 Francesco Pintus
Francesco Pintus Noemi Giordano
Noemi Giordano Daniela Francesca Giachino
Daniela Francesca Giachino Giorgia Mandrile
Giorgia Mandrile- 1Department of Clinical and Biological Sciences, University of Turin, Orbassano, Italy
- 2Genetic Unit and Thalassemia Centre, San Luigi University Hospital, Orbassano, Italy
Kidney Stone Disease (KSD) has a high prevalence (approximately 10%) and high recurrence risk: almost half of stone former patients will experience recurrence within 5–10 years. To date, KSD is managed mostly surgically with a heavy burden on the healthcare system and numerous invasive procedures for the patients. In the past years a genetic basis in KSD has been increasingly recognized, with a heritability rate reaching 50%. Through Genome-Wide Association Studies (GWAS) and Next-Generation Sequencing (NGS) several genetic causes of recurrent nephrolithiasis have been untangled, paving the way to new therapies and prevention strategies, through precision medicine-based approaches. Many loci with more than 200 unique genes have been associated with KSD susceptibility thanks to GWAS, even though the development of a polygenic risk score is still in progress. Moreover, today, about 40 genes linked to monogenic disease that are involved in kidney stones have been identified, leading to a precise diagnosis in cases that were previously considered idiopathic. Despite these advancements, genetic testing in kidney stone formers remains underutilized and inconsistently available. The absence of clear diagnostic guidelines, standardization, and widespread awareness, combined with lack of perceived benefit, has left the decision to test largely at the discretion of individual physicians. This paper reviews the updated evidences in KSD genetics and suggest a diagnostic algorithm aimed to increase the diagnostic rate of genetic stones, allowing a personalized treatment and, in turn, a higher disease-free survival for the patients and a more efficient allocation of resources, analyzing the cost-effectiveness of genetic testing in urolithiasis. Besides, it will provide a further look to promising prospects in the field of prevention methods for kidney stones.
1 Introduction
Kidney Stone Disease (KSD) prevalence is currently rising worldwide reaching approximately 10% in the US, Europe, and Asia (1). Half of stone formers will experience recurrence within 5–10 years and 75% within 20 years, this leading to an increased risk of chronic kidney disease (CKD) and kidney failure (2).
Calcium stones are reported in 94% of first-time stone former patients, followed by uric acid (4.8% of first-time stone formers), while brushite and struvite compositions are rare (0.9%) and cystine very rare (0.1%). Among common calcium stones, calcium oxalate (76%) is the most common composition (3).
Stone formation may have an underlying genetic base which can therefore explain a high risk of recurrence and thus needs to be investigated for better prevention and treatment.
Indeed, in the last years has emerged that KSD has an important genetic basis with a heritability approximately of 50%, as observed in twin studies (46% for female twins and 62% for male twins) (4, 5). The pathogenesis of KSD remains unclear in many cases, but it is likely due to both genetic and environmental factors. In the past decades, numerous Genome-Wide Association Studies (GWAS) and New Generation Sequencing (NGS) studies demonstrated a genetic cause in several cohort of nephrolithiasis patients, paving the way for new possible opportunities of therapy and prevention through precision medicine-based strategies (2, 6–9), likely reducing the burden on the healthcare system and offering new treatments besides surgical management of KSD (10, 11).
2 Guidelines
The guidelines on urolithiasis from the European Association of Urology (EAU) and from American Urological Association (AUA) (12, 13) suggest attending a first step analysis, with no distinction between low- and high-risk patients, on all first-time stone formers and repeated in case of early recurrence after complete stone clearance intervention, early recurrence while under prevention therapy or late recurrence after long stone-free period. In this first step biochemical work-up of urine and blood and, when possible, stone composition analysis are suggested.
Once the results are obtained, the patient is assigned to a low-risk or high-risk group for stone formation based on the stone type. Only high-risk stone formers undergo specific metabolic evaluation by biochemical analysis of two consecutive 24 h-urine samples.
2.1 Biochemical mechanisms of stone formation
Kidney stone formation is a complex and multi-mechanistic process involving several interconnected mechanisms, influenced by chemical, physiological and anatomical conditions that act synergistically (14).
Under certain circumstances, protective mechanisms against crystallization may be altered and promote the formation of stones (e.g., matrix proteins that normally inhibit the aggregation of calcium salts). Receptors and enzyme systems such as the calcium sensing receptor (CaSR), vasopressin V2, angiotensin II and aldosterone, as well as inflammatory processes, can represent other critical factors in stone pathogenesis, transforming a protective balance into a risk for stone formation. These pathways can be altered because of genetic variants and may represent potential therapeutic targets for the development of new personalized approaches in the prevention and treatment of kidney stones (15).
2.2 Urine metabolic evaluation
For individuals at high risk of stone formation, the International Urolithiasis Alliance (IAU) recommends a comprehensive metabolic evaluation as well as EAU and AUA (12, 13, 16), to provide the clinician with tools to tailor nutritional and pharmacological therapies by identifying specific metabolic alterations in urine. Metabolic assessment requires 24-h urine collections (one or two samples) on a random diet, measuring at least pH, volume, calcium, uric acid, sodium, citrate, oxalate, potassium, and creatinine. The EAU and AUA recommends two consecutive samples, though spot samples may be used when 24-h collection is not feasible, particularly in children (12, 13, 17). The presence of metabolic alterations in absence of other clinical explanations should indicate a molecular analysis in order to evaluate the presence of a genetic disease.
2.3 Risk stratification
Currently, formal guidelines for risk stratification and genetic testing have not yet been established.
According to Gefen et al., risk indicators for genetic forms that can be used as genetic testing criteria are those associated with nephrocalcinosis and higher rates of CKD in children (younger age, low serum bicarbonate, hypophosphatemia, hypercalcemia, and developmental delay) (18).
Payne et al. Analyzed a panel of 41 genes associated with monogenic or multifactorial diseases with increased risk of nephrolithiasis in KSD patients and identified as significant predictive factors for genetic variants family history of stones (55.6%), first episode of KSD before age 18 (66.7%), recurrent stone formers, need for both medical and surgical management prior to testing (88.9%). Moreover, important indicators for genetic evaluation were 24-h urine alterations (hypercalciuria without sodium abuse, severe hyperoxaluria), or clinical suspicion for distal renal tubular acidosis (19).
Other criteria in children may include growth retardation, impaired kidney filtration, abnormal tubular function (such as acidosis, alkalosis, or electrolyte/mineral imbalances), and systemic manifestations in the eyes or bones (20).
For what concerns family history, Saha et al. showed that children carrying pathogenic variants were typically younger at symptoms onset. Additionally, only a minority of cases had family history, because 81% cases had homozygous mutations with autosomal recessive syndromes, highlighting the fact that genetic testing should be considered even when family history is absent (21). Halbritter et al. observed that recessive mutations were more frequently found among children and in congenital disease, whereas dominant conditions usually manifested later in life (22).
Therefore, the EAU and AUA criteria for identifying high-risk stone formers, which include patients with early onset urolithiasis (particularly in children and teenagers), familial stone formation, recurrent stone formation, recent stone episodes, and specific stone compositions such as brushite, uric acid, urate, or infection stones (12, 13) could also be considered amongst the inclusion criteria for genetic testing (Table 1).
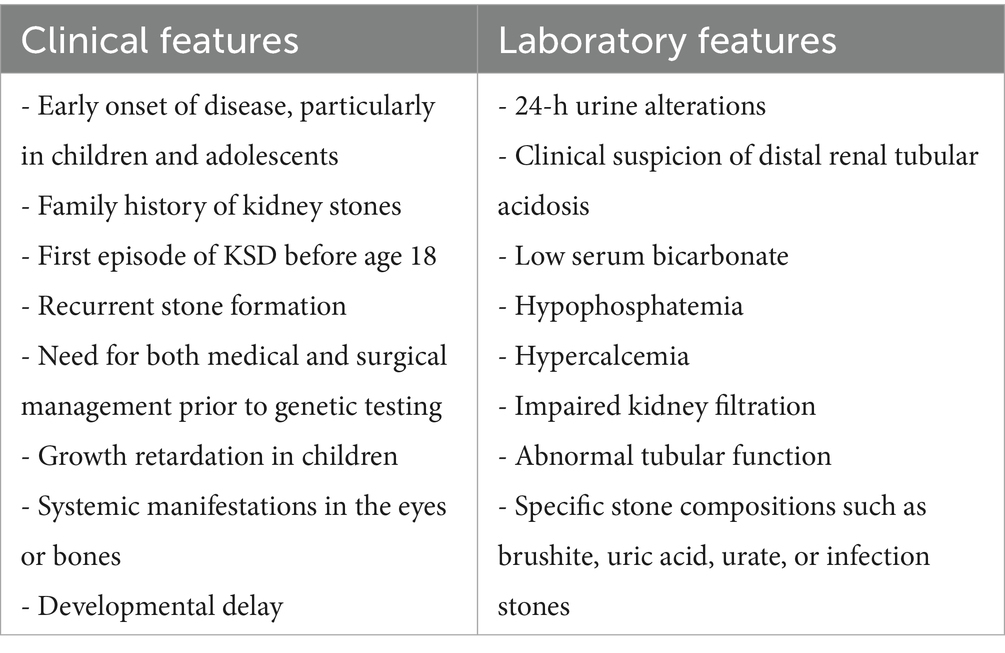
Table 1. Findings suggestive for a genetic basis of KS.
3 Genetic testing
Several studies underline the clinical importance of genetic testing in kidney stone formers: a missed diagnosis can easily lead to kidney failure, even after a kidney transplant (23) and in the majority of the cases the kidney stones are just the first symptom of presentation of a more complex disease.
Cogal et al. in a cohort of 703 families suspected of Primary Hyperoxaluria and a cohort of 111 families suspected of Dent Disease found by targeted NGS a monogenic cause of stone formation different from the initial hypothesis in 29/285 (10.2%) and 16/59 (27.1%) respectively (24). The most common biallelically mutated gene was CLDN16 (8 families) that together with CLDN19 is responsible for Familial Hypomagnesemia with Hypercalciuria and Nephrocalcinosis, variants were also found in SLC34A1 and SLC34A3 (5 families), accounting for Hypophosphatemic/Hypercalciuric Stone Formation with Bone Defects. Among the other monogenic diseases identified there were Infantile Hypercalcemia due to 24-Hydroxylase Deficiency (CYP24A1), Adenine Phosphoribosyl-transferase deficiency (APRT), Bartter Syndrome, Distal Renal Tubular Acidosis, Autosomal Dominant Hypocalcemia (CASR) and Dominant Fanconi Syndrome (HNF4A) (24). Amlie-Wolf et al. (25) observed 28/86 pathogenic/likely pathogenic (P/LP) variants in a cohort of patients from a nephrology unit, 27/28 (96%) variants led to a change in medical management of the patient and allowed cascade testing and management of the families. In 2021 Jayasinghe et al. (26) analyzed 204 patients with a suspected monogenic cause of kidney disease with exome sequencing (WES) and a molecular diagnosis was found in 80/204 patients (34% with a P/LP variant). Among the 80 positive results, 27/80 had a confirmed diagnosis (34%), 22 had a clarified diagnosis (28%), and 31 had a reclassified diagnosis (39%), allowing a better management of the patient and a more personalized treatment (26). Andregg et al. (27) conducted a WES analysis in 901 individuals, of which 787 were kidney stone formers, 23 of them had a mendelian form of KSD and 68 had a pathogenic/likely pathogenic that not corresponded to a mendelian form but likely predisposed to nephrolithiasis. In 5 (21.5%) of the 23 mendelian form, WES established a new or corrected a priori diagnosis.
These are just some of the studies where a molecular diagnosis shows a high impact on the management and treatment of not just the patients themselves but also of their families.
Therefore, genetic testing in KSD could be a useful tool to predict the prognosis and the onset of other clinical features, preventing unnecessary procedures and relapses, allowing individualized treatment that are increasingly available for monogenic kidney disorders (see section 4), and offering genetic counseling for family members.
Although the availability of genetic panels for kidney stones is increasing worldwide, access varies significantly across different Countries. This contributes to the global underutilization of genetic testing in clinical practice, beside the lack of standardized guidelines (28–31).
Therefore, it is necessary to establish how to select patients eligible for genetic testing (32, 33).
3.1 Monogenic diseases
The use of genetic testing in KSD patients currently leads to a diagnostic yield of 12–21% in children and young adults (<25 years old) and 1–11% in adults (>25 years old) (33), where the broad ranges depend on the selected cohort and on how stringent are the criteria used for assessing the variants pathogenicity. From these studies the most frequent genes harboring pathogenic/likely pathogenic variants are those of the solute carrier families, associated with several different monogenic diseases that increase the susceptibility to KSD, followed by Primary Hyperoxaluria genes and some members of the Claudin family (6, 18, 23–28, 34–37).
There are more than 30 genes linked to monogenic diseases that imply a higher risk of stone formation (Supplementary Table S1) (38, 39). Most of the severe syndromic and congenital forms have a recessive transmission, while milder conditions are often associated with dominant pathogenetic variants. In fact, according to recent research, for some recessive conditions heterozygous individuals can no longer be considered as simple healthy carriers, because they present renal calcifications more frequently than wildtype subjects, although to a lesser extent than individuals with biallelic variants (22).
Two significant examples of monogenic diseases that cause stone formation are Dent’s disease and cystinuria.
Dent’s disease (DD) is characterized by low-molecular-weight proteinuria, hypercalciuria, nephrolithiasis and nephrocalcinosis. Two genes cause the disease: CLCN5 (Type 1 DD) and OCRL (Type 2 DD). Type 2 DD may also present with extra renal symptoms, including mild cognitive impairment, hypotonia and cataracts, resulting in Lowe syndrome (40, 41).
Cystinuria is caused by mutations in SLC3A1 and SLC7A9 genes, leading to defective cystine transport that result in stone formation typically beginning in the first two decades of life, with higher severity in males. Cystine is an amino acid filtered by the kidneys and reabsorbed in the proximal tubule through a complex of two proteins encoded by SLC3A1 and SCL7A9. Mutations in either protein impair cystine reabsorption, leading to elevated urinary excretion. Due to cystine’s poor solubility in acidic urine, this causes stone formation, primarily in the distal tubule (42). Cystinuria is well-suited for gene therapy, mainly because of the disease’s autosomal recessive nature and manageable gene sizes (SLC3A1 2.3 kb, SLC7A9 1.8 kb), also, the core of the pathogenic mechanisms is located in the proximal tubule, which is easily accessible. Studies in albino mice using CRISPR/Cas9 have shown promising results (7).
3.2 GWAS and WES
GWAS recognized several genetic loci and common genetic variations (present in >1% of population) associated with KSD (43–51). As an example, Hao et al. (52) conducted the largest GWAS comprising 17,969 individuals and 702,230 controls prioritized 223 unique candidate genes, some of them identified in more studies (Supplementary Table S2). Most of these genes related to kidney physiology, as DGKH (a component of the intracellular CaSR-signaling pathway), CLDN14 (an integral membrane protein, component of tight junction helping decreasing the paracellular permeability to cations like calcium), SLC34A1 (sodium and phosphate transporter), CYP24A1 (variants lead to elevated 25-hydroxyvitamin D, increasing the reabsorption of calcium in kidney), ALPL (hydrolyzes extracellular inorganic pyrophosphate, which normally inhibits calcium oxalate and calcium phosphate crystallization in the urine). A significant role in nephrolithiasis predisposition is mediated by CASR and TRPV5 (epithelial calcium channel transient receptor) a long-standing candidate gene for KSD and hypercalciuria. Of note are the solute carrier and the claudin families, with many different members, whose pathogenic variants are found in inherited kidney disease, and with common variants often highlighted by GWAS as susceptibility. Another example of complex pathogenic mechanism is UMOD (uromodulin, an inhibitor of crystallization of calcium in urine), whose pathogenic variants are responsible of medullary cystic kidney disease, but some common variants have been seen to facilitate the expression of TRPV5 (22), therefore having a protective effect against nephrolithiasis.
Despite the large data available, the clinical use of polygenic scores for kidney diseases is limited by: (i) the scarcity of large GWAS, (ii) the low cross-ancestry transferability (Most GWAS are on European, excluding non-European population-specific variants, thus resulting in reduced predictive accuracy. E.g. the APOL1 risk locus is not detectable in European as the risk alleles are present exclusively on African haplotypes), and (iii) variable study quality (number of subjects, quality of imputation, the extent of ancestry bias control and the control of technical artifact). Further research is needed to enhance polygenic prediction by increasing sample diversity and improving score applicability across populations.
As an example of the utility of Polygenic risk score (PRS) and Genomic-wide Polygenic Score (GPS) there is the prediction of kidney failure, that frequently rely on creatinine levels and Estimated Glomerular Filtration Rate (eGFR), which are also affected by non-renal factors such as muscle creatine production. Existing population bases-GWAS for creatinine and eGFR are more robust and statistically significant than those for CKD, due to considerably limited population available for CKD studies than the general population, where eGFR and creatinine can be examined in relation to associated genes. Consequently, PRS and GPS specific to nephrolithiasis hold significant limitations and need larger, more diverse association studies for effective application (53). Nevertheless, integrating the use of PRS and GPS with family history and biochemical parameters could help in highlighting the high-risk patients. Moreover, the interplay between monogenic variants and polygenic background should not be forgotten; indeed, the GPS for CKD was a significant predictor of kidney disease in individuals with ADPKD and COL4A-associated nephropathy variants. Among carriers of ADPKD variants, those in the top tertile of the GPS distribution had a 54-fold increased risk of CKD compared with that of non-carriers, whereas those in the bottom tertile had only a 3-fold increased risk compared to that of non-carriers (53).
4 New stone prevention methods
Preventive strategies are selected based on etiology, stone type, and cost-effectiveness. Main preventive measures include fluid intake (>2.5 L/day), lifestyle changes (body weight control, adequate hydration during physical activity), and dietary management (controlled intake of calcium, sodium, and animal proteins, reduced ingestion of oxalate-rich foods).
The protective effects of bioactive components are still to be explored, except for coffee which has been shown to be protective. Thiazides are indicated in people with recurrent calcium stones, and alkaline citrate for a variety of stones. Allopurinol is recommended in some forms of hyperuricosuria. Bacterial eradication and antibiotic use may be important in certain situations, but they must be closely monitored due to the possibility of antimicrobial resistance. Finally, probiotics may help in prevention, but further research is needed to prove their efficacy (54).
Recent research has identified various protein receptors and enzymes as potential therapeutic targets in urolithiasis, including calcium-sensing receptors, vasopressin V2 receptors, and angiotensin II receptors, which regulate calcium excretion, water reabsorption, and electrolyte balance. By focusing on particular pathways involved in crystal formation, aggregation, and adhesion, these receptor-targeted therapies present promising prospects for precision medicine approaches, especially in preventing stone recurrence (55).
While hyperhydration remains the foundation of prevention, understanding genetic etiology could help personalize preventive strategies.
Primary Hyperoxaluria type 1 (PH1) serves as a prime example of how understanding the genetic basis of the disease has led to targeted therapy directly addressing the pathogenesis. PH1 is caused by pathogenic variants in the AGXT gene, encoding for the liver enzyme alanine glyoxylate aminotransferase (AGT), leading to excess oxalate production (56). Pyridoxine acts as a cofactor for AGT enzyme, increasing its activity and reducing oxalate production in specific AGXT variants (57). Lumasiran is a RNA interference therapeutic agent administered subcutaneously and directed at the liver. In patients with PH1, Lumasiran reduces hepatic oxalate production and increases glycolate concentrations by degrading the messenger RNA that encodes AGT (8). Other examples are the specific therapy in patients with CYP24A1 gene variants comprises azole drugs (e.g., ketoconazole and fluconazole), which inhibit cytochrome P450 enzymes, leading to normalization of biochemical parameters (58, 59). Another approach is to induce CYP3A4 activity with the drug rifampicin, providing an alternative method for the inactivation of active vitamin D metabolites (60). For SLC34A1 gene variants, the specific therapy consists of oral phosphorus supplementation, which leads to rapid correction of hypophosphatemia, normalization of calcium metabolism, and significant clinical improvement, as demonstrated by weight gain in patients. Sodium chloride supplementation may also be necessary to correct hypercalcemia in patients who also present with polyuria and polydipsia (61). For Cystinuria, cystine-binding drugs such as Tiopronin and D-penicillamine, are effective in reducing free urine cystine levels (62). Moreover, l-Cystine diamides, inhibitors of l-cystine crystallization (63), and α-Lipoic acid that inhibits cystine stone formation (64) have shown great promise in the treatment of Cystinuria but are still under evaluation. Early diagnosis of Adenine phosphoribosyltransferase (APRT) deficiency allows for treatment with allopurinol, which inhibits xanthine oxidase and reduces 2,8-DHA formation, potentially preventing, arresting, or even reversing kidney failure in both native and transplanted kidneys (65). In clinical trials Febuxostat showed better results than allopurinol in reducing DHA excretion in the prescribed doses (66).
Thus, early genetic diagnosis can enable targeted therapies, sparing kidney-liver transplantation to the patients and long-term complication (67).
5 Conclusion
Despite the KSD genetic predisposition is not yet fully understood, the present data are sufficient to recommend genetic testing of a stone-gene panel in high-risk KSD patients. In conclusion, Red Flags to recognize genetic forms in KSD patients can be: recurrent nephrolithiasis, first episode of KSD before the age of 18 years old, nephrocalcinosis, family history of stones, 24-h urine alterations, impaired kidney filtration, abnormal tubular function and syndromic conditions (Figure 1). The clinical utility of genetic diagnosis goes far beyond therapeutic applications only. It includes pre-symptomatic identification of monogenic forms before the development of irreversible renal damage, support for clinical management through more targeted monitoring, detection of recessive disorder heterozygous carriers who may be at increased risk, and utilization of tailored preventive interventions based on the individual genetic pattern. Furthermore, it allows for counseling of high-risk relatives, offers the possibility of prenatal diagnosis in the most severe cases, and directs future research toward new treatments this broader vision of the utility of genetic diagnostics highlights the importance of integrating genetic testing into the diagnostic workup of patients with recurrent renal lithiasis or with clinical features suggestive of hereditary forms.
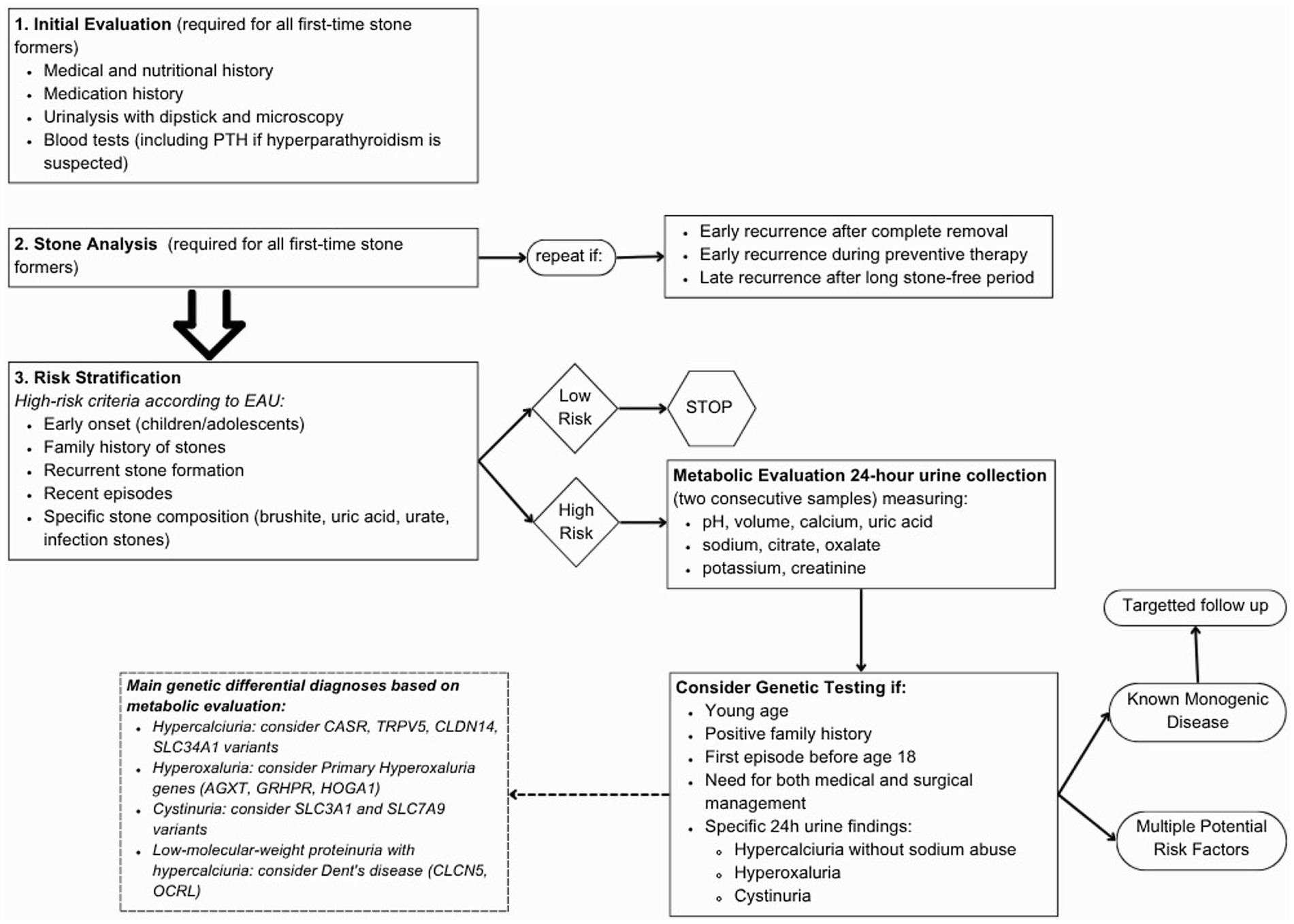
Figure 1. Flow chart to guide genetic testing; see Supplementary Table S1 for description of the diseases associated to each gene (67).
Undoubtedly, genetics will not be the only key to better understanding. Indeed, multi-omic studies have consistently shown that kidney stone patients exhibit gut and urinary dysbiosis with reduced oxalate-degrading bacteria (e.g., O. formigenes, Lactobacillus, Bifidobacterium) and enrichment of pathogens (e.g., Streptococcus, E. coli), alongside metabolic alterations involving tryptophan, fatty acids, and oxalic acid (68–70). These changes correlate with inflammation, oxidative stress, and urinary metabolite shifts (e.g., pyroglutamic acid, FAHFA), supporting the need of biomarker discovery and microbiome-targeted therapies such as probiotics and FMT (Fecal Microbiota Transplantation) (68, 69).
Author contributions
FP: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. NG: Conceptualization, Data curation, Writing – original draft, Writing – review & editing. DG: Writing – review & editing. GM: Conceptualization, Investigation, Methodology, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1631281/full#supplementary-material
References
1. Singh, P, Harris, PC, Sas, DJ, and Lieske, JC. The genetics of kidney stone disease and nephrocalcinosis. Nat Rev Nephrol. (2022) 18:224–40. doi: 10.1038/s41581-021-00513-4
2. Hill, F, and Sayer, JA. Precision medicine in renal stone-formers. Urolithiasis. (2019) 47:99–105. doi: 10.1007/s00240-018-1091-5
3. Singh, P, Enders, FT, Vaughan, LE, Bergstralh, EJ, Knoedler, JJ, Krambeck, AE, et al. Stone composition among first-time symptomatic kidney stone formers in the community. Mayo Clin Proc. (2015) 90:1356–65. doi: 10.1016/j.mayocp.2015.07.016
4. Goldfarb, DS, Avery, AR, Beara-Lasic, L, Duncan, GE, and Goldberg, J. A twin study of genetic influences on nephrolithiasis in women and men. Kidney Int Rep. (2019) 4:535–40. doi: 10.1016/j.ekir.2018.11.017
5. Howles, SA, and Thakker, RV. Genetics of kidney stone disease. Nature Reviews Urology Nature Research. (2020) 17:407–21. doi: 10.1038/s41585-020-0332-x
6. Huang, L, Qi, C, Zhu, G, Ding, J, Yuan, L, Sun, J, et al. Genetic testing enables a precision medicine approach for nephrolithiasis and nephrocalcinosis in pediatrics: a single-center cohort. Mol Gen Genomics. (2022) 297:1049–61. doi: 10.1007/s00438-022-01897-z
7. Peek, JL, and Wilson, MH. Gene therapy for kidney disease: targeting cystinuria In: Curr Opin Nephrol Hypertens. (2022) 31:175–9. doi: 10.1097/MNH.0000000000000768
8. Garrelfs, SF, Frishberg, Y, Hulton, SA, Koren, MJ, O’Riordan, WD, Cochat, P, et al. Lumasiran, an RNAi therapeutic for primary hyperoxaluria type 1. N Engl J Med. (2021) 384:1216–26. doi: 10.1056/NEJMoa2021712
9. D’ambrosio, V, and Ferraro, PM. Lumasiran in the management of patients with primary hyperoxaluria type 1: from bench to bedside. Int J Nephrol Renovasc Dis. (2022) 15:197–206. doi: 10.2147/IJNRD.S293682
10. Pearle, MS, Calhoun, EA, and Curhan, GC. Urologic Diseases of America Project. Urologic diseases in America project: urolithiasis. J Urol. (2005) 173:848–57. doi: 10.1097/01.ju.0000152082.14384.d7
11. Geraghty, RM, Cook, P, Walker, V, and Somani, BK. Evaluation of the economic burden of kidney stone disease in the UK: a retrospective cohort study with a mean follow-up of 19 years. BJU Int. (2020) 125:586–94. doi: 10.1111/bju.14991
13. Pearle, MS, Goldfarb, DS, Assimos, DG, Curhan, G, Denu-Ciocca, CJ, Matlaga, BR, et al. Medical management of kidney stones: AUA guideline. J Urol. (2014) 192:316–24. doi: 10.1016/j.juro.2014.05.006
14. Rodgers, AL. Physicochemical mechanisms of stone formation. Urolithiasis. (2017) 45:27–32. doi: 10.1007/s00240-016-0942-1
15. Negri, AL, and Spivacow, FR. Kidney stone matrix proteins: role in stone formation. World J Nephrol. (2023) 12:21–8. doi: 10.5527/wjn.v12.i2.21
16. Zhong, W, Osther, P, Pearle, M, Choong, S, Mazzon, G, Zhu, W, et al. International Alliance of urolithiasis (IAU) guideline on staghorn calculi management. World J Urol. (2024) 42:189. doi: 10.1007/s00345-024-04816-6
17. Akram, M, Jahrreiss, V, Skolarikos, A, Geraghty, R, Tzelves, L, Emilliani, E, et al. Urological guidelines for kidney stones: Overview and comprehensive update, J Clin Med. (2024) 13:1114. doi: 10.3390/jcm13041114
18. Gefen, AM, Sethna, CB, Cil, O, Perwad, F, Schoettler, M, Michael, M, et al. Genetic testing in children with nephrolithiasis and nephrocalcinosis. Pediatr Nephrol. (2023) 38:2615–22. doi: 10.1007/s00467-023-05879-0
19. Payne, NG, Boddu, SP, Wymer, KM, Heidenberg, DJ, Van Der Walt, C, Mi, L, et al. The use of genetic testing in nephrolithiasis evaluation: a retrospective review from a multidisciplinary kidney stone clinic. Urology. (2024) 193:20–6. doi: 10.1016/j.urology.2024.07.029
20. Langman, CB. A rational approach to the use of sophisticated genetic analyses of pediatric stone disease. Kidney Int. (2018) 93:15–8. doi: 10.1016/j.kint.2017.08.023
21. Saha, A, Kapadia, SF, Vala, KB, and Patel, HV. Clinical utility of genetic testing in Indian children with kidney diseases. BMC Nephrol. (2023) 24:212. doi: 10.1186/s12882-023-03240-z
22. Halbritter, J. Genetics of kidney stone disease—Polygenic meets monogenic. Nephrologie et Therapeutique. (2021) 17:S88–894. doi: 10.1016/j.nephro.2020.02.003
23. Stephan, R, and Hoppe, B. Genetic kidney stones disease in adults. 39, Nephrology Dialysis Transplantation (2024). p. 1381–1383. doi: 10.1093/ndt/gfae099
24. Cogal, AG, Arroyo, J, Shah, RJ, Reese, KJ, Walton, BN, Reynolds, LM, et al. Comprehensive genetic analysis reveals complexity of monogenic urinary stone disease. Kidney Int Rep. (2021) 6:2862–84. doi: 10.1016/j.ekir.2021.08.033
25. Amlie-Wolf, L, Baker, L, Hiddemen, O, Thomas, M, Burke, C, Gluck, C, et al. Novel genetic testing model: a collaboration between genetic counselors and nephrology. Am J Med Genet A. (2021) 185:1142–50. doi: 10.1002/ajmg.a.62088
26. Jayasinghe, K, Stark, Z, Kerr, PG, Gaff, C, Martyn, M, Whitlam, J, et al. Clinical impact of genomic testing in patients with suspected monogenic kidney disease. Genet Med. (2021) 23:183–91. doi: 10.1038/s41436-020-00963-4
27. Anderegg, MA, Olinger, EG, Bargagli, M, Geraghty, R, Taylor, L, Nater, A, et al. Prevalence and characteristics of genetic disease in adult kidney stone formers. Nephrology Dialysis Transplantation. (2024) 39:1426–41. doi: 10.1093/ndt/gfae074
28. Spasiano, A, Treccani, M, De Tomi, E, Malerba, G, Gambaro, G, and Ferraro, PM. Characteristics and yield of modern approaches for the diagnosis of genetic causes of kidney stone disease. Genes. (2024) 15:1470. doi: 10.3390/genes15111470
29. Mrug, M, Bloom, MS, Seto, C, Malhotra, M, Tabriziani, H, Gauthier, P, et al. Genetic testing for chronic kidney diseases: clinical utility and barriers perceived by nephrologists. Kidney Med. (2021) 3:1050–6. doi: 10.1016/j.xkme.2021.08.006
30. Knoers, N, Antignac, C, Bergmann, C, Dahan, K, Giglio, S, Heidet, L, et al. Genetic testing in the diagnosis of chronic kidney disease: recommendations for clinical practice. Nephrology Dialysis Transplantation. (2022) 37:239–54. doi: 10.1093/ndt/gfab218
31. Becherucci, F, Landini, S, Palazzo, V, Cirillo, L, Raglianti, V, Lugli, G, et al. A clinical workflow for cost-saving high-rate diagnosis of genetic kidney diseases. J Am Soc Nephrol. (2023) 34:706–20. doi: 10.1681/ASN.0000000000000076
32. Granhøj, J, Tougaard, B, Lildballe, DL, and Rasmussen, M. Family history is important to identify patients with monogenic causes of adult-onset chronic kidney disease. Nephron. (2022) 146:49–57. doi: 10.1159/000518175
33. Geraghty, R, Lovegrove, C, Howles, S, and Sayer, JA. Role of genetic testing in kidney stone disease: a narrative review. Curr Urol Rep. (2024) 25:311–23. doi: 10.1007/s11934-024-01225-5
34. Wuttke, M, König, E, Katsara, MA, Kirsten, H, Farahani, SK, Teumer, A, et al. Imputation-powered whole-exome analysis identifies genes associated with kidney function and disease in the UK biobank. Nat Commun. (2023) 14:1287. doi: 10.1038/s41467-023-36864-8
35. Giusti, F, Marini, F, Al-alwani, H, Marasco, E, Garagnani, P, Khan, AA, et al. A novel heterozygous mutation c.1627G>T (p.Gly543Cys) in the SLC34A1 gene in a male patient with recurrent nephrolithiasis and early onset osteopenia: a case report. Int J Mol Sci. (2023) 24:17289. doi: 10.3390/ijms242417289
36. Liu, Y, Ge, Y, Zhan, R, Zhao, Z, Li, J, and Wang, W. Identification of mutations in 15 nephrolithiasis-related genes leading to a molecular diagnosis in 85 Chinese pediatric patients. Pediatr Nephrol. (2023) 38:3645–61. doi: 10.1007/s00467-023-06028-3
37. Schönauer, R, Scherer, L, Nemitz-Kliemchen, M, Hagemann, T, Hantmann, E, Seidel, A, et al. Systematic assessment of monogenic etiology in adult-onset kidney stone formers undergoing urological intervention–evidence for genetic pretest probability. Am J Med Genet C Semin Med Genet. (2022) 190:279–88. doi: 10.1002/ajmg.c.31991
38. Halbritter, J, Baum, M, Hynes, AM, Rice, SJ, Thwaites, DT, Gucev, ZS, et al. Fourteen monogenic genes account for 15% of nephrolithiasis/nephrocalcinosis. J Am Soc Nephrol. (2015) 26:543–51. doi: 10.1681/ASN.2014040388
39. Braun, DA, Lawson, JA, Gee, HY, Halbritter, J, Shril, S, Tan, W, et al. Prevalence of monogenic causes in pediatric patients with nephrolithiasis or nephrocalcinosis. Clin J Am Soc Nephrol. (2016) 11:664–72. doi: 10.2215/CJN.07540715
40. Diéguez, L, Pilco, M, Butori, S, Kanashiro, A, Balaña, J, Emiliani, E, et al. Dent’s disease: a cause of monogenic kidney stones and Nephrocalcinosis. J Pers Med. (2024) 14:623. doi: 10.3390/jpm14060623
41. Ehlayel, AM, and Copelovitch, L. Update on dent disease. Pediatr Clin N Am. (2019) 66:169–78. doi: 10.1016/j.pcl.2018.09.003
42. D’Ambrosio, V, Capolongo, G, Goldfarb, D, Gambaro, G, and Ferraro, PM. Cystinuria: an update on pathophysiology, genetics, and clinical management. Pediatr Nephrol. (2022) 37:1705–11. doi: 10.1007/s00467-021-05342-y
43. Thorleifsson, G, Holm, H, Edvardsson, V, Walters, GB, Styrkarsdottir, U, Gudbjartsson, DF, et al. Sequence variants in the CLDN14 gene associate with kidney stones and bone mineral density. Nat Genet. (2009) 41:926–30. doi: 10.1038/ng.404
44. Gudbjartsson, DF, Holm, H, Indridason, OS, Thorleifsson, G, Edvardsson, V, Sulem, P, et al. Association of variants at UMOD with chronic kidney disease and kidney stones-role of age and comorbid diseases. PLoS Genet. (2010) 6:1–9. doi: 10.1371/journal.pgen.1001039
45. Oddsson, A, Sulem, P, Helgason, H, Edvardsson, VO, Thorleifsson, G, Sveinbjörnsson, G, et al. Common and rare variants associated with kidney stones and biochemical traits. Nat Commun. (2015) 6:7975. doi: 10.1038/ncomms8975
46. Urabe, Y, Tanikawa, C, Takahashi, A, Okada, Y, Morizono, T, Tsunoda, T, et al. A genome-wide association study of nephrolithiasis in the Japanese population identifies novel susceptible loci at 5q35.3, 7p14.3, and 13q14.1. PLoS Genet. (2012) 8:e1002541. doi: 10.1371/journal.pgen.1002541
47. Benonisdottir, S, Kristjansson, RP, Oddsson, A, Steinthorsdottir, V, Mikaelsdottir, E, Kehr, B, et al. Sequence variants associating with urinary biomarkers. Hum Mol Genet. (2019) 28:1199–211. doi: 10.1093/hmg/ddy409
48. Howles, SA, Wiberg, A, Goldsworthy, M, Bayliss, AL, Gluck, AK, Ng, M, et al. Genetic variants of calcium and vitamin D metabolism in kidney stone disease. Nat Commun. (2019) 10:5175. doi: 10.1038/s41467-019-13145-x
49. Tanikawa, C, Kamatani, Y, Terao, C, Usami, M, Takahashi, A, Momozawa, Y, et al. Novel risk loci identified in a genome-wide association study of urolithiasis in a Japanese population. J Am Soc Nephrol. (2019) 30:855–64. doi: 10.1681/ASN.2018090942
50. Wuttke, M, Li, Y, Li, M, Sieber, KB, Feitosa, MF, Gorski, M, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat Genet. (2019) 51:957–72. doi: 10.1038/s41588-019-0407-x
51. Chen, WC, Chen, YC, Chen, YH, Liu, TY, Tsai, CH, and Tsai, FJ. Identification of novel genetic susceptibility loci for calcium-containing kidney stone disease by genome-wide association study and polygenic risk score in a Taiwanese population. Urolithiasis. (2024) 52:94. doi: 10.1007/s00240-024-01577-0
52. Hao, X, Shao, Z, Zhang, N, Jiang, M, Cao, X, Li, S, et al. Integrative genome-wide analyses identify novel loci associated with kidney stones and provide insights into its genetic architecture. Nat Commun. (2023) 14:7498. doi: 10.1038/s41467-023-43400-1
53. Khan, A, and Kiryluk, K. Polygenic scores and their applications in kidney disease. Nat Rev Nephrol. (2024) 21:24–38. doi: 10.1038/s41581-024-00886-2
54. Peerapen, P, and Thongboonkerd, V. Kidney Stone Prevention. Adv Nutr. (2023) 14:555–69. doi: 10.1016/j.advnut.2023.03.002
55. Haque, Z, Taleuzzaman, M, Jamal, R, Al-Qahtani, NH, and Haque, A. Targeting protein receptors and enzymes for precision management of urolithiasis: a comprehensive review. Eur J Pharmacol. (2024) 981:176904. doi: 10.1016/j.ejphar.2024.176904
56. Fargue, S, and Acquaviva, BC. Primary hyperoxaluria type 1: pathophysiology and genetics. Clinical Kidney J. (2022) 15:I4–8. doi: 10.1093/ckj/sfab217
57. Gupta, A, Somers, MJG, and Baum, MA. Treatment of primary hyperoxaluria type 1. Clinical Kidney J. (2022) 15:I9–I13. doi: 10.1093/ckj/sfab232
58. Marks, J, Debnam, ES, and Unwin, RJ. Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol. (2010) 299:285–96. doi: 10.1152/ajprenal.00508.2009
59. Tebben, PJ, Milliner, DS, Horst, RL, Harris, PC, Singh, RJ, Wu, Y, et al. Hypercalcemia, hypercalciuria, and elevated calcitriol concentrations with autosomal dominant transmission due to CYP24A1 mutations: effects of ketoconazole therapy. J Clin Endocrinol Metab. (2012) 97:E423–7. doi: 10.1210/jc.2011-1935
60. Hawkes, CP, Li, D, Hakonarson, H, Meyers, KE, Thumme, KE, and Levine, MA. CYP3A4 induction by rifampin: an alternative pathway for vitamin D inactivation in patients with CYP24A1 mutations. J Clin Endocrinol Metab. (2017) 102:1440–6. doi: 10.1210/jc.2016-4048
61. Kurnaz, E, Savaş Erdeve, Ş, Çetinkaya, S, and Aycan, Z. Rare cause of infantile hypercalcemia: a novel mutation in the SLC34A1 gene. Horm Res Paediatr. (2019) 91:278–84. doi: 10.1159/000492899
62. Dolin, DJ, Asplin, JR, Flagel, L, Grasso, M, and Goldfarb, DS. Effect of Cystine-Binding Thiol Drugs on Urinary Cystine Capacity in Patients with Cystinuria. J Endourol. (2005) 19
63. Hu, L, Albanyan, H, Yang, J, Tan, X, Wang, Y, Yang, M, et al. Structure-activity relationships and pharmacokinetic evaluation of L-cystine diamides as L-cystine crystallization inhibitors for cystinuria. Med Chem Res. (2024) 33:1384–407. doi: 10.1007/s00044-024-03228-w
64. Servais, A, Thomas, K, Dello Strologo, L, Sayer, JA, Bekri, S, Bertholet-Thomas, A, et al. Cystinuria: clinical practice recommendation. Kidney Int. (2021) 99:48–58. doi: 10.1016/j.kint.2020.06.035
65. Rajput, P, Virani, Z, and Shah, B. Crystalline nephropathy due to APRT deficiency: a preventable cause of renal and renal allograft failure. Indian J Nephrol. (2020) 30:290–2. doi: 10.4103/ijn.IJN_106_19
66. Edvardsson, VO, Runolfsdottir, HL, Thorsteinsdottir, UA, Inger, IM, Oddsdottir, GS, Eiriksson, F, et al. Comparison of the effect of allopurinol and febuxostat on urinary 2,8-dihydroxyadenine excretion in patients with adenine phosphoribosyltransferase deficiency (APRTd): a clinical trial. Eur J Intern Med. (2018) 48:75–9. doi: 10.1016/j.ejim.2017.10.007
67. Policastro, LJ, Saggi, SJ, Goldfarb, DS, and Weiss, JP. Personalized intervention in monogenic stone formers. J Urol. (2018) 199:623–32. doi: 10.1016/j.juro.2017.09.143
68. Gao, H, Lin, J, Xiong, F, Yu, Z, Pan, S, and Huang, Y. Urinary microbial and metabolomic profiles in kidney stone disease. Front Cell Infect Microbiol. (2022) 12:953392. doi: 10.3389/fcimb.2022.953392
69. Pei, X, Liu, M, and Yu, S. How is the human microbiome linked to kidney stones? Front Cell Infect Microbiol. (2025) 15:1602413. doi: 10.3389/fcimb.2025.1602413
Keywords: kidney stone, genetic testing, polygenic risk score, genetic predisposition, hereditary kidney stone disease
Citation: Pintus F, Giordano N, Giachino DF and Mandrile G (2025) Genetics of kidney stones and the role of genetic testing in prevention: a guide for urologists. Front. Med. 12:1631281. doi: 10.3389/fmed.2025.1631281
Edited by:
Bo Xiao, Tsinghua University, ChinaReviewed by:
Becky Mingyao Ma, Queen Mary Hospital, Hong Kong SAR, ChinaMira Keddis, Mayo Clinic, United States
Copyright © 2025 Pintus, Giordano, Giachino and Mandrile. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giorgia Mandrile, Z2lvcmdpYS5tYW5kcmlsZUB1bml0by5pdA==
†These authors have contributed equally to this work and share first authorship