 Jiale Li1†
Jiale Li1† Bangchuan Hu
Bangchuan Hu- 1The Second Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, China
- 2Emergency and Critical Care Center, ICU, Zhejiang Provincial People’s Hospital (Affiliated People’s Hospital, Hangzhou Medical College), Hangzhou, Zhejiang, China
There is a growing body of evidence indicating that the stimulation of one organ can significantly influence the functioning of another. For instance, intestinal complications are frequently observed during respiratory diseases, and conversely, pulmonary complications can arise during intestinal diseases—a phenomenon referred to as lung-gut crosstalk. Patients suffering from mechanical ventilator-induced lung injury, chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), and other pulmonary conditions have been shown to experience gastrointestinal dysfunction and related disorders. Similarly, individuals with inflammatory bowel disease (IBD) have also been found to develop pulmonary complications. However, these studies are not enough to fully explain the mechanism of lung-intestinal crosstalk, and more potential mechanisms need to be explored and further elucidated. In this paper, we summarize recent research advancements regarding lung-intestinal interactions in the context of pulmonary and intestinal diseases, analyzing the potential mechanisms of lung-intestinal crosstalk from the perspectives of respiratory mechanics, inflammation, and microbiota. Additionally, we review evidence suggesting that adipokines may play a role in lung-gut interactions, and we propose new avenues for investigating the mechanisms underlying these interactions.
1 Introduction
As early as 1968, Warwick introduced the concept of lung-gut interaction, providing significant evidence for this phenomenon (1). With advancements in technology and improvements in clinical practices, it has been observed that patients with mechanical ventilation, lung injury, and lung diseases such as chronic obstructive pulmonary disease (COPD) and acute respiratory distress syndrome (ARDS) exhibit a higher incidence of gastrointestinal disorders. Conversely, respiratory diseases have also been identified in patients suffering from inflammatory bowel disease (2). These findings indicate a profound physiological and pathological relationship between the respiratory system and the intestine. Despite the increasing body of evidence elucidating lung-gut interaction, the underlying mechanisms remain poorly understood. Several population-based and observational studies have revealed an increased prevalence of respiratory diseases among patients with IBD, including ulcerative colitis (UC) and Crohn’s disease (CD), even in non-hospitalized settings. For instance, a nationwide cohort study reported that individuals with IBD had higher risks of developing asthma, bronchiectasis, and COPD, independent of smoking status and other confounders (3, 4). Conversely, individuals with chronic respiratory diseases such as asthma and COPD also exhibit higher incidences of IBD compared to the general population (5, 6). These findings indicate that the lung-gut axis interaction is not limited to critically ill or ventilated patients but may exist as a broader phenomenon affecting immunologically and microbially susceptible populations. The lung–gut axis has been increasingly recognized as a crucial mediator of pulmonary immune responses. Recent studies have elucidated the bidirectional interactions between gut microbiota and pulmonary inflammation in various diseases including lung cancer, asthma, and infections (7, 8). Such evidence underscores the importance of further investigating the mechanisms linking these two systems. In this paper, the authors analyze the mechanisms linking pulmonary and intestinal diseases to enhance our understanding of lung-intestinal interaction. Additionally, we explore the role of adipokines in this interaction, offering new insights into its effects.
2 Evidence for the existence of lung-gut crosstalk
To build the foundation for understanding the lung-gut axis, we first summarize clinical and experimental evidence demonstrating bidirectional interactions between pulmonary and intestinal systems. The lungs and intestines originate from the anterior region of the intestinal endoderm. Although they are anatomically separate, stimulation of one organ affects the other compartments, suggesting important communication between the gut and lungs.
2.1 From the lung to gut
The lung can influence intestinal function through multiple pathways, particularly under conditions of acute or chronic respiratory disease. Various lung diseases, including Ventilator-Induced Lung Injury (VILI), COPD, and respiratory tract infections caused by viruses or bacteria, can lead to gastrointestinal disease or dysfunction (9–11). VILI refers to the acute lung damage caused or worsened by mechanical ventilation. This type of injury can occur during both invasive and non-invasive ventilation and has been shown to play a significant role in the morbidity and mortality of critically ill patients (12). The primary mechanisms responsible for ventilator-induced lung injury are alveolar overdistention (volutrauma), barotrauma, atelectotrauma, and inflammation (biotrauma). Additional contributing factors include adverse heart-lung interactions, deflation-related injuries, and effort-induced injuries (13). While mechanical ventilation causes lung damage, it is also accompanied by changes in intestinal diseases. In a previous retrospective analysis, a study included a total of 242 patients, with 113 (46.7%) experiencing gastrointestinal bleeding. Among these patients, 86 had coffee ground material or positive occult blood in nasogastric aspirates, 12 had positive occult blood in stools, 5 had hematemesis, 2 had hematochezia, and 2 had both hematemesis and hematochezia. Clinically significant bleeding was diagnosed in eight patients (3.3%) with gastrointestinal bleeding. The majority of patients (67.3%) developed gastrointestinal bleeding within the first 48 h of mechanical ventilation, and 80% experienced it within the first 2 weeks (11).
However, the causes of COPD are often associated with long-term smoking and inhaling air pollutants. Its main features include persistent airflow obstruction and respiratory symptoms. Smoking is considered an independent risk factor for COPD and various gastrointestinal diseases such as Crohn’s disease (CD) (14). Multiple studies have shown that smoking may increase the risk of Crohn’s disease by three times (15). In a recent study, smoking was found to be associated with several functional gastrointestinal disorders and functional symptoms such as functional bloating, functional abdominal pain, functional diarrhea, and functional constipation (16). A population-based cohort study by Ekbom et al. (17) revealed that COPD patients had a 2.72% higher risk of developing CD and a 1.83% higher risk of developing ulcerative colitis (UC) compared to the control group (17). Additionally, a previous questionnaire-based prospective study showed that the highest proportion of patients with maximal gastrointestinal (GI) symptoms was found in the “GOLD-4” group of patients with COPD and the “uncontrolled” group of patients with asthma. From this, we can see that not only does lung-gut crosstalk exist, but the severity of symptoms in one system (bowel) is highly consistent with the severity of the other system (lung) (15).
In intensive care units, sepsis is the main indirect cause of acute lung injury and acute respiratory distress syndrome (18). Although the pathogenesis of sepsis, acute lung injury (ALI), and ARDS is complex and multifactorial, there are still some phenomena that support the interaction between lung and gut. In clinical cases, the most common source of sepsis is the lung (19). But during antibiotic treatment, the intestinal flora is also affected, resulting in microbial imbalance. Rosa discovered that Vancomycin treatment for acute Pseudomonas aeruginosa pneumonia in mice can lead to intestinal dysbacteriosis, causing an increase in Proteus and a decrease in Bacteroides, along with inflammatory changes in the intestinal tract (20). Following fecal microbiota transplantation, the susceptible and tissue injury phenotypes were reversed in mice. Additionally, pulmonary allergic responses impact the composition of the intestinal microbiota (21). In experimental influenza infection, it has been observed that IFN-γ produced by lung-derived CCR9 + CD4 + T cells alter the gut microbiota composition and induce intestinal immune injury (22). Furthermore, pulmonary IFN-Is production promotes the depletion of obligate anaerobic bacteria and the proliferation of proteobacteria in the gut, leading to significant intestinal dysregulation (23). Therefore, lung inflammation directly influences the structure of the intestinal bacterial community, exacerbating lung inflammation.
2.2 From the gut to lung
Conversely, gastrointestinal inflammation, especially in IBD, has been shown to contribute to pulmonary manifestations, highlighting the bidirectional nature of the lung-gut axis. When it comes to the impact of intestinal diseases on the lungs, inflammatory bowel disease is a very typical condition that causes lung dysfunction. IBD is a term used to describe chronic recurring inflammation in the gastrointestinal tract, with the major types being Crohn’s disease (CD) and ulcerative colitis (UC). CD is identified by transmural non-continuous and non-caseating granulomatous inflammation that can occur anywhere in the gastrointestinal tract but is most frequently found in the terminal ileum (24, 25). On the other hand, UC is characterized by continuous inflammation starting in the rectum and spreading upwards. Unlike CD, UC inflammation only affects the mucosa and submucosa, specifically in the colon (26, 27). Studies suggest that 40–60% of patients with IBD exhibit subclinical lung complications, which can be detected through changes in pulmonary function tests and high-resolution tomographic imaging (HRCT) (28). With the increasing use of bronchoscopy and histological examinations, a growing number of patients with IBD are being identified as having potential pulmonary dysfunction even in the absence of clear symptoms (29, 30). Various aspects of pulmonary involvement in IBD have been documented in the literature. For instance, Camus observed that out of 33 patients with ulcerative colitis (UC) or Crohn’s disease (CD), 60% exhibited bronchiectasis, inflammatory/obliterative small airway lesions, or upper airway narrowing (31). In a review conducted by Desai, it was found that bronchiectasis was present in 45% of IBD cases with large airway involvement (32). Furthermore, a population-based cohort study revealed that individuals with IBD, specifically UC and CD, had a higher prevalence of bronchiectasis (46%), pulmonary vasculitis, and interstitial pneumonia (52%), lung nodules (35%), pulmonary fibrosis (16%), and asthma (5.5%) compared to those without IBD (33). A recent survey based on a questionnaire of 13,499 participants showed that IBD patients, particularly those with ulcerative colitis and women, had a higher prevalence of asthma and respiratory symptoms (34). Therefore, from the perspective of lung disease caused by IBD, the phenomenon of gut-lung interaction has obvious manifestations.
2.3 Hypoxia and intestinal injury in pulmonary disease
Hypoxia and hypoxemia associated with pulmonary diseases represent a crucial pathway mediating lung-gut interaction. Systemic hypoxia can disrupt intestinal homeostasis by impairing epithelial oxygenation, leading to ATP depletion, increased intestinal epithelial apoptosis, and compromised barrier function. Hypoxia-inducible factors (HIFs), particularly HIF-1α, play dual roles in maintaining mucosal integrity and mediating inflammation, depending on the duration and severity of hypoxic stress (35, 36). Moreover, oxygen deprivation alters the gut microbial ecology, favoring pro-inflammatory species and increasing the risk of bacterial translocation (37, 38). These mechanisms explain the occurrence of gastrointestinal dysfunction in patients with severe respiratory diseases, even in the absence of mechanical ventilation.
3 Mechanisms of pulmonary disease in lung-gut crosstalk: patients with mechanical ventilation
3.1 Impact of mechanical ventilation from a mechanistic point of view
Beyond epidemiologic observations, mechanistic studies help elucidate how mechanical ventilation physiologically influences gut function. Mechanical ventilation involves the use of a ventilator, a medical device frequently utilized in intensive care units to support respiratory function, optimize the exchange of gases, and prevent hypoxia and hypercapnia. Several experimental studies in humans and animals have explored the MV-intestinal interactions from a mechanistic perspective. Positive pressure ventilation increases intrathoracic pressure, leading to decreased venous return, cardiac preload, and cardiac output, ultimately resulting in hypotension. Animal experiments have shown that splanchnic blood flow decreases in tandem with reductions in cardiac output induced by mechanical ventilation, even when mean arterial pressure is maintained (39). Additionally, gastric mucosal ischemia due to hypoperfusion is a common cause of gastrointestinal bleeding in mechanically ventilated patients. A retrospective study of 283 ICU patients ventilated for over 48 h found that peak inspiratory pressure ≥ 30 cm H2O was an independent risk factor for gastrointestinal bleeding in this population (11). However, PEEP can autonomously impact regional perfusion, with a particular focus on hindering splanchnic perfusion. This hindrance in splanchnic perfusion may lead to compromised gut barrier function, resulting in intestinal hyperpermeability, bacterial translocation, and ultimately facilitating the onset of multiple organ failure (MOF) (Figure 1) (40). Experimental studies have demonstrated a dose-dependent relationship between PEEP and splanchnic blood flow. Splanchnic blood flow reduction is typically limited at PEEP levels below 10 cm H2O but becomes more pronounced within the range of 15–20 cm H2O (41).
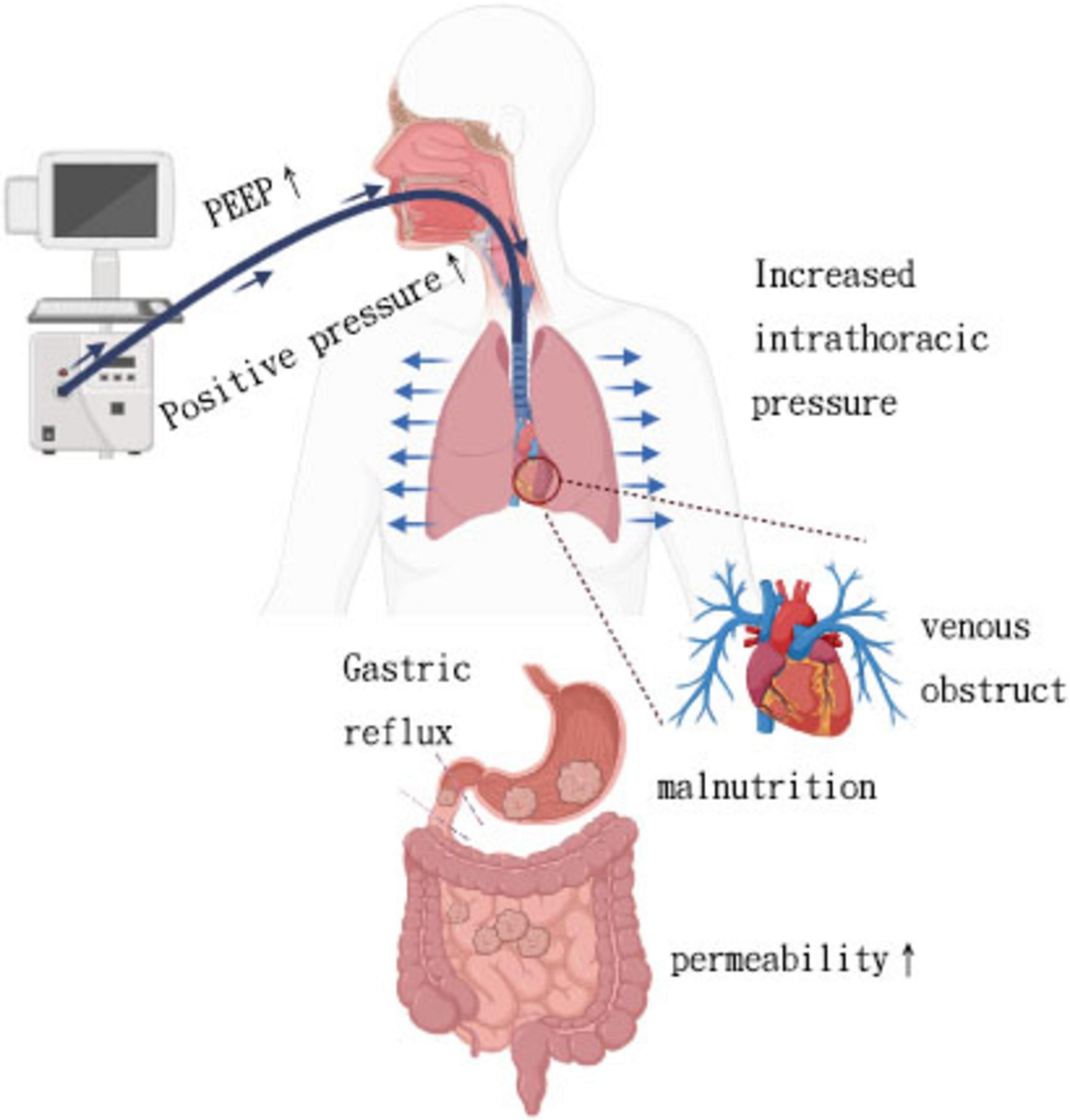
Figure 1. Interaction between mechanical ventilation and its effects on both lung and gut.
Positive pressure ventilation with high PEEP, while improving arterial oxygenation, can lead to blocked venous return, causing gastrointestinal tract congestion, edema, and feeding intolerance. In a recent retrospective study of 97 adult patients, Ryuichiro et al. found that high central venous pressure (CVP) during the first week after left ventricular assist device implantation was an independent risk factor associated with decreased enteral nutrition (EN) calorie intake (42).
The transmission of thoracic-abdominal pressure results in an increase in intra-abdominal pressure due to mechanical ventilation. Previous studies have also indicated that ventilatory-related pressure parameters may exacerbate gastrointestinal dysfunction by impacting intra-abdominal pressure (43, 44). Controlled mechanical ventilation results in increased internal abdominal pressure, causing the diaphragm to ascend. Prolonged mechanical ventilation can result in phrenic atrophy. Animal studies have demonstrated that even short durations of controlled mechanical ventilation, such as 18 h, can lead to diaphragm atrophy and weakness (45). This reduced ability of the diaphragm to contract may result in reflux of stomach contents, increasing the risk of gastroesophageal reflux for patients. Training the diaphragm of COVID-19 patients has been shown in previous studies to significantly reduce the risk of gastroesophageal reflux (46). Meanwhile, the possibility of gastrointestinal flora entering the lungs decreases.
In addition, commonly used drugs that support mechanical ventilation, such as opioids and sedatives (especially benzodiazepines), may reduce gastrointestinal motility and venous reflux through mechanisms such as venodilation or reduced response to vasopressors (47).
3.2 Impact of mechanical ventilation from an immunity point of view
The effects of mechanical ventilation extend beyond hemodynamics and include profound immunologic consequences, both locally in the lung and systemically, with implications for gut barrier integrity. Mechanical ventilation not only supplies the patient with oxygen, but it can also lead to lung injury (LI) due to increased inflation transpulmonary pressures (barotrauma), excessive alveolar distension (volutrauma), and the repetitive opening and closing of alveoli (atelectrauma). In addition to causing direct structural changes, these mechanical forces can also initiate an intricate inflammatory process, leading to inflammation at both local and systemic levels (biotrauma) (13). This inflammation has the potential to spread to other organs and systems, worsening multiple organ dysfunction and ultimately increasing mortality. Mechanical ventilation can potentially induce an inflammatory response and lead to lung damage, despite its effectiveness in maintaining gas exchange. These inflammatory processes may spread through the bloodstream, affecting organs outside the lungs and contributing to multiple organ failure (MOF).
It is believed that by increasing the permeability between alveoli and blood vessels, mechanical ventilation plays a role in expanding the inflammatory response beyond the lungs, ultimately causing damage to distant organs and impacting mortality rates (48). The inflammatory response induced by mechanical ventilation (MV), also known as biotrauma, is attributed to two distinct pathophysiologic mechanisms.
The first mechanism involves direct cellular trauma that disrupts cell walls, leading to the release of cytokines both locally at the alveolar level and systemically (49). The second mechanism, termed “mechanotransduction”, describes how cells detect mechanical forces through mechanosensation and convert them into cellular signaling events through mechanotransduction. In vitro studies have shown that a majority of pulmonary cells can produce cytokines in response to cyclic stretch (50). Mechanical injury triggers danger-associated molecular patterns (DAMPs) release, recruiting immune cells and inducing pro-inflammatory cytokine production. This, along with signaling cascade activation in alveolar epithelial and vascular endothelial cells from overstretching, as well as dysregulation of the neuroinflammatory reflex, results in a robust systemic inflammatory response. Prolonged exposure to harmful mechanical forces, especially high tidal volume MV, exacerbates this inflammatory process, leading to heightened alveolar-capillary barrier permeability and increased susceptibility to further lung damage (51).
Previous experimental studies have demonstrated that mechanical ventilation can induce the release of several pro-inflammatory mediators such as IL-1β, IL-6, IL-8, TNF-α, C-X-C motif ligand 1 (CXCL1), and CXCL10 (52). Meanwhile, elevated levels of TNF-α, IL-1β, and IL-6 as part of the inflammatory/sepsis cascade have been found to affect the permeability of tight junctions (TJ) in the intestine (53).
A team demonstrated that mechanical ventilation (MV) increases plasma TNF levels and gut permeability. They also found that administering a TNF-neutralizing antibody intravenously reversed gut hyperpermeability (54). Previous studies have highlighted the negative impact of TNF-α on the integrity of the intestinal epithelial barrier, either alone or in combination with other proinflammatory cytokines in various experimental models and cell culture settings (55–58). The mechanisms through which TNF-α disrupts the intestinal epithelial barrier involve multiple processes, such as inducing apoptosis in intestinal epithelial cells, changing the lipid composition of cell membranes, activating MLCK via calcium-calmodulin, promoting myosin light chain (MLC) phosphorylation by increasing MLCK protein expression, and reducing TJ protein expression. Particularly, the MLCK-mediated MLC phosphorylation pathway is crucial in the development of TNF-α-induced dysfunction in the intestinal epithelial barrier (59). In addition, IL-6 is a multifunctional inflammatory cytokine shown to participate in local and systemic inflammatory responses in the intestine, with pleiotropic effects. Whether IL-6 plays a protective role or disrupts the intestinal barrier is controversial (53). Recently, Ding used an experimental mice model of VILI to show increased levels of TNF-α, IL-1β, and IL-6 in both serum and gut tissues, measured by enzyme-linked immunosorbent assay (ELISA) (60). Additionally, Imai found that harmful ventilatory strategies, like high TV and zero PEEP, significantly increased epithelial cell apoptosis in the kidneys and small intestine, which correlated with organ dysfunction (61).
3.3 Impact of mechanical ventilation from a microbiome point of view
Another critical dimension of lung-gut interaction lies in the disruption of the respiratory and gastrointestinal microbiomes under mechanical ventilation. Utilizing culture-independent molecular techniques, researchers have identified the presence of non-pathogenic anaerobes such as Prevotella, Veillonella, and Fusobacterium in the normal alveoli of humans, which likely originate from the oropharyngeal flora (62–64). These lower levels of bacteria are believed to contribute to the maintenance of the ecological balance within the lungs (65).
In certain critical situations, patients in the ICU may require various interventions, including mechanical ventilation, antibiotic therapy, continuous blood purification, and immunosuppressive regimens (66). These interventions have the potential to impact the microbial composition and diversity of these patients (67–69). Changes in the lung microbiome due to critical illness are closely linked to both bacterial translocation, systemic, and local inflammation (62, 67, 70–72). The commensal microbial populations in the lungs may be displaced by potential pathogens from other ecosystems, such as the gastrointestinal tract and the skin (62, 73). In the study of BALF from ARDS patients, Kyo found an increase in lung bacterial burden as shown by elevated 16S rRNA gene copy numbers. They also noted a decrease in alpha diversity among ARDS patients, affecting both copy numbers and the relative abundance of betaproteobacteria (74). Additionally, patients with LI show higher levels of gut-associated bacteria in their lung microbiome, including species from the Enterobacteriaceae family, which have been linked to the development of ARDS (71).
Observational studies have suggested that gut microbes were not present in the upper airways of intubated individuals with ARDS or in septic mice, indicating bacterial translocation rather than aspiration. Panzer emphasizes that patients who undergo mechanical ventilation (MV) may experience early lung dysbiosis, which is accompanied by a significant rise in inflammatory mediators (IL-6, IL-8). This situation predisposes patients to the development of ARDS later (70). The movement of bacteria may occur through gut-draining lymphatics, the portal system, or systemic circulation (75, 76).
Limited data exists on how lung dysbiosis may impact the gut microbiome in patients with LI/ARDS. The compromised permeability of the alveoli-capillary membrane in LI/ARDS caused by epithelial or endothelial damage may contribute to gut-lung bacterial translocation (77). Critical illness can also alter environmental conditions that support gut bacterial growth, affecting the reproductive rates of microbial community members (67). In pulmonary infection diseases, microorganisms originating from the lungs trigger a pro-inflammatory response in intestinal epithelial cells, causing an increase in TJ permeability and damage to intestinal mucosal integrity. This ultimately leads to gastrointestinal injury and the development of MODS (78, 79).
There is increasing evidence suggesting that alterations in the lung microbiome can impact gut integrity, and the translocation of gut microbiota may worsen lung infections. However, there are significant gaps in knowledge regarding how lung dysbiosis during mechanical ventilation specifically influences the gut microbiome. Further research is crucial to uncover these mechanisms and their importance in clinical settings.
4 Potential mechanisms linking inflammatory bowel disease to pulmonary dysregulation
4.1 Dysbiosis of intestinal microbiota and inflammation in IBD
Typically, dysbiosis in patients with IBD is associated with changes in increased pro-inflammatory bacteria as well as reduced protective bacteria. In patients with Crohn’s disease (CD), intestinal dysbiosis, characterized by reduced diversity of Firmicutes species, is thought to occur before disease onset (80–82). A specific reduction in the relative abundance of the species Dialister invisus and Faecalibacterium prausnitzii from the Firmicutes phylum, as well as Bifidobacterium adolescentis from the Actinobacteria phylum, is observed in patients with Crohn’s disease (CD) compared to healthy controls. In contrast, there is an increase in the mucolytic species Ruminococcus gnavus and Ruminococcus torques, also belonging to the Firmicutes phylum, among CD patients (80, 83). Common experimental mouse models support the importance of the gut microbiota in the development of IBD, such as protein 2 (NOD2) deficient in the nucleotide-binding oligomerization domain, which typically has ileitis under standard feeding conditions. However, disease penetrance is reduced in certain pathogen-free environments (84–86) (Figure 2).
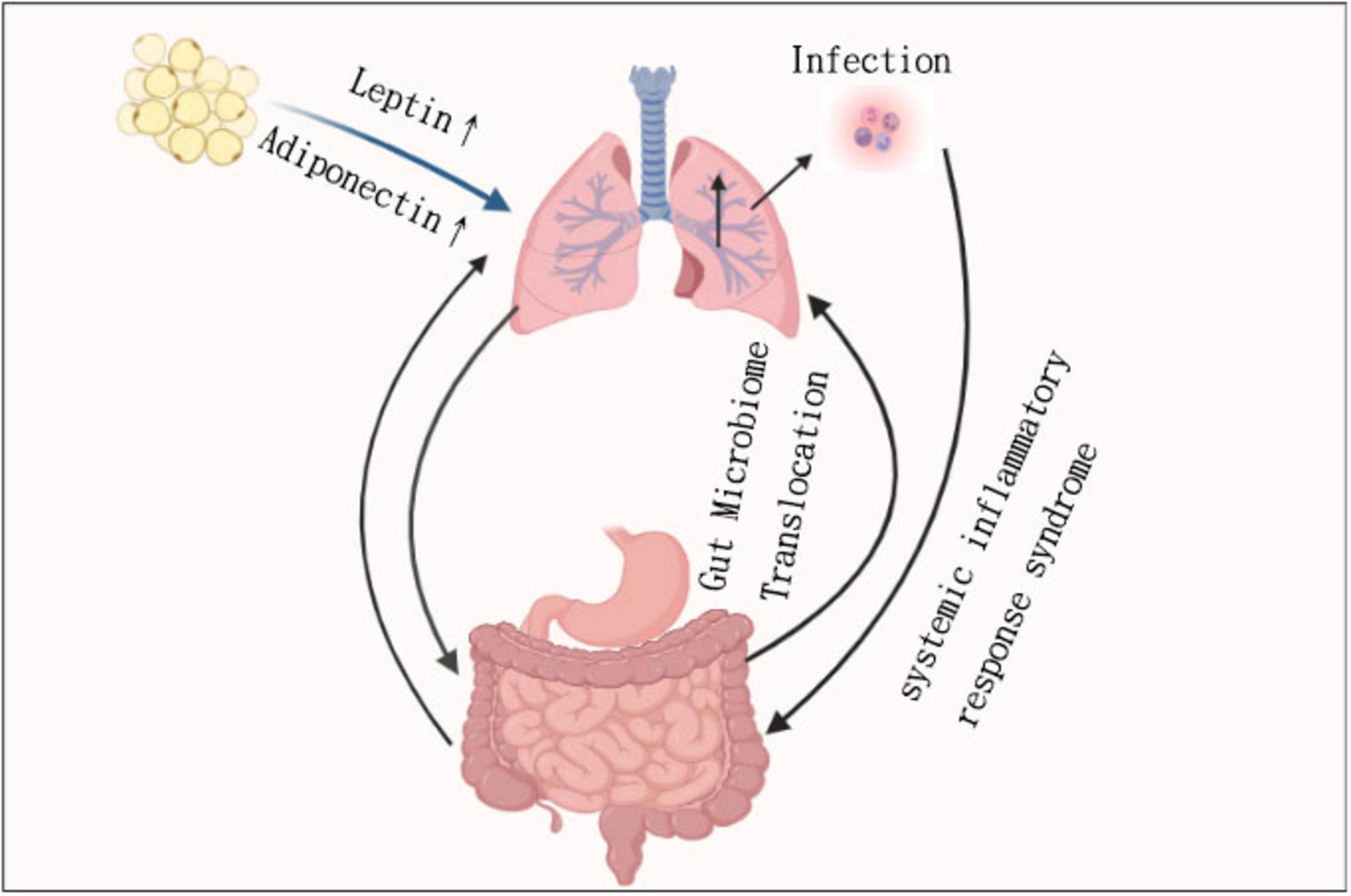
Figure 2. The core pathways between lung inflammation and gut permeability, including dysbiosis and IBD risk.
Adherent invasive Escherichia coli (AIEC) is an organism commonly associated with CD. This bacterium can exploit weaknesses in host defenses, such as impaired bacterial recognition, thereby reducing intracellular killing capacity and allowing AIEC dissemination (87). AIEC can survive and multiply in macrophages without causing cell death. This process also promotes increased secretion of the proinflammatory tumor necrosis factor alpha (TNF-α), which plays a role in Crohn’s disease (88).
A study in an inducible model of ileitis found that severe ileitis was associated with a shift in microbial communities. This shift transitioned from populations predominantly composed of species within the Firmicutes phylum to those largely represented by species within the Proteobacteria phylum. The translocation of AIEC is associated with dysbiosis that resembles that observed in patients with CD. Toxoplasma gondii induces a significant degree of dysbiosis with AIEC invasion being pronounced in mice deficient in the ileitis susceptibility gene NOD2, while it is notably reduced in mice lacking the pro-inflammatory CC chemokine receptor 2 (CCR2). This model illustrates the mechanisms of resistance to enteritis (89). Furthermore, in alternative models of ileitis susceptibility, dysbiosis has been shown to precede the development of ileitis (90).
Similar to CD, UC is characterized by a decrease in the number of Firmicutes species and an increase in the number of Proteobacteria species. UC is also related to the interaction of genetic susceptibility, inflammation, and microbial disturbances (91).
4.2 Intestinal dysbiosis influences lung health
Although direct experimental evidence remains limited, emerging research suggests that alterations in the intestinal microbiota may impact pulmonary immunity and inflammation. These proposed gut-lung connections are primarily derived from animal studies and correlational data in human cohorts, highlighting the need for further mechanistic research. Studies demonstrate that when antibiotic treatment eradicates gut bacteria, mice exhibit increased susceptibility to pneumonia and respiratory infections. In the absence of gut microbiota, alveolar macrophages experience transcriptomic changes that result in diminished phagocytic activity and reduced bacterial clearance (92, 93). Through changes in circulating inflammatory cytokines and changes in the airway of the intestinal microbiota, it has been demonstrated that intestinal microbiota dysbiosis affects the composition of the respiratory microbiota. This has only been demonstrated in models of severe sepsis, but not yet in COPD.
One study showed that segmented filamentous bacteria (SFB) in the gut induce Th17 responses and IL-22 production in the lungs and prevent pneumococcal respiratory infections by reducing bacterial numbers and lung inflammation (94). In a mouse model of sepsis, more abundant gut bacteria were observed in the lungs, and the communities found in the gut and lungs were more similar compared to sham mice (70). A meta-analysis of 16 studies found that Helicobacter pylori infection is associated with an increased risk of COPD (95). Helicobacter pylori is a bacterium that resides on the lining of the human stomach. During acute exacerbations of COPD, the gut microbiota exhibits a significant reduction in bacterial classes defined by operational taxonomic units (OTUs) (9). Although the gut microbiota has not been extensively studied in COPD, smoking—an independent risk factor for the condition—exhibits a strong association with COPD. Additionally, smoking is linked to dysbiosis in the fecal microbiota of patients with Crohn’s disease (CD), characterized by an increased relative abundance of Bacteroides and Prevotella (96).
4.3 Immunological crosstalk in the lung–gut axis
Immunological signaling is central to the bidirectional communication between the lungs and the gut. Mucosal barriers in both organs are densely populated by innate and adaptive immune cells, including macrophages, dendritic cells (DCs), innate lymphoid cells (ILCs), and T lymphocytes, which play crucial roles in maintaining homeostasis and orchestrating inflammatory responses (97, 98).
During respiratory infections or mechanical injury (e.g., mechanical ventilation), activated alveolar macrophages release cytokines such as IL-6, TNF-α, and IL-1β, which may enter systemic circulation and influence intestinal immune tone (99, 100). Similarly, in IBD, elevated levels of IL-17, IL-23, and other pro-inflammatory cytokines contribute to lung injury and immune cell infiltration in the airway.
Furthermore, the gut microbiota indirectly shapes pulmonary immunity by modulating the differentiation and migration of immune cells. Short-chain fatty acids (SCFAs) derived from gut commensals promote regulatory T cell (Treg) development and can suppress excessive inflammation in the lung (101, 102). Disruption of the microbiome, therefore, may impair mucosal immune tolerance and promote pathological inflammation in both organs.
Of particular interest is the role of the gut–lung axis in systemic immune priming. Studies have demonstrated that antigen-presenting cells in the gut can prime T cells that later home to the lungs during allergic asthma or bacterial pneumonia, highlighting a shared mucosal immunological network (103).
Collectively, these findings support that the lung–gut axis functions as an integrated mucosal immune circuit, capable of driving both protective and pathogenic outcomes depending on the immune context and disease stage.
4.4 Alterations in the lung microbiome in pulmonary diseases
While much attention has focused on gut microbiota, the composition of the lung microbiome also plays a critical role in the lung–gut axis. Unlike the sterile lung paradigm once believed, it is now established that healthy lungs harbor low-biomass but functionally significant microbial communities.
In disease states, the pulmonary microbiota undergoes notable alterations. In COPD and asthma, there is a shift toward increased Proteobacteria (e.g., Haemophilus influenzae) and decreased diversity (104). In ARDS, lung microbiota shows enrichment in gut-associated taxa, suggesting possible translocation and systemic dissemination during severe lung injury (105). Although gut-derived bacteria have been detected in the lungs of ARDS patients, whether these microbes represent active colonization or transient presence remains unclear. Moreover, direct evidence from metagenomic or metabolomic studies evaluating their functional impact on pulmonary immunity is currently limited, underscoring the need for further multi-omics investigations in this area.
These microbial changes can modulate local and systemic immunity. For instance, enrichment of pathogenic microbes in the lung can promote pro-inflammatory cytokine production, which in turn affects gut permeability and immune tone (106). Conversely, dysbiosis of lung microbiota may reduce regulatory immune signals, leading to enhanced susceptibility to gut inflammation.
Moreover, interactions between pulmonary microbiota and gut microbiota may be mediated through shared metabolites (e.g., SCFAs, bile acids) or systemic immune circuits involving dendritic cells and regulatory T cells (107).
Understanding how lung microbiota shifts in response to infections and chronic inflammation is essential to fully elucidate the bidirectional crosstalk along the lung–gut axis.
5 Emerging roles of adipokines in lung–gut crosstalk: hypotheses and early evidence
In addition to mechanical and inflammatory factors, adipose-derived signaling molecules—adipokines—have emerged as potential mediators of lung-gut crosstalk. Adipose tissue is now increasingly recognized as a hormonally active organ capable of influencing both local and systemic inflammation. While the role of adipokines in systemic inflammatory diseases is relatively well-established, their specific contribution to lung–gut crosstalk remains largely hypothetical. Here, we summarize selected findings from human and animal studies that may offer insight into potential mechanisms, while acknowledging that direct evidence remains limited. The endocrine function of adipose tissue depends on its ability to secrete hormones, factors, and protein signals (termed “adipokines”) that are responsible for metabolic and pro-inflammatory effects. In obese patients, the elevated release of pro-inflammatory adipokines has a detrimental impact on lung health. The influence of lung-related inflammatory factors on intestinal function has been previously elucidated, making it crucial to explore the role of adipokines in the lung-intestinal interaction. This discussion will enhance our understanding of the mechanisms underlying the lung-intestinal interplay (108, 109).
Since the discovery of adipokines, adipose tissue has been identified as a central node in inter-organ crosstalk networks that mediate regulation of the respiratory system in multiple organs and tissue types (110). Adipokines are the mediators between adipose tissue and target organs and is a key factor in metabolic disorders caused by obesity (111). Dysfunctional adipose tissue releases inflammatory adipokines in morbid obesity, leading to increased risks of pulmonary dysfunction, resulting in low-grade systemic inflammation (112).
5.1 Leptin and adiponectin: linking adipose dysfunction to pulmonary immunity
Leptin and adiponectin, two of the most abundant adipokines, exert profound regulatory effects on pulmonary immune responses and inflammation. Acting through both systemic and local mechanisms—including modulation of surfactant production, macrophage activation, and Treg/Th17 balance—these adipokines bridge the metabolic status of adipose tissue with the development and progression of various respiratory diseases. Understanding their opposing roles is essential for elucidating how adipose dysfunction contributes to lung pathology, especially in obesity-related conditions such as asthma, COPD, and COVID-19 (109, 113–116).
Leptin, a hormone secreted by adipose cells, has its receptors expressed both centrally in the hypothalamus and peripherally in various tissues (117, 118). Additionally, leptin can be produced in the lungs by adipose-like lipofibroblasts, where it plays a crucial role in the regulation of pulmonary surfactant production (119). It has been demonstrated that there is a significant association between genetic variants of the leptin receptor and the decline in lung function among patients with COPD (116). Furthermore, the relationship between leptin and the pathogenesis of lung disease is influenced by excess adipose tissue. Research indicates that serum leptin levels are strongly correlated with both body fat and lung function, and that elevated leptin levels contribute to obesity hypoventilation syndrome (OHS) (120).
Adiponectin is a hormone secreted by adipose tissue that plays a crucial role in the regulation of whole-body energy homeostasis. In contrast to leptin, adiponectin levels are observed to be reduced in individuals with impaired lung function and obesity (121–123).
Both leptin and adiponectin have a direct regulatory effect on adipose tissue function (117, 124) and new biomarker indicators of dysfunctional adipose tissue have been proposed to use the ratio of adiponectin to leptin serum levels (125). There is evidence that dysfunction of adipose tissue caused by obesity and systemic inflammation can cause airway inflammation and lead to lung disease (126). Multiple studies have shown an inverse correlation with the adiponectin/leptin serum ratio in patients with pulmonary dysfunction (127). These findings suggest that altering the adipokine secretion spectrum produced by adipose dysfunction in obese subjects may contribute to the decline of lung function in COVID-19 patients. Similar associations were observed in obese asthmatic patients compared with obese subjects without asthma, in which, in addition to increases in serum markers of inflammation and alterations in alveolar macrophage function, adiponectin/leptin mRNA expression ratio is also observed to be lower in adipose tissue. Bariatric surgery in obese patients with asthma has been shown to reverse inflammation and restore macrophage function, indicating a connection between excess adipose tissue, the pathogenesis of lung disease, and lung immune function (126).
Previous studies have shown that leptin and adiponectin have a direct regulatory effect on lung inflammation and immune function (128–130). Indeed, adiponectin-related adipoR1 has been observed to be expressed in immune regulatory T cells (T cells) in the lungs, regulating immune responses during lung inflammation (131). Regulatory T cells (Tregs) play a crucial role in suppressing immune responses, and their development is closely linked to helper T17 (Th17) cells, which can enhance immune responses and inflammation. Therefore, the balance between these two cell populations is essential for the regulation of the immune system. In vivo studies in mice have demonstrated that the activation of adipoR1 in lung Tregs promotes their proliferation and activity, thereby effectively suppressing immune responses and inflammation (128). Leptin has been shown in animal and in vitro models to exert opposing effects on regulatory T cells (Tregs) and Th17 cells, potentially impacting inflammatory balance in both pulmonary and intestinal tissues. However, direct confirmation of these effects in the context of lung–gut interaction is still lacking (129). Furthermore, the administration of leptin to Th2 immune cells in the lungs of mice enhanced the production of pro-inflammatory cytokines including IL-4, IL-5, and IL-13, while concurrently reducing allergen-induced inflammation in leptin-deficient mice (130, 131). These findings indicate that obesity-related dysfunction of adipose tissue may modify the adipokine profile, specifically the adiponectin/leptin ratio. This alteration could result in an imbalance in the Treg/Th17 ratio, an enhancement of Th2 responses, and an elevation in proinflammatory cytokine secretion, all of which may contribute to an increased risk of respiratory diseases (132).
5.2 Immunological functions of leptin and adiponectin in intestinal inflammation
Characteristic alterations in mesenteric fat associated with Crohn’s disease, a subtype of IBD, indicate that mesenteric fat may have a significant immunomodulatory role in the pathogenesis of this condition. In Crohn’s disease, mesenteric fat proliferates and encircles inflamed regions of the small intestine, suggesting that this adipose tissue compartment directly contributes to the disease process (133).
5.2.1 Leptin and T cell–mediated gut inflammation
Leptin was initially believed to function solely as a satiety factor (134, 135). However, research conducted by Lord and colleagues demonstrated that the adipokine leptin plays a crucial role in promoting T cell proliferation, which results in an increase in type 1 helper T cells (TH1) while simultaneously inhibiting the production of TH2 cytokines (136). Furthermore, studies using experimental autoimmune encephalomyelitis models provide compelling evidence for the regulatory role of leptin in autoimmune diseases (137, 138).
Previous studies have measured systemic leptin concentrations during acute experimental colitis. In the current study, it was found that plasma leptin concentrations were elevated in patients with TNBS-induced colitis and indomethacin-induced ileitis, and these levels correlated with disease severity. Notably, this increase in leptin is transient, with concentrations returning to baseline over time (139). Additionally, an independent study has demonstrated significantly increased leptin expression in mesenteric adipose tissue in patients diagnosed with Crohn’s disease (140, 141).
Previous studies have demonstrated that leptin-deficient ob/ob mice exhibit protection against dextran sulfate sodium (DSS)-induced colitis, whereas administration of leptin renders these mice susceptible to the disease (142). Furthermore, the researchers established a T cell transfer model of colitis, which indicated that the stimulatory effects of leptin play a crucial role in this model, significantly delaying the onset of colitis (143). Additional data show that OB/OB mice are protected in TH1 (trinitrobenzene sulfonic acid-TNBS) or TH2 (oxazolone) cell-driven models, which emphasizes that leptin’s T cell stimulating ability is critical for the observed effects (144).
In addition to its effects on epithelial cells, leptin also modulates intestinal inflammation through T cell studies by Sitharaman et al., which showed that inflamed epithelial cells produce leptin and release it into the intestinal lumen. The presence of luminal leptin leads to the activation of the NF-κB pathway in intestinal epithelial cells. Intestinal inflammation and epithelial wall damage will occur after rectal application of leptin (145).
5.2.2 Adiponectin: dual roles in gut immune regulation
Adiponectin, as an important adipokine, is increased in inflammatory bowel disease. The study conducted by Yamamoto et al. examined the expression of adiponectin in the adipose tissue of patients with Crohn’s disease. The findings indicated that adiponectin expression in the adipose tissue of these patients was positively correlated with adiponectin levels in the same tissue. Furthermore, fat content was found to be upregulated in patients with Crohn’s disease compared to those with colitis or healthy controls (146).
Although some experimental models (DSS and TNBS) have shown that adiponectin can induce the production of pro-inflammatory cytokines in the colon (147). For example, globular adiponectin mediated pro-proliferative as well as pro-inflammatory effects through activation of extracellular-signal regulated kinase (ERK), p38 mitogen-activated protein kinase (MAPK), and NF-κB signaling on colonic epithelial cells (148). However, Nishihara verified that adiponectin protects against inflammation, which may be mediated through direct anti-inflammatory effects on colon epithelial cells (149).
A recent study demonstrated that treatment with adiponectin-deficient DSS resulted in more severe colitis, which was associated with an increase in activated B cells in the colon as well as elevated levels of pro-inflammatory cytokines such as IL-1β, IL-4, and IL-6, and enhanced STAT3 signaling. Gene knockout animals exhibited reduced epithelial cell proliferation and increased cellular stress. In in vitro experiments, adiponectin was shown to reverse these effects. These findings support the notion that adiponectin remains stable within the intestinal environment (150).
5.3 Adipokine signaling pathways linking gut and lung inflammation
Intestinal stability is essential for maintaining host health and preventing IBD. The Wnt/β-Catenin signaling pathway is highly active within the intestinal microenvironment, playing a crucial role in the renewal process of intestinal epithelial cells. In IBD patients, the expression of genes associated with the Wnt/β-catenin signaling pathway is significantly increased, and disruptions in this pathway’s function can induce T cell inflammation, thereby promoting the occurrence of colitis (151).
With increased expression in the Wnt/β-catenin pathway, the Wnt protein levels rise, leading to the activation of the related Wnt/Wingless signaling pathway. Activation of Wnt signaling has been shown to reduce triglyceride (TG) accumulation and increase free fatty acid (FFA) levels by promoting lipolysis and reducing fat production (152).
Following lipolysis, lipids stored within adipocytes are released into the bloodstream, where they undergo further processing and stimulate the secretion of leptin. It is clear from the preceding discussion that leptin significantly influences lung-related diseases. Although the Wnt/β-catenin signaling pathway has been implicated in causing pulmonary inflammatory damage (153), limited research exists on whether this pathway affects lung function through adipokines. While adiponectin levels are markedly elevated in IBD patients, adiponectin does not appear to be directly associated with Wnt-related signaling pathways and may independently inhibit inflammation progression. However, limited literature exists regarding adipokine-mediated signaling pathways in the context of lung diseases leading to intestinal diseases. Consequently, further investigation is warranted to clarify the role of adipokines in lung-intestinal interactions. Future studies should explore the mechanisms by which adipokines influence microflora translocation in lung-intestinal interactions (Figure 3).
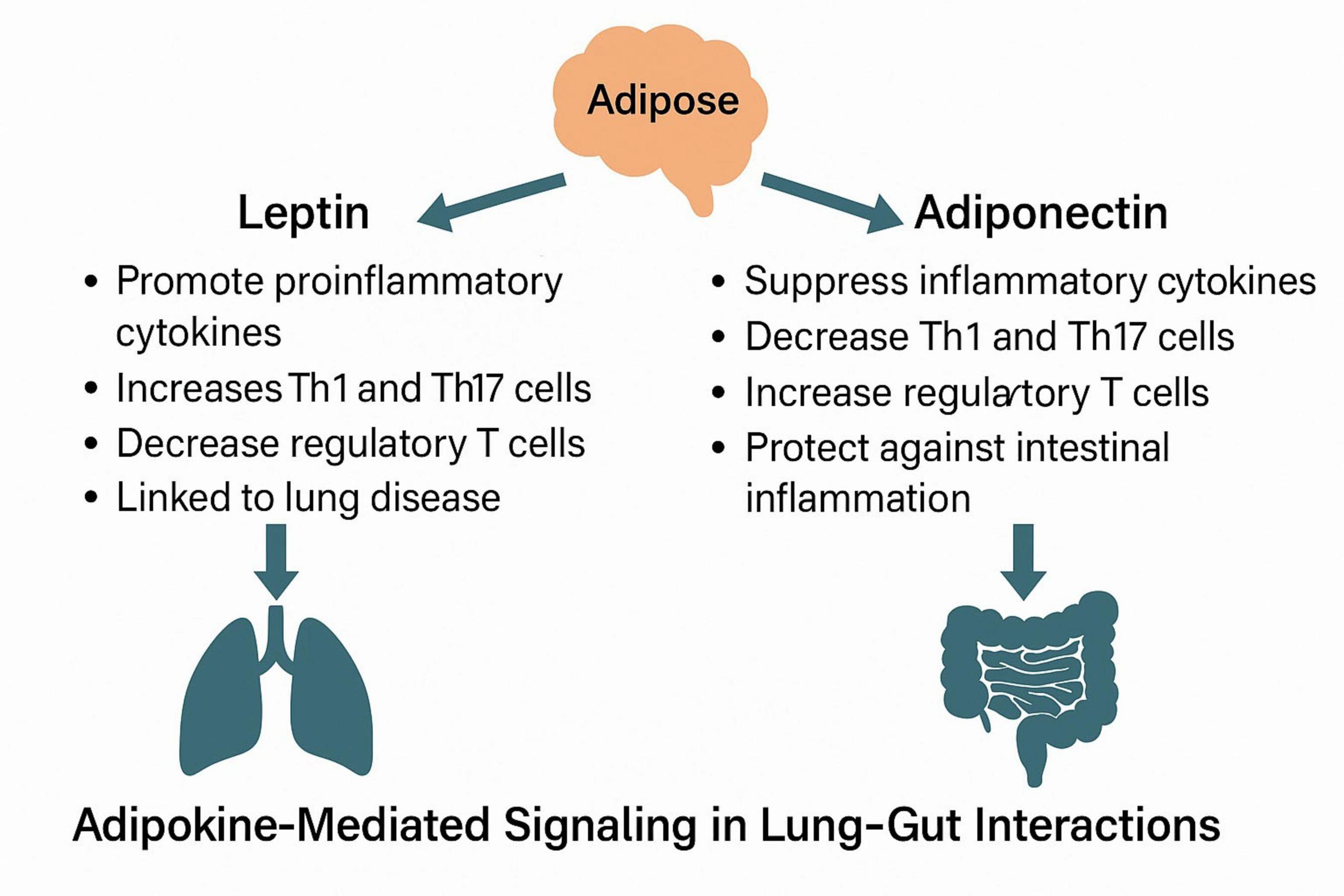
Figure 3. Immunomodulatory roles of leptin and adiponectin in lung–gut crosstalk.
5.4 Limitations and future research directions
While emerging findings suggest that adipokines such as leptin and adiponectin may play immunomodulatory roles in both pulmonary and gastrointestinal contexts, definitive evidence linking these molecules to lung–gut crosstalk remains limited. Most current data are derived from observational human studies, experimental colitis models, or indirect respiratory outcomes in obesity research. Future investigations should aim to directly assess how adipokines influence epithelial barrier function, cytokine signaling, and microbial composition across both organ systems, using integrative multi-omics and targeted genetic models. Furthermore, clinical studies stratifying patients by adipokine profiles and disease comorbidities may offer insight into translational relevance. Moreover, modulation of gut microbiota has been proposed as a potential therapeutic strategy for pulmonary diseases. Zhang et al. summarized probiotic and prebiotic interventions that alleviate airway inflammation, and Li et al. proposed that reshaping gut microbiota may improve response to immunotherapy in lung cancer patients (7, 8). These insights underscore the translational potential of targeting the gut–lung axis. Given the central roles of leptin and adiponectin in lung and gut inflammation, potential therapeutic strategies may include leptin receptor antagonists, adiponectin receptor agonists (e.g., AdipoRon), and agents that modulate the adiponectin/leptin ratio. These approaches have shown promise in preclinical models and may offer novel interventions in lung–gut axis–related diseases, especially in the context of obesity-driven inflammation.
6 Conclusion and outlook
The lungs and intestines both originate from the foregut region of the endoderm, which contributes to their functional similarities. Different pathogens elicit comparable immune responses, thereby further supporting the theory of lung-gut interaction. The primary mechanism underlying this interaction focuses on bacterial translocation and the corresponding immune system response.
While mechanical ventilation-induced lung injury and inflammatory bowel disease may complement the potential for pulmonary and intestinal diseases, the underlying causes and consequences of lung and intestinal dysbiosis remain poorly understood. Furthermore, given the widespread distribution of adipose tissue in the human body, adipokines play a significant role in mediating lung-intestinal interactions by influencing the dynamics of lung and intestinal microbiota and immune responses. Additionally, the mechanisms of lung-intestinal interaction associated with other types of lung and intestinal diseases are anticipated to be further elucidated in future research. The role of adipokines in lung-intestinal interactions was further investigated by categorizing patients with varying degrees of obesity and malnutrition.
Author contributions
JL: Conceptualization, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. YC: Conceptualization, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. BH: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (No. 82272188).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Moran G, Gordon M, Sinopolou V, Radford S, Darie A, Vuyyuru S, et al. British Society of Gastroenterology guidelines on inflammatory bowel disease in adults. Gut. (2025) 74:s1–101. doi: 10.1136/gutjnl-2024-334395
3. Vutcovici M, Bitton A, Ernst P, Kezouh A, Suissa S, Brassard P. Inflammatory bowel disease and risk of mortality in COPD. Eur Respir J. (2016) 47:1357–64. doi: 10.1183/13993003.01945-2015
4. Brassard P, Vutcovici M, Ernst P, Patenaude V, Sewitch M, Suissa S, et al. Increased incidence of inflammatory bowel disease in Québec residents with airway diseases. Eur Respir J. (2015) 45:962–8. doi: 10.1183/09031936.00079414
5. Peng Y, Liao W, Su C, Chen H, Hsia T, Chu C, et al. Association of inflammatory bowel disease with asthma risk: a nationwide cohort study. Allergy Asthma Proc. (2015) 36:e92–8. doi: 10.2500/aap.2015.36.3869
6. Friedman S, Garvik O, Nielsen J, Jølving L, Andersen M, Nørgård B. The consequences of preterm birth in the children of mothers with inflammatory bowel disease: a nationwide cohort study. Inflamm Bowel Dis. (2025) 3:izaf010. doi: 10.1093/ibd/izaf010
7. Zhang D, Li S, Wang N, Tan H, Zhang Z, Feng Y. The cross-talk between gut microbiota and lungs in common lung diseases. Front Microbiol. (2020) 11:301. doi: 10.3389/fmicb.2020.00301
8. Li R, Li J, Zhou X. Lung microbiome: new insights into the pathogenesis of respiratory diseases. Signal Transduct Target Ther. (2024) 9:19. doi: 10.1038/s41392-023-01722-y
9. Sun Z, Zhu Q, Shen Y, Yan T, Zhou X. Dynamic changes of gut and lung microorganisms during chronic obstructive pulmonary disease exacerbations. Kaohsiung J Med Sci. (2020) 36:107–13. doi: 10.1002/kjm2.12147
10. Liu S, Zhao Y, Feng X, Xu H. SARS-CoV-2 infection threatening intestinal health: a review of potential mechanisms and treatment strategies. Crit Rev Food Sci Nutr. (2023) 63:12578–96. doi: 10.1080/10408398.2022.2103090
11. Chu Y, Jiang Y, Meng M, Jiang J, Zhang J, Ren H, et al. Incidence and risk factors of gastrointestinal bleeding in mechanically ventilated patients. World J Emerg Med. (2010) 1:32–6.
12. Ak AK, Anjum F. Ventilator-Induced Lung Injury (VILI), StatPearls. Treasure Island, FL: StatPearls Publishing LLC (2024).
13. Tremblay L, Slutsky A. Ventilator-induced injury: from barotrauma to biotrauma. Proc Assoc Am Physicians. (1998) 110:482–8.
14. Birrenbach T, Böcker U. Inflammatory bowel disease and smoking: a review of epidemiology, pathophysiology, and therapeutic implications. Inflamm Bowel Dis. (2004) 10:848–59. doi: 10.1097/00054725-200411000-00019
15. Ojha U, Singh D, Choudhari O, Gothi D, Singh S. Correlation of severity of functional gastrointestinal disease symptoms with that of asthma and chronic obstructive pulmonary disease: a multicenter study. Int J Appl Basic Med Res. (2018) 8:83–8. doi: 10.4103/ijabmr.IJABMR_258_17
16. Lundström O, Manjer J, Ohlsson B. Smoking is associated with several functional gastrointestinal symptoms. Scand J Gastroenterol. (2016) 51:914–22. doi: 10.1080/00365521.2016.1174878
17. Ekbom A, Brandt L, Granath F, Löfdahl C, Egesten A. Increased risk of both ulcerative colitis and Crohn’s disease in a population suffering from COPD. Lung. (2008) 186:167–72. doi: 10.1007/s00408-008-9080-z
18. Bellani G, Laffey J, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. (2016) 315:788–800. doi: 10.1001/jama.2016.0291
19. Wang J, Li F, Sun R, Gao X, Wei H, Li L, et al. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat Commun. (2013) 4:2106. doi: 10.1038/ncomms3106
20. Rosa C, Pereira J, Cristina de Melo Santos N, Brancaglion GA, Silva EN, Tagliati CA, et al. Vancomycin-induced gut dysbiosis during Pseudomonas aeruginosa pulmonary infection in a mice model. J Leukoc Biol. (2020) 107:95–104. doi: 10.1002/JLB.4AB0919-432R
21. Vital M, Harkema J, Rizzo M, Tiedje J, Brandenberger C. Alterations of the murine gut microbiome with age and allergic airway disease. J Immunol Res. (2015) 2015:892568. doi: 10.1155/2015/892568
22. Wang J, Li F, Wei H, Lian Z, Sun R, Tian Z. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J Exp Med. (2014) 211:2397–410. doi: 10.1084/jem.20140625
23. Deriu E, Boxx G, He X, Pan C, Benavidez S, Cen L, et al. Influenza virus affects intestinal microbiota and secondary Salmonella infection in the gut through type i interferons. PLoS Pathog. (2016) 12:e1005572. doi: 10.1371/journal.ppat.1005572
24. Gajendran M, Loganathan P, Catinella A, Hashash JG. A comprehensive review and update on Crohn’s disease. Dis Mon. (2018) 64:20–57. doi: 10.1016/j.disamonth.2017.07.001
25. Savin Z, Kivity S, Yonath H, Yehuda S. Smoking and the intestinal microbiome. Arch Microbiol. (2018) 200:677–84. doi: 10.1007/s00203-018-1506-2
26. Head K, Jurenka J. Inflammatory bowel disease Part 1: ulcerative colitis–pathophysiology and conventional and alternative treatment options. Altern Med Rev. (2003) 8:247–83.
27. Khor B, Gardet A, Xavier R. Genetics and pathogenesis of inflammatory bowel disease. Nature. (2011) 474:307–17. doi: 10.1038/nature10209
28. Vutcovici M, Brassard P, Bitton A. Inflammatory bowel disease and airway diseases. World J Gastroenterol. (2016) 22:7735–41. doi: 10.3748/wjg.v22.i34.7735
29. Bourdillon A, Hajek M, Wride M, Lee M, Lerner M, Kohli N. Correlations of radiographic and endoscopic observations in subglottic stenosis. Ann Otol Rhinol Laryngol. (2022) 131:724–9. doi: 10.1177/00034894211042768
30. Majima S, Wakahara K, Iwano S, Kinoshita F, Nakamura M, Hashimoto N, et al. Airway involvement in inflammatory bowel disease: inflammatory bowel disease patients have bronchial wall thickening. Respir Investig. (2022) 60:713–9. doi: 10.1016/j.resinv.2022.06.003
31. Camus P, Piard F, Ashcroft T, Gal A, Colby T. The lung in inflammatory bowel disease. Medicine (Baltimore). (1993) 72:151–83. doi: 10.1097/00005792-199372030-00003
32. Desai D, Patil S, Udwadia Z, Maheshwari S, Abraham P, Joshi A. Pulmonary manifestations in inflammatory bowel disease: a prospective study. Indian J Gastroenterol. (2011) 30:225–8. doi: 10.1007/s12664-011-0129-1
33. Pemmasani G, Loftus E, Tremaine W. Prevalence of pulmonary diseases in association with inflammatory bowel disease. Dig Dis Sci. (2022) 67:5187–94. doi: 10.1007/s10620-022-07385-z
34. Kisiel M, Sedvall M, Malinovschi A, Franklin K, Gislason T, Shlunssen V, et al. Inflammatory bowel disease and asthma. results from the RHINE study. Respir Med. (2023) 216:107307. doi: 10.1016/j.rmed.2023.107307
35. Karhausen J, Furuta G, Tomaszewski J, Johnson R, Colgan S, Haase V. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. (2004) 114:1098–1096. doi: 10.1172/JCI21086
36. Kelly C, Zheng L, Campbell E, Saeedi B, Scholz C, Bayless A, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. (2015) 17:662–71. doi: 10.1016/j.chom.2015.03.005
37. Fleischer R, Velling M, Peters W, Peterka T, Franke F, Vymyslická P, et al. Invasive fascioloides magna infections impact gut microbiota in a definitive host in Europe. Int J Parasitol Parasites Wildl. (2024) 25:101024. doi: 10.1016/j.ijppaw.2024.101024
38. Onghena L, Heldens A, De Paepe K, Antwi M, Van Vlierberghe H, Raevens S, et al. The double-edged sword of metabolic and bariatric surgery: extending the biliary limb can trigger bacterial translocation, sepsis, and liver inflammation – an experimental study. Int J Surg. (2025). doi: 10.1097/JS9.0000000000002502 [Epub ahead of print].
39. Haldén E, Jakobson S, Janerås L, Norlén K. Effects of positive end-expiratory pressure on cardiac output distribution in the pig. Acta Anaesthesiol Scand. (1982) 26:403–8. doi: 10.1111/j.1399-6576.1982.tb01789.x
40. Bruhn A, Hernandez G, Bugedo G, Castillo L. Effects of positive end-expiratory pressure on gastric mucosal perfusion in acute respiratory distress syndrome. Crit Care. (2004) 8:R306–11. doi: 10.1186/cc2905
41. Beyer J, Conzen P, Schosser R, Messmer K. The effect of PEEP ventilation on hemodynamics and regional blood flow with special regard to coronary blood flow. Thorac Cardiovasc Surg. (1980) 28:128–32. doi: 10.1055/s-2007-1022063
42. Abe R, Matsumoto A, Sakaguchi R, Toda K, Sawa Y, Uchiyama A, et al. Perioperative enteral nutrition after left ventricular assist device implantation. Nutr Metab Insights. (2018) 11:1178638818810393. doi: 10.1177/1178638818810393
43. Puiac C, Szederjesi J, Lazar A, Almasy E, Rad P, Puscasiu L. Influence of ventilation parameters on intraabdominal pressure. J Crit Care Med (Targu Mures). (2016) 2:80–4. doi: 10.1515/jccm-2016-0016
44. Torquato J, Lucato J, Antunes T, Barbas C. Interaction between intra-abdominal pressure and positive-end expiratory pressure. Clinics (Sao Paulo). (2009) 64:105–12. doi: 10.1590/s1807-59322009000200007
45. Powers S, Shanely R, Coombes J, Koesterer T, McKenzie M, Van Gammeren D, et al. Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol (1985). (2002) 92:1851–8. doi: 10.1152/japplphysiol.00881.2001
46. Widjanantie S, Syam A, Nusdwinuringtyas N, Susanto A, Hidayat R, Kekalih A, et al. Effects of modified diaphragmatic training on gastroesophageal reflux disease questionnaire score, diaphragmatic excursion, and maximum inspiratory pressure in adults with gastroesophageal reflux disease after COVID-19: a single-blinded randomized control. Acta Med Indones. (2023) 55:269–76.
47. Mutlu G, Mutlu E, Factor P. GI complications in patients receiving mechanical ventilation. Chest. (2001) 119:1222–41. doi: 10.1378/chest.119.4.1222
48. Slutsky A, Tremblay L. Multiple system organ failure. is mechanical ventilation a contributing factor? Am J Respir Crit Care Med. (1998) 157:1721–5. doi: 10.1164/ajrccm.157.6.9709092
49. Plötz F, Slutsky A, van Vught A, Heijnen C. Ventilator-induced lung injury and multiple system organ failure: a critical review of facts and hypotheses. Intensive Care Med. (2004) 30:1865–72. doi: 10.1007/s00134-004-2363-9
50. Vlahakis N, Schroeder M, Limper A, Hubmayr R. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol. (1999) 277:L167–73. doi: 10.1152/ajplung.1999.277.1.L167
51. Madahar P, Beitler JR. Emerging concepts in ventilation-induced lung injury. F1000Res. (2020) 9:F1000 Faculty Rev-222. doi: 10.12688/f1000research.20576.1
52. Chen L, Xia H, Shang Y, Yao S. Molecular mechanisms of ventilator-induced lung injury. Chin Med J (Engl). (2018) 131:1225–31. doi: 10.4103/0366-6999.226840
53. Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi M, Ereifej L, et al. Interleukin-6 modulation of intestinal epithelial tight junction permeability is mediated by JNK pathway activation of claudin-2 gene. PLoS One. (2014) 9:e85345. doi: 10.1371/journal.pone.0085345
54. Guery B, Welsh D, Viget N, Robriquet L, Fialdes P, Mason C, et al. Ventilation-induced lung injury is associated with an increase in gut permeability. Shock. (2003) 19:559–63. doi: 10.1097/01.shk.0000070738.34700.bf
55. Clayburgh D, Musch M, Leitges M, Fu Y, Turner J. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J Clin Invest. (2006) 116:2682–94. doi: 10.1172/JCI29218
56. Marchiando A, Shen L, Graham W, Weber C, Schwarz B, Austin J, et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J Cell Biol. (2010) 189:111–26. doi: 10.1083/jcb.200902153
57. Fischer A, Gluth M, Pape U, Wiedenmann B, Theuring F, Baumgart D. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. (2013) 304:G970–9. doi: 10.1152/ajpgi.00183.2012
58. Feng Y, Teitelbaum D. Tumour necrosis factor–induced loss of intestinal barrier function requires TNFR1 and TNFR2 signalling in a mouse model of total parenteral nutrition. J Physiol. (2013) 591:3709–23. doi: 10.1113/jphysiol.2013.253518
59. Ye D, Ma T. Cellular and molecular mechanisms that mediate basal and tumour necrosis factor-alpha-induced regulation of myosin light chain kinase gene activity. J Cell Mol Med. (2008) 12:1331–46. doi: 10.1111/j.1582-4934.2008.00302.x
60. Ding N, Xiao H, Zhen L, Li H, Zhang Z, Ge J, et al. Systemic cytokines inhibition with Imp7 siRNA nanoparticle ameliorates gut injury in a mouse model of ventilator-induced lung injury. Biomed Pharmacother. (2023) 165:115237. doi: 10.1016/j.biopha.2023.115237
61. Imai Y, Parodo J, Kajikawa O, de Perrot M, Fischer S, Edwards V, et al. Injurious mechanical ventilation and end-organ epithelial cell apoptosis and organ dysfunction in an experimental model of acute respiratory distress syndrome. JAMA. (2003) 289:2104–12. doi: 10.1001/jama.289.16.2104
62. Kelly B, Imai I, Bittinger K, Laughlin A, Fuchs B, Bushman F, et al. Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome. (2016) 4:7. doi: 10.1186/s40168-016-0151-8
63. Marsland B, Trompette A, Gollwitzer E. The gut-lung axis in respiratory disease. Ann Am Thorac Soc. (2015) 12:S150–6. doi: 10.1513/AnnalsATS.201503-133AW
64. Shukla S, Budden K, Neal R, Hansbro P. Microbiome effects on immunity, health and disease in the lung. Clin Transl Immunol. (2017) 6:e133. doi: 10.1038/cti.2017.6
65. O’Dwyer D, Dickson R, Moore B. The lung microbiome, immunity, and the pathogenesis of chronic lung disease. J Immunol. (2016) 196:4839–47. doi: 10.4049/jimmunol.1600279
66. Martin-Loeches I, Dickson R, Torres A, Hanberger H, Lipman J, Antonelli M, et al. The importance of airway and lung microbiome in the critically ill. Crit Care. (2020) 24:537. doi: 10.1186/s13054-020-03219-4
67. Dickson R. The microbiome and critical illness. Lancet Respir Med. (2016) 4:59–72. doi: 10.1016/S2213-260000427-0
68. Agudelo-Ochoa G, Valdés-Duque B, Giraldo-Giraldo N, Jaillier-Ramírez A, Giraldo-Villa A, Acevedo-Castaño I, et al. Gut microbiota profiles in critically ill patients, potential biomarkers and risk variables for sepsis. Gut Microbes. (2020) 12:1707610. doi: 10.1080/19490976.2019.1707610
69. Szychowiak P, Villageois-Tran K, Patrier J, Timsit J, Ruppé É. The role of the microbiota in the management of intensive care patients. Ann Intensive Care. (2022) 12:3. doi: 10.1186/s13613-021-00976-5
70. Dickson R, Singer B, Newstead M, Falkowski N, Erb-Downward J, Standiford T, et al. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. (2016) 1:16113. doi: 10.1038/nmicrobiol.2016.113
71. Panzer A, Lynch S, Langelier C, Christie J, McCauley K, Nelson M, et al. Lung microbiota is related to smoking status and to development of acute respiratory distress syndrome in critically ill trauma patients. Am J Respir Crit Care Med. (2018) 197:621–31. doi: 10.1164/rccm.201702-0441OC
72. Zakharkina T, Martin-Loeches I, Matamoros S, Povoa P, Torres A, Kastelijn J, et al. The dynamics of the pulmonary microbiome during mechanical ventilation in the intensive care unit and the association with occurrence of pneumonia. Thorax. (2017) 72:803–10. doi: 10.1136/thoraxjnl-2016-209158
73. Akrami K, Sweeney D. The microbiome of the critically ill patient. Curr Opin Crit Care. (2018) 24:49–54. doi: 10.1097/MCC.0000000000000469
74. Kyo M, Nishioka K, Nakaya T, Kida Y, Tanabe Y, Ohshimo S, et al. Unique patterns of lower respiratory tract microbiota are associated with inflammation and hospital mortality in acute respiratory distress syndrome. Respir Res. (2019) 20:246. doi: 10.1186/s12931-019-1203-y
75. Bingula R, Filaire M, Radosevic-Robin N, Bey M, Berthon J, Bernalier-Donadille A, et al. Desired turbulence? Gut-lung axis, immunity, and lung cancer. J Oncol. (2017) 2017:5035371. doi: 10.1155/2017/5035371
76. Deitch E. Gut lymph and lymphatics: a source of factors leading to organ injury and dysfunction. Ann N Y Acad Sci. (2010) 1207:E103–11. doi: 10.1111/j.1749-6632.2010.05713.x
77. Zhou X, Liao Y. Gut-lung crosstalk in sepsis-induced acute lung injury. Front Microbiol. (2021) 12:779620. doi: 10.3389/fmicb.2021.779620
78. Reintam Blaser A, Poeze M, Malbrain M, Björck M, Oudemans-van Straaten H, Starkopf J, et al. Gastrointestinal symptoms during the first week of intensive care are associated with poor outcome: a prospective multicentre study. Intensive Care Med. (2013) 39:899–909. doi: 10.1007/s00134-013-2831-1
79. Alverdy J, Krezalek M. Collapse of the microbiome, emergence of the pathobiome, and the immunopathology of sepsis. Crit Care Med. (2017) 45:337–47. doi: 10.1097/CCM.0000000000002172
80. Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. (2011) 60:631–7. doi: 10.1136/gut.2010.223263
81. Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. (2006) 55:205–11. doi: 10.1136/gut.2005.073817
82. Kang S, Denman S, Morrison M, Yu Z, Dore J, Leclerc M, et al. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. (2010) 16:2034–42. doi: 10.1002/ibd.21319
83. Martinez-Medina M, Aldeguer X, Gonzalez-Huix F, Acero D, Garcia-Gil L. Abnormal microbiota composition in the ileocolonic mucosa of Crohn’s disease patients as revealed by polymerase chain reaction-denaturing gradient gel electrophoresis. Inflamm Bowel Dis. (2006) 12:1136–45. doi: 10.1097/01.mib.0000235828.09305.0c
84. Kim S, Tonkonogy S, Albright C, Tsang J, Balish E, Braun J, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. (2005) 128:891–906. doi: 10.1053/j.gastro.2005.02.009
85. Biswas A, Liu Y, Hao L, Mizoguchi A, Salzman N, Bevins C, et al. Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci USA. (2010) 107:14739–44. doi: 10.1073/pnas.1003363107
86. Maharshak N, Packey C, Ellermann M, Manick S, Siddle J, Huh E, et al. Altered enteric microbiota ecology in interleukin 10-deficient mice during development and progression of intestinal inflammation. Gut Microbes. (2013) 4:316–24. doi: 10.4161/gmic.25486
87. Mukhopadhya I, Hansen R, El-Omar E, Hold GL. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. (2012) 9:219–30. doi: 10.1038/nrgastro.2012.14
88. Glasser A, Boudeau J, Barnich N, Perruchot M, Colombel J, Darfeuille-Michaud A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect Immun. (2001) 69:5529–37. doi: 10.1128/IAI.69.9.5529-5537.2001
89. Craven M, Egan C, Dowd S, McDonough S, Dogan B, Denkers E, et al. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn’s disease. PLoS One. (2012) 7:e41594. doi: 10.1371/journal.pone.0041594
90. Dobranowski P, Tang C, Sauvé J, Menzies S, Sly L. Compositional changes to the ileal microbiome precede the onset of spontaneous ileitis in SHIP deficient mice. Gut Microbes. (2019) 10:578–98. doi: 10.1080/19490976.2018.1560767
91. Frank D, St Amand A, Feldman R, Boedeker E, Harpaz N, Pace N. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. (2007) 104:13780–5. doi: 10.1073/pnas.0706625104
92. Chen L, Chen P, Hsu C. Commensal microflora contribute to host defense against Escherichia coli pneumonia through toll-like receptors. Shock. (2011) 36:67–75. doi: 10.1097/SHK.0b013e3182184ee7
93. Schuijt T, Lankelma J, Scicluna B, de Sousa e Melo F, Roelofs JJ, de Boer JD, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut. (2016) 65:575–83. doi: 10.1136/gutjnl-2015-309728
94. Gauguet S, D’Ortona S, Ahnger-Pier K, Duan B, Surana N, Lu R, et al. Intestinal microbiota of mice influences resistance to Staphylococcus aureus pneumonia. Infect Immun. (2015) 83:4003–14. doi: 10.1128/IAI.00037-15
95. Wang F, Liu J, Zhang Y, Lei P. Association of Helicobacter pylori infection with chronic obstructive pulmonary disease and chronic bronchitis: a meta-analysis of 16 studies. Infect Dis (Lond). (2015) 47:597–603. doi: 10.3109/00365548.2014.989539
96. Benjamin J, Hedin C, Koutsoumpas A, Ng S, McCarthy N, Prescott N, et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm Bowel Dis. (2012) 18:1092–1090. doi: 10.1002/ibd.21864
97. Ogulur I, Mitamura Y, Yazici D, Pat Y, Ardicli S, Li M, et al. Type 2 immunity in allergic diseases. Cell Mol Immunol. (2025) 22:211–42. doi: 10.1038/s41423-025-01261-2
98. Hou Q, Huang J, Ayansola H, Masatoshi H, Zhang B. Intestinal stem cells and immune cell relationships: potential therapeutic targets for inflammatory bowel diseases. Front Immunol. (2021) 11:623691. doi: 10.3389/fimmu.2020.623691
99. Wei X, Qian W, Narasimhan H, Chan T, Liu X, Arish M, et al. Macrophage peroxisomes guide alveolar regeneration and limit SARS-CoV-2 tissue sequelae. Science. (2025) 387:eadq2509. doi: 10.1126/science.adq2509
100. Li B, Liu J, He W, Zhou Y, Zhao M, Xia C, et al. Inhibition of macrophage inflammasome assembly and pyroptosis with GC-1 ameliorates acute lung injury. Theranostics. (2025) 15:2360–74. doi: 10.7150/thno.101866
101. Hong Y, Cui J, Xu G, Li N, Peng G. Intestinal IL-17 family orchestrates microbiota-driven histone deacetylation and promotes Treg differentiation to mediate the alleviation of asthma by Ma-Xing-Shi-Gan decoction. Phytomedicine. (2025) 142:156656. doi: 10.1016/j.phymed.2025.156656
102. Perez Umana E, Mendes E, Casaro M, Lazarini M, Oliveira F, Sperling A, et al. Exogenous acetate mitigates later enhanced allergic airway inflammation in a menopausal mouse model. Front Cell Infect Microbiol. (2025) 15:1543822. doi: 10.3389/fcimb.2025.1543822
103. Fink L, Zeuthen L, Ferlazzo G, Frøkiaer H. Human antigen-presenting cells respond differently to gut-derived probiotic bacteria but mediate similar strain-dependent NK and T cell activation. FEMS Immunol Med Microbiol. (2007) 51:535–46. doi: 10.1111/j.1574-695X.2007.00333.x
104. Welp A, Bomberger J. Bacterial community interactions during chronic respiratory disease. Front Cell Infect Microbiol. (2020) 10:213. doi: 10.3389/fcimb.2020.00213
105. Tang Y, Liu B, Ma A, Wang B, Xiong H, Zhou Y, et al. Lung microbiota: a new hope for treating acute respiratory distress syndrome? Front Microbiol. (2025) 16:1586949. doi: 10.3389/fmicb.2025.1586949
106. Meier P, Reina G, Lehner S, Jovic M, Kovacova M, Spitalsky Z, et al. The release and cytotoxicity of novel light inducible antimicrobial coatings after incineration. Ecotoxicol Environ Saf. (2025) 300:118421. doi: 10.1016/j.ecoenv.2025.118421
107. Parvez A, Jubyda F, Karmakar J, Jahan A, Akter N, Ayaz M, et al. Antimicrobial potential of biopolymers against foodborne pathogens: an updated review. Microb Pathog. (2025) 204:107583. doi: 10.1016/j.micpath.2025.107583
108. Brock J, Billeter A, Müller-Stich B, Herth F. Obesity and the lung: what we know today. Respiration. (2020) 99:856–66. doi: 10.1159/000509735
109. Jutant E, Tu L, Humbert M, Guignabert C, Huertas A. The thousand faces of leptin in the lung. Chest. (2021) 159:239–48. doi: 10.1016/j.chest.2020.07.075
110. Dixon A, Peters U. The effect of obesity on lung function. Expert Rev Respir Med. (2018) 12:755–67. doi: 10.1080/17476348.2018.1506331
111. Hornung F, Rogal J, Loskill P, Löffler B, Deinhardt-Emmer S. The inflammatory profile of obesity and the role on pulmonary bacterial and viral infections. Int J Mol Sci. (2021) 22:3456. doi: 10.3390/ijms22073456
112. McGillis J. White adipose tissue, inert no more! Endocrinology. (2005) 146:2154–6. doi: 10.1210/en.2005-0248
113. Salvator H, Grassin-Delyle S, Brollo M, Couderc L, Abrial C, Victoni T, et al. Adiponectin inhibits the production of TNF-α, IL-6 and chemokines by human lung macrophages. Front Pharmacol. (2021) 12:718929. doi: 10.3389/fphar.2021.718929
114. Thyagarajan B, Jacobs D, Smith L, Kalhan R, Gross M, Sood A. Serum adiponectin is positively associated with lung function in young adults, independent of obesity: the CARDIA study. Respir Res. (2010) 11:176. doi: 10.1186/1465-9921-11-176
115. Suzuki M, Makita H, Östling J, Thomsen L, Konno S, Nagai K, et al. Lower leptin/adiponectin ratio and risk of rapid lung function decline in chronic obstructive pulmonary disease. Ann Am Thorac Soc. (2014) 11:1511–9. doi: 10.1513/AnnalsATS.201408-351OC
116. Hansel N, Gao L, Rafaels N, Mathias R, Neptune E, Tankersley C, et al. Leptin receptor polymorphisms and lung function decline in COPD. Eur Respir J. (2009) 34:103–10. doi: 10.1183/09031936.00120408
117. Siegrist-Kaiser C, Pauli V, Juge-Aubry C, Boss O, Pernin A, Chin W, et al. Direct effects of leptin on brown and white adipose tissue. J Clin Invest. (1997) 100:2858–64. doi: 10.1172/JCI119834
118. Genchi V, D’Oria R, Palma G, Caccioppoli C, Cignarelli A, Natalicchio A, et al. Impaired leptin signalling in obesity: is leptin a new thermolipokine? Int J Mol Sci. (2021) 22:6445. doi: 10.3390/ijms22126445
119. Torday J, Sun H, Wang L, Torres E, Sunday M, Rubin L. Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am J Physiol Lung Cell Mol Physiol. (2002) 282:L405–10. doi: 10.1152/ajplung.2002.282.3.L405
120. Phipps P, Starritt E, Caterson I, Grunstein R. Association of serum leptin with hypoventilation in human obesity. Thorax. (2002) 57:75–6. doi: 10.1136/thorax.57.1.75
121. Gavrila A, Chan J, Yiannakouris N, Kontogianni M, Miller L, Orlova C, et al. Serum adiponectin levels are inversely associated with overall and central fat distribution but are not directly regulated by acute fasting or leptin administration in humans: cross-sectional and interventional studies. J Clin Endocrinol Metab. (2003) 88:4823–31. doi: 10.1210/jc.2003-030214
122. Miller M, Cho J, Pham A, Ramsdell J, Broide D. Adiponectin and functional adiponectin receptor 1 are expressed by airway epithelial cells in chronic obstructive pulmonary disease. J Immunol. (2009) 182:684–91. doi: 10.4049/jimmunol.182.1.684
123. Jasinski-Bergner S, Büttner M, Quandt D, Seliger B, Kielstein H. Adiponectin and its receptors are differentially expressed in human tissues and cell lines of distinct origin. Obes Facts. (2017) 10:569–83. doi: 10.1159/000481732
124. Fu Y, Luo N, Klein R, Garvey W. Adiponectin promotes adipocyte differentiation, insulin sensitivity, and lipid accumulation. J Lipid Res. (2005) 46:1369–79. doi: 10.1194/jlr.M400373-JLR200
125. Frühbeck G, Catalán V, Rodríguez A, Ramírez B, Becerril S, Salvador J, et al. Involvement of the leptin-adiponectin axis in inflammation and oxidative stress in the metabolic syndrome. Sci Rep. (2017) 7:6619. doi: 10.1038/s41598-017-06997-0
126. Sideleva O, Suratt B, Black K, Tharp W, Pratley R, Forgione P, et al. Obesity and asthma: an inflammatory disease of adipose tissue not the airway. Am J Respir Crit Care Med. (2012) 186:598–605. doi: 10.1164/rccm.201203-0573OC
127. Di Filippo L, De Lorenzo R, Sciorati C, Capobianco A, Lorè N, Giustina A, et al. Adiponectin to leptin ratio reflects inflammatory burden and survival in COVID-19. Diabetes Metab. (2021) 47:101268. doi: 10.1016/j.diabet.2021.101268
128. Ramos-Ramírez P, Malmhäll C, Tliba O, Rådinger M, Bossios A. Adiponectin/AdipoR1 axis promotes IL-10 release by human regulatory T cells. Front Immunol. (2021) 12:677550. doi: 10.3389/fimmu.2021.677550
129. Bruzzaniti S, Bocchino M, Santopaolo M, Calì G, Stanziola A, D’Amato M, et al. An immunometabolic pathomechanism for chronic obstructive pulmonary disease. Proc Natl Acad Sci USA. (2019) 116:15625–34. doi: 10.1073/pnas.1906303116
130. Zheng H, Zhang X, Castillo E, Luo Y, Liu M, Yang X. Leptin enhances TH2 and ILC2 responses in allergic airway disease. J Biol Chem. (2016) 291:22043–52. doi: 10.1074/jbc.M116.743187
131. Ramos-Ramírez P, Malmhäll C, Johansson K, Adner M, Lötvall J, Bossios A. Lung regulatory T cells express adiponectin receptor 1: modulation by obesity and airway allergic inflammation. Int J Mol Sci. (2020) 21:8990. doi: 10.3390/ijms21238990
132. Palma G, Sorice G, Genchi V, Giordano F, Caccioppoli C, D’Oria R, et al. Adipose tissue inflammation and pulmonary dysfunction in obesity. Int J Mol Sci. (2022) 23:7349. doi: 10.3390/ijms23137349
133. Weidinger C, Ziegler J, Letizia M, Schmidt F, Siegmund B. Adipokines and their role in intestinal inflammation. Front Immunol. (2018) 9:1974. doi: 10.3389/fimmu.2018.01974
134. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman J. Positional cloning of the mouse obese gene and its human homologue. Nature. (1994) 372:425–32. doi: 10.1038/372425a0
135. Friedman J, Halaas J. Leptin and the regulation of body weight in mammals. Nature. (1998) 395:763–70. doi: 10.1038/27376
136. Lord G, Matarese G, Howard J, Baker R, Bloom S, Lechler R. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. (1998) 394:897–901. doi: 10.1038/29795
137. Matarese G, Di Giacomo A, Sanna V, Lord G, Howard J, Di Tuoro A, et al. Requirement for leptin in the induction and progression of autoimmune encephalomyelitis. J Immunol. (2001) 166:5909–16. doi: 10.4049/jimmunol.166.10.5909
138. Sanna V, Di Giacomo A, La Cava A, Lechler R, Fontana S, Zappacosta S, et al. Leptin surge precedes onset of autoimmune encephalomyelitis and correlates with development of pathogenic T cell responses. J Clin Invest. (2003) 111:241–50. doi: 10.1172/JCI16721
139. Barbier M, Cherbut C, Aubé A, Blottière H, Galmiche J. Elevated plasma leptin concentrations in early stages of experimental intestinal inflammation in rats. Gut. (1998) 43:783–90. doi: 10.1136/gut.43.6.783
140. Zulian A, Cancello R, Micheletto G, Gentilini D, Gilardini L, Danelli P, et al. Visceral adipocytes: old actors in obesity and new protagonists in Crohn’s disease? Gut. (2012) 61:86–94. doi: 10.1136/gutjnl-2011-300391
141. Siegmund B. Mesenteric fat in Crohn’s disease: the hot spot of inflammation? Gut. (2012) 61:3–5. doi: 10.1136/gutjnl-2011-301354
142. Siegmund B, Lehr H, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology. (2002) 122:2011–25. doi: 10.1053/gast.2002.33631
143. Siegmund B, Sennello J, Jones-Carson J, Gamboni-Robertson F, Lehr H, Batra A, et al. Leptin receptor expression on T lymphocytes modulates chronic intestinal inflammation in mice. Gut. (2004) 53:965–72. doi: 10.1136/gut.2003.027136
144. Batra A, Okur B, Glauben R, Erben U, Ihbe J, Stroh T, et al. Leptin: a critical regulator of CD4+ T-cell polarization in vitro and in vivo. Endocrinology. (2010) 151:56–62. doi: 10.1210/en.2009-0565
145. Sitaraman S, Liu X, Charrier L, Gu L, Ziegler T, Gewirtz A, et al. Colonic leptin: source of a novel proinflammatory cytokine involved in IBD. FASEB J. (2004) 18:696–8. doi: 10.1096/fj.03-0422fje
146. Yamamoto K, Kiyohara T, Murayama Y, Kihara S, Okamoto Y, Funahashi T, et al. Production of adiponectin, an anti-inflammatory protein, in mesenteric adipose tissue in Crohn’s disease. Gut. (2005) 54:789–96. doi: 10.1136/gut.2004.046516
147. Fayad R, Pini M, Sennello J, Cabay R, Chan L, Xu A, et al. Adiponectin deficiency protects mice from chemically induced colonic inflammation. Gastroenterology. (2007) 132:601–14. doi: 10.1053/j.gastro.2006.11.026
148. Ogunwobi O, Beales I. Adiponectin stimulates proliferation and cytokine secretion in colonic epithelial cells. Regul Pept. (2006) 134:105–13. doi: 10.1016/j.regpep.2006.02.001
149. Nishihara T, Matsuda M, Araki H, Oshima K, Kihara S, Funahashi T, et al. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology. (2006) 131:853–61. doi: 10.1053/j.gastro.2006.06.015
150. Obeid S, Wankell M, Charrez B, Sternberg J, Kreuter R, Esmaili S, et al. Adiponectin confers protection from acute colitis and restricts a B cell immune response. J Biol Chem. (2017) 292:6569–82. doi: 10.1074/jbc.M115.712646
151. Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett. (2014) 345:235–41. doi: 10.1016/j.canlet.2013.07.032
152. Liu M, Hemba-Waduge R, Li X, Huang X, Liu T, Han X, et al. WNT/Wingless signaling promotes lipid mobilization through signal-induced transcriptional repression. Proc Natl Acad Sci USA. (2024) 121:e2322066121. doi: 10.1073/pnas.2322066121
Keywords: lung-gut crosstalk, ventilator-induced lung injury, inflammatory bowel disease, immunity, microbiome, adipokines
Citation: Li J, Chen Y and Hu B (2025) The intricate interactions between the lungs and gut in patients: unraveling the crosstalk mechanism. Front. Med. 12:1624907. doi: 10.3389/fmed.2025.1624907
Received: 08 May 2025; Accepted: 08 July 2025;
Published: 30 July 2025.
Edited by:
David Dora, Semmelweis University, HungaryReviewed by:
Amit Kumar, University of Maryland, United StatesAnett Hudák, Pharmacoidea Ltd., Hungary
Copyright © 2025 Li, Chen and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bangchuan Hu, NDM0ODkwMDM3QHFxLmNvbQ==
†These authors have contributed equally to this work