Abstract
This case report explores the consequences of ruxolitinib via inhibition janus kinase 1 (JAK1) and JAK2 pathways in the context of fungal defense in a patient diagnosed with pulmonary coccidioidomycosis during ruxolitinib therapy for polycythemia vera. The patient experienced a relapse of pulmonary coccidioidomycosis after antifungal treatment was discontinued while continuing ruxolitinib use. This case illustrates the heightened risk of discontinuing antifungal therapy in endemic regions, emphasizing the critical need for continued monitoring. Furthermore, this case underscores the vital role of the JAK1 and JAK2 signaling cascade, particularly the interferon-gamma (INF-γ)-JAK1 and JAK2-signal transducer and activator of transcription 1 (STAT) axis, in antifungal defense. Recent studies have revealed that the loss of function in JAK1 (but not JAK2), leads to impaired macrophage activation and reduced T-helper 1 (Th1) cell responses, thereby compromising the body's ability to fight off dimorphic fungi, such as Coccidioides. Other proposed fungal immune mechanisms in the JAK-STAT pathway are discussed. Clinicians tailoring JAK inhibitor treatment options for patients must be aware of the INF-γ-JAK1-STAT pathway's pivotal role in antifungal defense.
Introduction
The Janus kinase-signal transducer and activator of transcription (JAK-STAT) pathway is a well-known cornerstone in immune regulation, orchestrating a variety of cellular responses via cytokine signaling. Inhibition of JAK1 and JAK2 can compromise the immune system's ability to defend against infections. This case report explores the consequences of inhibiting JAK1 and JAK2 via ruxolitinib, particularly in the context of fungal defense in a patient diagnosed with pulmonary coccidioidomycosis.
Ruxolitinib, a selective JAK1 and JAK2 inhibitor, is frequently used to treat polycythemia vera (PV), myelofibrosis, and graft-vs.-host disease (GVHD). While effective for controlling disease progression, its immunosuppressive effects can increase susceptibility to infections. Notably, fungal infections were not prominently featured in early clinical trials (1, 2). However, more recent studies and case reports have documented occurrences of invasive fungal infections in patients treated with ruxolitinib (3, 4).
We present a case of a 60-year-old male residing in Arizona who developed pulmonary coccidioidomycosis during ruxolitinib therapy. Following treatment with itraconazole, symptoms and radiologic imaging improved, and antifungal therapy was discontinued after 18 months of treatment. A year later, the patient redeveloped pulmonary coccidioidomycosis with continued ruxolitinib treatment. This case illustrates the potential consequences of discontinuing antifungal therapy, raising concerns about prophylaxis, and underscoring the critical role of the JAK1 and JAK2 signaling cascade, particularly the interferon gamma (IFN-γ)-JAK1 and JAK2-STAT1 axis, in antifungal defense (3, 5–7). Informed consent was affirmed and obtained by the patient, with data privacy safeguards.
Case presentation
A 60-year-old male residing in Arizona with a history of PV on ruxolitinib therapy presented with a four-week history of productive cough with whitish sputum and intermittent fever. A chest computed tomography (CT) scan demonstrated a left lower lobe consolidation accompanied by satellite nodules and mediastinal lymphadenopathy (Figure 1A). He was initially treated with oseltamivir and levofloxacin by his primary care physician, without clinical improvement. Serologic testing returned positive for both Coccidioides IgM and IgG by immunoassay, with a complement fixation (CF) titer of 1:4. His diagnosis of pulmonary coccidioidomycosis was confirmed via a fungal sputum culture, and antifungal therapy with fluconazole was initiated; however, due to persistent symptoms after 2 weeks, he was transitioned to itraconazole. Itraconazole, a strong CYP3A4 inhibitor, is known to increase ruxolitinib blood levels. Given the potential for drug–drug interaction between itraconazole and ruxolitinib, the patient's hematologist reduced the patient's ruxolitinib dose by 50% from per oral (PO) 10 mg twice daily (BID) to PO 5 mg BID, with monthly monitoring of the patient's complete blood count, which remained within normal limits. The patient's serum itraconazole and hydroxyitraconazole levels remained within therapeutic range (>1 mcg/mL) throughout the patient's clinical course while on the antifungal, which was measured every 6 months.
Figure 1
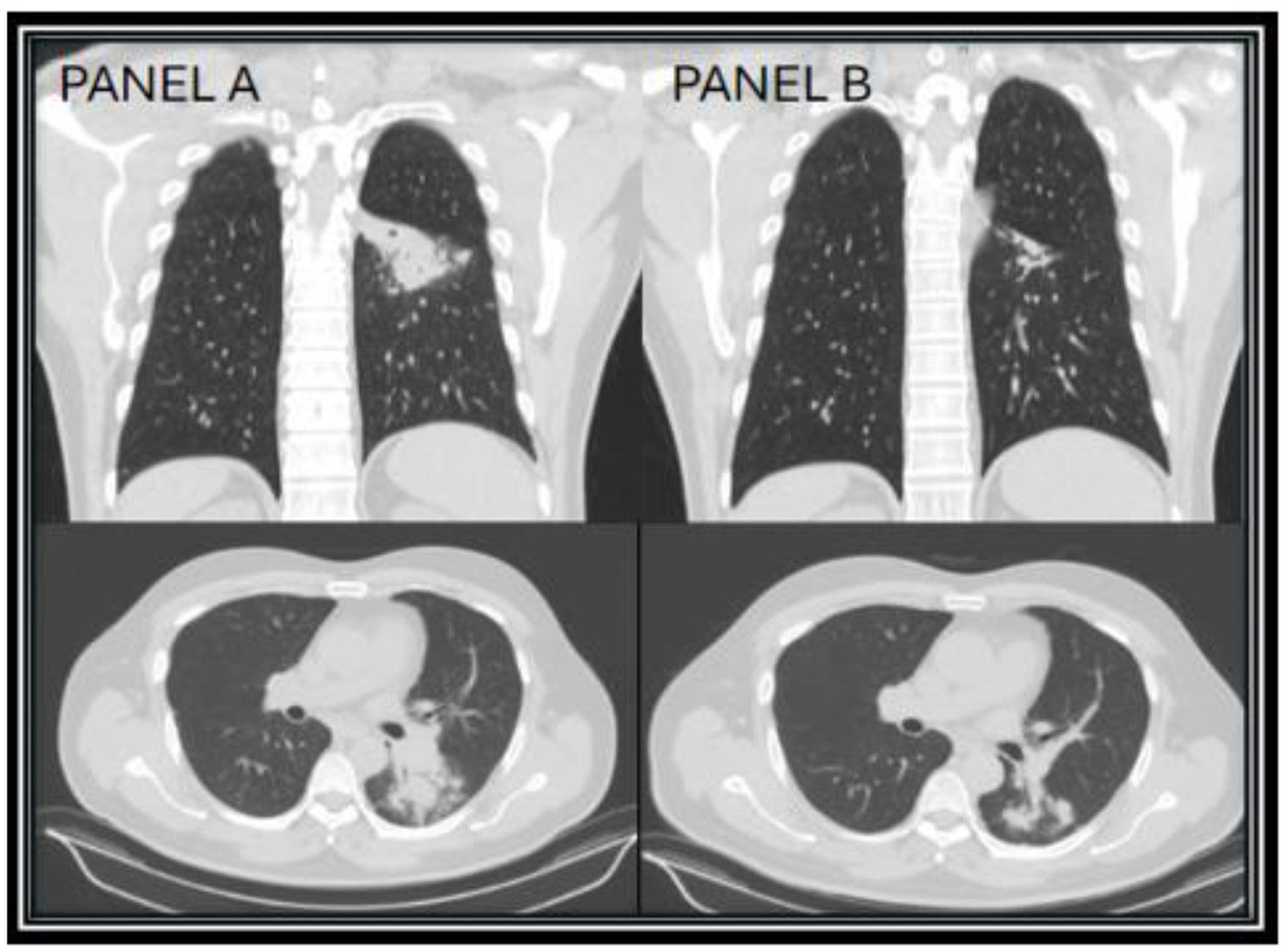
CT chest images taken 1 month after diagnosis (year zero) of Coccidioides infection showing the superior left lower lobe infiltrate (A), and repeat CT chest taken 17 months on itraconazole showing improvement of the superior left lower lobe infiltrate (B).
After discussions with infectious disease and hematology regarding continued treatment of ruxolitinib in the setting of an active infection, the patient decided to continue ruxolitinib in conjunction with itraconazole, due to the patient's excellent disease control of his PV on ruxolitinib. On repeat chest CT done at 17 months of treatment, the patient's left lower lobe consolidation showed near resolution, with small residual fibrotic changes (Figure 1B). At the patient's follow up visit, he endorsed complete resolution of symptoms, with clinical improvement over 18 months. Due to presumed disease control, no elevation in CF titers (1:4), and proper IDSA recommended treatment length in immunocompromised patients, itraconazole was discontinued and the patient was counseled to call their provider with any relapse of symptoms (8). Additionally, 2 months later, the ruxolitinib dose was increased to PO 10 mg BID.
However, the patient expressed concerns about a new onset dry cough a year later. He was urged to present to the clinic for a clinical workup, but he did not follow up for 6 months. When he presented to the clinic, he continued to endorse persistent symptoms of dry cough and shortness of breath. A CT chest revealed a new right upper lobe infiltrate, and the patient's Coccidioides CF titer had spiked to 1:64 (Figure 2). Recognizing this as a relapse of pulmonary coccidioidomycosis, the patient was promptly restarted on itraconazole, which resulted in dramatic resolution of symptoms within 2 weeks. With initiation of itraconazole, the patient's ruxolitinib dose was again reduced to PO 5 mg BID. Follow-up testing showed a steady decline in CF titers to 1:4, confirming the effectiveness of continued antifungal therapy. Repeat CT imaging revealed resolution of right upper lobe infiltrate after 2 years of itraconazole use (Figure 3). At the time of publication of this paper, the patient has continued to maintain effective disease control on itraconazole and ruxolitinib and will continue with indefinite antifungal treatment.
Figure 2
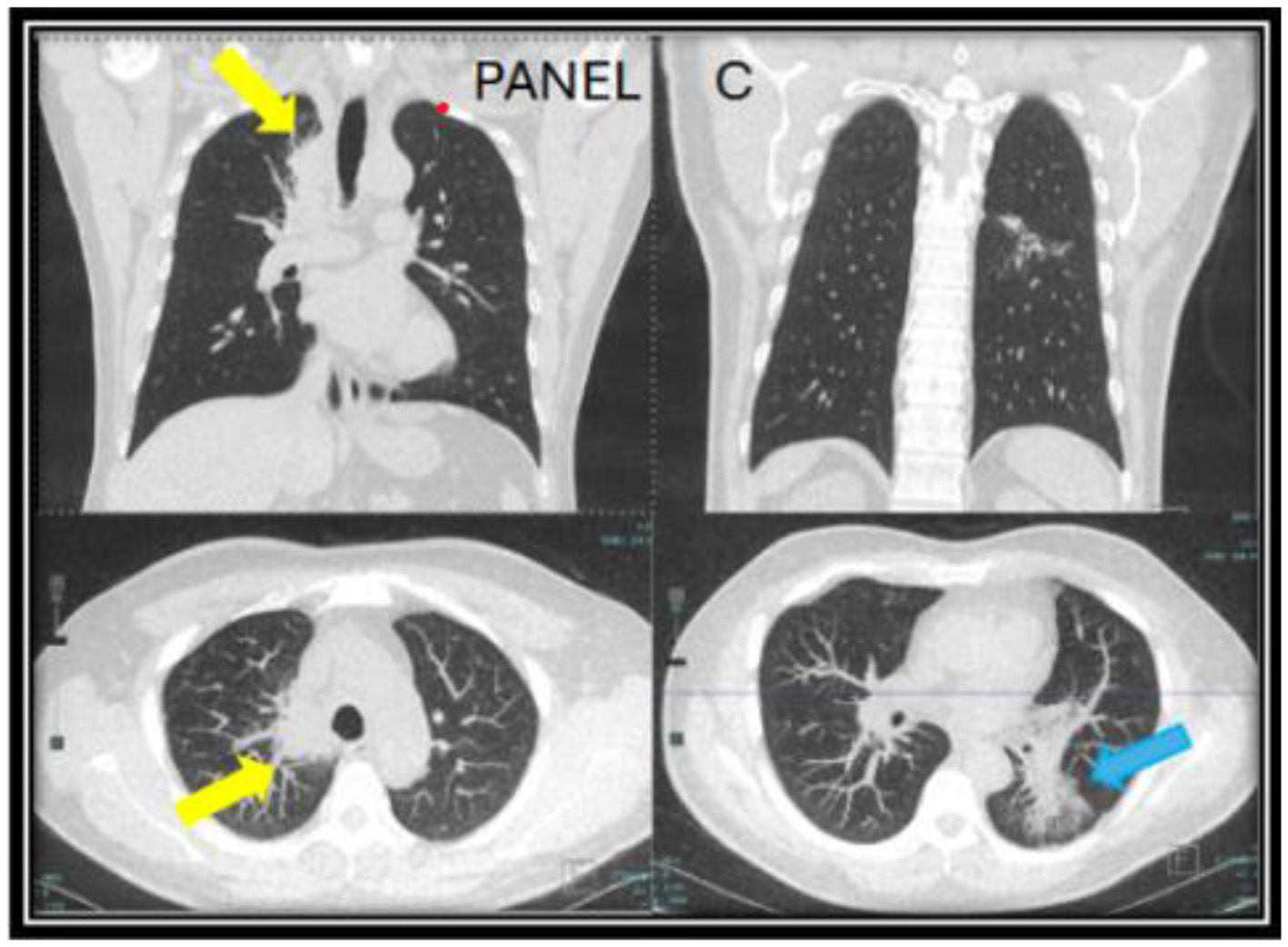
CT chest images taken a year after of being off of itraconazole (year two) when the patient developed a new persistent cough associated with a rise in Coccidioides complement fixation titers. A new mass-like consolidation of the medial right lung apex measuring 5.1 × 2.9 × 0.8 cm (yellow arrow) was noted. The superior segment of the left lower lobe continued to improve (blue arrow). The nodular consolidation has decreased, and mild scarring and bronchiectasis now persisted in this area (C).
Figure 3
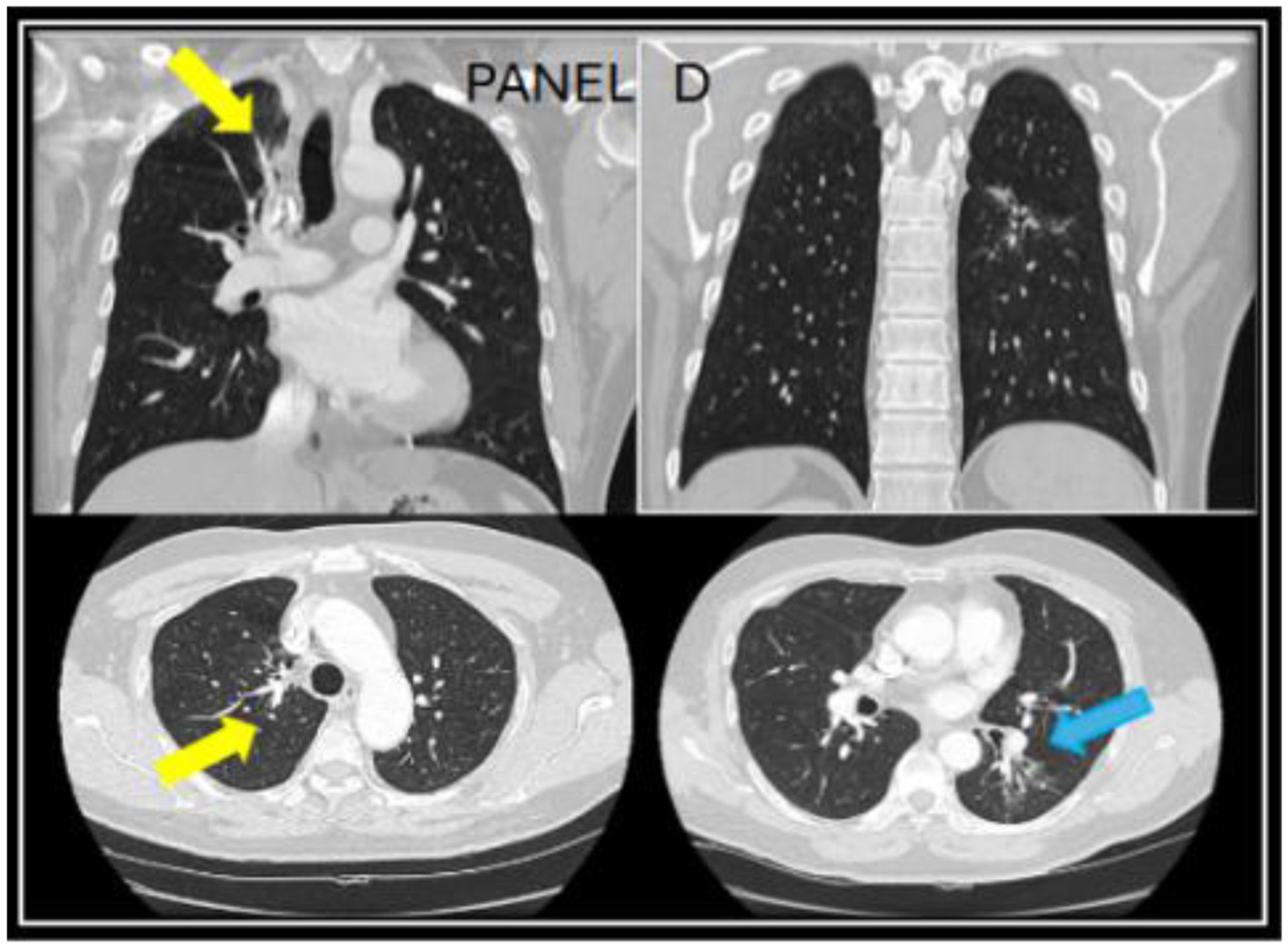
A follow up CT chest done at year four while being on long-term itraconazole showed resolution of the right upper lung infiltrate (yellow arrows) and residual reticulated bronchiectasis in the left lower lobe [blue arrow in (D)].
Of note, the patient had no other concomitant risks for immunosuppression during his clinical course. He had no episodes of absolute lymphopenia, leukopenia or neutropenia, which was monitored every 6 months. Hepatic function was monitored via hepatic panel, and hepatic synthetic function (albumin, prothrombin time, international normalized ratio) which remained within normal limits, measured every 6 months. Renal function was monitored via basic metabolic panel which remained within normal limits, measured every 6 months. The patient did not have diabetes, which was monitored via hemoglobin A1c yearly and fasting blood sugar, and was not on any other immunosuppressant medications. Patient's clinical course is depicted in a timeline in Figure 4.
Figure 4
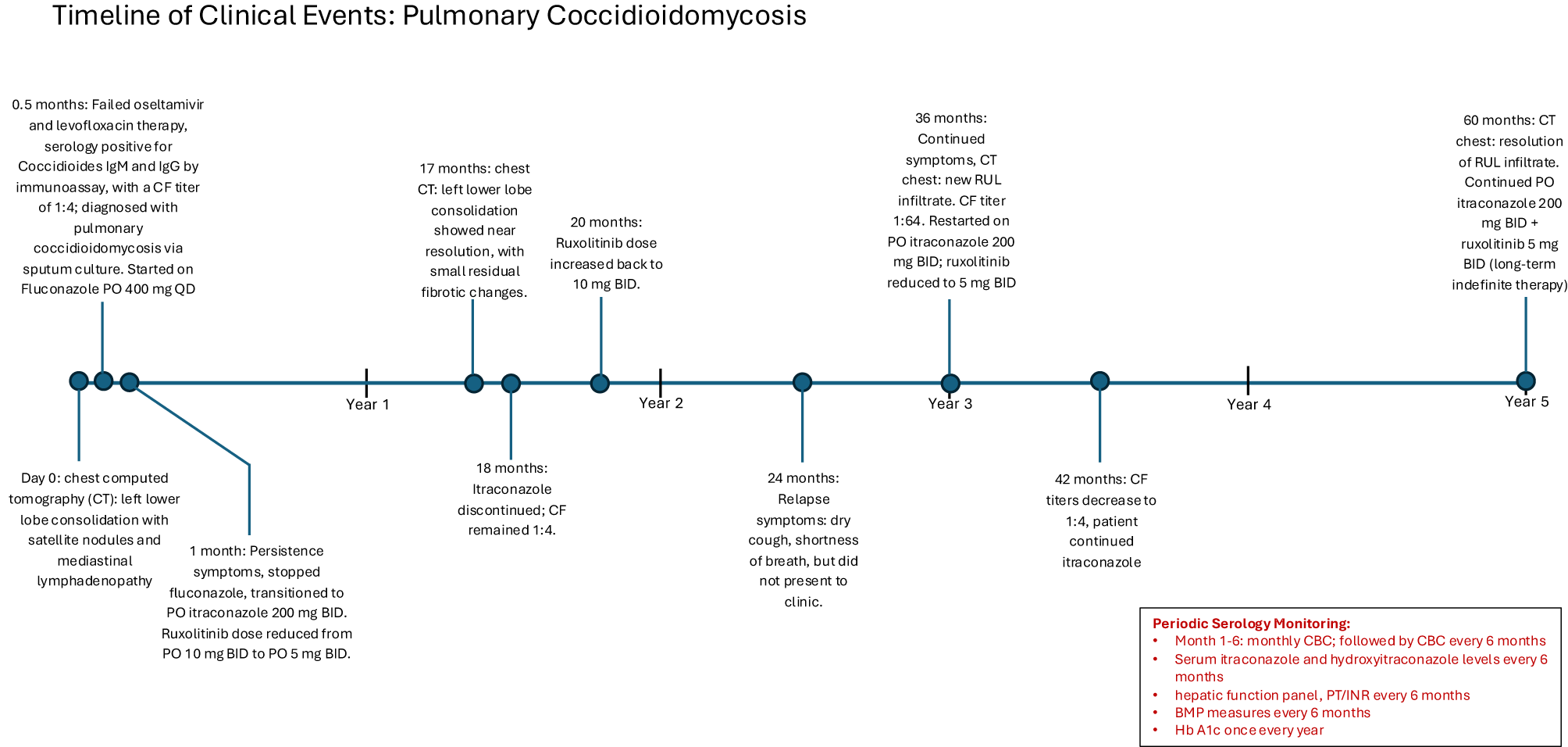
A timeline of the patient's clinical course.
Discussion
The JAK-STAT signaling pathway plays a pivotal role in immune regulation, particularly in modulating responses to infections. In the RESPONSE phase 3 clinical trial for ruxolitinib, it was reported that infections occurred in 41.8% of patients taking ruxolitinib vs. 36.9% in the standard therapy group for patients with PV (1). The most frequently associated infections in patients taking ruxolitinib include urinary tract infections (4.7–24.6%), pneumonia (5.3–13.1%), herpes zoster (1.9–11.5%), and sepsis (1.3–7.9%) (9). Other infections, such as tuberculosis, toxoplasmosis, progressive multifocal leukoencephalopathy, and hepatitis B reactivation, have also been reported (10, 11). In the RESPONSE-1 and RESPONSE-2 clinical trials, opportunistic fungal infections were not significantly different between patients taking ruxolitinib and those in the control group (2, 12). In a systematic review conducted by Lussana et al., patients taking ruxolitinib were found to have an increased risk of herpes zoster infection but did not report an increased risk of fungal infections (10). Nonetheless, numerous cases in literature identify patients on ruxolitinib with disseminated or severe opportunistic fungal infections, such as cryptococcal pneumonia, Pneumocystis jirovecii, and Rhizomucor pulmonary infections (4, 13–16). Chiu et al. investigated a systematic review identifying 28 patients with PV or myelofibrosis treated with ruxolitinib who experienced invasive fungal infections. The most common pathogens were Cryptococcus (46%), Coccidioides (11%), and Pneumocystis jirovecii (11%). In most cases, ruxolitinib was discontinued permanently (76%) or put on hold (18%). Additionally, no patient had received antifungals prior to invasive fungal infection diagnosis (3). These cases underscore the increased susceptibility to fungal pathogens in patients on JAK1 and JAK2 inhibitors (Table 1). Patient diagnoses of mycotic infections in these various studies used serologic testing and sensitivities based off of the IDSA guidelines CDC reports, and large cohort studies (Appendix Tables A1, A2).
Table 1
| Mycosis | JAK inhibitor involved | Reported clinical presentation | Relevant references |
|---|---|---|---|
| Coccidioidomycosis | Ruxolitinib (JAK1/JAK2) | 8 cases: 4 pulmonary coccidioidomycosis, 3 multisite disseminated to skin, bone, liver, and spleen, 1 skin coccidioidomycosis | (4) |
| STAT3 GOF treated with ruxolitinib (JAK1/JAK2) | Disseminated coccidiomycosis | (40) | |
| STAT1 GOF treated with ruxolitinib (JAK1/JAK2) | Disseminated coccidiomycosis to L medial rectus muscle | (44) | |
| Ruxolitinib (JAK1/JAK2) | 1 patient: unspecified clinical presentation | (16) | |
| Ruxolitinib (JAK1/JAK2) | Pulmonary coccidiomycosis | Current Case Report (Priessnitz et al.) | |
| Upadacitinib (JAK1) | unspecified | (30, 31) | |
| Histoplasmosis | Ruxolitinib (JAK1/JAK2) | Disseminated histoplasmosis with fevers, pulmonary nodules | (3) |
| 1 patient: unspecified clinical presentation | (16) | ||
| Cryptococcosis | Ruxolitinib (JAK1/JAK2) | Disseminated cryptococcal pneumonia | (3) |
| STAT3 GOF treated with ruxolitinib (JAK1/JAK2) | 1 patient with Cryptococcal pneumonia | (40) | |
| Ruxolitinib (JAK1/JAK2) | 1 patient: unspecified clinical presentation | (16) | |
| upadacitinib (JAK1) | Unspecified | (26) | |
| Pneumocystis jirovecii pneumonia | Ruxolitinib (JAK1/JAK2), Upadacitinib (JAK1) | Severe pneumonia requiring hospitalization | (3) |
| STAT3 GOF treated with ruxolitinib (JAK1/JAK2) | 2 patients: Severe pneumonia requiring hospitalization | (40) | |
| Ruxolitinib (JAK1/JAK2) | 3 patients: unspecified clinical presentation | (16) | |
| Upadacitinib (JAK1) | Unspecified | (26) | |
| Upadacitinib (JAK1) | Unspecified | (30, 31) | |
| Aspergillosis | Ruxolitinib (JAK1/JAK2) | 5 patients | (16) |
| upadacitinib (JAK1) | Unspecified | (30, 31) | |
| Candidiasis | Ruxolitinib (JAK1/JAK2) | 1 patient | (14) |
| Ruxolitinib (JAK1/JAK2) | 6 patients with Candidiasis: unspecified clinical presentation | (16) | |
| dermatophytosis | STAT1 GOF treated with ruxolitinib (JAK1/JAK2) | bilateral feet, acutely worsened on treatment and resistant to all antifungal medications | (44) |
Reported endemic mycoses associated with JAK1/JAK2 inhibitors and clinical presentations.
When reviewing the literature for patients on JAK1 and JAK2 inhibitors that developed coccidioidomycosis, a case series by Kusne et al. documents eight cases of coccidioidomycosis in patients receiving ruxolitinib in endemic regions of the San Joaquin Valley of California and Arizona. Three of these patients had primary pulmonary coccidioidomycosis prior to ruxolitinib, with treatment with antifungals that were continued after initiation of ruxolitinib, resulting in no further disease. In the other five cases, coccidioidomycosis developed after the initiation of ruxolitinib. All five patients presented with symptoms, including bilateral pulmonary nodules or extrathoracic dissemination to the skin, bones, liver, and spleen. In all five cases, appropriate antifungal treatment was started, and ruxolitinib was discontinued indefinitely. One of these patients had a history of coccidioidomycosis, who was not on immunosuppression, and had been successfully treated for 1 year. Five years after antifungal treatment, the patient started ruxolitinib therapy with negative Coccidioides CF titers. However, two years into ruxolitinib treatment, the patient developed multisite disseminated coccidioidomycosis, necessitating the discontinuation of ruxolitinib and initiation of antifungal therapy (4). Similarly, both the patient described by Kusne as well as the patient in our case developed a relapse of pulmonary Coccidioides infection while on ruxolitinib. However, unlike other cases, our patient successfully continued ruxolitinib during both the initial infection and the relapse, achieving resolution with antifungal therapy. The relapse following antifungal discontinuation highlights the risks of stopping therapy in endemic regions. In the literature, there is ongoing debate as to whether the increased risk of fungal infections in patients taking ruxolitinib is attributable to the underlying hematologic disease (PV or myelofibrosis) or the immunosuppressive effects of the drug itself. Our case supports the latter, as the patient's PV remained largely stable throughout the treatment course, while the relapse of pulmonary coccidioidomycosis occurred following the cessation of antifungal therapy (3).
The current consensus for JAK1 and JAK2 inhibitors does not support antifungal prophylaxis (8). Clinicians should be made aware of the heightened risk of relapse of fungal infections in patients on JAK1 and JAK2 inhibitors. Specifically, our case highlights the critical importance of regularly screening for fungal infections, particularly in patients residing in endemic regions. This case suggests that there may be a role for continued long-term antifungal prophylaxis, particularly in high-risk patients who have experienced a relapse while on JAK inhibitors. However, the decision to implement prophylaxis must carefully balance the benefits of infection prevention against the potential risks, including drug interactions, azole resistance, and medication-induced organ damage.
JAK-STAT proposed immune regulation of dimorphic fungi
Our case suggests that an increased risk for dimorphic fungal infections, such as Coccidioides, in endemic areas is associated with ruxolitinib-mediated inhibition of the JAK1 and JAK2 pathways, compared to normal exposure risk alone. This section will review proposed JAK-STAT immune mechanisms that may contribute to heightened risk of infections with dimorphic fungi with Figures 5, 6 also highlighting the various pathways.
Figure 5
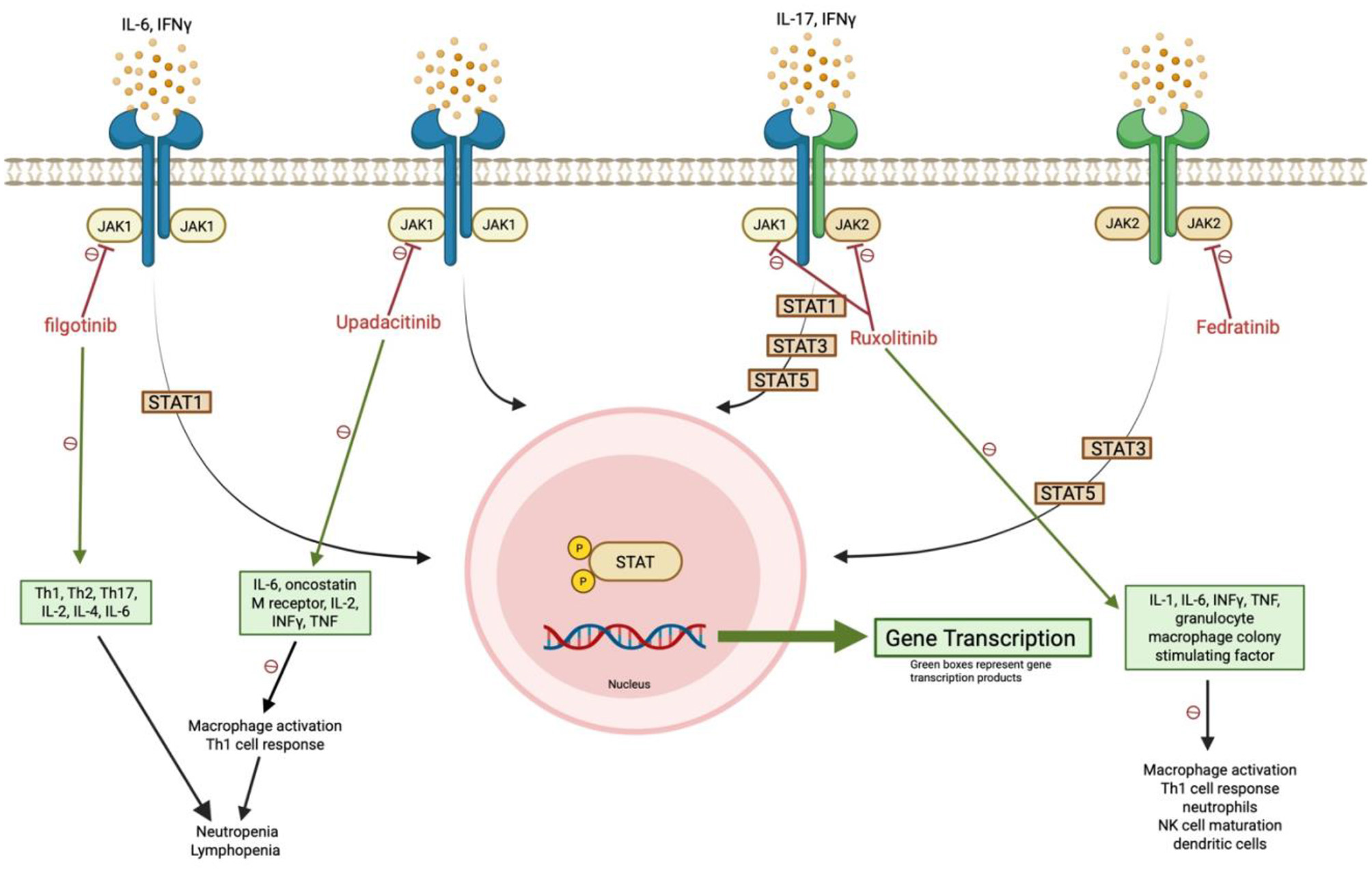
JAK1/JAK2 inhibition disrupts antifungal immune signaling. Ruxolitinib and other selective JAK inhibitors impair STAT phosphorylation and downstream gene transcription, reducing Th1 responses, macrophage activation, and innate immune cell function. These effects, particularly from JAK1 blockade, may compromise defense against dimorphic fungi such as Coccidioides. Green boxes indicate affected gene transcription products.
Figure 6
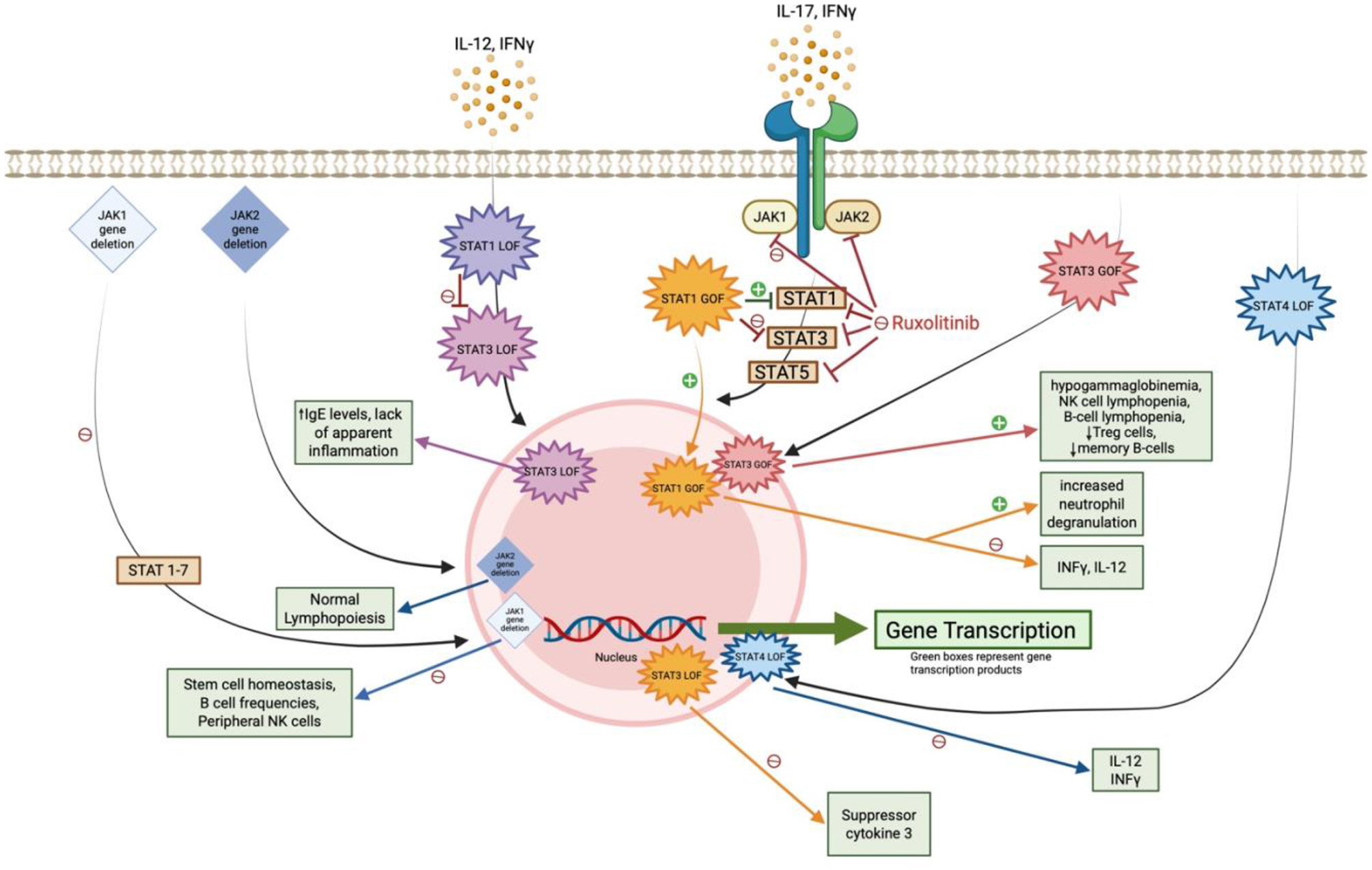
Impact of JAK-STAT gene mutations and ruxolitinib on antifungal immunity. This diagram illustrates how JAK1/JAK2 gene deletions and STAT gain- or loss-of-function (GOF/LOF) mutations influence downstream immune signaling. Green boxes denote key transcription products involved in antifungal defense; disruptions in these pathways contribute to increased susceptibility to endemic mycoses.
Ruxolitinib's mechanism of action involves inhibition of JAK1 and JAK2, which prevents the tyrosine phosphorylation of the various seven STAT proteins. This blockade disrupts their transport to the nucleus, thereby impeding downstream gene transcription of cytokine proliferation (3, 6, 17–19). JAK1 and JAK2 signaling, particularly through INF-γ-JAK-STAT pathways, is essential for activating macrophages, dendritic cells, and T-helper 1 (Th1) cell responses, which are crucial for controlling fungal pathogens like Coccidioides (3, 5–7). This pathway has downstream effects on cytokines such as IL-1, IL-6, INF-γ, tumor necrosis factor-alpha (TNF-α), and granulocyte-macrophage colony-stimulating factor, all of which recruit neutrophils and mononuclear phagocytes (3, 17, 19, 20). Additionally, recent studies suggest that ruxolitinib may impair the maturation and function of natural killer (NK) cells and dendritic cells (17). Animal studies involving loss of function (LOF) or gain of function (GOF) gene mutations targeting the JAK or STAT pathways have identified specific fungal immunity signaling cascades critical for immune cell activation and function. However, the precise mechanism by which the JAK-STAT pathway controls Coccidioides remains unclear. A study of primary immune deficiency cases involving disseminated coccidioidomycosis found two patients had STAT3 LOF mutations, one had IFN-γ receptor-1 deficiency, three had IL-12 receptor LOF mutations, and two had STAT1 GOF mutations. These findings suggest that the IL-12/IFN-γ axis and STAT3-mediated immunity play a role in protection against coccidiomycosis (21).
JAK1 inhibition's role on fungal immunity
Looking deeper into the JAK inhibitor pathways, recent studies have suggested that the loss of function in JAK1, in particular, leads to impaired macrophage activation and reduced Th1 cell responses, thereby compromising the body's ability to combat dimorphic fungi, such as Coccidioides (17). In contrast, selective inhibition of JAK2 alone does not appear to have the same profound impact on fungal immunity. This concept is highlighted by a case of a 59-year-old woman with myelofibrosis who was treated with ruxolitinib and developed disseminated histoplasmosis. The infection improved after discontinuation of ruxolitinib and proper antifungal treatment. As fedratinib, a selective JAK2 inhibitor is approved for use in myelofibrosis (but not for PV as in our case), the patient was switched to fedratinib without experiencing a relapse of histoplasmosis or other fungal infections (3). While selective JAK1 or JAK2 inhibitors are not used for treatment of PV, this case underscores the importance of these pathways in antifungal immunity. Further studies in mouse models support this notion. Inducible deletion of JAK1, which blocks phosphorylation of all STAT proteins, significantly disrupts stem cell homeostasis and reduces B cell frequencies (22, 23). Similarly, NK cell-specific deletion of JAK1 severely reduces peripheral NK cell populations (24). In contrast, inducible deletion of JAK2 does not impact lymphopoiesis, and NK cell-specific JAK2 deficiency does not interfere with NK-cell homeostasis (22, 24, 25).
Moreover, newly approved selective JAK1 inhibitors, such as upadacitinib and filgotinib, drug-reported safety profile discloses an increased risk for invasive fungal infections, such as cryptococcosis and pneumocystis (upadacitinib only) and endemic mycosis infections, as well as an increased risk of neutropenia and lymphopenia (26, 27). Filgotinib inhibits Th1, Th2, and Th17 differentiation, as well as cytokines IL-2, IL-4, and IL-6, which play critical roles in immunity (28, 29). Robust Th1 and Th17 immune response have been shown to be crucial for combating Coccidioides, and inhibition of both have been associated with chronic and disseminated coccidioidomycosis (17, 28). Upadacitinib inhibits cytokines such as IL-6, oncostatin M receptor, IL-2, IFNγ, and TNF and is involved in immune cell migration and inflammatory responses (28). In a review article, an increased risk of pneumocystis carinii pneumonia was observed in 6.1% of patients treated with upadacitinib 30 mg BID over 84 weeks. Endemic coccidioidomycosis was reported in 0.2% of patients receiving upadacitinib 6–12 mg BD over 72 weeks, and aspergillosis was found in 0.15% of patients receiving upadacitinib 15 mg OD over 48 weeks. These findings raise important questions regarding the immunological behavior of selective JAK1 inhibition (30, 31). Notably, the selective JAK1 inhibitor, itacitinib, was discontinued after phase 3 clinical trials due to its ineffectiveness in treatment of GVHD, and there are no current data on the risks of fungal infections with this drug (32). This suggests that the JAK1-STAT pathway plays a pivotal role in defense against fungal infections.
STAT gene mutation's role on fungal immunity
In a comprehensive review of JAK-STAT defects and immune dysregulation conducted by Chaimowitz et al., an increased risk of invasive fungal infections was noted due to various immune mechanisms. In comparison to our case, potential immune dysregulation resulting from the inhibition of JAK-STAT pathways will be discussed below.
STAT3 LOF
Inhibition of JAK1 prevents the phosphorylation of STAT3, leading to reductions in STAT3 downstream gene transcription. This phenotype can resemble that identified patients with an autosomal dominant STAT3 LOF mutation, also called hyper-IgE syndrome (33). These patients present with elevated IgE levels (>2,000 U/mL), recurrent pneumonia and recurrent staphylococcal skin abscesses with a lack of apparent inflammation (34). They often develop bronchiectasis and pneumatoceles and are at a higher risk for fungal infections such as Aspergillus, requiring prophylaxis with itraconazole. It is also noted that endemic mycoses frequently cause disseminated disease including histoplasmosis, Coccidioides, and Cryptococcus, requiring prophylaxis for high-risk exposure in all patients identified with a STAT3 LOF gene mutation (35, 36). Alternatively, discussed earlier in this case report, Chiu et al. described a patient who developed disseminated histoplasmosis while on the selective JAK1 and JAK2 inhibitor, ruxolitinib. The histoplasmosis is completely resolved with a proper antifungal treatment course and switching treatment to the selective JAK2 inhibitor fedratinib (3). Fedratinib's immune mechanism is thought to inhibit the phosphorylation of STAT3 and STAT5, which could correlate with a STAT3 LOF gene mutation, challenging the idea that endemic mycoses are associated with STAT3 LOF mutations (28).
STAT3 GOF
Although the JAK1 pathway is not correlated with increased phosphorylation of STAT3, patients with a STAT3 GOF mutation were susceptible to severe infections. These include viral infections, nontuberculous mycobacterial infections, fungal infections, and opportunistic infections, often accompanied by presentations of hypogammaglobulinemia, NK cell lymphopenia, T cell lymphopenia, B-cell lymphopenia, decreased in regulatory T cells and decreased memory B cells (36, 37). It has been noted that patients with T cell lymphopenia are at higher risk of viral and/or fungal infections (38, 39). In a review by Forbes et al., it was reported that 16 patients with STAT3 GOF mutations were treated with ruxolitinib to treat their immune defects. Interestingly, one patient developed disseminated coccidioidomycosis while being treated on ruxolitinib. The dose of ruxolitinib was reduced, although the patient ultimately died 6 months later due to the progression of disseminated coccidioidomycosis (40). This highlights that STAT3 GOF mutations may also contribute to dimorphic fungal immunity pathways.
STAT1 GOF
Although inhibition of the JAK1 pathway is not correlated with increased phosphorylation of STAT1, novel research reported that in patients with STAT1 GOF gene mutations, despite treatment with ruxolitinib for improvement of their chronic mucocutaneous candidiasis (CMC), patients still had an increased risk for disseminated endemic mycoses (41, 42). Patients with STAT1 GOF mutations present with CMC due to impaired IL-17 differentiation, leading to a dysregulation in STAT1/STAT3 signaling. This results in increased STAT1 gene transcription and a reduction in the expression of STAT3 dependent genes, such as suppressor of cytokine 3 (SOCS3), which contributes to reduced antifungal immunity (36, 43–45). Selective JAK1 and JAK2 inhibition with ruxolitinib has been shown to improve the dysregulation of increased phosphorylation of STAT1, leading to enhanced activation of T cells and B cells, leading to increased activation of CD45, CD52, and CD99, with partial restoration of NK cell cytotoxic function. These effects are highly effective in treating severe CMC and restoring the patient's immune system (36, 45–48).
However, Zimmerman et al. reported that two of six patients with STAT1 GOF mutations, who were treated with ruxolitinib due to their gene mutation, had disseminated fungal infections. One patient, who had severe disseminated coccidioidomycosis before starting ruxolitinib, acutely worsened after five months of ruxolitinib treatment, which required the cessation of ruxolitinib. Prior to ruxolitinib treatment, the patient's Th17 levels were normal, and she had very low CD4+, CD8+, and NK cells (41). The second patient experienced severe dermatophytosis to bilateral feet that was resistant to all antifungal treatments. After 4 weeks of treatment with ruxolitinib, the dermatophytosis worsened, requiring cessation of the drug (44).
In a study by Sampaio et al., 12% of patients with STAT1 GOF mutations were found to have disseminated Coccidioides or Histoplasmosis. Further investigation revealed that these mutations caused aberrant regulation of INF-γ and IL-12 signaling, leading to suppressed immune responses to disseminated mycobacteria and dimorphic yeast (5). Additionally, an in vivo experiment showed that neutrophils from STAT1 GOF mutation patients are highly primed for a pro-inflammatory state, characterized by cytokine release, degranulation, and transcription. Treatment with ruxolitinib in these patients had little to no effect and may even exacerbate the neutrophil pro-inflammatory state. This suggests that the SAT1 GOF neutrophil immune pathway operates through a separate pathway than ligand-induced STAT1 phosphorylation. Although endemic mycosis infections are not typically associated with a pro-inflammatory neutrophilic state, this immune pathway play a role in such infections (49). This raises that while ruxolitinib treatment can repair the immune defects causing CMC in STAT1 GOF mutations, it does not alleviate the immune defects responsible for invasive fungal diseases. The specifics of this immune pathway remain unclear.
STAT4 LOF
In a case report of a Brazilian patient who is heterozygous for STAT4 LOF, the patient presented with paracoccidioidomycosis (50). It appears that an autosomal dominant STAT4 LOF leads to impaired IL-12-dependent IFN-γ immunity, resulting in an increased risk of paracoccidioidomycosis (36).
JAK inhibitors fungal immunosuppression management in endemic areas
This case highlights the critical role of the JAK1 and JAK2 signaling pathways in antifungal defense, particularly through the INF-γ-JAK1-STAT axis. While ruxolitinib is effective in managing hematologic disorders, it underscores the potential to increase susceptibility to fungal infections, such as coccidioidomycosis, especially in endemic regions. As the U.S. Food and Drug Administration expands approval for JAK inhibitor treatments for numerous diseases, clinicians must be aware of the increased risk of invasive fungal infections in JAK1 inhibitors. A key takeaway is the importance of regular screening for fungal infections in patients on JAK inhibitors. This case also suggests that the increased risk of fungal infections is closely related to the immunosuppressive effects of ruxolitinib rather than the underlying hematological, autoimmune, or malignant condition.
However, limitations include the reliance on individual case reports and a lack of large-scale, randomized studies examining fungal infection risks with JAK inhibitors. Future research should focus on delineating the specific immune pathways affected by coccidioidomycosis immunity within the JAK-STAT pathway, considering whether these immune pathways increase the risk of endemic fungal infections compared to the standard exposure risk. Since these endemic fungal infections are specific to a certain geographical region, clinical trials should be conducted in these areas to better stratify actual risk. Additionally, exploration of alternative treatment strategies or more selective JAK inhibitors is needed to mitigate fungal risks. Further studies are needed to determine the efficacy and safety of antifungal prophylaxis in this patient population, balancing the risks of adverse outcomes with the benefits of infection prevention.
Conclusion
This case demonstrates the critical role of JAK1 and JAK2 gene transcription in antifungal defense, particularly in patients receiving ruxolitinib therapy, a JAK1 and JAK2 inhibitor, for PV or myelofibrosis. The patient's relapse of pulmonary coccidioidomycosis after discontinuing itraconazole while still on ruxolitinib highlights the need for continuous monitoring and a discussion on the risks and benefits of continuing antifungal treatment. Although, identification and observations from similar cases would strengthen this clinical hypothesis. Furthermore, clinicians should regularly screen patients taking JAK inhibitors in endemic regions for coccidioidomycosis. This case underscores the crucial role JAK1 inhibition plays in antifungal defense and raises the question of whether more selective JAK inhibitors, with avoidance of JAK1 inhibition, could mitigate fungal risks while maintaining disease control. Further research is needed to clarify best practices for managing antifungal prophylaxis in patients on JAK inhibitors in endemic areas.
Statements
Author contributions
JP: Supervision, Methodology, Data curation, Investigation, Conceptualization, Resources, Visualization, Validation, Writing – original draft, Writing – review & editing, Project administration, Formal analysis, Funding acquisition. TC: Formal analysis, Writing – original draft, Project administration, Methodology, Data curation, Validation, Investigation, Writing – review & editing. NN: Investigation, Conceptualization, Supervision, Validation, Writing – review & editing, Data curation, Writing – original draft, Formal analysis.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FMD declared a shared affiliation with the authors JP and NN to the handling editor at the time of review.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
JAK, Janus Kinase; STAT, Janus kinase-signal transducer and activator of transcription; IFN-γ, interferon-gamma; Th1, T-helper 1; PV, polycythemia vera; GVHD, graft-vs.-host disease; CT, Computed tomography; CF, complement fixation; PO, per oral; BID, twice daily; TNF-α, tumor necrosis factor-alpha; NK, natural killer; LOF, loss of function; GOF, gain of function; SOCS3, suppressor of cytokine 3.
References
1.
Vannucchi AM Kiladjian JJ Griesshammer M Masszi T Durrant S Passamonti F et al . Ruxolitinib vs. standard therapy for the treatment of polycythemia vera. N Engl J Med. (2015) 372:426–35. 10.1056/NEJMoa1409002
2.
Passamonti F Palandri F Saydam G Callum J Devos T Guglielmelli P et al . Ruxolitinib vs. best available therapy in inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): 5-year follow up of a randomised, phase 3b study. Lancet Haematol. (2022) 9:e480–92. 10.1016/S2352-3026(22)00102-8
3.
Chiu CY John TM Matsuo T Wurster S Hicklen RS Khattak RR et al . Disseminated histoplasmosis in a patient with myelofibrosis on ruxolitinib: a case report and review of the literature on ruxolitinib-associated invasive fungal infections. J Fungi. (2024) 10:264. 10.3390/jof10040264
4.
Kusne Y Kimes KE Feller FF Patron R Banacloche JG Blair JE et al . Coccidioidomycosis in patients treated with ruxolitinib. Open Forum Infect Dis. (2020) 7:ofaa167. 10.1093/ofid/ofaa167
5.
Sampaio EP Hsu AP Pechacek J Bax HI Dias DL Paulson ML et al . Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. (2013) 131:1624–34. 10.1016/j.jaci.2013.01.052
6.
Reinwald M Silva JT Mueller NJ Fortún J Garzoni C de Fijter JW et al . ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: an infectious diseases perspective (Intracellular signaling pathways: tyrosine kinase and mTOR inhibitors). Clin Microbiol Infect. (2018) 24:S53–s70. 10.1016/j.cmi.2018.02.009
7.
Tamiya T Kashiwagi I Takahashi R Yasukawa H Yoshimura A . Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol. (2011) 31:980–5. 10.1161/ATVBAHA.110.207464
8.
Galgiani JN Ampel NM Blair JE Catanzaro A Geertsma F Hoover SE et al . 2016 Infectious Diseases Society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. (2016) 63:e112–46. 10.1093/cid/ciw360
9.
Elli EM Baratè C Mendicino F Palandri F Palumbo GA . Mechanisms underlying the anti-inflammatory and immunosuppressive activity of ruxolitinib. Front Oncol. (2019) 9:1186. 10.3389/fonc.2019.01186
10.
Lussana F Cattaneo M Rambaldi A Squizzato A . Ruxolitinib-associated infections: a systematic review and meta-analysis. Am J Hematol. (2018) 93:339–47. 10.1002/ajh.24976
11.
Peng Y Meng L Hu X Han Z Hong Z . Tuberculosis in patients with primary myelofibrosis during ruxolitinib therapy: case series and literature review. Infect Drug Resist. (2020) 13:3309–16. 10.2147/IDR.S267997
12.
Kiladjian JJ Zachee P Hino M Pane F Masszi T Harrison CN et al . Long-term efficacy and safety of ruxolitinib vs. best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol. (2020) 7:e226–37. 10.1016/S2352-3026(19)30207-8
13.
Sylvine P Thomas S Pirayeh E . Infections associated with ruxolitinib: study in the French Pharmacovigilance database. Ann Hematol. (2018) 97:913–4. 10.1007/s00277-018-3242-8
14.
Polverelli N Breccia M Benevolo G Martino B Tieghi A Latagliata R et al . Risk factors for infections in myelofibrosis: role of disease status and treatment. A multicenter study of 507 patients. Am J Hematol. (2017) 92:37–41. 10.1002/ajh.24572
15.
Verstovsek S Mesa RA Gotlib J Gupta V DiPersio JF Catalano J V et al . Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. (2017) 10:55. 10.1186/s13045-017-0417-z
16.
Gold JAW Tolu SS Chiller T Benedict K Jackson BR . Incidence of invasive fungal infections in patients initiating ibrutinib and other small molecule kinase inhibitors-United States, July 2016-June 2019. Clin Infect Dis. (2022) 75:334–7. 10.1093/cid/ciab1026
17.
Little JS Weiss ZF Hammond SP . Invasive fungal infections and targeted therapies in hematological malignancies. J Fungi. (2021) 7:1058. 10.3390/jof7121058
18.
Davis JS Ferreira D Paige E Gedye C Boyle M . Infectious complications of biological and small molecule targeted immunomodulatory therapies. Clin Microbiol Rev. (2020) 33:e00035-19. 10.1128/CMR.00035-19
19.
Ruiz-Camps I Aguilar-Company J . Risk of infection associated with targeted therapies for solid organ and hematological malignancies. Ther Adv Infect Dis. (2021) 8:2049936121989548. 10.1177/2049936121989548
20.
Romani L . Immunity to fungal infections. Nat Rev Immunol. (2011) 11:275–88. 10.1038/nri2939
21.
Odio CD Marciano BE Galgiani JN Holland SM . Risk factors for disseminated coccidioidomycosis, United States. Emerg Infect Dis. (2017) 23:308–11. 10.3201/eid2302.160505
22.
Klein K Stoiber D Sexl V Witalisz-Siepracka A . Untwining anti-tumor and immunosuppressive effects of JAK inhibitors-a strategy for hematological malignancies?Cancers. (2021) 13:2611. 10.3390/cancers13112611
23.
Witalisz-Siepracka A Klein K Prinz D Leidenfrost N Schabbauer G Dohnal A et al . Loss of JAK1 drives innate immune deficiency. Front Immunol. (2018) 9:3108. 10.3389/fimmu.2018.03108
24.
Kleppe M Spitzer MH Li S Hill CE Dong L Papalexi E et al . Jak1 integrates cytokine sensing to regulate hematopoietic stem cell function and stress hematopoiesis. Cell Stem Cell. (2017) 21:489–501.e7. 10.1016/j.stem.2017.08.011
25.
Park SO Wamsley HL Bae K Hu Z Li X Choe SW et al . Conditional deletion of Jak2 reveals an essential role in hematopoiesis throughout mouse ontogeny: implications for Jak2 inhibition in humans. PLoS ONE. (2013) 8:e59675. 10.1371/journal.pone.0059675
26.
AbbVie Inc (2024) . RINVOQ [package insert]. North Chicago, IL: AbbVie Inc.
27.
Gilead Sciences & European Medicines Agency . Jyseleca, INN-filgotinib, Gilead Sciences, European Medicines Agency (2022). Available from: www.jyseleca.eu (Retrieved January 15, 2025).
28.
Hu X Li J Fu M Zhao X Wang W . The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther. (2021) 6:402. 10.1038/s41392-021-00791-1
29.
Van Rompaey L Galien R van der Aar EM Clement-Lacroix P Nelles L Smets B et al . Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. (2013) 191:3568–77. 10.4049/jimmunol.1201348
30.
Bălănescu A . Pascual-Ramos V Connell C Gold D Chen A Shi H Shapiro A Pope J Schulze-Koops H. CG Incidence of infections in patients aged ≥ 50 years with RA and ≥ 1 additional cardiovascular risk factor: results from a phase 3b/4 randomized safety study of tofacitinib vs. TNF inhibitors. Am Coll Rhematol. (2021) 73:1684. 10.1136/annrheumdis-2021-220991
31.
Winthrop K Isaacs J Calabrese L Mittal D Desai S Barry J et al . Opportunistic infections associated with Janus kinase inhibitor treatment for rheumatoid arthritis: A structured literature review. Semin Arthritis Rheum. (2023) 58:152120. 10.1016/j.semarthrit.2022.152120
32.
Taylor NP . Incyte sinks as itacitinib fails pivotal test in acute GVHD. In: Fierce Biotech (2020). Available online at: https://www.fiercebiotech.com/biotech/incyte-sinks-as-itacitinib-fails-pivotal-test-acute-gvhd (Accessed September 8, 2025).
33.
Minegishi Y Saito M Tsuchiya S Tsuge I Takada H Hara T et al . Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. (2007) 448:1058–62. 10.1038/nature06096
34.
Hsu AP Davis J Puck JM . STAT3 hyper IgE syndrome. In:AdamMPMirzaaGMPagonRAetal., editors, GeneReviews® (2020). Seattle (WA): University of Washington.
35.
Tsilifis C Freeman AF Gennery AR . STAT3 Hyper-IgE Syndrome-an Update and Unanswered Questions. J Clin Immunol. (2021) 41:864–80. 10.1007/s10875-021-01051-1
36.
Chaimowitz NS Smith MR Forbes Satter LR . JAK/STAT defects and immune dysregulation, and guiding therapeutic choices. Immunol Rev. (2024) 322:311–28. 10.1111/imr.13312
37.
Haapaniemi EM Kaustio M Rajala HL van Adrichem AJ Kainulainen L Glumoff V et al . Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood. (2015) 125:639–48. 10.1182/blood-2014-04-570101
38.
Vogel TP Leiding JW Cooper MA Forbes Satter LR . STAT3 gain-of-function syndrome. Front Pediatr. (2022) 10:770077. 10.3389/fped.2022.770077
39.
Leiding JW Vogel TP Santarlas VGJ Mhaskar R Smith MR Carisey A et al . Monogenic early-onset lymphoproliferation and autoimmunity: natural history of STAT3 gain-of-function syndrome. J Allergy Clin Immunol. (2023) 151:1081–95. 10.1016/j.jaci.2022.09.002
40.
Forbes LR Vogel TP Cooper MA Castro-Wagner J Schussler E Weinacht KG et al . Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol. (2018) 142:1665–9. 10.1016/j.jaci.2018.07.020
41.
Zimmerman O Rösler B Zerbe CS Rosen LB Hsu AP Uzel G et al . Risks of ruxolitinib in STAT1 gain-of-function-associated severe fungal disease. Open Forum Infect Dis. (2017) 4:ofx202. 10.1093/ofid/ofx202
42.
Bazan-Socha S Gradzikiewicz A Celińska-Lowenhoff M Matyja-Bednarczyk A Maciołek A Babol-Pokora K . Chronic mucocutaneous candidiasis, pancytopenia, and systemic mycosis in a patient with STAT1 gene mutation ineffectively treated with ruxolitinib. Cent Eur J Immunol. (2022) 47:92–4. 10.5114/ceji.2022.114884
43.
Liu L Okada S Kong XF Kreins AY Cypowyj S Abhyankar A et al . Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. (2011) 208:1635–48. 10.1084/jem.20110958
44.
Zimmerman O Olbrich P Freeman AF Rosen LB Uzel G Zerbe CS et al . STAT1 gain-of-function mutations cause high total STAT1 levels with normal dephosphorylation. Front Immunol. (2019) 10:1433. 10.3389/fimmu.2019.01433
45.
Toubiana J Okada S Hiller J Oleastro M Lagos Gomez M Aldave Becerra JC et al . Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. 10.1182/blood-2015-11-679902
46.
Kayaoglu B Kasap N Yilmaz NS Charbonnier LM Geckin B Akcay A et al . Stepwise reversal of immune dysregulation due to STAT1 gain-of-function mutation following ruxolitinib bridge therapy and transplantation. J Clin Immunol. (2021) 41:769–79. 10.1007/s10875-020-00943-y
47.
Borgström EW Edvinsson M Pérez LP Norlin AC Enoksson SL Hansen S et al . Three adult cases of STAT1 gain-of-function with chronic mucocutaneous candidiasis treated with JAK inhibitors. J Clin Immunol. (2023) 43:136–50. 10.1007/s10875-022-01351-0
48.
Vargas-Hernández A Mace EM Zimmerman O Zerbe CS Freeman AF Rosenzweig S et al . Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. (2018) 141:2142–2155.e5. 10.1016/j.jaci.2017.08.040
49.
Parackova Z Vrabcova P Zentsova I Sediva A Bloomfield M . Neutrophils in STAT1 gain-of-function have a pro-inflammatory signature which is not rescued by JAK inhibition. J Clin Immunol. (2023) 43:1640–59. 10.1007/s10875-023-01528-1
50.
Schimke LF Hibbard J Martinez-Barricarte R Khan TA de Souza Cavalcante R Borges de . Oliveira Junior E, et al. Paracoccidioidomycosis associated with a heterozygous STAT4 mutation and impaired IFN-γ immunity. J Infect Dis. (2017) 216:1623–34. 10.1093/infdis/jix522
51.
U .S. Centers for Disease Control and Prevention. Clinical Overview of Valley Fever. U.S. Atlanta, GA: Centers for Disease Control and Prevention (2025). Available from: https://www.cdc.gov/valley-fever/hcp/clinical-overview/index.html (Accessed 8 September 2025).
52.
Saubolle MA McKellar PP Sussland D . Epidemiologic, clinical, and diagnostic aspects of coccidioidomycosis. J Clin Microbiol. (2007) 45:26–30. 10.1128/JCM.02230-06
53.
Wheat LJ Freifeld AG Kleiman MB Baddley JW McKinsey DS Loyd JE et al . Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America. Clin Infect Dis. (2007) 45:807–25. 10.1086/521259
54.
U .S. Centers of Disease Control and Prevention. Clinical Overview of Histoplasmosis. Atlanta, GA: U.S. Centers of Disease Control and Prevention (2025). Available from: https://www.cdc.gov/histoplasmosis/hcp/clinical-overview/index.html#:~:text=develop%20disseminated%20histoplasmosis.-,Diagnosis,on%20clinical%20manifestation%20and%20severity (Accessed 8 September 2025).
55.
Connolly PA Durkin MM Lemonte AM Hackett EJ Wheat LJ . Detection of histoplasma antigen by a quantitative enzyme immunoassay. Clin Vaccine Immunol. (2007) 14:1587–91. 10.1128/CVI.00071-07
56.
Swartzentruber S LeMonte A Witt J Fuller D Davis T Hage C et al . Improved detection of Histoplasma antigenemia following dissociation of immune complexes. Clin Vaccine Immunol. (2009) 16:320–2. 10.1128/CVI.00409-08
57.
Shikanai-Yasuda MA Mendes RP Colombo AL Queiroz-Telles F Kono ASG Paniago AMM et al . Brazilian guidelines for the clinical management of paracoccidioidomycosis. Rev Soc Bras Med Trop. (2017) 50:715–40. 10.1590/0037-8682-0230-2017
58.
Perfect JR Dismukes WE Dromer F Goldman DL Graybill JR Hamill RJ et al . Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of america. Clin Infect Dis. (2010) 50:291–322. 10.1086/649858
59.
Vidal JE Boulware DR . Lateral flow assay for cryptococcal antigen, an important advance to improve the continuum of HIV care and reduce cryptococcal meningitis-related mortality. Rev Inst Med Trop São Paulo. (2015) 57:38–45. 10.1590/S0036-46652015000700008
60.
Ibrahim A Chattaraj A Iqbal Q Anjum A Rehman MEU Aijaz Z et al . Pneumocystis jiroveci pneumonia: a review of management in Human Immunodeficiency Virus (HIV) and non-HIV immunocompromised patients. Avicenna J Med. (2023) 13:23–34. 10.1055/s-0043-1764375
61.
Tasaka S . Recent advances in the diagnosis and management of pneumocystis pneumonia. Tuberc Respir Dis. (2020) 83:132–40. 10.4046/trd.2020.0015
62.
Son HJ Sung H Park SY Kim T Lee HJ Kim SM et al . Diagnostic performance of the (1-3)-β-D-glucan assay in patients with Pneumocystis jirovecii compared with those with candidiasis, aspergillosis, mucormycosis, and tuberculosis, and healthy volunteers. PLoS ONE. (2017) 12:e0188860. 10.1371/journal.pone.0188860
63.
Patterson TF Thompson 3rd GR Denning DW Fishman JA Hadley S Herbrecht R et al . Practice guidelines for the diagnosis and management of aspergillosis: 2016 update by the infectious diseases society of America. Clin Infect Dis. (2016) 63:e1–60. 10.1093/cid/ciw326
64.
Maertens J Verhaegen J Lagrou K Van Eldere J Boogaerts M . Screening for circulating galactomannan as a noninvasive diagnostic tool for invasive aspergillosis in prolonged neutropenic patients and stem cell transplantation recipients: a prospective validation. Blood. (2001) 97:1604–10. 10.1182/blood.V97.6.1604
Appendix
Table A1
| Coccidioidomycosis serologic test | Sensitivity (%) | Specificity (%) | Notes |
|---|---|---|---|
| Complement fixation (CF) antibody titer | 70–90% (disseminated) | ~90% | Rising CF titers correlate with disease activity; can remain elevated for months (8, 51) |
| Immunodiffusion (ID) IgM and IgG | IgM: ~75% (acute), IgG: ~80–85% | >95% | IgM appears early; IgG indicates ongoing or disseminated infection (8, 51) |
| EIA (enzyme immunoassay) IgG/IgM | >90% combined | ~85–95% | Most sensitive early test; occasional false positives (8, 51, 52) |
Confirmatory serologic testing for coccidioidomycosis.
Sensitivity and specificity are approximate ranges based on published data: IDSA guidelines, CDC reports, and large cohort studies. Performance varies by patient population (immunosuppressed vs. immunocompetent) and disease stage.
Table A2
| Mycotic infection | Serologic test | Sensitivity (%) | Specificity (%) | Notes |
|---|---|---|---|---|
| Histoplasmosis | Complement fixation (CF) antibody titer | 70–90% (subacute/chronic) | ~90% | Lower sensitivity in acute pulmonary infection (53–55) |
| Immunodiffusion (ID) M and H bands | ~70% | >95% | M band = acute/chronic infection; H band = active infection (53, 55, 56) | |
| Histoplasma antigen (urine/serum) | Urine: ~90% (disseminated AIDS) | ~95% | Preferred in disseminated disease; cross-reactivity with other fungi (Blastomyces, Paracoccidioides) (53–55) | |
| Paracoccidioidomycosis | Immunodiffusion | ~80–90% | >95% | Regional variability in test performance (57) |
| Complement fixation | ~70–85% | ~90% | Used less frequently (57) | |
| Cryptococcosis | Serum cryptococcal antigen (CrAg, latex agglutination or lateral flow) | >95% (disseminated) | >95% | High sensitivity in HIV/AIDS; preferred test (58, 59) |
| Pneumocystis jirovecii | Serum Beta-D-Glucan | ~90% | ~80% | Not specific for Pneumocystis; positive in other fungal infections (60–62) |
| PCR (BAL fluid) | >95% | ~90% | Preferred for diagnosis; no serologic antibody testing available (60–62) | |
| Aspergillosis | Serum galactomannan antigen | ~70% (invasive disease) | ~85–90% | False positives with certain antibiotics (e.g., piperacillin/tazobactam) (63, 64) |
| Beta-D-Glucan | ~80% | ~80% | Non-specific; positive in multiple fungal infections (63, 64) |
Confirmatory serologic testing for other endemic and opportunistic mycoses.
Sensitivity and specificity are approximate ranges based on published data: IDSA guidelines, CDC reports, and large cohort studies. Performance varies by patient population (immunosuppressed vs. immunocompetent) and disease stage.
Summary
Keywords
JAK1 and JAK2 inhibitors, ruxolitinib, coccidioidomycosis, coccidioidomycosis/immunology, JAK-STAT signaling pathway, antifungal immunity
Citation
Priessnitz JK, Colore T and Nicolasora N (2025) A case displaying the importance of JAK1 and JAK2 gene transcription in antifungal defense against coccidioidomycosis. Front. Med. 12:1643068. doi: 10.3389/fmed.2025.1643068
Received
12 June 2025
Accepted
05 September 2025
Published
14 October 2025
Volume
12 - 2025
Edited by
Sam Donta, Falmouth Hospital, United States
Reviewed by
Edith Sánchez-Paredes, National Autonomous University of Mexico, Mexico
Fariba Mirbod Donovan, The University of Arizona, United States
Updates
Copyright
© 2025 Priessnitz, Colore and Nicolasora.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nelson Nicolasora nelson.nicolasora@bannerhealth.comJennifer K. Priessnitz jennifer.priessnitz@gmail.com
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.