 Anna Morgul
Anna Morgul Margarita Sharova
Margarita Sharova Vladimir Kenis
Vladimir Kenis Maria Orlova1
Maria Orlova1- 1Research Centre for Medical Genetics, Moscow, Russia
- 2H. Turner National Medical Research Center for Children’s Orthopedics and Trauma Surgery of the Ministry of Health of the Russian Federation, Saint Petersburg, Russia
Acroscyphodysplasia (ASD) is an ultra-rare skeletal dysplasia characterized by severe brachydactyly, metaphyseal scaphoid knee deformities, growth retardation, and intellectual disability. To date, only seven cases of ASD have been reported, all associated with missense variants in the PDE4D gene. We report a 7-year-old girl with ASD features, including midface hypoplasia, severe growth retardation (−4.81 Shwachman–Diamond syndrome (SDS) height), progressive postnatal development of “cup-shaped” knee metaphyses, and unilateral humeral bowing, demonstrating mosaic growth plate involvement. Whole-genome sequencing revealed a novel PDE4D missense variant (c.934C>T, p. Leu312Phe) in the upstream conserved region 2 (UCR2) autoinhibitory domain, which is distinct from known acrodysostosis-associated variants. Expanding the clinical and radiological characteristics, as well as the mutation spectrum of PDE4D-related ASD, is crucial for understanding syndrome variability, aiding in earlier detection, and improving recurrence risk assessment.
Introduction
Acroscyphodysplasia (ASD; Online Mendelian Inheritance in Man (OMIM) catalog number: 250215) is an ultra-rare autosomal dominant form of skeletal dysplasia characterized by metaphyseal scaphoid deformity of the knee joints, severe brachydactyly with cone-shaped epiphyses, distinct growth retardation, facial dysostosis with nasal hypoplasia, and intellectual disability (1, 2). The term metaphyseal ASD was introduced by Verloes et al. (3). The authors consolidated clinical data from four patients, two of whom had been previously reported by Bellini et al. and Jequier et al. They identified distinct radiological features, including a characteristic morphology of the knee joints characterized by cup-shaped metaphyses with embedded epiphyses (termed “skypho,” derived from the Greek word for “cup”) and micromelia of the lower limbs. The prefix “acro-” denotes the involvement of the distal parts of the limbs (3–5). ASD has since been reported as a phenotypic feature of acrodysostosis type 2 (associated with PRKAR1A and PDE4D genes) and pseudohypoparathyroidism (associated with GNAS gene) (6).
The etiopathogenesis of ASD is not yet fully elucidated. To date, only seven cases of ASD have been reported, all associated with missense variants in the PDE4D gene, which is also implicated in the etiology of acrodysostosis type 2, with or without hormonal resistance (OMIM # 614613) (1, 2, 6, 7). Previous reports have suggested that the PDE4D gene may be responsible for ASD, representing a more severe form of skeletal dysplasia encompassing acrodysostosis. The protein product of the PDE4D gene is cAMP-specific phosphodiesterase 4D (PDE4D), a key component of the GPCR-cAMP-PKA signaling pathway, which mediates cellular hormonal control and regulates hypertrophic differentiation of growth plate chondrocytes (8, 9). Given the limited number of reported PDE4D-related ASD cases, accumulating data on new ASD cases will contribute to a deeper understanding of its clinical manifestations, pathogenic mechanisms, and optimization of diagnostic approaches.
In this study, we analyzed the clinical and radiological characteristics of ASD in a 7-year-old girl carrying a novel de novo heterozygous variant in the PDE4D gene.
Materials and methods
A comprehensive examination was conducted on a 7-year-old female proband presenting with phenotypic features of skeletal dysplasia, developmental delay, and facial dysmorphisms. To refine the diagnosis, a comprehensive clinical-genealogical analysis was performed, along with X-rays of the spine, hip joints, and long bones of the extremities, as well as molecular genetic testing.
Written informed consent was obtained from the proband’s parents. The study was performed in accordance with the Declaration of Helsinki and approved by the Institutional Review Board of the Research Centre for Medical Genetics, Moscow, Russia.
Blood samples were collected from the proband and his unaffected parents, and genomic DNA was extracted by standard methods using a Wizard Genomic DNA Purification Kit (Promega, WI, United States).
Whole-genome sequencing was performed using a DNBSEQ-G400 instrument in paired-end mode (2 × 150 bp), with an average on-target coverage of 30×. Library preparation was carried out using the MGIEasy FS PCR-Free DNA Library Prep Set (BGI, Beijing, China). Bioinformatic analysis was performed using an in-house software pipeline, as previously described [PubMed Identifier (PMID): 29504900], with modifications. In brief, quality control of raw reads was conducted using FastQC (version 0.11.5), followed by read mapping to the hg19 human genome assembly using minimap2 (version 2.24-r1122). Alignments were sorted, and duplicates were marked using the Picard Toolkit (version 2.18.14). Base recalibration and variant calling were performed using GATK3.8. Variant annotation was performed using the ANNOVAR tool (version 2018-04-16). Copy number variation (CNV) and structural variation (SV) analyses were performed using Manta (version 1.6.0). Further filtering was based on functional consequences and population frequencies, according to the American College of Medical Genetics and Genomics (ACMG) recommendations (10), as well as clinical relevance determined using the Human Phenotype Ontology database. The complementary DNA (cDNA) and protein positions in the PDE4D gene corresponded to transcript NM_001104631.2. Identified PDE4D variants were validated by Sanger sequencing according to standard protocols, using a 3500xL Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific, MA, United States).
Clinical case
The proband was a 7-year-old girl who underwent clinical evaluation due to hyperactivity, delayed motor and speech development, short stature, obesity, and difficulties with independent ambulation. The parents were healthy, non-consanguineous, and had three older healthy daughters and one older healthy son. Prenatal ultrasound during the final weeks of gestation revealed a 2-week intrauterine growth retardation. The child was delivered by cesarean section at 40–41 weeks of gestation with a birth weight of 2,600 g (−2.09 SDS) and a length of 48 cm (−1.07 SDS). Craniofacial dysmorphisms, including hypertelorism and a saddle nose, were noted at birth. Brachydactyly of both hands and feet became obvious by 4 months of age. Due to the facial phenotype and brachydactyly, achondroplasia was suspected, and a screen for variants in the FGFR3 gene was performed, yielding no positive outcome. Standard cytogenetic testing demonstrated a normal female karyotype 46, XX.
The patient exhibited global developmental delay, with the following milestones: head control at 9 months, independent sitting at 18 months, and ambulation at 20 months of age. Expressive language was limited to vocalizations without meaningful speech. Receptive language skills were partially impaired (inconsistent responses to names and commands). Stereotypic hand-flapping movements were observed during periods of excitation. The patient remained dependent on spoon-feeding and lacked toilet training skills. Recurrent upper respiratory infections were documented in early childhood. During 1–6 years of age, the patient experienced febrile seizures (2–3 annual episodes). Video-EEG monitoring showed no epileptiform activity. Progressive weight gain led to a class-IV obesity diagnosis by 6 years of age. Progressive weight gain became clinically evident after 4 years of age, culminating in a diagnosis of stage-IV obesity by 6 years of age. Orthopedic evaluation revealed radiographic features suggestive of Cerebral, Ocular, Dental, Auricular, Skeletal (CODAS) syndrome, including bifid distal femoral morphology and nasal hypoplasia. Ophthalmological examination demonstrated retinal angiopathy. Brain magnetic resonance imaging (MRI) findings included corpus callosum hypoplasia, periventricular leukopathy (localized to the posterior horns of the lateral ventricles), Arnold–Chiari malformation type I, and hyperostosis of the calvarial bones (Figure 1C). Cardiac evaluation identified a 2.5-mm foramen ovale. Abdominal ultrasonography confirmed left renal lumbar ectopia.
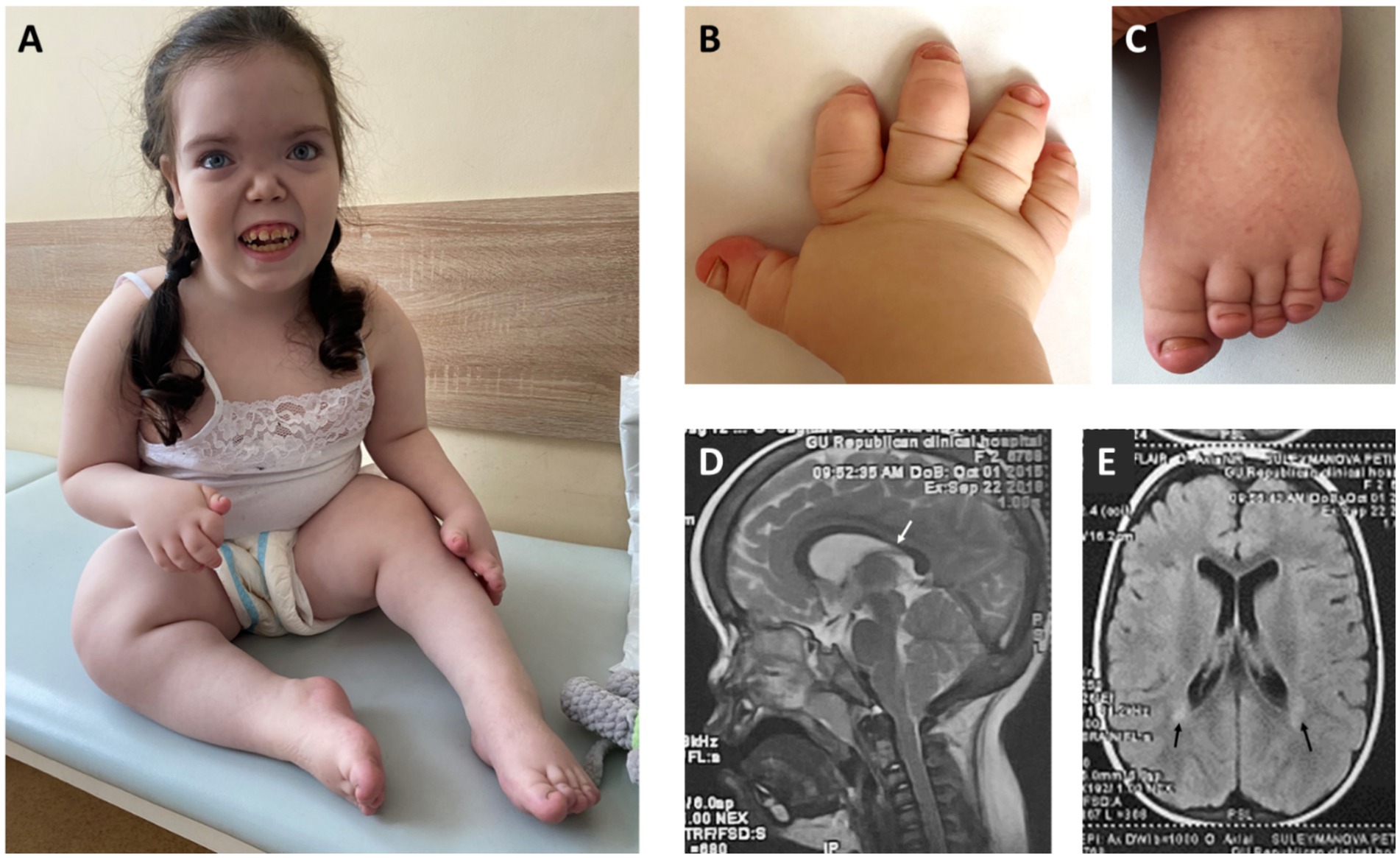
Figure 1. (A) - Facial features: broad forehead with prominent frontal bossing, hypertelorism, low nasal bridge, midface hypoplasia, long smooth philtrum, tented upper lip, protruding upper incisors, enamel hypoplasia, open mouth. Obesity, growth retardation with micromelia, knee joint contractures. (B,C) – Severe brachydactyly with nail hypoplasia. (D) – Midsagittal view of brain MRI – thinning of the isthmus of corpus callosum (white arrow). (E) – Axial view of brain MRI – periventricular leukomalacia adjacent to the external angles of the lateral ventricles (black arrows).
Clinical evaluation at 7 years of age revealed severe growth retardation (height 95 cm, −4.79 SDS), excessive body weight (30 kg, body mass index (BMI) + 4.17 SDS), and a trend toward macrocephaly (head circumference 54 cm, +1.93 SDS). The patient exhibited facial features such as flat face, severe midface hypoplasia, flat nasal bridge, hypertelorism, telecanthus, and disproportionate skeletal features, such as rhizomelic limb shortening and brachydactyly of hands and feet, with marked shortening of distal phalanges and nail hypoplasia (Figures 1A–C). Neurological examination demonstrated flexion contractures of the knee joints, generalized muscle hypotonia, and an abnormal waddling gait characterized by knee flexion posture and forward trunk inclination. Brain MRI findings included thinning of the isthmus of the corpus callosum and periventricular leukomalacia (Figures 1D,E).
The most striking radiological findings were demonstrated in the knees and hands. Interestingly, typical epi-metaphyseal deformity was not presented in the early radiographs—distal femoral epiphyseal growth plates were not deformed, and the shape of epiphyses was not changed at 3 months of age. At 3 years of age, anteroposterior radiographs of the knees already documented noticeable cone-shaped epiphyses of both distal femora and proximal tibiae, which progressed to the cupped metaphyseal deformity (typical scypho-deformity of the knees) at 7 years of age. Radiographs of the hips demonstrated normal radiographic parameters and intact growth plates. Spine radiographs show mild scoliosis and thoracolumbar kyphosis; interpedicular distance at L3–L5 remains unchanged. Severe bowing of the left humerus and valgus deformity of the elbow were found on the left side, with a relatively normal right upper limb. Short metacarpals and phalanges (mostly middle phalanges) and cone-shaped epiphyses of metacarpal bones were found on the radiographs of the hands (Figure 2).
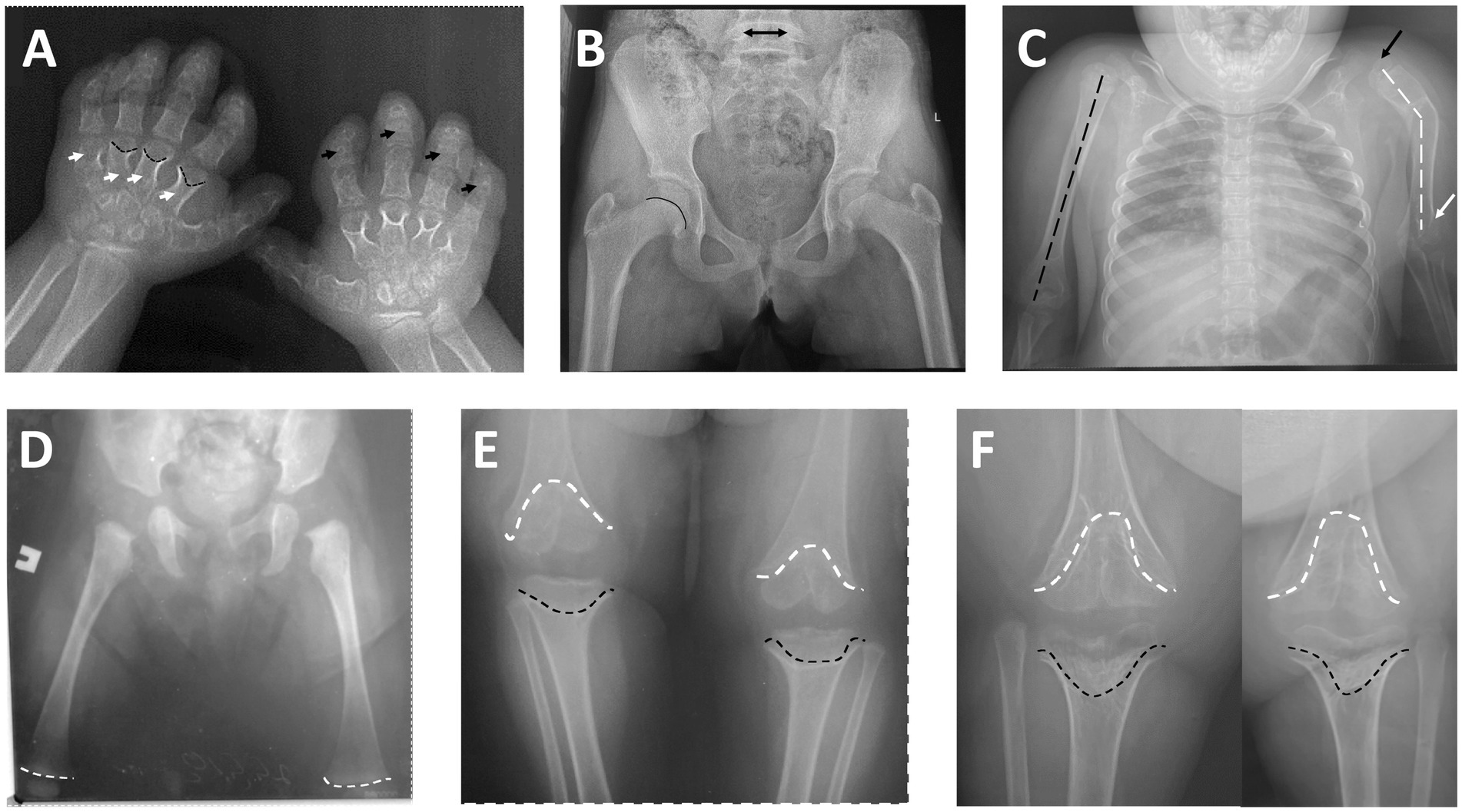
Figure 2. (A) Posteroanterior radiograph of the hands: short metacarpals (white arrows) and phalanges—mostly middle phalanges (black arrows), cone-shaped epiphyses of metacarpal bones (black broken lines). (B) Anteroposterior radiograph of the hips (at 7 years of age)—normal radiographic parameters, intact growth plate (black line) and normal interpedicular distance of L5 (black arrow). (C) Anteroposterior radiograph of the chest and upper limbs: normal axis of the right humerus (black broken line) with intact shoulder and elbow; severe axial deformity of the left humerus (black broken lines), subluxation of the left shoulder (black arrow), and deformity of the left elbow (white arrow). (D) Anteroposterior radiograph of the hips and femoral bones (age 3 months)—notice normal distal femoral growth plates (white broken lines). (E) Anteroposterior radiograph of the knee joints (at 3 years of age): remarkable epi-metaphyseal deformity (cone-shaped) of both distal femur (white broken lines) and proximal tibia (black broken lines). (F) Anteroposterior radiograph of the knee joints (at 7 years of age): progress of the epi-metaphyseal deformity (wedge-shaped) of both distal femur (white broken lines) and proximal tibia (black broken lines).
Biochemical parameters of mineral metabolism, including total serum calcium, inorganic serum phosphorus, parathyroid hormone, and alkaline phosphatase levels, were found to be within the established reference ranges. However, a deficiency of vitamin D-25-OH was identified, with a concentration of 24.96 ng/mL, which falls below the normal range of 30–100 ng/mL.
Whole-genome sequencing identified a novel pathogenic variant in exon 7 of the PDE4D gene (c.934C>T), resulting in a heterozygous missense substitution, p. Leu312Phe. The identified variant was confirmed by Sanger sequencing in the proband’s DNA but was not present in the parents’ DNA, confirming it as de novo. This variant has been classified as likely pathogenic based on the ACMG guidelines (PM2, PM5, and PM1) (10).
Discussion
ASD is an ultra-rare skeletal dysplasia characterized by distinctive radiological findings such as cup-shaped metaphyses with embedded epiphyses (particularly evident in the knees), combined with characteristic clinical features including acrodysostosis-like brachydactyly and nasal hypoplasia, which together constitute the primary diagnostic criteria for this condition (2). Furthermore, short stature and intellectual disability have been consistently documented in all reported cases. In the presented case, a 7-year-old girl exhibited pronounced phenotypic and radiological features—including midface and nasal hypoplasia, metaphyseal cupping, brachydactyly, severe short stature (−4.81 SDS), and obesity—suggestive of a rare skeletal dysplasia with intellectual disability. Given the overlapping clinical manifestations with other hereditary disorders, the diagnosis of ASD was challenging due to its rarity. Thus, the presence of cognitive impairment combined with coarse facial features, obesity, short stature, and brachydactyly initially raised suspicion for CHOPS syndrome (OMIM catalog number: 616368). However, the proband lacked commonly associated cardiac and pulmonary anomalies, and radiographic skeletal findings were inconsistent with this diagnosis (11). In addition, the metaphyseal changes observed in the proband’s knee joints showed partial similarity to the characteristic ‘bifid’ appearance of the distal femur noted in CODAS syndrome (OMIM: 600373), which similarly presents with developmental delay, short stature, midface and nasal hypoplasia, and other congenital anomalies (12). Ultimately, whole-genome sequencing led to a definitive diagnosis by identifying a novel de novo heterozygous missense variant in exon 7 of the PDE4D gene (c.934C>T, p.Leu312Phe) that had not been previously reported. Most known pathogenic variants in this gene, which primarily cause acrodysostosis type 2, are missense mutations distributed throughout the coding sequence. However, previously described missense variants in ASD cases are located in the N-conserved domains UCR1 and upstream conserved region 2 (UCR2) (Figure 3B). These domains interact with the catalytic domain of the adjacent monomer to form an autoinhibitory domain that partially occludes the catalytic site (13, 15). The novel variant we detected results in the substitution of leucine (with its non-polar alkyl R-group) by phenylalanine (containing an aromatic R-group) at position 312 (p.Leu312Phe) (Figure 3A), located within the conserved UCR2 domain of PDE4D (Figure 3B).
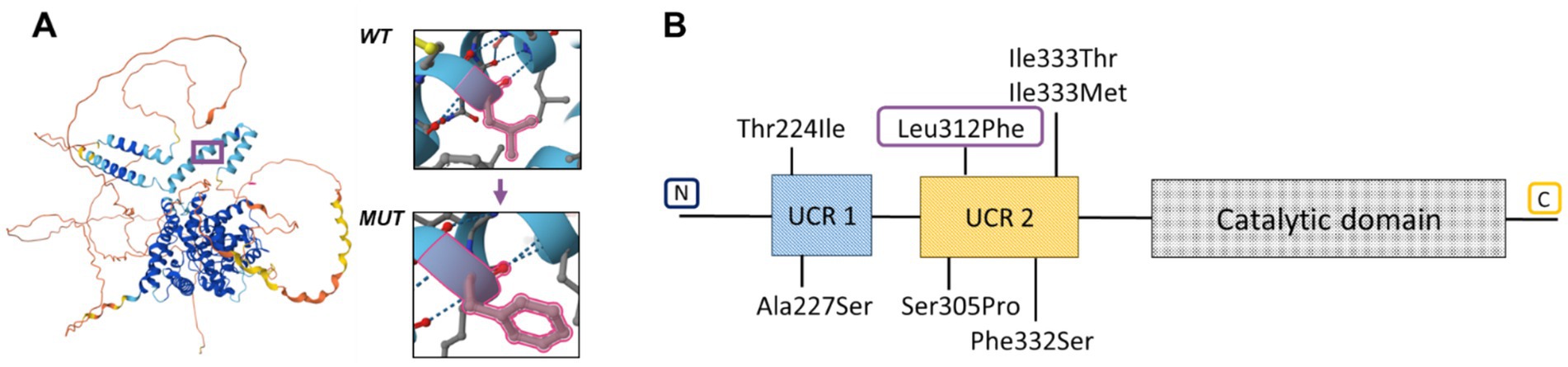
Figure 3. (A) Structure of protein PDE4D with amino acid substitution. (B) Genotype of patients with acroscyphodysplasia in PDE4D.
It should be noted that another substitution at the same position (c.934C>G), resulting in a leucine-to-valine change (p.Leu312Val), was previously reported by Briet et al. (14). Similar to leucine, valine contains a non-polar aliphatic side chain. The authors demonstrated that this variant, along with three other variants, led to increased activation of PDE4D3 expressed in Chinese hamster ovary cells, thereby attenuating PTHR-cAMP-PKA signaling during development. The PDE4D isoform is highly expressed in the brain, particularly in the hippocampus. Mice lacking the PDE4D gene show enhanced long-term potentiation, which is associated with improved learning, memory, and even neural regeneration (16). This particular variant was identified in a 42-year-old woman who presented with severe intellectual disability, marked brachydactyly, brachymetatarsia, and brachymetacarpia, short stature (139 cm − 3.74 SDS), thickened bones, and obesity. Vitamin D supplementation in this patient normalized previously elevated parathyroid hormone (PTH) levels, that is, 107 pg./mL (normal range 10–65). Despite the phenotypic severity, the report did not include descriptions of facial phenotype or detailed radiographic findings of the knee joints.
Previous reports on ASD patients have demonstrated that the disorder incorporates acrodysostosis-like features such as more severe brachydactyly, brachymetatarsia, and brachymetacarpia with cone-shaped epiphyses, along with characteristic metaphyseal cup-shaped deformities of the knees. The phenotype consistently includes shortened and abnormally curved diaphyses of long bones, significant postnatal growth retardation (ranging from −3 to −8.8 SDS), delayed walking onset, and progressive flexion contractures of the knee joints. Notably, two reported ASD cases exhibited severe midface hypoplasia with nasal stenosis, resulting in clinically significant respiratory and swallowing difficulties, obstructive sleep apnea, and chronic otitis media (1, 6). In our reported case, the 7-year-old girl exhibited severe acrodysostosis with cone-shaped epiphyses of metacarpal bones and phalanges, combined with knee cup-shaped metaphyses containing embedded epiphyses, along with bowed left humerus and profound growth retardation (−4.81 SDS).
The general spectrum of the radiological changes found in the proband is typical for the condition and resembles the previously published data (1). Among the interesting radiological findings, a typical cupped metaphyseal deformity was not present in the early radiographic images. The previous publication did not present a series of radiographs demonstrating the natural history of radiological changes. The earliest published radiograph of the lower limbs at 7 months of age (6) demonstrates an obvious abnormality of the growth plates. In our patient, a radiograph obtained at 3 months of age revealed intact growth plates and a normal shape of the epiphyses. During the 3–7 year age range, cupped metaphyseal deformity progressed to typical scypho-deformity. Interestingly, radiographs of the hips were basically normal; the same pattern of radiologically normal hips can be noted in the previous publications (1–3, 6). Severe bowing of the left humerus and valgus deformity of the elbow secondary to the growth disturbances were found on the left side, demonstrating a mosaic pattern of damage. This unilateral humerus bowing was also noticed in the patient with ASD (6). In contrast, short metacarpals and phalanges were symmetrical and equally distributed among the fingers.
Our findings, demonstrating the postnatal origin of the scypho-deformity in ACD, are quite interesting and unique. It is unclear which factor initiates permanent damage to the growth plates, especially taking into account the mosaic (unilateral and/or asymmetrical) pattern. This unusual segmental distribution and atypical local pattern, characterized by predominantly central growth plate arrest leading to scypho-deformity, led to misdiagnosis with sequelae of neonatal sepsis and meningococcemia (6). Further studies can help to clarify the underlying mechanism of this selectivity.
Thinning of the isthmus of the corpus callosum and periventricular leukomalacia found on the brain MRI are within the spectrum of non-specific changes that can be found both in congenital and acquired CNS pathologies responsible for developmental delay and movement disorders (17).
The patient presented with a complete absence of speech, lack of social interaction, hyperexcitability, stereotypic hand-flapping movements, inability for self-care, and severely impaired wide-based gait with knee flexion and forward trunk inclination. Marked midface hypoplasia and nasal hypoplasia were present since birth, exacerbated by recurrent acute respiratory infections and febrile episodes. Notably, the patient showed no hormonal resistance. Literature review reveals that only two reported ASD cases demonstrated mild parathyroid hormone resistance, with one additional case showing peripheral hypothyroidism, while no other endocrine abnormalities have been documented (1, 2, 6).
To date, only six missense variants in the PDE4D gene have been identified in ultra-rare ASD cases, including our reported one (1, 2, 6, 7) (Table 1). Recently reported two ASD cases with GNAS variants further suggest genetic heterogeneity of this disorder (1). These findings indicate that ASD likely represents part of a broader spectrum of diseases characterized by overlapping clinical features and dysregulation of the intracellular cAMP-dependent signaling pathway. This disease continuum may encompass both forms of acrodysostosis (caused by PRKAR1A and PDE4D variants), Albright hereditary osteodystrophy, and pseudohypoparathyroidism (caused by GNAS variants).
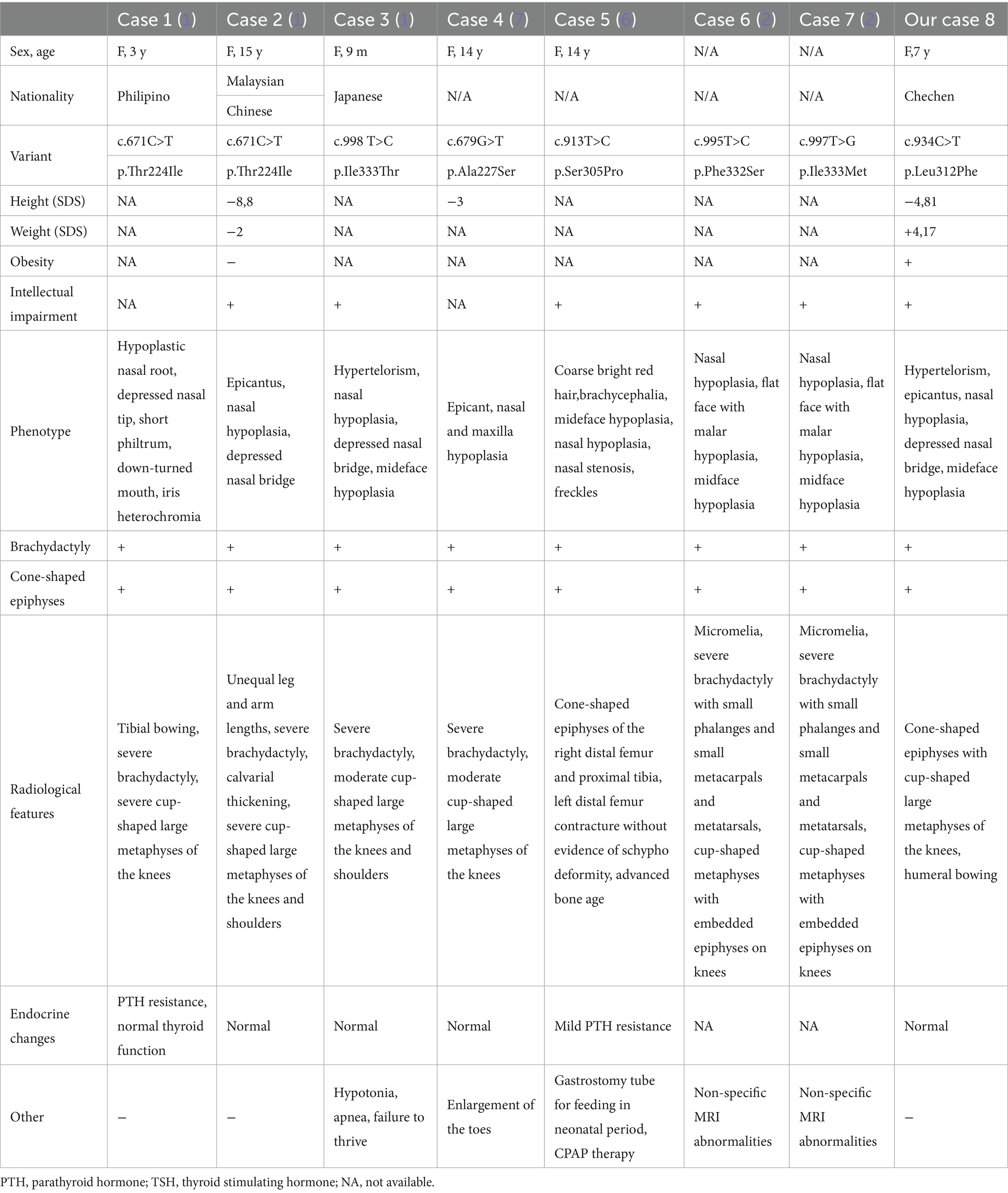
Table 1. Phenotype and genotype of patient with acroscyphodysplasia with variants in PDE4D gene.
Our reported case significantly contributes to expanding both the phenotypic and genotypic spectrum of ASD, while providing critical evidence for its recognition as a distinct nosological entity within the classification of genetic skeletal disorders. The detailed clinical and molecular characterization of this patient enhances the current understanding of genotype–phenotype correlations and the molecular mechanisms underlying skeletal development, particularly those mediated by cAMP signaling. This case underscores the importance of comprehensive molecular analysis in atypical presentations of skeletal dysplasia and contributes to the evolving taxonomy of genetic disorders involving cAMP signaling defects. The accumulation of such carefully characterized cases will be essential for establishing a definitive diagnostic pathway and understanding the full clinical spectrum of ASD.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of the Research Center for Medical Genetics, Russia (protocol code 2021-3, 12 March 2021). Written informed consent was obtained from the parents for the publication of any images or data included in this article.
Author contributions
AM: Writing – review & editing, Investigation, Data curation, Formal analysis, Conceptualization. MS: Conceptualization, Supervision, Writing – original draft, Validation, Visualization. VK: Writing – review & editing, Validation. MO: Writing – review & editing, Investigation, Data curation, Formal analysis. OR: Investigation, Writing – review & editing, Formal analysis, Data curation. TM: Conceptualization, Data curation, Formal analysis, Writing – review & editing, Investigation, Supervision, Methodology.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The research was carried out within the state assignment of the Ministry of Science and Higher Education of the Russian Federation for the Research Centre for Medical Genetics (RCMG).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mitsui, T, Kim, O-H, Hall, CM, Offiah, A, Johnson, D, Jin, D-K, et al. Acroscyphodysplasia as a phenotypic variation of pseudohypoparathyroidism and acrodysostosis type 2. Am J Med Genet A. (2014) 164:2529–34. doi: 10.1002/ajmg.a.36669
2. Michot, C, Le Goff, C, Blair, E, Blanchet, P, Capri, Y, Gilbert-Dussardier, B, et al. Expanding the phenotypic spectrum of variants in PDE4D/PRKAR1A: from acrodysostosis to acroscyphodysplasia. Eur J Hum Genet. (2018) 26:1611–22. doi: 10.1038/s41431-018-0135-1
3. Verloes, A, Le, MM, Farriaux, J-P, and Maroteaux, P. Metaphyseal acroscyphodysplasia. Clin Genet. (1991) 39:362–9. doi: 10.1111/j.1399-0004.1991.tb03043.x
4. Bellini, FBM. Su un caso di disostosi periferica. [A case of peripheral dysostosis.]. Minerva Pediatr. (1966) 18:106–10.
5. Jequier, S, Bellini, F, and Mackenzie, DA. Metaphyseal chondrodysplasia with ectodermal dysplasia. Skeletal Radiol. (1981) 7:107–12. doi: 10.1007/BF00347374
6. Kartalias, K, Gillies, AP, Penã, MT, Estrada, A, Bulas, DI, Ferreira, CR, et al. Fourteen-year follow-up of a child with acroscyphodysplasia with emphasis on the need for multidisciplinary management: a case report. BMC Med Genet. (2020) 21:189. doi: 10.1186/s12881-020-01127-6
7. Linglart, A, Fryssira, H, Hiort, O, Holterhus, P-M, Perez de Nanclares, G, Argente, J, et al. PRKAR1A and PDE4D mutations cause Acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab. (2012) 97:E2328–38. doi: 10.1210/jc.2012-2326
8. Loid, P, Pekkinen, M, Reyes, M, Mustila, T, Viljakainen, H, Jüppner, H, et al. GNAS, PDE4D, and PRKAR1A mutations and GNAS methylation changes are not a common cause of isolated early-onset severe obesity among Finnish children. Front Pediatr. (2020) 8:145. doi: 10.3389/fped.2020.00145
9. Mika, D, and Conti, M. PDE4D phosphorylation: a coincidence detector integrating multiple signaling pathways. Cell Signal. (2016) 28:719–24. doi: 10.1016/j.cellsig.2015.11.001
10. Richards, S, Aziz, N, Bale, S, Bick, D, Das, S, Gastier-Foster, J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
11. Raible, SE, Mehta, D, Bettale, C, Fiordaliso, S, Kaur, M, Medne, L, et al. Clinical and molecular spectrum of CHOPS syndrome. Am J Med Genet A. (2019) 179:1126–38. doi: 10.1002/ajmg.a.61174
12. Royer-Bertrand, B, Castillo-Taucher, S, Moreno-Salinas, R, Cho, TJ, Chae, JH, Choi, M, et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci Rep. (2015) 5:154. doi: 10.1038/srep17154
13. Xie, M, Blackman, B, Scheitrum, C, Mika, D, Blanchard, E, Lei, T, et al. The upstream conserved regions (UCRs) mediate homo-and hetero-oligomerization of type 4 cyclic nucleotide phosphodiesterases (PDE4s). Biochem J. (2014) 459:539–50. doi: 10.1042/BJ20131681
14. Briet, C, Pereda, A, Le Stunff, C, Motte, E, de Dios Garcia-Diaz, J, de Nanclares, GP, et al. Mutations causing acrodysostosis-2 facilitate activation of phosphodiesterase 4D3. Hum Mol Genet. (2017) 26:3883–94. doi: 10.1093/hmg/ddx271
15. Cedervall, P, Aulabaugh, A, Geoghegan, KF, McLellan, TJ, and Pandit, J. Engineered stabilization and structural analysis of the autoinhibited conformation of PDE4. Proc Natl Acad Sci USA. (2015) 112:E1414–22. doi: 10.1073/pnas.1419906112
16. Rutten, K, Misner, DL, Works, M, Blokland, A, Novak, TJ, Santarelli, L, et al. Enhanced long-term potentiation and impaired learning in phosphodiesterase 4D-knockout (PDE4D−/−) mice. Eur J Neurosci. (2008) 28:625–32. doi: 10.1111/j.1460-9568.2008.06349.x
Keywords: acroscyphodysplasia, PDE4D , scaphoid knee deformities, brachydactyly, growth retardation
Citation: Morgul A, Sharova M, Kenis V, Orlova M, Ryzhkova O and Markova T (2025) Case Report: The widening genetic and phenotypic spectrum of ultra-rare PDE4D-related acroscyphodysplasia. Front. Med. 12:1623593. doi: 10.3389/fmed.2025.1623593
Edited by:
Hao Zhang, The Affiliated Hospital of Qingdao University, ChinaReviewed by:
Patrizia Ferretti, University College London, United KingdomMelissa Schepers, University of Hasselt, Belgium
Copyright © 2025 Morgul, Sharova, Kenis, Orlova, Ryzhkova and Markova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margarita Sharova, c2hhcm92YUBtZWQtZ2VuLnJ1