Abstract
Sickle cell disease (SCD) is the most common monogenic disorder, including a group of autosomal recessive hemoglobinopathies characterized by hemoglobin polymerization and sickling of red blood cells when low oxygen concentrations are present. SCD has a growing public health significance, affecting nearly 8 million people globally, with a high prevalence observed in Sub-Saharan Africa and Mediterranean countries. Improved understanding of SCD is essential, particularly given recent migratory flows that have contributed to an increase in the number of affected individuals in Europe and Italy. An early diagnosis is crucial to start the appropriate therapy to ensure the patients with the best outcome and improved quality of life, but clinical signs of SCD are often not easily recognized as symptoms are nonspecific and difficult to frame within the context of a congenital hemolytic disease. Given the availability of simple and multiple diagnostic tools, a simplified approach based on red-flags can facilitate the diagnostic suspicion in clinical practice to promptly identify individuals to be referred to specialized centers. The present narrative review aims to discuss the main clinical features, diagnostic tools of SCD, and provide practical illness scripts to facilitate the approach of non SCD-expert healthcare professionals to its diagnosis. Patient’s history, including ethnicity, region of origin, familial cases of SCD and other congenital or unexplained anemias, previous clinical manifestations, remain fundamental in guiding diagnostic suspicion of SCD, together with a few crucial lab parameters. The implementation of screening projects is essential to ensure early diagnosis and rapid access to care for affected individuals.
1 Introduction
Diagnosis is the cornerstone of providing safe, efficient, and effective medical care. A physician’s ability to diagnose a patient’s illness—that is, arrive at an explanation for a patient’s health problem—is one of the hallmarks of medical expertise and is fundamental to assigning correct and effective treatments. The most promising and effective way to improve the outcomes of the diagnostic process is to improve the education of health professionals (1).
Despite increased attention paid to sickle cell disease (SCD) and the presence of national guidelines and recommendations, the awareness of clinical suspicion signs and diagnostic tools may still be improved among non SCD-dedicated physicians, that are often first-line care providers, including but not limited to emergency room (ER) physicians, internists and pediatricians (2). Prenatal screening is not available in every country, contributing to diagnostic and referral delay (3). In Italy only a small number of local newborn screening programs for SCD have been reported, including those launched in Modena, Ferrara, Novara, and Pordenone, following the presence of carriers due to immigration (4), the pilot screenings in Padova and Monza (5), the protocols for the screening of carriers for thalassemia and hemoglobinopathies in women between 13 and 50 years in Sicily (6, 7), and early diagnosis project involving the pediatricians in Campania (8), which showed that screening using point-of-care tests by primary care is feasible and effective for early detection in at-risk children (9). Unfortunately, some of these programs were discontinued due to lack of funding or the end of the project and Italy still remains without a universal screening campaign (10). This discontinuity may contribute to delayed diagnoses, missed early intervention, and an increased risk of preventable complications among affected individuals. Avoidable delays are the most burdensome consequences of the not optimal knowledge of the disease and its diagnostic management. Importantly, the availability of multiple diagnostic tools for SCD (4, 11), and their relative appropriateness to different clinical settings may be a source of complexity.
This scenario suggests that a simplified approach may be helpful in clinical practice for non-SCD experts. It has been suggested that physicians can enhance their diagnostic performance establishing new connections between their knowledge and specific clinical encounters, thereby enabling stronger associations between clinical features and the knowledge retained in memory (1, 12). For instance, clinicians often rely on a mental model developed through experience to efficiently recognize and reason through medical conditions. This model can be defined as the illness script, a structured model that helps to retrieve clinical knowledge during diagnosis. According to Bowen’s model, a main step in the diagnostic reasoning is the definition of “illness scripts” developed by physicians, based on their exposure to patients, and include key clinical elements that help prompt diagnostic suspicion (12).
The present narrative review aims to discuss the SCD clinical features with a critical approach leading to an SCD script, to facilitate the retaining of those features in mind, thus increasing the confidence in suspecting a SCD diagnosis when appropriate. To this aim we will present a practical SCD script applicable to adults and children, for both acute and chronic setting, considering it a possible daily practice tool for non-SCD experts.
We will provide a clinical-oriented overview, aligned with current guidelines and including diagnostic approaches and tools, to help non-SCD expert physicians for a timely suspicion of SCD and diagnostic reasoning, hence increasing the rate of diagnosis and referral.
2 Sickle cell disease: pathophysiology and global prevalence
Sickle cell disorders are a group of monogenic conditions that include the most common hemoglobinopathies worldwide, together with thalassemia, inherited in an autosomal recessive fashion (13). Homozygosity for the abnormal hemoglobin variant S (HbS) is the most common type of SCD and causes the most severe clinical phenotype, termed sickle cell anemia (Figure 1); however, patients with SCD may display other genotypes due to combination of the HbS gene with other non-HbS variants such as HbC, HbE, and HbD or to the many types of HbS-β-thalassemia (14). The severity of SCD can vary depending on the patient’s genetic background, as different Hb variants influence the polymerization of HbS. The residual expression of fetal Hb (HbF) in postnatal red blood cells (RBCs) can interfere with the polymerization of HbS, reducing the tendency of RBCs to sickle. Moreover, the simultaneous presence of mutations in the alpha-globin gene (alpha-chain defects) in patients with HbS, can attenuate the clinical manifestations and severity of SCD again through polymerization interference (13, 15). The inheritance of both normal adult HbA and HbS is defined as sickle cell trait (HbAS) (16), and provides a survival advantage against death from malaria, thus explaining the higher incidence of SCD in countries where this mosquito-borne disease is particularly widespread (17). The sickle cell trait usually does not trigger a clinical phenotype except under extreme hypoxic conditions (18).
Figure 1

Molecular and structural differences between normal hemoglobin (HbA) and sickle hemoglobin (HbS). (A) structure and subunits of HbA found in healthy red blood cells. The HbA molecule consists of two α-chains and two β-chains, assembled into a globular structure, allowing red blood cells to maintain a round, flexible shape for efficient oxygen transport. The genetic sequence on chromosome 11 that encodes the β-chain is shown, with no mutations present. (B) Structure and impact of sickle hemoglobin (HbS) which differs from HbA because of a substitution of valine for glutamic acid at position 6 (Glu6Val) in the β-chain. This single-point mutation leads to hemoglobin polymerization under low-oxygen conditions, causing red blood cells to adopt a rigid, sickle shape. These deformed cells can obstruct blood flow. The genetic sequence on chromosome 11 highlights this mutation (C20A). Created in BioRender (2024) https://BioRender.com/n07g051.
SCD has a large and growing global public health significance. According to the latest estimates, the number of newborns with SCD increased from 453,000 to 515,000 between 2000 and 2021, likely attributed also to improvements in diagnosis (3). Of these births, 76.5% were of the HbSS and HbSβ0 genotypes. All-age global SCD prevalence raised by 41.4% in 2021, with 7.74 million cases (3). The highest SCD disability burden is currently concentrated in western and central sub-Saharan Africa, Mediterranean area and India (3). In Europe, the prevalence of the disease is heterogeneous, from 0.4 (range 0.3–0.5) to 18.8 (16.5–21.1) per 100,000 population in Netherlands and Greece, respectively (3). In Italy, the prevalence of 4.4 (3.8–5.0) per 100,000 general population has been estimated in 2021, but is reported to be much higher in the subset of sub-Saharan origin (3, 19, 20). Recent real-world data indicate that the Italian SCD population comprises two distinctly different subgroups: the native SCD patients, whose more frequent genotype is HbSβ, and non-native SCD patients, mainly Africans or Afro-Americans whose more frequent genotype is HbSS (20).
Patients with SCD have on average a reduced life expectancy compared to the general population. Although this may vary depending on individual factors and healthcare access, estimates suggest a reduction of up to 20 years in some high-income settings, with an even greater gap in low-income countries. This difference is likely due to more difficult in access to medical services in low-income countries. In middle- and high-income countries, improved survival has been achieved through early diagnosis via newborn screening, prophylactic antibiotics, hydroxyurea therapy, and accessible care programs (13, 21).
Early diagnosis of SCD allows for risk mitigation and early treatment intervention. Universal newborn screening programs for SCD are currently available only in some countries worldwide, including USA. For what concerns Europe, the screening map is quite heterogeneous, with some countries, including Italy, not yet offering a universal or targeted newborn screening program for SCD, despite recommendations of national guidelines (22, 23).
3 Main complications, characteristics, and timing in SCD
The morbidity of SCD is progressive throughout the life span. SCD complications can have an acute onset or a chronic progression, with exacerbations of chronic complications. In addition, some SCD complications are more frequent at specific life stages, while others can occur across multiple life stages or persist throughout life (21). Besides the reduced life expectancy, the quality of life is also poor in this patient population (14, 24). Tables 1 and 2 summarize some of the most frequent acute and chronic complications that could arise in pediatric and adult patients presenting to the ER or to healthcare assistance and that could be suggestive of undiagnosed SCD.
Table 1
| Manifestation | Cause | Features | Diagnostic approach | Ref |
|---|---|---|---|---|
| ACS | Pulmonary infiltrate caused by Streptococcus pneumoniae, Mycoplasma pneumoniae and Chlamydia pneumoniae Unknown cause in some cases |
|
Complete lab investigations, blood gas analysis, ECG and thorough physical inspection, imaging tests Defined as the presence of new infiltrate at imaging together with at least one of the following symptoms: fever, chest pain, dyspnea, desaturation |
(21, 22, 25, 37–40) |
| Acute splenic sequestration/acute anemia | Splenic sequestration of sickled RBCs |
|
Medical history, evaluation of clinical presentation and complete lab investigations, blood culture | (21, 22, 38) |
| Acute recurrent pain-VOC | Microvascular obstruction leading to impaired oxygen supply to the periphery and ischemia–reperfusion injury, oxidative stress, inflammation, endothelial dysfunction |
|
Medical history (including site, intensity and kind of pain and other associated symptoms), lab and imaging investigations, also aimed to rule out any possible surgical emergency Attention to all patient-reported information, including perceived changes in functional capacity and overall quality of life |
(22, 38, 41–43) |
| Infections | Functional asplenia contributes to the severe infections from S. pneumoniae, Haemophilus influenzae, Neisseria meningitidis |
|
Medical history (including vaccination and antibiotic prophylaxis) and ethnical origin, rapid evaluation of clinical presentation (fever) and complete lab investigations, blood count and culture | (16, 21, 22) |
| Osteomyelitis | Inflammation of bone secondary to Staphylococcus aureus infection |
|
Complete lab investigations; culture from a sample of either bone, synovial fluid, or blood, to be integrated by radiological data | (44) |
| Priapism | Circulation obstruction in the microvessels of the corpora cavernosa |
|
Doppler ultrasound, blood gas analysis, intracavernous pressure have been used to better define the prognostic factors | (21, 22, 45, 46) |
| Stroke | Sickle cell-related vasculopathy of major cerebral arteries or complete occlusion |
|
Evaluation of acute neurologic symptoms Neuro-imaging (MRI, non-contrast CT, angio-MRI) should be quickly performed in case of acute neurological symptoms Non-contrast CT can rule out hemorrhagic stroke and may show ischemic stroke; CT scan might not detect ischemic stroke within the first 6 h |
(21, 22, 38) |
| Venous thromboembolism | Endothelial dysfunction, hemolysis, increased levels of procoagulant factors, VOC |
|
Medical history, physical examination (pain, swelling, warmth, or discoloration), laboratory tests, and imaging studies | (21) |
Main potential acute clinical manifestations of SCD in pediatric and adult patients and appropriate diagnostic tools and parameters.
ACS, acute chest syndrome; CT, computed tomography; ECG, electrocardiogram; Hb, hemoglobin; Htc, hematocrit; MRI, magnetic resonance imaging; RBC, red blood cells; SCD, sickle cell disease; VOC, vaso-occlusive crisis.
Table 2
| Description | Cause | Features | Diagnostic approach | References |
|---|---|---|---|---|
| Avascular necrosis | Vaso-occlusion leading to infarction of the articular surfaces and head of long bones |
|
MRI | (16, 47, 48) |
| Chronic anemia | Persistent decrease of Hb levels due to repeated hemolysis, with compensation sign |
|
Complete blood investigation including reticulocyte count, LDH and bilirubin, peripheral blood smear | (11, 49) |
| Chronic pain | Bone infarction, chronic osteomyelitis, leg ulcers, avascular necrosis of the femoral or humeral head |
|
Multidimensional assessment (location, type, and pattern of pain), functional limitations patient reported outcomes | (21, 43, 50) |
| Gallstone disease | Increased hemolysis |
|
Abdominal ultrasound | (21, 51) |
| Leg ulcers | Vaso-occlusion and hypoxia of the skin High hemolytic rate, low Hb May develop without injury or underlying infection |
|
Clinical evaluation, physical examination, laboratory testing | (16) |
| Ophthalmic disease | Sickling of erythrocytes and increased blood viscosity in HbSC individuals (higher Hb levels) leads to retinal ischemia and scarring Peripheral retinal arteriolar occlusions |
|
Ophthalmic evaluation | (16, 51) |
| Pulmonary hypertension | Increased pulmonary pressure (≥25 mmHg), likely due to increased blood viscosity, intravascular hemolysis inducing vascular dysfunction |
|
Right heart catheterization Non-invasive estimation by Doppler echocardiography |
(16, 52, 53) |
| Renal dysfunction | HbS polymerization in the renal medulla |
|
Evaluation of GFR and proteinuria | (16, 52, 54) |
Main potential chronic clinical manifestations of SCD in pediatric and adult patients and appropriate diagnostic tools and parameters.
Hb, hemoglobin; HbS, hemoglobin S; HbSC, heterozygous for hemoglobin S and C; HbSS, homozygous for HbS; MRI, magnetic resonance imaging; GFR, glomerular filtration rate.
4 Facilitating SCD diagnostic suspicion
Non-SCD dedicated physicians, including ER physicians, may play a critical role in early detection of undiagnosed SCD and prompt treatment of its complications. It is a pleonasm saying that this critical role has its roots in the diagnostic suspect and is realized by a diagnostic plan (25).
Here, we propose SCD illness scripts, summarizing the predisposing conditions, the pathophysiological insult and the clinical consequences of SCD, in children and adults both in the acute and chronic setting. The SCD scripts aim to facilitate physicians recalling SCD as a possible diagnosis when encountering some clinical features. If SCD scripts are kept in mind, all the following information would be easily recalled and considered in the diagnostic process (Figure 2).
Figure 2
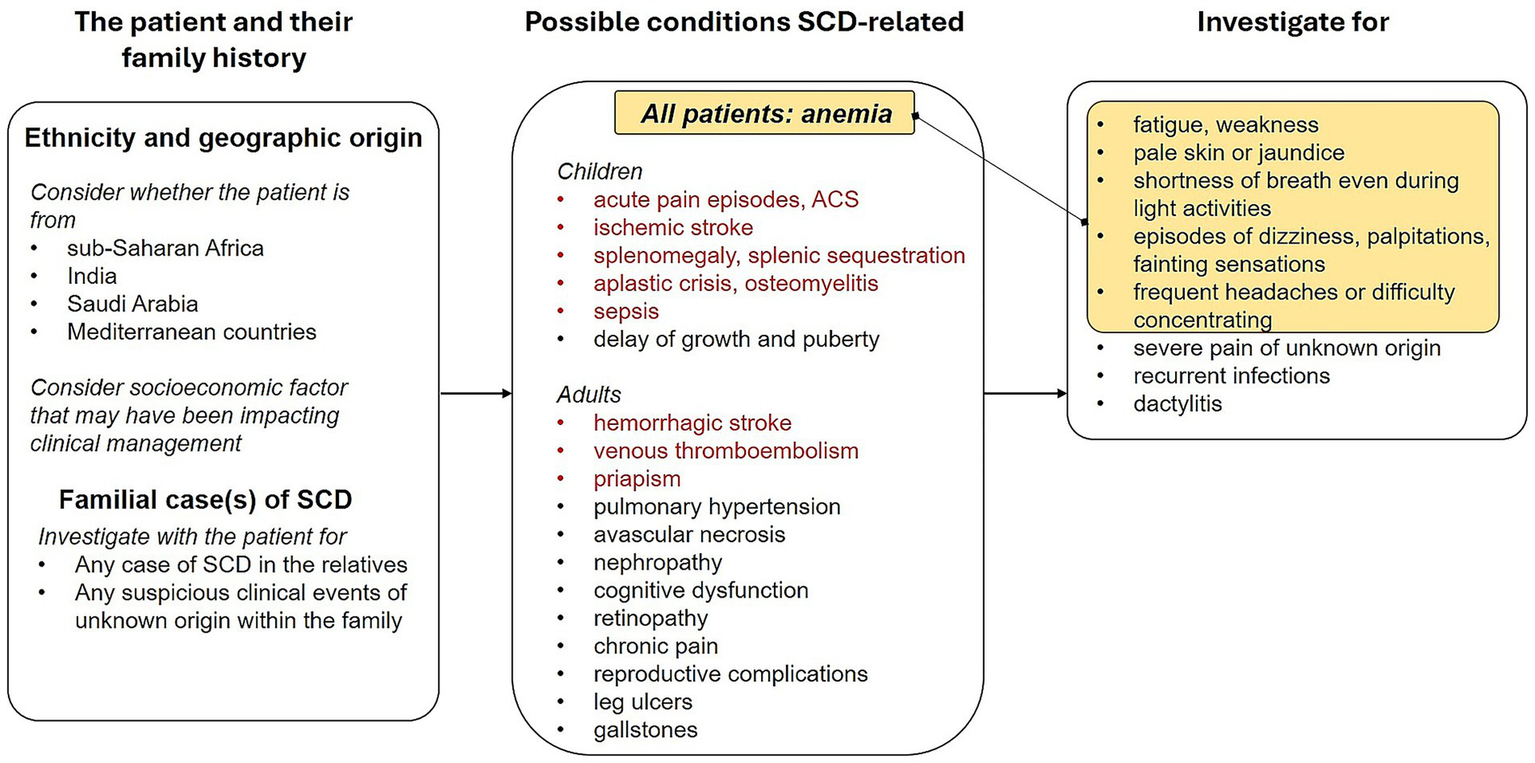
Script for diagnostic suspect of sickle cell disease in both pediatric and adult patients.
4.1 Ethnicity and origins
Particular attention should be paid to patient’s ethnicity and geographic origin, considering the SCD is particularly common among people whose ancestors come from western and central sub-Saharan Africa, India, Saudi Arabia and Mediterranean countries and that migration and mobility increased the frequency of this genetic condition also in the American and European continents (3). In the Italian population, two phenotypes of patients with similar proportions have been identified by a retrospective study involving 34 hemoglobinopathies centers: children of African descent, typically HbSS, and adults of Caucasian descent, that are most frequently HbS/β genotype (20).
4.2 Family history
The presence of any familial case of either diagnosed SCD or clinical events ascribable to SCD and thalassemic trait should be investigated as well in case of a diagnostic suspect of this hemoglobinopathy, considering that SCD is an autosomal disorder with a recessive Mendelian inheritance requiring both parents to carry the Hb mutation (26).
4.3 Information about clinical conditions possibly due to SCD
The collection of information about frequency and timing of complications during a patient’s life course in a comprehensive history intake is no doubt helpful in SCD diagnostic pathway. Importantly, a huge number of varied conditions may emerge from the archetypal SCD pathogenic triad: red blood cell sickling, hemolysis and vaso-occlusion. Regarding the timing of SCD complications during the life course, acute pain episodes (due to vaso-occlusive crises, VOCs), acute chest syndrome (ACS) and ischemic stroke can occur at any life stage, although more frequent in younger patients. Importantly, SCD patients can exhibit a delay of growth and puberty during childhood and adolescence due to ongoing hemolytic anemia and due to the increased metabolic demands. In adulthood, hemorrhagic stroke, leg ulcers, pulmonary hypertension and reproductive complications can occur more frequently. Aplastic crisis, osteomyelitis, splenic sequestration and infarction and sepsis are more frequent in childhood and adolescence, while avascular necrosis, cognitive dysfunction, chronic pain, priapism, nephropathy, retinopathy, gallstones, and venous thromboembolism are generally more frequent in adulthood (Table 1) (21). Vaso-occlusion, with associated ischemia, is also responsible for ACS and avascular necrosis, while hemolysis-related endothelial dysfunction underlies also pulmonary hypertension, priapism, stroke and leg ulcers (21).
4.4 Clinical examination
With particular reference to pediatric patients, specific elements to be considered during physical exam are pallor (assessed by examining the palms of the hands and mucous membranes in non-Caucasian individuals), jaundice, and splenomegaly (less frequent with increasing age) (22). Attention should be paid to recurrent episodes of unexplained, atraumatic severe pain (in particular bone, abdominal and wandering pain), chronic hemolytic anemia, recurrent infections (mainly lung and bone infections), priapism episodes, and dactylitis characterized by painful swelling in the fingers and toes, all symptoms possibly consequent to sickling and hemolysis of RBCs (21, 22). Some clinical manifestations, including aplastic crisis, osteomyelitis, stroke, splenic sequestration and bronchopneumonia, could be indicative of SCD onset in children. The detection of hemolytic anemia (elevated LDH and bilirubin, reduced haptoglobin) in association with nonspecific complications (systemic inflammatory disease, pancreatitis, septic arthritis) should raise a stronger suspicion of SCD, considered that these manifestations are not pathognomonic signs of SCD, and differential diagnoses should be considered as well (22).
4.5 Variability
One of the major challenges in SCD diagnosis is the clinical variability of this disease. Despite the hereditary pattern, the severity and age of onset of SCD clinical manifestations vary among patients, under the influence of a combination of genetic, epigenetic and environmental factors (16, 27), including the genotype of globin genes and the presence of genetic polymorphisms that act as modifiers on globin genes (13, 16, 21, 27). Another relevant determinant, is the socioeconomic factor that, intertwined with the geographic area of origin and, hence, with the genetic background, influences the access to diagnosis and treatment (3) therefore modulating the clinical features.
5 Diagnostic approaches and tools for SCD
In addition to a complete medical history and physical examination, and to adequate imaging studies to evaluate any complication, a wide range of laboratory methodologies should be applied to diagnose SCD. As mentioned above, the availability of multiple diagnostic tools for SCD and their relative appropriateness to different clinical settings may be a source of complexity not easily managed by non SCD-experts. In Table 3 and Figure 3 we summarize the main diagnostics characteristics and the analytical approach they use.
Table 3
| Test | Parameters measured | Technique | Timing | Ref |
|---|---|---|---|---|
| Routine hematologic investigation | Hb concentration, RBC count, Reticulocyte count Indirect bilirubin LDH Haptoglobin WBC count Mean cellular volume Mean corpuscular Hb packed cell volume Peripheral blood smear for RBC morphology |
Automatic cell counting Staining Microscopy |
In case of clinical suspect | (55–59) |
| Determination of Hb variants | Identification and quantification of HbA, HbS, HbF | Electrophoresis Iso-electric focusing High performance liquid chromatography Mass spectrometry |
Newborn screening and in every case of clinical suspect | (26, 38, 60) |
| Molecular screening | Single nucleotide polymorphisms to detect specific SCD-related mutations | Multiplex PCR, ARMS, ASA, MLPA | Population-specific mutation detection Confirmation of a diagnosis (either prenatal, newborn, or adult) In case of atypical hematologic parameters In any case of clinical concern, when available data are unsure or inconsistent with clinical evidence |
(31, 56, 61) |
Summary of diagnostic tests for hematologic evaluation, techniques, and clinical applications.
Figure 3
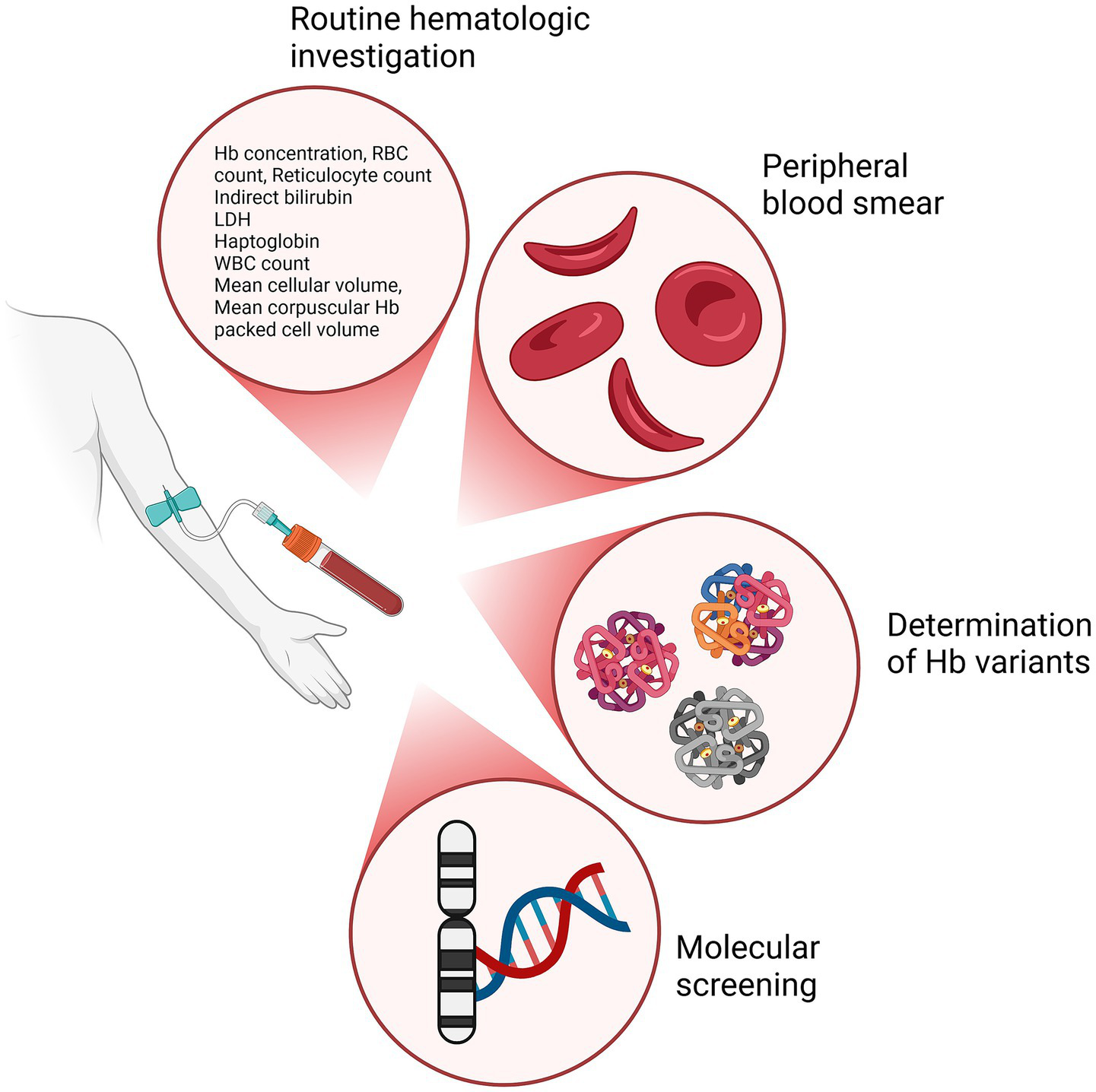
Diagnostic techniques for SCD investigation and the relative outcomes. Primary diagnostic approaches used in the evaluation of SCD: routine hematologic investigation to assess key hematological parameters, peripheral blood smear to assess the shape, size, and morphology of RBCs, determination of Hb variants identifying different Hb types, and molecular screening to identify mutations in the DNA, particularly on genes related to Hb production. Created in BioRender (2024) https://BioRender.com/c90x086.
6 Discussion
Epidemiological data report an increased prevalence of SCD worldwide between 2000 and 2021 and a concomitant increase in mortality rate (3), underscoring the critical need for early diagnosis and management of this condition. As non-SCD-expert clinicians (especially in ERs, pediatrics, general medicine) often do not suspect SCD, it is crucial to raise awareness of key presenting features, such as anemia accompanied by pain in patients from high-risk ethnic backgrounds, and to include SCD in the differential diagnosis. Identification of SCD at an early stage and appropriate referral to a specialized center enables the proactive management of symptoms, minimizing the frequency and severity of painful crises and the onset of other complications (28, 29). Implementation of newborn screening programs as in US or UK (30, 31), currently lacking in Italy could potentially reduce the missed early diagnoses. Regular monitoring of children with SCD for early identification of complications, education of patients and their families to recognizing symptoms, understanding treatment options, and adhering to care plans can lead to a better management of the disorder (3, 4, 32).
The early identification of the people potentially at risk is challenging for clinicians who encounter SCD infrequently given to SCD’s complexity, as the clinical symptoms may overlap with other conditions (31, 33). Despite SCD’s variability, there are key clinical features that can help to raise diagnostic suspicion even among non-experts. These features include a relevant ethnicity and geographical origin (such as sub-Saharan Africa, India, the Middle East, and the Mediterranean), a family history of the disease, and hemolytic anemia, which should be not mistaken for anemia due to a chronic condition or inflammation, in a patient experiencing an acute event. Recognizing these features as part of a “core set” could prompt non-SCD specialists to consider SCD earlier in their diagnostic process. Due to the extensive migratory fluxes, SCD individuals and SCD-trait carriers are now common in European countries, including Italy (20), thus the awareness of the key clinical features associated to SCD should be shared across Italian non SCD-referral centers and especially at the territorial levels.
In addition to relevant ethnicity, family history, and hemolytic anemia, frequent or recurrent presentations of symptoms should raise high clinical suspicion to investigate SCD as a unifying diagnosis. In children, the striking example is cerebrovascular injuries that can occur upon VOCs and can lead to ischemia (21), resulting in long-term cognitive impairments (34). These symptoms may not be readily associated with SCD but should raise suspicion when occurring in at-risk populations. Transcranial Doppler screening evaluates the velocity of intracranial circulation which is a marker for risk of ischemic strokes and can be performed in specialized SCD centers, although limited access to this test is reported in Europe and Italy (21, 35).
Recurrent pain episodes of unknown origin are additional clinical features that should raise diagnostic suspicion (16). The presence of these atypical and often disparate clinical events should strengthen the suspicion of SCD and lead clinicians to consider the disorder as a part of their differential diagnoses.
Acute complications such as severe pain crises, stroke, or splenic sequestration, are clinical signs that should be considered for diagnosis in pediatric patients, while adult cases may feature more often chronic complications (21).
Education on the hallmarks of SCD and adherence to established guidelines could help to improve diagnostic accuracy and prevent recurrence of common errors in clinical practice, reducing missed or delayed diagnoses (31, 33). The lack of use of structured illness scripts to guide suspicion and diagnosis can limit the proper identification of the patient. The integration of SCD scripts “−concise summaries of core clinical features and an overview of diagnostic steps as we presented here− into clinical training, medical software, and ER triage protocols can help non-expert clinicians to recognize SCD’s warning signs and could help to standardize the initial approach to suspected SCD cases, facilitating a more structured, systematic evaluation. With such tools, non-experts could be more confident in identifying signs suggestive of SCD and could promptly refer the patient to the closest specialized center.
The establishment of clear and standardized protocols for referral based on key clinical indicators can improve the poor communication between first line health care professionals and specialized centers, to improve the access to comprehensive care thus ameliorating patient outcomes and quality of life (36).
7 Conclusion
Practical tips, specific training and knowledge for a rapid diagnosis of SCD are of key importance for non SCD-expert healthcare professionals. Avoidable diagnostic and referral delay continue to be burdensome consequences of this still inadequate knowledge of SCD. Practical tools, such as the proposed SCD scripts, favoring the diagnostic suspect, seem valuable and may find a place in this scenario. The diagnostic reasoning may be further strengthened by established competence about available diagnostic tools and currently available tests and their applicability in the clinical practice, to favor non-SCD expert facing with SCD diagnosis.
Statements
Author contributions
GP: Conceptualization, Writing – original draft, Writing – review & editing, Methodology. SB: Conceptualization, Project administration, Methodology, Writing – original draft, Writing – review & editing. CG: Conceptualization, Methodology, Writing – review & editing, Project administration, Writing – original draft. FA: Conceptualization, Methodology, Writing – review & editing, Writing – original draft. SP: Conceptualization, Methodology, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Medical writing support was provided by Amalia Forte (PhD) at Health Publishing & Services Srl and was funded by Pfizer. Editorial support was provided by Barbara Bartolini at Health Publishing & Services Srl and was funded by Pfizer.
Conflict of interest
SB and CG are Pfizer employees. FA, GP, and SP were paid consultants to Pfizer in connection with the development of this manuscript.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Olson APJ Graber ML . Improving diagnosis through education. Acad Med. (2020) 95:1162–5. doi: 10.1097/acm.0000000000003172
2.
Tanabe P Reddin C Thornton VL Todd KH Wun T Lyons JS . Emergency department sickle cell assessment of needs and strengths (ED-SCANS), a focus group and decision support tool development project. Acad Emerg Med. (2010) 17:848–58. doi: 10.1111/j.1553-2712.2010.00779.x
3.
Collaborators GSCD . Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000-2021: a systematic analysis from the global burden of disease study 2021. Lancet Haematol. (2023) 10:e585–99. doi: 10.1016/s2352-3026(23)00118-7
4.
Mosca A Paleari R Palazzi G Pancaldi A Iughetti L Venturelli D et al . Screening for sickle cell disease: focus on newborn investigations. Clin Chem Lab Med. (2024) 62:1804–13. doi: 10.1515/cclm-2024-0478
5.
Colombatti R Martella M Cattaneo L Viola G Cappellari A Bergamo C et al . Results of a multicenter universal newborn screening program for sickle cell disease in Italy: a call to action. Pediatr Blood Cancer. (2019) 66:e27657. doi: 10.1002/pbc.27657
6.
Decreto della Regione Sicilia n. 2357 del . (2003). Available online at: https://pti.regione.sicilia.it/portal/page/portal/PIR_PORTALE/PIR_LaStrutturaRegionale/PIR_AssessoratoSalute/PIR_AreeTematiche/PIR_Epidemiologia/PIR_RESTETalassemie/decreto%20talassemia.pdf (Accessed December 18, 2003)
7.
Decreto della Regione Sicilia n. 103 del (2016). Available online at: https://pti.regione.sicilia.it/portal/page/portal/PIR_PORTALE/PIR_LaStrutturaRegionale/PIR_AssessoratoSalute/PIR_DipartimentoOsservatorioEpidemiologico/PIR_Infoedocumenti/PIR_DecretiDipartimentoASOE/PIR_Decreti2016/PIR_Gennaio2016/DDG%20N.%20103%20SERV%206.pdf (Accessed January 25, 2016)
8.
Rare OM . Anemia falciforme, avviato in Campania un progetto di diagnosi precoce (2024). Available online at: https://www.osservatoriomalattierare.it/malattie-rare/anemia-falciforme/21152-anemia-falciforme-avviato-in-campania-un-progetto-di-diagnosi-precoce
9.
Casale M Scianguetta S Palma T Pinfildi L Vallefuoco G Capellupo MC et al . Screening for sickle cell disease by point-of-care tests in Italy: pilot study on 1000 at risk children. Eur J Pediatr. (2025) 184:157. doi: 10.1007/s00431-025-05988-y
10.
Rare OM . Drepanocitosi, la comunità scientifica chiede screening universale, terapie per tutti i pazienti e sostegno alla ricerca (2023). Available online: https://bit.ly/4i6ZBbj
11.
Williams TN Thein SL . Sickle cell anemia and its phenotypes. Annu Rev Genomics Hum Genet. (2018) 19:113–47. doi: 10.1146/annurev-genom-083117-021320
12.
Bowen JL . Educational strategies to promote clinical diagnostic reasoning. N Engl J Med. (2006) 355:2217–25. doi: 10.1056/NEJMra054782
13.
Kirkham JK Estepp JH Weiss MJ Rashkin SR . Genetic variation and sickle cell disease severity: a systematic review and meta-analysis. JAMA Netw Open. (2023) 6:e2337484. doi: 10.1001/jamanetworkopen.2023.37484
14.
Piel FB Steinberg MH Rees DC . Sickle cell disease. N Engl J Med. (2017) 377:305. doi: 10.1056/NEJMc1706325
15.
Hardouin G Magrin E Corsia A Cavazzana M Miccio A Semeraro M . Sickle cell disease: from genetics to curative approaches. Annu Rev Genomics Hum Genet. (2023) 24:255–75. doi: 10.1146/annurev-genom-120122-081037
16.
Houwing ME de Pagter PJ van Beers EJ Biemond BJ Rettenbacher E Rijneveld AW et al . Sickle cell disease: clinical presentation and management of a global health challenge. Blood Rev. (2019) 37:100580. doi: 10.1016/j.blre.2019.05.004
17.
Mano RM Kuona P Misihairabgwi JM . Determination of birth prevalence of sickle cell disease using point of care test HemotypeSC™ at Rundu hospital, Namibia. BMC Pediatr. (2024) 24:323. doi: 10.1186/s12887-024-04805-z
18.
Tsaras G Owusu-Ansah A Boateng FO Amoateng-Adjepong Y . Complications associated with sickle cell trait: a brief narrative review. Am J Med. (2009) 122:507–12. doi: 10.1016/j.amjmed.2008.12.020
19.
Russo G De Franceschi L Colombatti R Rigano P Perrotta S Voi V et al . Current challenges in the management of patients with sickle cell disease - a report of the Italian experience. Orphanet J Rare Dis. (2019) 14:120. doi: 10.1186/s13023-019-1099-0
20.
Graziadei G De Franceschi L Sainati L Venturelli D Masera N Bonomo P et al . Transfusional approach in multi-ethnic sickle cell patients: real-world practice data from a multicenter survey in Italy. Front Med. (2022) 9:832154. doi: 10.3389/fmed.2022.832154
21.
Kavanagh PL Fasipe TA Wun T . Sickle cell disease: a review. JAMA. (2022) 328:57–68. doi: 10.1001/jama.2022.10233
22.
Oncologia AIE, Pediatrica, rosso" GdLPdg Perrotta-Giovanna CS (2023) LINEE-GUIDA per la GESTIONE DELLA MALATTIA DREPANOCITICA in ETA’PEDIATRICA in ITALIA Available at: https://www.aieop.org/web/5419/ (Accessed September 7, 2025).
23.
EMOGLOBINOPATIE SITE . Diagnostica di I e II livello delle Emoglobinopatie. Buone Pratiche SITE (2022).
24.
Colombatti R Casale M Russo G . Disease burden and quality of life of in children with sickle cell disease in Italy: time to be considered a priority. Ital J Pediatr. (2021) 47:163. doi: 10.1186/s13052-021-01109-1
25.
Porter M . Rapid fire: sickle cell disease. Emerg Med Clin North Am. (2018) 36:567–76. doi: 10.1016/j.emc.2018.04.002
26.
Bain BJ Daniel Y Henthorn J de la Salle B Hogan A Roy NBA et al . Significant haemoglobinopathies: a guideline for screening and diagnosis: a British Society for Haematology guideline: a British Society for Haematology guideline. Br J Haematol. (2023) 201:1047–65. doi: 10.1111/bjh.18794
27.
Steinberg MH Sebastiani P . Genetic modifiers of sickle cell disease. Am J Hematol. (2012) 87:795–803. doi: 10.1002/ajh.23232
28.
Dexter D McGann PT . Saving lives through early diagnosis: the promise and role of point of care testing for sickle cell disease. Br J Haematol. (2022) 196:63–9. doi: 10.1111/bjh.17678
29.
Colombatti R Perrotta S Samperi P Casale M Masera N Palazzi G et al . Organizing national responses for rare blood disorders: the Italian experience with sickle cell disease in childhood. Orphanet J Rare Dis. (2013) 8:169. doi: 10.1186/1750-1172-8-169
30.
El-Haj N Hoppe CC . Newborn screening for SCD in the USA and Canada. Int J Neonatal Screen. (2018) 4:36. doi: 10.3390/ijns4040036
31.
Daniel Y Elion J Allaf B Badens C Bouva MJ Brincat I et al . Newborn screening for sickle cell disease in Europe. Int J Neonatal Screen. (2019) 5:15. doi: 10.3390/ijns5010015
32.
Hoyt CR Hurwitz S Varughese TE Yaeger LH King AA . Individual-level behavioral interventions to support optimal development of children with sickle cell disease: a systematic review. Pediatr Blood Cancer. (2023) 70:e30178. doi: 10.1002/pbc.30178
33.
Bulgin D Tanabe P Asnani M Royal CDM . Twelve tips for teaching a comprehensive disease-focused course with a global perspective: a sickle cell disease example. Med Teach. (2019) 41:275–81. doi: 10.1080/0142159X.2017.1420151
34.
Sahu T Pande B Sinha M Sinha R Verma HK . Neurocognitive changes in sickle cell disease: a comprehensive review. Ann Neurosci. (2022) 29:255–68. doi: 10.1177/09727531221108871
35.
Voi V Gutierrez-Valle V Cuzzubbo D McMahon C Casale M Manu Pereira MDM et al . Limited access to transcranial Doppler screening and stroke prevention for children with sickle cell disease in Europe: results of a multinational EuroBloodNet survey. Pediatr Blood Cancer. (2024) 71:e31190. doi: 10.1002/pbc.31190
36.
Horiuchi SS Zhou M Snyder A Paulukonis ST . Hematologist encounters among Medicaid patients who have sickle cell disease. Blood Adv. (2022) 6:5128–31. doi: 10.1182/bloodadvances.2022007622
37.
Paul RN Castro OL Aggarwal A Oneal PA . Acute chest syndrome: sickle cell disease. Eur J Haematol. (2011) 87:191–207. doi: 10.1111/j.1600-0609.2011.01647.x
38.
Barriteau CM McNaull MA . Sickle cell disease in the emergency department: complications and management. Clin Pediatr Emerg Med. (2018) 19:103–9. doi: 10.1016/j.cpem.2018.05.005
39.
Koehl JL Koyfman A Hayes BD Long B . High risk and low prevalence diseases: acute chest syndrome in sickle cell disease. Am J Emerg Med. (2022) 58:235–44. doi: 10.1016/j.ajem.2022.06.018
40.
Vichinsky EP Styles LA Colangelo LH Wright EC Castro O Nickerson B . Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative study of sickle cell disease. Blood. (1997) 89:1787–92. doi: 10.1182/blood.V89.5.1787
41.
Tran H Gupta M Gupta K . Targeting novel mechanisms of pain in sickle cell disease. Hematol Am Soc Hematol Educ Program. (2017) 2017:546–55. doi: 10.1182/asheducation-2017.1.546
42.
Zaidi AU Glaros AK Lee S Wang T Bhojwani R Morris E et al . A systematic literature review of frequency of vaso-occlusive crises in sickle cell disease. Orphanet J Rare Dis. (2021) 16:460. doi: 10.1186/s13023-021-02096-6
43.
Darbari DS Sheehan VA Ballas SK . The vaso-occlusive pain crisis in sickle cell disease: definition, pathophysiology, and management. Eur J Haematol. (2020) 105:237–46. doi: 10.1111/ejh.13430
44.
Al Farii H Zhou S Albers A . Management of osteomyelitis in sickle cell disease: review article. J Am Acad Orthop Surg Glob Res Rev. (2020) 4:e20.00002–10. doi: 10.5435/JAAOSGlobal-D-20-00002
45.
Adeyoju A Olujohungbe A Morris J Yardumian A Bareford D Akenova A et al . Priapism in sickle-cell disease; incidence, risk factors and complications – an international multicentre study. BJU Int. (2002) 90:898–902. doi: 10.1046/j.1464-410x.2002.03022.x
46.
Mantadakis E Cavender JD Rogers ZR Ewalt DH Buchanan GR . Prevalence of priapism in children and adolescents with sickle cell anemia. J Pediatr Hematol Oncol. (1999) 21:518–22. doi: 10.1097/00043426-199911000-00013
47.
Alshurafa A Soliman AT De Sanctis V Ismail O Abu-Tineh M Hemadneh MKE et al . Clinical and epidemiological features and therapeutic options of avascular necrosis in patients with sickle cell disease (SCD): a cross-sectional study. Acta Biomed. (2023) 94:e2023198. doi: 10.23750/abm.v94i5.14603
48.
Casale M Toro G Porcelli F Quota A Rosso R Spadola V et al . Long-term outcomes of avascular necrosis in sickle cell disease using joint-specific patient-reported outcome measures: results from a multicentre study. Br J Haematol. (2024) 206:310–9. doi: 10.1111/bjh.19802
49.
Xu JZ Thein SL . Revisiting anemia in sickle cell disease and finding the balance with therapeutic approaches. Blood. (2022) 139:3030–9. doi: 10.1182/blood.2021013873
50.
Curtis SA Raisa BM Roberts JD Hendrickson JE Starrels J Lesley D et al . Non-crisis related pain occurs in adult patients with sickle cell disease despite chronic red blood cell exchange transfusion therapy. Transfus Apher Sci. (2022) 61:103304. doi: 10.1016/j.transci.2021.103304
51.
Allali S Taylor M Brice J de Montalembert M . Chronic organ injuries in children with sickle cell disease. Haematologica. (2021) 106:1535–44. doi: 10.3324/haematol.2020.271353
52.
Nguweneza A Oosterwyk C Banda K Nembaware V Mazandu G Kengne AP et al . Factors associated with blood pressure variation in sickle cell disease patients: a systematic review and meta-analyses. Expert Rev Hematol. (2022) 15:359–68. doi: 10.1080/17474086.2022.2043743
53.
Gladwin MT . Cardiovascular complications and risk of death in sickle-cell disease. Lancet. (2016) 387:2565–74. doi: 10.1016/S0140-6736(16)00647-4
54.
Yeruva SL Paul Y Oneal P Nouraie M . Renal failure in sickle cell disease: prevalence, predictors of disease, mortality and effect on length of hospital stay. Hemoglobin. (2016) 40:295–9. doi: 10.1080/03630269.2016.1224766
55.
Saad STO Gilli SO . Hemoglobin sβ thalassemia, SC disease and SD disease: clinical and laboratorial aspects In: CostaFFConranN, editors. Sickle cell Anemia: From basic science to clinical practice. Cham: Springer International Publishing (2016). 319–37.
56.
Harteveld CL Achour A Arkesteijn SJG Ter Huurne J Verschuren M Bhagwandien-Bisoen S et al . The hemoglobinopathies, molecular disease mechanisms and diagnostics. Int J Lab Hematol. (2022) 44:28–36. doi: 10.1111/ijlh.13885
57.
Nouraie M Lee JS Zhang Y Kanias T Zhao X Xiong Z et al . The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica. (2013) 98:464–72. doi: 10.3324/haematol.2012.068965
58.
Acharya V Prakasha K . Computer aided technique to separate the red blood cells, categorize them and diagnose sickle cell anemia. J Eng Sci Technol Rev. (2019) 12:67–80. doi: 10.25103/jestr.122.10
59.
Alvarez O Montague NS Marin M O'Brien R Rodriguez MM . Quantification of sickle cells in the peripheral smear as a marker of disease severity. Fetal Pediatr Pathol. (2015) 34:149–54. doi: 10.3109/15513815.2014.987937
60.
Arishi WA Alhadrami HA Zourob M . Techniques for the detection of sickle cell disease: a review. Micromachines. (2021) 12. doi: 10.3390/mi12050519
61.
Waterfall CM Cobb BD . Single tube genotyping of sickle cell anaemia using PCR-based SNP analysis. Nucleic Acids Res. (2001) 29:519, 119e–1119e. doi: 10.1093/nar/29.23.e119
Summary
Keywords
sickle cell disease, diagnosis, medical history, illness script, diagnostic tools, clinical script
Citation
Palazzi G, Benemei S, Gallucci C, Arcioni F and Perrotta S (2025) Sickle cell disease: suspect, check, diagnose—practical tips for non-SCD experts to suspect and diagnose SCD in low-prevalence European settings. Front. Med. 12:1646682. doi: 10.3389/fmed.2025.1646682
Received
13 June 2025
Accepted
31 August 2025
Published
16 September 2025
Volume
12 - 2025
Edited by
Imo J. Akpan, Columbia University, United States
Reviewed by
Margo Rollins, Children’s Healthcare of Atlanta at Egleston, United States
Vaishnavi Rathod, Interfaith Medical Center, United States
Updates
Copyright
© 2025 Palazzi, Benemei, Gallucci, Arcioni and Perrotta.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Benemei, silvia.benemei@pfizer.com
†These authors share last authorship
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.