 Fang Sun
Fang Sun Zhenzhen Wu
Zhenzhen Wu Zhenze Yu
Zhenze Yu- Department of Dermatology, Affiliated Aoyang Hospital of Jiangsu University, Zhangjiagang, China
Epidermolysis bullosa (EB) is a heterogeneous group of hereditary skin diseases caused by mutations in structural proteins at the dermal-epidermal junction. Dystrophic epidermolysis bullosa (DEB), one of its main types, is characterized by recurrent pruritic blisters, bullae, atrophy, and scarring, often accompanied by nail dystrophy. Dystrophic epidermolysis bullosa pruriginosa, also known as epidermolysis bullosa pruriginosa (EBP), is a rare clinical subtype of DEB. In addition to the common manifestations of skin blisters and ulcers, patients with EBP also present severe pruritus. Traditional treatments for EBP have limited efficacy. In this study, we report the case of a 59-year-old male patient with EBP who showed significant improvement in skin lesions and pruritus after 10 months of treatment with tofacitinib, a pan-JAK inhibitor. This case highlights the potential of JAK inhibitors in treating EBP, although long-term safety requires further investigation.
Introduction
Epidermolysis bullosa (EB) encompasses a group of genetically determined skin disorders characterized by extreme skin fragility and the formation of blisters upon minor trauma (1). Based on the cleavage level at the dermal-epidermal junction, EB is classified into four main types: epidermolysis bullosa simplex, junctional EB, dystrophic EB (DEB), and Kindler syndrome (2, 3). DEB is characterized by recurrent episodes of pruritic blisters, which gradually progress to atrophy and scarring (4). Clinical onset may occur in adulthood and is frequently accompanied by nail dystrophy of varying severity (4). Dystrophic epidermolysis bullosa pruriginosa, also known as epidermolysis bullosa pruriginosa (EBP), is a rare clinical subtype of DEB. In addition to the common manifestations of skin blisters and ulcers, patients with EBP also present a unique feature, namely severe pruritus (5). The disease is inherited in an autosomal dominant or recessive pattern, and the causative gene is COL7A1, which encodes type VII collagen, a crucial component of anchoring fibrils at the dermal-epidermal junction (6). Diagnosis typically involves a combination of clinical presentation, histopathological examination, and comprehensive genetic assessment. However, traditional therapeutic approaches, including topical corticosteroids, tacrolimus, thalidomide, and systemic immunosuppressants, have shown limited effectiveness. Recently, biologic drugs and small-molecule inhibitors have expanded the treatment options for this disease (7, 8).
Case presentation
A 59-year-old male presented to our department with nodules, scars, and severe pruritus on both lower extremities that had persisted for more than 30 years. The patient reported the onset of pruritus on both lower legs approximately 30 years earlier, with no identifiable cause; systemic conditions commonly associated with pruritus were excluded. After scratching, blisters formed and subsequently ruptured, gradually evolving into nodules and scars. Despite seeking medical advice at other hospitals, a definitive diagnosis was not established, and topical medications were ineffective. Over time, the number of skin lesions increased, and the pruritus became intense. There was no family history of similar skin diseases.
On physical examination, numerous brownish-red nodules the size of soybeans and linear scars were densely distributed on the extensor surfaces of both lower extremities. Some lesions had scales, scratch marks, and crusts, and a few dried blisters were also observed. Scattered brownish-red nodules were present on both thighs (Figure 1A). The toenails showed dystrophic changes such as onychatrophy, onycholysis, onychorrhexis, and even anonychia (Figure 1B). The visual analog scale (VAS) score for pruritus was 8/10.
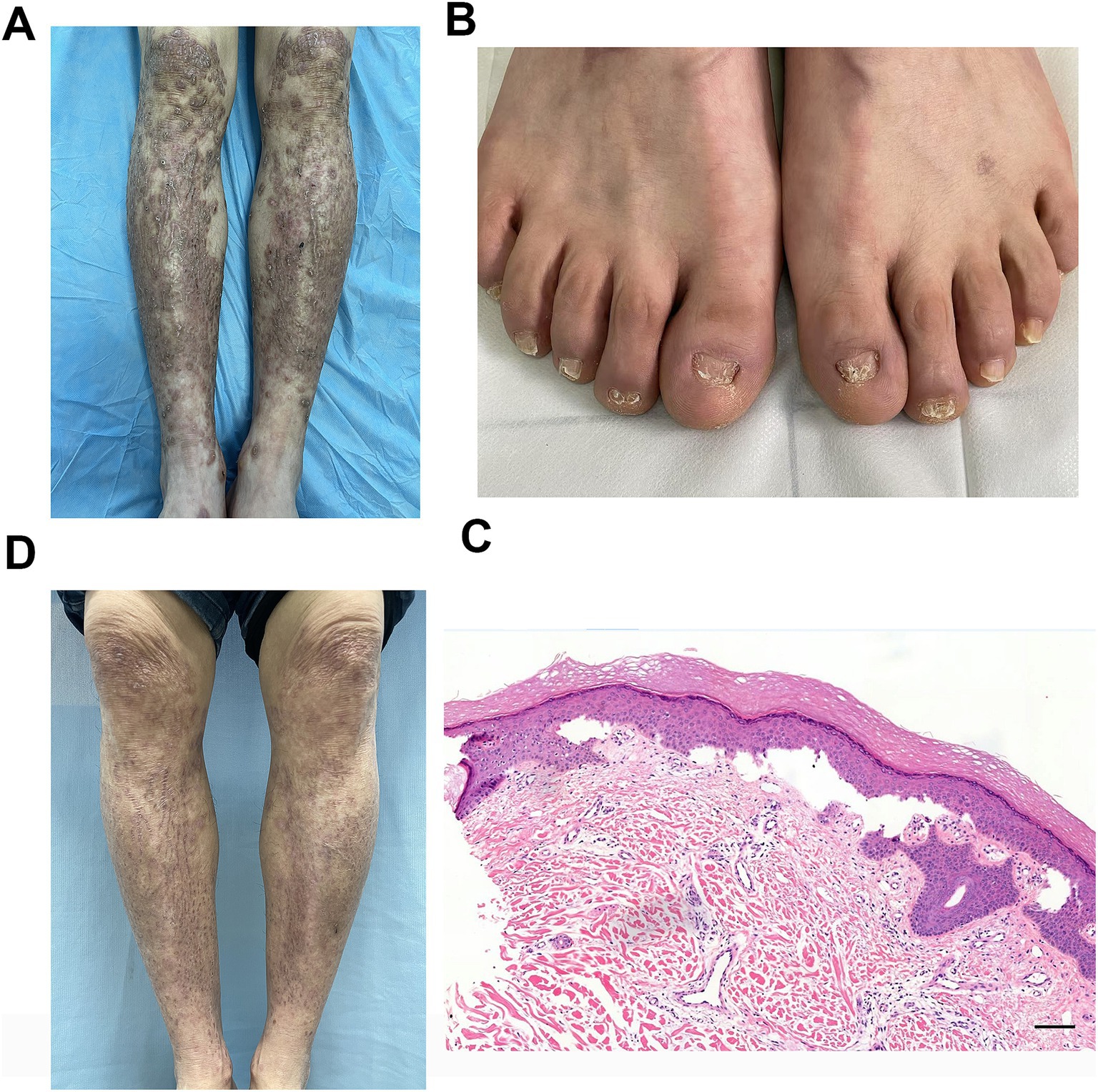
Figure 1. (A) Typical skin lesions of brownish-red nodules the size of soybeans and linear scars on the pretibial area. (B) Characteristic dystrophic nail changes included onychatrophy, onycholysis, onychorrhexis, and anonychia. (C) Histopathological examination of pretibial skin lesions revealed hyperkeratosis of the epidermis, thickening of the granular layer, mild acanthosis, dermo-epidermal cleavage, significant proliferation of small blood vessels in the dermal papillary layer, and a substantial infiltration of lymphocytes, plasma cells, and eosinophils around the vessels (H&E staining, scale bar = 100 μm). (D) Marked improvement of the pretibial area at the 10-month follow-up visit.
Histopathological examination of a skin biopsy revealed hyperkeratosis of the epidermis, thickening of the granular layer, mild acanthosis, dermo-epidermal cleavage, significant proliferation of small blood vessels in the dermal papillary layer, and a substantial infiltration of lymphocytes, plasma cells, and eosinophils around the vessels (Figure 1C). Given the patient’s clinical manifestations, differential diagnoses including pemphigus and pemphigoid were initially considered. To rule out these autoimmune blistering disorders, serological testing for pathogenic antibodies was performed, which revealed negative results for desmoglein 1 (Dsg1), desmoglein 3 (Dsg3), bullous pemphigoid 180 (BP180), and bullous pemphigoid 230 (BP230). Thus, pemphigus and pemphigoid were excluded from the final diagnosis. Additionally, genetic testing revealed a heterozygous missense mutation in COL7A1: c.8201G>A (p.Gly2734Asp). Based on the clinical manifestations, immunological and histopathological findings, and genetic results, a diagnosis of EBP was made. The patient was prescribed tofacitinib at a dose of 5 mg orally twice daily with no topical medications administered. After 10 months of treatment, follow-up showed marked improvement in skin lesions (Figure 1D) and pruritus (VAS score: 1/10). Safety monitoring remained normal throughout therapy. Monitored items included clinical evaluation for infections (including screening for tuberculosis, hepatitis B, hepatitis C, HIV, and syphilis) and laboratory tests for liver function, renal function, complete blood count, lipid profile, and tumor markers.
Discussion
In this study, we report a case of a patient with EBP harboring a heterozygous mutation in the COL7A1 gene, specifically c.8201G>A (p.Gly2734Asp). This mutation has been previously documented in the Polish DEB population (9) and is currently curated in the Human Gene Mutation Database (HGMD) (10). This variant probably falls into the category of autosomal dominantly inherited mutations. The proband’s father, a carrier of the c.8201G>A (p.Gly2734Asp) variant, displayed transient toenail loss, and this sign is potentially linked to EB (9). However, research on this specific mutation remains limited. Its impacts on the structure and function of the COL7A1 gene, which encodes the type VII collagen protein, have not yet been fully elucidated. The present study further supports that mutations at this locus can lead to EBP.
Pruritus is the most common and challenging symptom to manage in EBP patients. It initiates a vicious cycle of itching, scratching, and wound formation. Currently, the exact pathophysiology of pruritus in EB remains incompletely understood. The primary goals of treatment are to relieve pruritus and improve skin lesions. Several therapeutic approaches, including emollients, topical corticosteroids, oral antihistamines, gabapentin, thalidomide, and immunosuppressants, have been reported in the literature, but they often yield unsatisfactory results.
Some studies have reported successful treatment of DEB with dupilumab (7, 8), a human monoclonal antibody targeting interleukin (IL)-4 and IL-13, suggesting that DEB may be driven by Th2-mediated immune mechanisms (11–13). Another study examining the wound transcriptome profile of patients with EB revealed abnormally elevated expression of inflammation-related genes in EB lesions. These include IL-1A, IL-1B, IL-6, and IL-8, as well as components of the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway, compared with normal skin from the same patients and with healthy controls (14). In contrast, JAK inhibitors broadly suppress the JAK–STAT pathway, which is used by multiple cytokines, including IL-4, IL-13, and IL-31, thereby exerting a more comprehensive antipruritic effect (15). Tofacitinib, a pan-JAK inhibitor, primarily inhibits JAK1 and JAK3, with weaker inhibitory effects on JAK2 and TYK2 (16). In this study, tofacitinib demonstrated significant therapeutic efficacy in treating EBP. To the best of our knowledge, this is among the first reported cases of EBP treated with tofacitinib, further supporting its therapeutic potential (17, 18). Previous studies have also shown that oral JAK inhibitors (such as baricitinib and upadacitinib) may be more effective than dupilumab, but adverse reactions should be carefully monitored (8, 19). To place our case in the broader context of targeted therapies for DEB, we summarize the key characteristics of biologic drugs and JAK inhibitors reported for its management (Table 1).
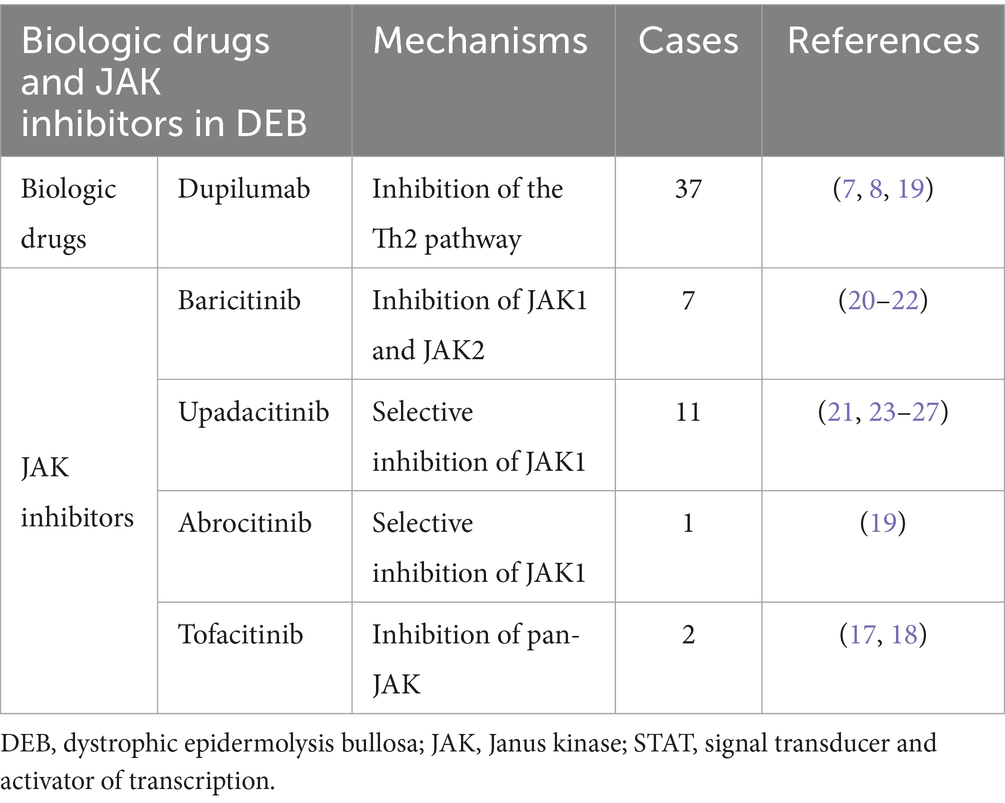
Table 1. Biologic drugs and JAK inhibitors used in DEB.
Conclusion
JAK inhibitors, such as tofacitinib, appear to be effective treatment options for EBP. Given the need for continuous treatment in EBP patients, the long-term safety of JAK inhibitors requires further investigation. Additional rigorous studies are needed to validate these efficacy observations and to develop comprehensive safety guidelines for the long-term use of JAK inhibitors in EBP patients.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The requirement of ethical approval for the studies involving humans was waived by the Ethics Committee of Aoyang Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
FS: Conceptualization, Writing – original draft, Investigation, Data curation. ZW: Conceptualization, Investigation, Writing – original draft. ZY: Writing – review & editing, Data curation, Investigation, Conceptualization.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bardhan, A, Bruckner-Tuderman, L, Chapple, ILC, Fine, JD, Harper, N, Has, C, et al. Epidermolysis bullosa. Nat Rev Dis Primers. (2020) 6:78. doi: 10.1038/s41572-020-0210-0
2. Koller, U, and Bauer, JW. Emerging DNA & RNA editing strategies for the treatment of epidermolysis bullosa. J Dermatolog Treat. (2024) 35:2391452. doi: 10.1080/09546634.2024.2391452
3. Danescu, S, Negrutiu, M, and Has, C. Treatment of epidermolysis bullosa and future directions: a review. Dermatol Ther. (2024) 14:2059–75. doi: 10.1007/s13555-024-01227-8
4. Kaznoski, I, Ghareeb, E, and Zinn, Z. Dominant dystrophic epidermolysis bullosa across three distinct skin types. J Am Acad Dermatol. (2025) 93:1346–59. doi: 10.1016/j.jaad.2025.05.1449
5. Kim, WB, Alavi, A, Pope, E, and Walsh, S. Epidermolysis bullosa pruriginosa: case series and review of the literature. Int J Low Extrem Wounds. (2015) 14:196–9. doi: 10.1177/1534734615572469
6. Lwin, SM. Decoding COL7A1: a genetic roadmap to prognostication and personalized medicine in recessive dystrophic epidermolysis bullosa. Br J Dermatol. (2025) 192:794–5. doi: 10.1093/bjd/ljaf056
7. Dou, L, Chen, F, He, W, Wang, J, Wang, C, Chen, X, et al. Efficacy and safety of dupilumab monotherapy in patients with dystrophic epidermolysis bullosa: a retrospective study of 8 cases. J Dermatol. (2025) 52:1159–65. doi: 10.1111/1346-8138.17753
8. Ling, S, and Dou, X. Real-world experience with JAK inhibitors and dupilumab in dystrophic epidermolysis bullosa: a case report and literature review. Acta Derm Venereol. (2025) 105:adv43588. doi: 10.2340/actadv.v105.43588
9. Wertheim-Tysarowska, K, Sobczynska-Tomaszewska, A, Kowalewski, C, Kutkowska-Kazmierczak, A, Wozniak, K, Niepokoj, K, et al. Novel and recurrent COL7A1 mutation in a Polish population. Eur J Dermatol. (2012) 22:23–8. doi: 10.1684/ejd.2011.1583
10. Stenson, PD, Mort, M, Ball, EV, Chapman, M, Evans, K, Azevedo, L, et al. The human gene mutation database HGMD®: optimizing its use in a clinical diagnostic or research setting. Hum Genet. (2020) 139:1197–207. doi: 10.1007/s00439-020-02199-3
11. De Gregorio, C, Ramos-Gonzalez, G, Morales-Catalan, B, Ezquer, F, and Ezquer, M. Paw skin as a translational model for investigating fibrotic and inflammatory wound healing defects in recessive dystrophic epidermolysis bullosa. Int J Mol Sci. (2025) 26:4281. doi: 10.3390/ijms26094281
12. Quintana-Castanedo, L, Sanchez-Ramon, S, Maseda, R, Illera, N, Perez-Conde, I, Molero-Luis, M, et al. Unveiling the value of C-reactive protein as a severity biomarker and the IL4/IL13 pathway as a therapeutic target in recessive dystrophic epidermolysis bullosa: a multiparametric cross-sectional study. Exp Dermatol. (2024) 33:e15146. doi: 10.1111/exd.15146
13. Samuelov, L. Dystrophic epidermolysis bullosa: from disease biology to biologic therapy. Br J Dermatol. (2024) 191:159–60. doi: 10.1093/bjd/ljae182
14. Onoufriadis, A, Proudfoot, LE, Ainali, C, Torre, D, Papanikolaou, M, Rayinda, T, et al. Transcriptomic profiling of recessive dystrophic epidermolysis bullosa wounded skin highlights drug repurposing opportunities to improve wound healing. Exp Dermatol. (2022) 31:420–6. doi: 10.1111/exd.14481
15. Hou, PC, Aala, W Jr, Tu, WT, McGrath, JA, and Hsu, CK. Real-world experience of using dupilumab and JAK inhibitors to manage pruritus in epidermolysis bullosa pruriginosa. Skin Health Dis. (2024) 4:e445. doi: 10.1002/ski2.445
16. Wang, QX, He, HY, Niu, YL, and Fang, S. Refractory eosinophilic pustular folliculitis treated with tofacitinib: a case series and literature review. Clin Exp Dermatol. (2025) 50:1196–200. doi: 10.1093/ced/llaf053
17. Zhou, X, Geng, J, Wang, M, Yang, J, Zou, J, and Li, W. Novel compound heterozygous mutations of the COL7A1 gene in a Chinese patient with recessive dystrophic epidermolysis bullosa pruriginosa and digestive symptoms successfully treated with tofacitinib. J Dermatol. (2024) 51:e8–e10. doi: 10.1111/1346-8138.16945
18. Chen, KJ, Fang, S, Ye, Q, and Jia, M. Successful use of tofacitinib in epidermolysis bullosa pruriginosa. Clin Exp Dermatol. (2022) 47:598–600. doi: 10.1111/ced.14998
19. Hui, HZ, Guo, HX, and Shi, BJ. Abrocitinib successfully treated dupilumab-resistant patients with rare dominant dystrophic epidermolysis bullosa with atopic dermatitis. Am J Ther. (2025) 32:e370–2. doi: 10.1097/MJT.0000000000001878
20. He, Z, Dong, Q, Xi, Y, and Zheng, R. Epidermolysis bullosa pruriginosa treated with baricitinib: a case report. Medicine. (2024) 103:e38854. doi: 10.1097/MD.0000000000038854
21. Kwon, IJ, Kim, SE, Kim, SC, and Lee, SE. Efficacy of oral JAK1 or JAK1/2 inhibitor for treating refractory pruritus in dystrophic epidermolysis bullosa: a retrospective case series. J Dermatol. (2024) 51:441–7. doi: 10.1111/1346-8138.17079
22. Jiang, X, Wang, H, Lee, M, and Lin, Z. Epidermolysis bullosa pruriginosa treated with baricitinib. JAMA Dermatol. (2021) 157:1243–4. doi: 10.1001/jamadermatol.2021.3174
23. Wu, X, Wang, Z, and Xu, Z. Successful use of upadacitinib in treating a child with epidermolysis bullosa pruriginosa: a case report and literature review. Pediatr Dermatol. (2025). doi: 10.1111/pde.15963
24. Lai, S, Lin, C, Guo, Z, Lai, Y, Xie, L, Wan, C, et al. A novel COL7A1 mutation in a patient with dystrophic epidermolysis bullosa. Successful treatment with upadacitinib. Clin Cosmet Investig Dermatol. (2025) 18:183–90. doi: 10.2147/CCID.S499144
25. Zhang, Z, and Lin, Z. Epidermolysis bullosa pruriginosa treated with upadacitinib. JAMA Dermatol. (2024) 160:1124–5. doi: 10.1001/jamadermatol.2024.2787
26. Guo, L, Wang, SF, Wang, HM, Zeng, YP, and Jin, HZ. A case of epidermolysis bullosa pruriginosa with a COL7A1 gene mutation successfully treated with upadacitinib. J Dermatol. (2025) 52:e5–7. doi: 10.1111/1346-8138.17239
Keywords: epidermolysis bullosa, dystrophic epidermolysis bullosa, dystrophic epidermolysis bullosa pruriginosa, JAK inhibitor, tofacitinib
Citation: Sun F, Wu Z and Yu Z (2025) Case Report: Effective treatment of dystrophic epidermolysis bullosa pruriginosa with tofacitinib. Front. Med. 12:1648732. doi: 10.3389/fmed.2025.1648732
Edited by:
Dennis Niebel, University Medical Center Regensburg, GermanyReviewed by:
Irene Rivera Ruiz, University Hospital Fundación Jiménez Díaz, SpainSorina Danescu, University of Medicine and Pharmacy Iuliu Hatieganu, Romania
Copyright © 2025 Sun, Wu and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenze Yu, eXp6X2FveWFuZ0AxNjMuY29t