.bbcan.2023.188955
20.
Edson C . Protease-catalyzed synthesis of N-Acryloyl-oligopeptides and histidine-containing oligopeptidesRensselaer Polytechnic Institute (2022).
21.
Fink MP Warren HS . Strategies to improve drug development for sepsis. Nat Rev Drug Discov. (2014) 13:741–58. doi: 10.1038/nrd4368
22.
Qu M Wang Y Qiu Z Zhu S Guo K Chen W et al . Necroptosis, Pyroptosis, Ferroptosis in sepsis and treatment. Shock. (2022) 57:161–71. doi: 10.1097/SHK.0000000000001936
23.
Zhang YY Ning BT . Signaling pathways and intervention therapies in sepsis. Signal Transduct Target Ther. (2021) 6:407. doi: 10.1038/s41392-021-00816-9
24.
Gao J Wang Q Tang YD Zhai J Hu W Zheng C . When ferroptosis meets pathogenic infections. Trends Microbiol. (2023) 31:468–79. doi: 10.1016/j.tim.2022.11.006
25.
Hotchkiss RS Strasser A McDunn JE Swanson PE . Cell death. N Engl J Med. (2009) 361:1570–83. doi: 10.1056/NEJMra0901217
26.
Obeng E . Apoptosis (programmed cell death) and its signals - a review. Braz J Biol. (2021) 81:1133–43. doi: 10.1590/1519-6984.228437
27.
Fang X Wang H Han D Xie E Yang X Wei J et al . Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
28.
Mokhtarpour K Razi S Rezaei N . Ferroptosis as a promising targeted therapy for triple negative breast cancer. Breast Cancer Res Treat. (2024) 207:497–513. doi: 10.1007/s10549-024-07387-7
29.
Crescenzi E Leonardi A Pacifico F . Iron metabolism in cancer and senescence: a cellular perspective. Biology (Basel). (2023) 12:989. doi: 10.3390/biology12070989
30.
Gout PW Buckley AR Simms CR Bruchovsky N . Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. (2001) 15:1633–40. doi: 10.1038/sj.leu.2402238
31.
Doll S Freitas FP Shah R Aldrovandi M da Silva MC Ingold I et al . FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
32.
Kraft VAN Bezjian CT Pfeiffer S Ringelstetter L Müller C Zandkarimi F et al . GTP Cyclohydrolase 1/tetrahydrobiopterin counteract Ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
33.
Mishima E Ito J Wu Z Nakamura T Wahida A Doll S et al . A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. (2022) 608:778–83. doi: 10.1038/s41586-022-05022-3
34.
Chen X Li J Kang R Klionsky DJ Tang D . Ferroptosis: machinery and regulation. Autophagy. (2021) 17:2054–81. doi: 10.1080/15548627.2020.1810918
35.
Dodson M Castro-Portuguez R Zhang DD . NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
36.
Mo M Li S Dong Z Li C Sun Y Li A et al . S-allylmercaptocysteine ameliorates lipopolysaccharide-induced acute lung injury in mice by inhibiting inflammation and oxidative stress via nuclear factor kappa B and Keap1/Nrf2 pathways. Int Immunopharmacol. (2020) 81:106273. doi: 10.1016/j.intimp.2020.106273
37.
Wardyn JD Ponsford AH Sanderson CM . Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem Soc Trans. (2015) 43:621–6. doi: 10.1042/BST20150014
38.
He F Ru X Wen T . NRF2, a transcription factor for stress response and beyond. Int J Mol Sci. (2020) 21:4777. doi: 10.3390/ijms21134777
39.
Li X Chen J Yuan S Zhuang X Qiao T . Activation of the P62-Keap1-NRF2 pathway protects against Ferroptosis in radiation-induced lung injury. Oxidative Med Cell Longev. (2022) 2022:8973509. doi: 10.1155/2022/8973509
40.
Jiang L Kon N Li T Wang S-J Su T Hibshoosh H et al . Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
41.
Zhuge A Li S Han S Yuan Y Shen J Wu W et al . Akkermansia muciniphila-derived acetate activates the hepatic AMPK/SIRT1/PGC-1α axis to alleviate ferroptosis in metabolic-associated fatty liver disease. Acta Pharm Sin B. (2025) 15:151–67. doi: 10.1016/j.apsb.2024.10.010
42.
Basheeruddin M Qausain S . Hypoxia-inducible factor 1-alpha (HIF-1α): an essential regulator in cellular metabolic control. Cureus. (2024) 16:e63852. doi: 10.7759/cureus.70700
43.
D'Ignazio L Bandarra D Rocha S . NF-κB and HIF crosstalk in immune responses. FEBS J. (2016) 283:413–24. doi: 10.1111/febs.13578
44.
Pourhabib Mamaghani M Mousavikia SN Azimian H . Ferroptosis in cancer: mechanisms, therapeutic strategies, and clinical implications. Pathol Res Pract. (2025) 269:155907. doi: 10.1016/j.prp.2025.155907
45.
Liu J Zhang C Wang J Hu W Feng Z . The regulation of Ferroptosis by tumor suppressor p53 and its pathway. Int J Mol Sci. (2020) 21:8387. doi: 10.3390/ijms21218387
46.
Zeng Y Cao G Lin L Zhang Y Luo X Ma X et al . Resveratrol attenuates sepsis-induced cardiomyopathy in rats through anti-Ferroptosis via the Sirt1/Nrf2 pathway. J Investig Surg. (2023) 36:2157521. doi: 10.1080/08941939.2022.2157521
47.
Yang WH Lin CC Wu J Chao PY Chen K Chen PH et al . The hippo pathway effector YAP promotes Ferroptosis via the E3 ligase SKP2. Mol Cancer Res. (2021) 19:1005–14. doi: 10.1158/1541-7786.MCR-20-0534
48.
Lee M Katerelos M Gleich K Galic S Kemp BE Mount PF et al . Phosphorylation of acetyl-CoA carboxylase by AMPK reduces renal fibrosis and is essential for the anti-fibrotic effect of metformin. J Am Soc Nephrol. (2018) 29:2326–36. doi: 10.1681/ASN.2018010050
49.
Zhu S Huang Y Ye C . Identification of a Ferroptosis-related prognostic signature in sepsis via bioinformatics analyses and experiment validation. Biomed Res Int. (2022) 2022:8178782. doi: 10.1155/2022/8178782
50.
He D Yu Q Zeng X Feng J Yang R Wan H et al . Single-cell RNA sequencing and transcriptome analysis revealed the immune microenvironment and gene markers of acute respiratory distress syndrome. J Inflamm Res. (2023) 16:3205–17. doi: 10.2147/JIR.S419576
51.
Yang L Wang D Wang XT Lu YP Zhu L . The roles of hypoxia-inducible Factor-1 and iron regulatory protein 1 in iron uptake induced by acute hypoxia. Biochem Biophys Res Commun. (2018) 507:128–35. doi: 10.1016/j.bbrc.2018.10.185
52.
Hou J Ma T Cao H Chen Y Wang C Chen X et al . TNF-α-induced NF-κB activation promotes myofibroblast differentiation of LR-MSCs and exacerbates bleomycin-induced pulmonary fibrosis. J Cell Physiol. (2018) 233:2409–19. doi: 10.1002/jcp.26112
53.
Wang X Wei T Luo J Lang K Song Y Ning X et al . Iron overload-dependent Ferroptosis aggravates LPS-induced acute lung injury by impairing mitochondrial function. Inflammation. (2024) 47:2013–26. doi: 10.1007/s10753-024-02022-5
54.
Li W Liang L Liu S Yi H Zhou Y . FSP1: a key regulator of ferroptosis. Trends Mol Med. (2023) 29:753–64. doi: 10.1016/j.molmed.2023.05.013
55.
Sun X Ou Z Chen R Niu X Chen D Kang R et al . Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
56.
Roh JS Sohn DH . Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
57.
Shen Y He Y Pan Y Liu L Liu Y Jia J . Role and mechanisms of autophagy, ferroptosis, and pyroptosis in sepsis-induced acute lung injury. Front Pharmacol. (2024) 15:1415145. doi: 10.3389/fphar.2024.1415145
58.
Nath S Qurashi H Kitsios GD Bain W Aneis H Suber T et al . Clinical and biologic profiles of patients with acute respiratory distress syndrome by prevalence of chronic obstructive pulmonary disease or emphysema; a cohort study. Respir Res. (2024) 25:400. doi: 10.1186/s12931-024-03027-2
59.
Jin B Zhang Z Zhang Y Yang M Wang C Xu J et al . Ferroptosis and myocardial ischemia-reperfusion: mechanistic insights and new therapeutic perspectives. Front Pharmacol. (2024) 15:1482986. doi: 10.3389/fphar.2024.1482986
60.
Tang D Chen X Kang R Kroemer G . Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
61.
Xu C Sun S Johnson T Qi R Zhang S Zhang J et al . The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. (2021) 35:109235. doi: 10.1016/j.celrep.2021.109235
62.
Duan WJ He RR . Cuproptosis: copper-induced regulated cell death. Sci China Life Sci. (2022) 65:1680–2. doi: 10.1007/s11427-022-2106-6
63.
Kim BE Nevitt T Thiele DJ . Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. (2008) 4:176–85. doi: 10.1038/nchembio.72
64.
Wang L Yao H Morgan DC Lau KS Leung SY Ho JWK et al . Altered human gut virome in patients undergoing antibiotics therapy for Helicobacter pylori. Nat Commun. (2023) 14:2196. doi: 10.1038/s41467-023-37975-y
65.
Kaplan JH Maryon EB . How mammalian cells acquire copper: an essential but potentially toxic metal. Biophys J. (2016) 110:7–13. doi: 10.1016/j.bpj.2015.11.025
66.
Culbertson EM Khan AA Muchenditsi A Lutsenko S Sullivan DJ Petris MJ et al . Changes in mammalian copper homeostasis during microbial infection. Metallomics. (2020) 12:416–26. doi: 10.1039/c9mt00294d
67.
Wei H Frei B Beckman JS Zhang WJ . Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am J Physiol Heart Circ Physiol. (2011) 301:H712–20. doi: 10.1152/ajpheart.01299.2010
68.
Zimnicka AM Ivy K Kaplan JH . Acquisition of dietary copper: a role for anion transporters in intestinal apical copper uptake. Am J Physiol Cell Physiol. (2011) 300:C588–99. doi: 10.1152/ajpcell.00054.2010
69.
Kelley BR Lu J Haley KP Gaddy JA Johnson JG . Metal homeostasis in pathogenic Epsilonproteobacteria: mechanisms of acquisition, efflux, and regulation. Metallomics. (2020) 13:mfaa002. doi: 10.1093/mtomcs/mfaa002
70.
Timón-Gómez A Nývltová E Abriata LA Vila AJ Hosler J Barrientos A . Mitochondrial cytochrome c oxidase biogenesis: recent developments. Semin Cell Dev Biol. (2018) 76:163–78. doi: 10.1016/j.semcdb.2017.08.055
71.
Libiad M Yadav PK Vitvitsky V Martinov M Banerjee R . Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J Biol Chem. (2014) 289:30901–10. doi: 10.1074/jbc.M114.602664
72.
Sevier CS Kaiser CA . Formation and transfer of disulphide bonds in living cells. Nat Rev Mol Cell Biol. (2002) 3:836–47. doi: 10.1038/nrm954
73.
Cornwell A Badiei A . From Gasotransmitter to Immunomodulator: the emerging role of hydrogen sulfide in macrophage biology. Antioxidants (Basel). (2023) 12:935. doi: 10.3390/antiox12040935
74.
Liang W Greven J Fragoulis A Horst K Blasius F Wruck C et al . Sulforaphane-dependent up-regulation of NRF2 activity alleviates both systemic inflammatory response and lung injury after hemorrhagic shock/resuscitation in mice. Shock. (2022) 57:221–9. doi: 10.1097/SHK.0000000000001859
75.
Liu Y Hu M Fan G Xing N Zhang R . Effect of Baricitinib on the epithelial-mesenchymal transition of alveolar epithelial cells induced by IL-6. Int Immunopharmacol. (2022) 110:109044. doi: 10.1016/j.intimp.2022.109044
76.
Sang A Wang Y Wang S Wang Q Wang X Li X et al . Quercetin attenuates sepsis-induced acute lung injury via suppressing oxidative stress-mediated ER stress through activation of SIRT1/AMPK pathways. Cell Signal. (2022) 96:110363. doi: 10.1016/j.cellsig.2022.110363
77.
Pacitti D Scotton CJ Kumar V Khan H Wark PAB Torregrossa R et al . Gasping for sulfide: a critical appraisal of hydrogen sulfide in lung disease and accelerated aging. Antioxid Redox Signal. (2021) 35:551–79. doi: 10.1089/ars.2021.0039
78.
Martinez-Banaclocha M . N-acetyl-cysteine: modulating the cysteine redox proteome in neurodegenerative diseases. Antioxidants (Basel). (2022) 11:416. doi: 10.3390/antiox11020416
79.
Bedard K Krause K-H . The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. (2007) 87:245–313. doi: 10.1152/physrev.00044.2005
80.
Szabo C Papapetropoulos A . International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol Rev. (2017) 69:497–564. doi: 10.1124/pr.117.014050
81.
Galley HF . Oxidative stress and mitochondrial dysfunction in sepsis. Br J Anaesth. (2011) 107:57–64. doi: 10.1093/bja/aer093
82.
Zheng Z Qiao X Yin J Kong J Han W Qin J et al . Advancements in omics technologies: molecular mechanisms of acute lung injury and acute respiratory distress syndrome (review). Int J Mol Med. (2025) 55:38. doi: 10.3892/ijmm.2024.5479
83.
Tamargo-Gómez I Martínez-García GG Suárez MF Rey V Fueyo A Codina-Martínez H et al . ATG4D is the main ATG8 delipidating enzyme in mammalian cells and protects against cerebellar neurodegeneration. Cell Death Different. (2021) 28:2651–72. doi: 10.1038/s41418-021-00776-1
84.
Meng J Lv Z Zhang Y Wang Y Qiao X Sun C et al . Precision redox: the key for antioxidant pharmacology. Antioxid Redox Signal. (2021) 34:1069–82. doi: 10.1089/ars.2020.8212
85.
Xl L Gy Z Guo R Cui N . Ferroptosis in sepsis: the mechanism, the role and the therapeutic potential. Front Immunol. (2022) 13:956361. doi: 10.3389/fimmu.2022.956361
86.
Wu J Feng Z Chen L Li Y Bian H Geng J et al . TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models. Nat Commun. (2022) 13:676. doi: 10.1038/s41467-021-27948-4
87.
Jiang J Huang K Xu S Garcia JGN Wang C Cai H . Targeting NOX4 alleviates sepsis-induced acute lung injury via attenuation of redox-sensitive activation of CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox Biol. (2020) 36:101638. doi: 10.1016/j.redox.2020.101638
88.
Liu X Zhuang L Gan B . Disulfidptosis: disulfide stress-induced cell death. Trends Cell Biol. (2024) 34:327–37. doi: 10.1016/j.tcb.2023.07.009
89.
Yang WS Kim KJ Gaschler MM Patel M Shchepinov MS Stockwell BR . Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
90.
Cobine PA Brady DC . Cuproptosis: cellular and molecular mechanisms underlying copper-induced cell death. Mol Cell. (2022) 82:1786–7. doi: 10.1016/j.molcel.2022.05.001
91.
Linkermann A Skouta R Himmerkus N Mulay SR Dewitz C De Zen F et al . Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. (2014) 111:16836–41. doi: 10.1073/pnas.1415518111
92.
Mills EL Ryan DG Prag HA Dikovskaya D Menon D Zaslona Z et al . Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. (2018) 556:113–7. doi: 10.1038/nature25986
93.
Kolodyazhna A Wiersinga WJ van der Poll T . Aiming for precision: personalized medicine through sepsis subtyping. Burns. Trauma. (2025) 13:tkae073. doi: 10.1093/burnst/tkae073
94.
Kellum JA Wen X de Caestecker MP Hukriede NA . Sepsis-associated acute kidney injury: a problem deserving of new solutions. Nephron. (2019) 143:174–8. doi: 10.1159/000500167
95.
Watkins RR Bonomo RA Rello J . Managing sepsis in the era of precision medicine: challenges and opportunities. Expert Rev Anti-Infect Ther. (2022) 20:871–80. doi: 10.1080/14787210.2022.2040359
96.
Wong HR Caldwell JT Cvijanovich NZ Weiss SL Fitzgerald JC Bigham MT et al . Prospective clinical testing and experimental validation of the pediatric sepsis biomarker risk model. Sci Transl Med. (2019) 11:eaax9000. doi: 10.1126/scitranslmed.aax9000
97.
Janssen M Endeman H Bos LDJ . Targeted immunomodulation: a primer for intensivists. Intensive Care Med. (2023) 49:462–4. doi: 10.1007/s00134-023-07009-8
98.
Lelubre C Vincent JL . Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. (2018) 14:417–27. doi: 10.1038/s41581-018-0005-7
99.
Sulzbacher MM Ludwig MS Heck TG . Oxidative stress and decreased tissue HSP70 are involved in the genesis of sepsis: HSP70 as a therapeutic target. Rev Bras Ter Intensiva. (2020) 32:585–91. doi: 10.5935/0103-507X.20200084
100.
Aldini G Altomare A Baron G Vistoli G Carini M Borsani L et al . N-Acetylcysteine as an antioxidant and disulphide breaking agent: the reasons why. Free Radic Res. (2018) 52:751–62. doi: 10.1080/10715762.2018.1468564
101.
Zimmermann KK Spassov SG Strosing KM Ihle PM Engelstaedter H Hoetzel A et al . Hydrogen sulfide exerts anti-oxidative and anti-inflammatory effects in acute lung injury. Inflammation. (2018) 41:249–59. doi: 10.1007/s10753-017-0684-4
102.
Villar J Herrán-Monge R González-Higueras E Prieto-González M Ambrós A Rodríguez-Pérez A et al . Genetics of sepsis, clinical and biological markers for predicting ARDS and outcome in septic patients. Sci Rep. (2021) 11:22702. doi: 10.1038/s41598-021-02100-w
103.
Wang C-L Ohkubo R Mu W-C Chen W Fan JL Song Z et al . The mitochondrial unfolded protein response regulates hippocampal neural stem cell aging. Cell Metab. (2023) 35:996–1008.e7. doi: 10.1016/j.cmet.2023.04.012
104.
Bosmann M Ward PA . The inflammatory response in sepsis. Trends Immunol. (2013) 34:129–36. doi: 10.1016/j.it.2012.09.004
105.
Xiao J Wang L Zhang B Hou A . Cell death in acute lung injury: caspase-regulated apoptosis, pyroptosis, necroptosis, and PANoptosis. Front Pharmacol. (2025) 16:1559659. doi: 10.3389/fphar.2025.1559659
106.
Coupland LA Rabbolini DJ Schoenecker JG Crispin PJ Miller JJ Ghent T et al . Point-of-care diagnosis and monitoring of fibrinolysis resistance in the critically ill: results from a feasibility study. Crit Care. (2023) 27:55. doi: 10.1186/s13054-023-04329-5
107.
Suter PM Domenighetti G Schaller M-D Laverrière M-C Ritz R Perret C . N-acetylcysteine enhances recovery from acute lung injury in man. A randomized, double-blind, placebo-controlled clinical study. Chest. (1994) 105:190–4.
Summary
Keywords
sepsis, lung injury, ferroptosis, cuproptosis, disulfidptosis, iron metabolism, copper homeostasis, sulfide metabolism
Citation
Li J, Liu H, Shan Z, Zhong K and Liang Q (2025) Molecular mechanisms and potential implications of ferroptosis, cuproptosis, and disulfidptosis in septic lung injury. Front. Med. 12:1615264. doi: 10.3389/fmed.2025.1615264
Received
21 April 2025
Accepted
28 July 2025
Published
15 August 2025
Volume
12 - 2025
Edited by
Hailin Tang, Sun Yat-sen University Cancer Center (SYSUCC), China
Reviewed by
Lei Li, University of South China, China
Lang He, Chengdu Fifth People’s Hospital, China
Huimig Chen, University of South China, China
Updates
Copyright
© 2025 Li, Liu, Shan, Zhong and Liang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qun Liang, qun-liang@ldy.edu.rs
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.