Abstract
Introduction:
Cantú syndrome (CS) is a rare genetic disorder caused by gain-of-function (GOF) mutations in the KCNJ8 (Kir6.1) or ABCC9 (SUR2) subunits of ATP-sensitive potassium (KATP) channels. CS is characterized by multisystem abnormalities such as cardiovascular defects, hypertrichosis, and skeletal malformations, but its impact on intestinal homeostasis remains poorly understood.
Methods:
We investigated the effects of CS-associated KATP channel overactivity on epithelial barrier integrity and tight junction (TJ) proteins using murine models. Heterozygous (SUR2wt/AV) and homozygous (SUR2AV/AV) SUR2(A478V) mutants, as well as Kir6.1(V65M) mice, were studied. mRNA and protein expression of Occludin, Claudin-1, and ZO-1 were analyzed, alongside histological and immunohistochemical assessments. Markers of apoptosis and survival, including caspase-3 activity and BCL2/BCL2L1 expression, were also evaluated.
Results:
GOF mutations in KATP channels caused significant dysregulation of TJ proteins. Occludin expression was increased in SUR2AV/AV mice but decreased in SUR2wt/AV and Kir6.1 mutants, while Claudin-1 and ZO-1 were consistently reduced across all models. Immunohistochemistry revealed disrupted TJ localization and reduced apical junctional integrity. Histological analyzes showed epithelial disorganization, smooth muscle hypertrophy, fibrosis, and inflammatory infiltration. These alterations were accompanied by increased caspase-3 activity and reduced BCL2 and BCL2L1 expression.
Discussion:
Our findings demonstrate that CS-associated KATP channel GOF mutations disrupt tight junction dynamics and induces structural remodeling of the colon. This establishes a novel link between KATP channel dysregulation, metabolic-epithelial interactions, and intestinal pathophysiology in CS. Furthermore, the results highlight potential therapeutic targets to mitigate barrier dysfunction, providing a basis for developing interventions to address gastrointestinal symptoms in CS.
Introduction
Cantú syndrome (CS, OMIM 239850), first described in 1982, is a rare multisystem disorder caused by gain-of-function (GOF) mutations in subunits of ATP-sensitive potassium (KATP) channels, specifically the pore-forming Kir6.1 (KCNJ8) and regulatory SUR2 (ABCC9) subunits (1–4). Clinically, CS is characterized by distinctive craniofacial dysmorphisms, cardiovascular anomalies, hypertrichosis, and skeletal malformations (5–7). Electrophysiological analyses have demonstrated that disease-associated mutations enhance channel activity via reduced MgATP-mediated inhibition and heightened MgADP sensitivity, leading to aberrant cellular excitability (2, 8–10). While no targeted treatments are currently available, diagnosis is facilitated by clinical evaluation and genetic testing. Prognosis is variable and largely determined by cardiovascular involvement, although many individuals experience favorable outcomes with appropriate care.
KATP channels function as metabolic sensors, linking intracellular nucleotide concentrations to membrane excitability. These octameric complexes consist of four inwardly rectifying Kir6.x subunits (Kir6.1/KCNJ8 or Kir6.2/KCNJ11) and four sulfonylurea receptor subunits (SUR1/ABCC8 or SUR2/ABCC9). Their tissue-specific configurations confer functional specialization across the pancreatic, cardiovascular, skeletal, and neural systems (11–14). Clinically, KATP channels are important pharmacological targets, with sulfonylureas and potassium channel openers used to treat conditions such as diabetes, angina, and hypertension (10, 15–17).
In the gastrointestinal tract, KATP channels regulate chloride secretion, influence smooth muscle contractility, and modulate neural reflexes, playing a role in disorders like inflammatory bowel disease and Hirschsprung's disease (18–23). Strong expression of KATP subunits has been observed in the human colon. Specifically, Kir6.2/SUR1 is predominantly localized to the colonic epithelium, modulating fluid transport through ATP-sensitive chloride secretion (20, 24, 25). In contrast, Kir6.1/SUR2B is enriched in colonic smooth muscle, influencing contractile tone in response to ATP/ADP dynamics (26–28). Within the enteric nervous system, Kir6.2/SUR1 is expressed in inhibitory neurons of the myenteric and submucosal plexuses, dampening excitability and modulating reflex pathways (29–31). Meanwhile, Kir6.1/SUR2B subunits localize to interstitial cells of Cajal (ICC), where they may support pacemaker activity and coordinate motility (32–36).
Rodent studies support a role for colonic KATP channels in chloride secretion and epithelial barrier maintenance, with functional interactions noted between these channels and hydrogen sulfide (H2S) signaling (37). Co-localization of Kir6.1/SUR2 with tight junction (TJ) proteins in the small intestine further suggests involvement in barrier integrity (20, 38). Tight junctions comprising transmembrane proteins such as claudins and occludin, and scaffold proteins including ZO-1, ZO-2, and ZO-3, regulate epithelial permeability and are frequently disrupted in intestinal inflammation (39–42). These observations indicate a potential role for KATP channel dysregulation in epithelial pathophysiology.
Our previous work demonstrated that CS-associated GOF mutations in murine KCNJ8 [Kir6.1(V65M)] and ABCC9 [SUR2(A478V)] recapitulate key human phenotypes, including profound skeletal muscle pathology and cardiovascular defects (43, 44). However, the impact of these mutations on intestinal homeostasis, particularly within the colonic epithelium and junctional architecture, remains insufficiently understood.
The Kir6.1(V65M) and SUR2(A478V) mutations likely perturb colonic physiology via distinct mechanisms. Kir6.1(V65M) may enhance channel open probability, promoting excessive potassium efflux and membrane hyperpolarization, impairing ion transport and TJ integrity. In contrast, SUR2(A478V) may decrease ATP sensitivity, resulting in constitutive channel activation, metabolic dysregulation, oxidative stress, and caspase-3-dependent apoptosis. These divergent mechanisms suggest mutation-specific effects on epithelial and barrier function. To investigate this, we examined murine models harboring heterozygous (SUR2wt/AV) and homozygous (SUR2AV/AV) SUR2(A478V) alleles, as well as Kir6.1(V65M) variants. Our study aimed to define the impact of aberrant KATP channel activity on the assembly, localization, and function of tight junction components within the colonic epithelium.
Results
Kir6.1- and SUR2-dependent KATP channels are expressed at the epithelial level and the muscular level in the colon
Histological analyses of human and murine colonic tissue reveal distinct localization of Kir6.1 and SUR2-gated KATP channels across epithelial and muscular compartments. Immunohistochemical staining demonstrates robust expression of Kir6.1 and SUR2 subunits within the smooth muscle layers, with pronounced enrichment in circular and longitudinal muscle fibers (Figures 1–3). These subunits are predominantly localized along the plasma membrane of smooth muscle cells, consistent with their role in modulating colonic motility through regulation of muscle tone and contractility in response to ATP and ADP fluctuations. Notably, Kir6.1 and SUR2 expression is also evident in ICC, which are distributed among smooth muscle fibers and concentrated within the myenteric and submucosal plexuses, suggesting their involvement in coordinating colonic pacemaker activity (34–36).
Figure 1

Immunohistochemical localization of KATP channel subunits Kir6.1 and SUR2 in normal human colon tissue. Representative immunohistochemical staining showing the expression of the KATP channel subunits Kir6.1 (A, A1) and SUR2 (B, B1) in histological sections of normal human colon. Images were acquired at low magnification [10×; (A, B)] to provide an overview of tissue architecture and at higher magnification [20×; (A1, B1)] to highlight cellular localization.
Figure 2

Immunohistochemical localization of KATP channel Kir6.1 subunit in normal mouse colon tissue. Representative immunohistochemical staining illustrating the distribution of KATP channel Kir6.1 subunit in histological sections of normal mouse colon at varying magnifications. (A) shows low-magnification imaging (10×) to provide an overview of overall tissue morphology. (B1, B2) (20×) and (C1, C2) (40×) present higher magnification views to resolve specific regional expression. At 20× and 40× magnification, Kir6.1 immunoreactivity is observed in distinct layers of the colon, including the lamina propria and muscularis (B1, C1), as well as within the crypt structures (B2, C2).
Figure 3
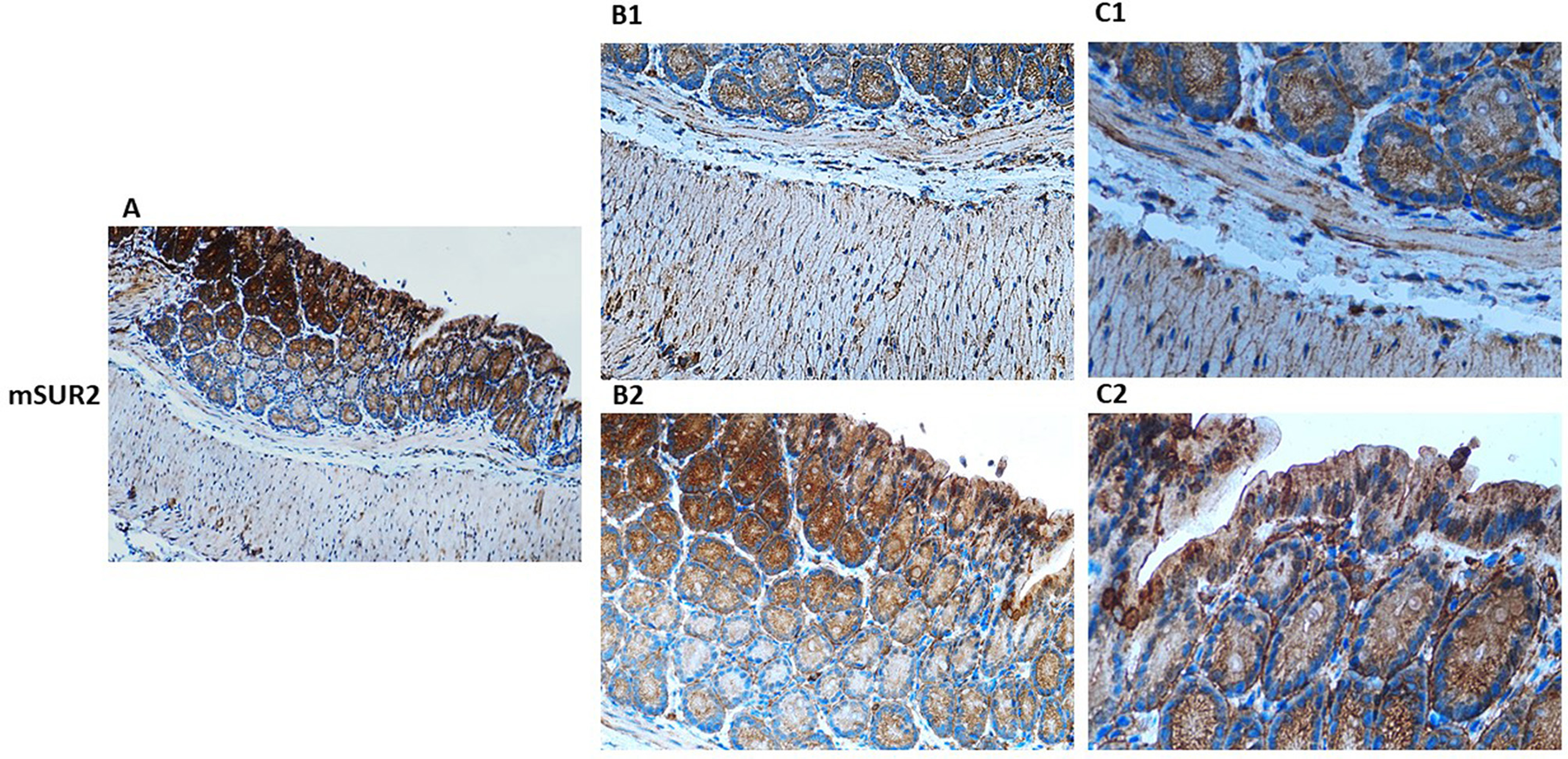
Immunohistochemical localization of KATP channel SUR2 subunit in normal mouse colon tissue. Representative immunohistochemical staining illustrating the distribution of KATP channel SUR2 subunit in histological sections of normal mouse colon at varying magnifications. (A) shows low-magnification imaging (10×) to provide an overview of overall tissue morphology. (B1, B2) (20×) and (C1, C2) (40×) present higher magnification views to resolve specific regional expression. At 20× and 40× magnification, Kir6.1 immunoreactivity is observed in distinct layers of the colon, including the lamina propria and muscularis (B1, C1), as well as within the crypt structures (B2, C2).
Within the epithelial compartment, Kir6.1 and SUR2 immunoreactivity is predominantly localized to the apical and basolateral membranes of enterocytes, with pronounced staining in the crypts of Lieberkühn (Figure 3). This distribution suggests a functional role in epithelial ion transport, particularly in chloride and water secretion, contributing to maintaining intestinal fluid homeostasis. Co-localization studies with Occludin and ZO-1 reveal the presence of Kir6.1 and SUR2 within tight junction complexes, implicating them in regulating epithelial barrier integrity. High-resolution imaging further demonstrates punctate staining along lateral cell membranes, suggesting potential interactions between KATP channels and junctional proteins, which may modulate paracellular permeability (20, 38).
Beyond epithelial and muscular compartments, Kir6.1 and SUR2 expression extends to neuronal structures of the enteric nervous system, with detectable levels in myenteric and submucosal ganglia. Within these regions, expression is particularly associated with inhibitory motor neurons, where KATP channel activation is linked to reduced excitability and modulation of enteric reflexes. This expression pattern aligns with electrophysiological studies implicating KATP channels in enteric neurotransmission (20). The spatially distinct distribution of Kir6.1 and SUR2 subunits across epithelium, smooth muscle, and neural plexuses underscores their multifaceted roles in colonic physiology, influencing both epithelial transport processes and neuromuscular regulation of peristalsis (Figures 1–3).
GOF CS mutations result in an alteration in the mRNA expression of tight junction proteins (Occludin, Claudin-1, and ZO-1) in colon tissue
Quantitative real-time PCR (qRT-PCR) analysis of colonic RNA extracted from CS and WT mice revealed significant changes in the mRNA expression profiles of key tight junction proteins, including Occludin, Claudin-1, and ZO-1, in mutant mice compared to controls. Specifically, the expression of Occludin, a critical tight junction protein responsible for regulating epithelial barrier integrity, was differentially regulated depending on the specific KATP mutation; Occludin expression was significantly (p < 0.05) upregulated in SUR2AV/AV mice compared to WT controls. In contrast, substantial downregulation of Occludin was observed in SUR2wt/AV and Kir6.1wt/VM mice compared to SUR2AV/AV mice, suggesting disruption in tight junction assembly and function. These alterations in Occludin expression may indicate an underlying increase in epithelial permeability, potentially contributing to the mutants' altered intestinal barrier function. Similarly, Claudin-1, another integral component of tight junctions known to play a role in maintaining the paracellular permeability barrier, showed a significant decrease in expression in SUR2AV/AV and SUR2wt/AV mice compared to WT mice. However, in Kir6.1wt/VM mice, Claudin-1 expression was significantly increased compared to SUR2AV/AV, suggesting potential compensatory mechanisms or differential regulation in this specific genetic background. These changes indicate potential defects in establishing tight junctions that could compromise epithelial barrier function in these mutant strains. Additionally, ZO-1, a critical scaffolding protein that links transmembrane tight junction proteins to the actin cytoskeleton, showed a significant (p < 0.05) decrease in mRNA expression in SUR2AV/AV, SUR2wt/AV, and Kir6.1wt/VM mice compared to WT controls. This reduction in ZO-1 transcript levels is consistent with structural and functional disruptions of tight junctions, which may further compromise the intestinal epithelium's integrity. These findings suggest a complex and multifactorial disruption of tight junction protein expression and assembly in mutant mice, potentially contributing to impaired epithelial permeability and barrier function. Results are expressed as mean ± SD (n = 3 biological replicates per group, each with three technical replicates). Statistical significance was assessed by one-way ANOVA with Tukey's post hoc test; exact p-values are reported in Figure 4.
Figure 4
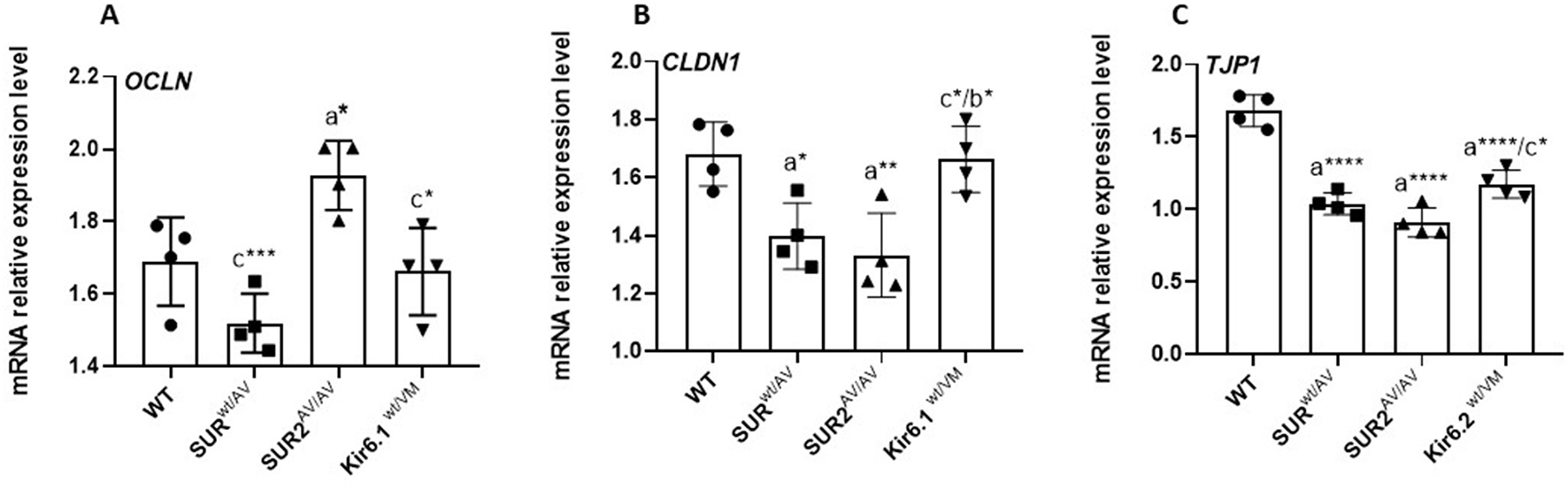
Quantitative RT-PCR analysis of tight junction gene expression. mRNA levels of OCLN(A), CLDN1(B), and TJP1 [ZO-1; (C)] in colon tissue from WT and CS mice (SUR2wt/AV, SUR2AV/AV, Kir6.1wt/VM). Results are expressed as mean ± SD of a minimum of three independent experiments with three replicates per experimental condition in each experiment (n. animals used for these evaluations: 4 animals per genotype). a: significance vs. WT, b: significance vs. SUR2wt/AV, c: significance vs. SUR2AV/AV, and d: significance (p < 0.05) vs. Kir6.1wt/VM. One-way ANOVA followed by Tukey's post hoc test was used to evaluate statistical significance; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Gain-of-function (GOF) mutations in the CS gene enhance transcriptional activation of the intrinsic apoptotic machinery in the colon
Gain-of-function (GOF) mutations in the CS gene selectively modulate the transcriptional profile of apoptotic regulators in the colon. RT–qPCR analysis revealed a significant (p < 0.05) downregulation of anti-apoptotic BCL2L2 and BCL2 mRNA levels in colonic tissue from SUR2wt/AV, SUR2AV/AV, and Kir6.1wt/VM mice compared to wild-type controls. In contrast, the pro-apoptotic gene BAX expression remained unchanged (p > 0.05) across all CS mutant models. This selective repression of anti-apoptotic transcripts, without a compensatory increase in pro-apoptotic signaling, suggests a disrupted apoptotic equilibrium that may underlie aberrant epithelial turnover and prolonged cell survival. These findings point to a previously unrecognized impact of CS GOF mutations on apoptotic homeostasis in the colon, with potential implications for tissue integrity and disease susceptibility (Figure 5).
Figure 5

mRNA expression levels of BCL2L1(A), BCL2(B), and BAX(C) in colon tissue of CS mice vs. WT mice. Results are expressed as mean ± SD of a minimum of three independent experiments with three replicates per experimental condition in each experiment (n. animals used for these evaluations: 4 animals per genotype). a: significance vs. WT, b: significance vs. SUR2wt/AV, c: significance vs. SUR2AV/AV, and d: significance (p < 0.05) vs. Kir6.1wt/VM. One-way ANOVA followed by Tukey's post hoc test was used to evaluate statistical significance; *P < 0.05, **P < 0.01, ***P < 0.001.
Gain-of-function mutations in the CS gene result in a transcriptional shift toward pro-apoptotic signaling in the colon, a process closely intertwined with oxidative stress responses. In colon tissue from SUR2wt/AV, SUR2AV/AV, and Kir6.1wt/VM mice, RT–qPCR analysis revealed significant upregulation of CASP9 and CASP3, central mediators of the mitochondrial (intrinsic) apoptotic pathway, along with reduced expression of the anti-apoptotic genes BCL2 and BCL2L2 (Bcl-xL). This molecular profile is consistent with increased susceptibility to mitochondrial outer membrane permeabilization and caspase activation in response to cellular stress (Figure 6).
Figure 6

mRNA expression levels of CASP3(A) and CASP9(B) in CS mice and WT mice's colon tissue. Results are expressed as mean ± SD of a minimum of three independent experiments with three replicates per experimental condition in each experiment (n. animals used for these evaluations: 4 animals per genotype). a: significance vs. WT, b: significance vs. SUR2wt/AV, c: significance vs. SUR2AV/AV, and d: significance (p < 0.05) vs. Kir6.1wt/VM. One-way ANOVA followed by Tukey's post hoc test was used to evaluate statistical significance; *P < 0.05, **P < 0.01, ***P < 0.001.
Gain-of-function CS mutations lead to the disruption of tight junction protein expression and epithelial barrier integrity
Immunohistochemical staining of colonic sections from CS and WT mice revealed significant alterations in the expression of tight junction proteins, particularly Occludin and Claudin-1. In WT mice, both proteins were strongly expressed at the apical junctions of epithelial cells, maintaining the characteristic organization of tight junctions. In contrast, CS mice exhibited a marked reduction in the expression of Occludin and Claudin-1, particularly at intercellular junctions. The staining intensity of Occludin was notably diminished, indicating a disruption in tight junction integrity, while Claudin-1 expression was similarly reduced, further suggesting impaired barrier function. This decrease in tight junction protein expression may underlie a structural and functional impairment of the epithelial barrier, potentially contributing to increased intestinal permeability in CS mice. These findings support the hypothesis that altered KATP channel activity plays a role in destabilizing tight junctions, which may exacerbate barrier dysfunction and intestinal inflammation in the mutant phenotype (Figure 7).
Figure 7

Representative histological analysis of the sample with hematoxylin-eosin (H&E) reaction and immunostaining for junction proteins (occludin, and Claudin-1) on colon sections of (a, a1) WT, (b, b1) SUR2wt/AV(c, c1) SUR2AV/AV and (d, d1) Kir6.1wt/VM mice at 20×. (n. animals used for these evaluations: 4 animals per genotype.
Gain-of-function CS mutations induce structural and functional alterations in the colonic epithelium and smooth muscle of CS mice
Histological examination of colonic sections from CS and WT mice revealed notable structural alterations in the colonic epithelial layer and smooth muscle of CS mice. CS mice disrupted the typical organization of tightly packed columnar cells with well-defined apical-basal polarity in the epithelial layer. The epithelium exhibited signs of disorganization, including irregular cell arrangement and a marked increase in intercellular spaces, indicative of a compromised epithelial barrier. This disruption is likely associated with the destabilization of tight junctions, as evidenced by the reduced expression of tight junction proteins, such as Occludin and Claudin-1. Moreover, the epithelial layer of CS mice showed increased cellular infiltration, indicative of an inflammatory response, which further supports the notion of epithelial dysfunction. Notably, sections from CS mice exhibited autolytic disintegration phenomena, likely a consequence of the inflammatory state, accompanied by intense caspase-3 immunoreactivity (Figure 8).
Figure 8

Representative histological analysis of the sample with hematoxylin-eosin (H&E) reaction and immunostain for casp-3 on colon sections of (a1, a2) WT, (b1, b2) SUR2AV/AV SUR2wt/AV and (c1, c2) Kir6.1wt/VM mice at 20× (left) and 40× (right; n. animal used for these evaluations: 3 animals per genotype).
CS mice displayed hypertrophic changes in the underlying smooth muscle layer characterized by increased muscle thickness and apparent disorganization of smooth muscle fibers. Smooth muscle cells appeared structurally disordered and showed signs of hyperplasia, particularly in the muscularis propria, most prominently in SURAV/AV mice. These structural changes were accompanied by increased fibrosis and heightened extracellular matrix deposition. This fibrotic remodeling suggests potential smooth muscle dysfunction, possibly contributing to impaired motility and contractility in CS mice (Table 1).
Table 1
| Pathological changes | WT | SUR2wt/AV | SUR2AV/AV | Kir6.1wt/VM |
|---|---|---|---|---|
| Diffuse inflammation | –/+ | ++ | +++ | +++ |
| Patchy/focal inflammation | – | ++ | +++ | + |
| Active cryptitis | – | +++ | +++ | +++ |
| Mucosal remodeling | – | +++ | +++ | +++ |
| Alteration of the musculature | – | ++ | ++ | +++ |
| Neutrophils in lamina propria | – | ++ | ++ | ++ |
A comparative histological analysis was performed to assess the severity and distribution of pathological changes in colonic tissue across genotypes, including WT and CS mice (SUR2wt/AV, SUR2AV/AV, Kir6.1wt/VM).
Overall, these structural alterations in the epithelial and smooth muscle layers of CS mice reflect a complex disruption of colonic architecture, likely contributing to observed physiological abnormalities such as increased intestinal permeability and impaired motility. These impairments may stem from altered KATP channel activity in CS mice, which exacerbates both epithelial and smooth muscle dysfunction, ultimately contributing to the pathophysiology of the mutant phenotype (Figure 8, Table 1). These alterations reflect a shared pathological signature similar to chronic inflammatory bowel disease, with the most severe manifestations in SUR2AV/AV and Kir6.1wt/VM mice.
Discussion
To our knowledge, this study provides the first comprehensive evidence that Kir6.1 and SUR2 subunits of KATP channels are strongly expressed in both human and murine colons, with predominant localization in the colonic epithelium. These findings suggest that KATP channels are crucial in maintaining intestinal barrier integrity, enabling selective nutrient absorption while preventing pathogen infiltration. Prior research has documented the presence of Kir6.1/SUR2 complexes in rodents' non-vascular smooth muscle and colonic epithelial cells, and their co-localization with TJ proteins in the small intestine (40, 43). Altogether, these data highlight the multifaceted role of KATP channels in epithelial and endothelial homeostasis.
KATP channels are well-known metabolic sensors that couple ATP/ADP ratios to ion fluxes and membrane potential, ensuring cellular adaptation to metabolic stressors such as hypoxia or ischemia (13, 16). They regulate apical ion transport in the colon, particularly chloride secretion, as shown in rodent models (26, 45).
We employed multiple murine models carrying gain-of-function (GOF) mutations in SUR2 [SUR2(A478V)] or Kir6.1 [Kir6.1(V65M)] to explore the impact of KATP channel hyperactivity. Heterozygous SUR2A478V animals showed moderate epithelial alterations, while homozygous mutants displayed more severe pathology, including smooth muscle hypertrophy, fibrosis, and inflammation, consistent with a dose-dependent GOF effect. Kir6.1V65M mice exhibited pronounced deficits in motility and neuromuscular coordination, which aligns with findings by York et al. (20), who demonstrated that GOF mutations impair gastrointestinal contractility. Pharmacological activation with pinacidil inhibited motility, whereas glibenclamide reversed transit deficits, especially in Kir6.1wt/VM mice.
In WT mice, epithelial homeostasis is maintained through cell turnover, TJ remodeling, and immune surveillance. In contrast, CS mice with GOF mutations in Kir6.1 or SUR2 displayed compromised barrier function and heightened inflammation (20). Histological and RT-qPCR analyses revealed that WT colons had normal expression of TJ proteins such as Occludin and Claudin-1, essential for controlling paracellular permeability. CS mice, however, exhibited significantly reduced expression of these proteins, confirmed by diminished immunoreactivity in tissue sections hallmarks of impaired barrier function. So, Occludin levels are differentially regulated depending on the specific KATP mutation: upregulated in SUR2AV/AV mice and downregulated in SUR2wt/AV and Kir6.1V65M mutants. These differential expression patterns may reflect mutation-specific effects on tight junction dynamics and compensatory responses, rather than uniform dysregulation.
Our data suggest that KATP channel hyperactivity compromises TJ integrity through converging mechanisms, notably membrane hyperpolarization and overproduction of reactive oxygen species (ROS) (29, 30). TJs are composed of transmembrane proteins (e.g., claudins, occludins), cytoplasmic scaffolding proteins (e.g., ZO-1, ZO-2, ZO-3), and actin cytoskeleton components (46–48). ROS-induced actin depolymerization can destabilize the junctional complex, increasing permeability (47, 49). Electrolyte imbalance and metabolic dysregulation in CS mice likely exacerbate TJ disruption.
GOF mutations in KATP subunits led to structural and molecular abnormalities, including epithelial disorganization, inflammation, and apoptosis. These defects were accompanied by increased caspase-3 activity, indicating widespread programmed cell death. Mechanistically, sustained potassium efflux and plasma membrane hyperpolarization elevate intracellular calcium, enhancing mitochondrial respiration and generating excess ROS. This oxidative stress damages TJ components, disrupts cytoskeletal anchoring, and activates inflammatory pathways. It also perturbs apoptotic regulation by decreasing BCL2/BCL-XL and upregulating BAX, further driving caspase-3-mediated cell death and tissue disintegration.
Additionally, KATP channels may directly interact with TJ components, as shown by the co-localization of Kir6.1 and SUR2 with Claudin-1, implicating a structural role in barrier maintenance.
Beyond epithelial cells, our localization studies revealed expression of Kir6.1 and SUR2 in colonic smooth muscle and ICC, suggesting involvement in neuromuscular coordination and pacemaker activity. This may underlie the motility impairments seen in CS models, especially in Kir6.1 mutant.
Overall, our findings point to KATP channel dysregulation as a central driver of intestinal dysfunction in CS via oxidative stress, inflammatory activation, and epithelial disruption. However, the precise molecular pathways, particularly calcium signaling and ROS, warrant further investigation.
This study provides novel insights into the role of KATP channels in CS-related intestinal pathology. Its strength lies in using multiple murine models and a multi-level analytical approach encompassing molecular, histological, and functional parameters. Identifying oxidative stress and apoptosis as mediators of TJ dysfunction highlights promising therapeutic targets.
Nevertheless, some limitations should be acknowledged. While murine models offer valuable mechanistic insights, interspecies differences in physiology and immune responses may limit translation to humans. Our analyses focused primarily on structural, molecular and histological features of colonic epithelial integrity. Functional permeability assays, such as Using chamber measurements or sugar absorption tests, were not performed, representing an important limitation. Such assays provide direct quantification of paracellular flux, electrolyte transport and barrier function, and would be essential to corroborate the molecular observations derived from frozen tissues, which are well suited for immunohistochemistry and histochemical analyses but not for functional studies. By concentrating on the colon, our work does not address potential contributions from other intestinal regions. Although the colon was selected owing to its established relevance in disease, the small intestine differs substantially in immune cell populations, microbial ecology, nutrient absorption and epithelial turnover, all of which may shape pathophysiology and responses to intervention. Further, direct assessments of intracellular calcium flux and reactive oxygen species generation would strengthen the mechanistic link between KATP channel hyperactivity, oxidative stress and tight junction disruption. Future studies will therefore aim to define the causal chain connecting KATP hyperactivity, calcium flux and ROS production, incorporating Ussing chamber assays and complementary measures of epithelial transport to validate the structural findings reported here. Finally, although CS is not generally associated with sex differences in cardiovascular function, possible sex-specific effects of CS mutations on intestinal physiology cannot be excluded, and forthcoming experiments in CS mice will address this question.
Despite these limitations, this work lays the foundation for future research to develop therapeutic strategies targeting KATP channels or downstream pathways to restore intestinal barrier function in CS and related conditions.
Materials and methods
Animal care and ethical statements
Given the current lack of evidence supporting sex-based differences in conditioned stimulus (CS) responses, our experiments were conducted exclusively using male mice to maintain consistency and reduce potential variability (8, 9). Despite this, we are aware that sex differences may influence epithelial and immune function, potentially affecting disease pathophysiology and therapeutic responses due to hormonal influences. Male knock-in mice carrying Kir6.1(V65M) (Kir6.1wt/VM), SUR2(A478V) heterozygous (SUR2wt/AV), and SUR2(A478V) homozygous (SUR2AV/AV) mutations mimicking human Cantú syndrome (CS) were generated via CRISPR/Cas9 gene editing at Washington University in St. Louis, USA, and subsequently transferred to Italy. Genotyping was performed as described by Huang et al. (8, 9). Animals were housed in groups of two to four per cage at the Animal Facility of the Department of Pharmacy-Drug Sciences, University of Bari, Italy. Housing conditions were maintained at 22 ± 1 °C with 50 ± 5% relative humidity and a 12:12 light/dark cycle, with ad libitum access to a standard laboratory diet and water. All experimental procedures adhered to European Directive 2010/63/EU on the protection of animals used for scientific purposes and were approved by the Institutional Animal Care and Use Committee (IACUC) of Washington University School of Medicine, as well as by the Italian Ministry of Health and the University of Bari's OPBA (Organization for Animal Health; protocol 8515-X/10, January 30, 2019).
Animal sacrifice and tissue harvesting
Animals were sacrificed by cervical dislocation under deep anesthesia with ZOLETIL 50/50 (40 mg/kg, i.p.). Tissues and organs were harvested under sterile conditions to prevent contamination. Organs were weighed and either snap-frozen in liquid nitrogen for protein extraction or embedded in OCT for immunofluorescence and immunohistochemistry, then stored at −80 °C. All procedures were conducted in Ringer's solution (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 0.5 mM CaCl2, 5 mM glucose, 10 mM MOPS, pH 7.2).
Total RNA isolation and RT-qPCR
According to the manufacturer's instructions, total RNA was isolated from colon tissue using TRIzol reagent (Invitrogen, Waltham, MA, USA). RNA purity and concentration were assessed with a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), while RNA integrity was confirmed by electrophoresis on a denaturing agarose gel. Complementary DNA (cDNA) was synthesized via reverse transcription. Gene expression levels were quantified by real-time quantitative PCR (RT-qPCR) using a 2 × SYBR Green master mix (Applied Biosystems, Foster City, CA, USA) following the manufacturer's guidelines. Primer sequences for RT-qPCR are listed in Table 2. Relative gene expression was determined by the ΔCT method, with GAPDH as the internal control for normalization.
Table 2
| Gene | Forward | Reverse |
|---|---|---|
| OCLN | CTCCCATCCGAGTTTCAGGT | GCTGTCGCCTAAGGAAAGAG |
| CLDN1 | GTTTGCAGAGACCCCATCAC | AGAAGCCAGGATGAAACCCA |
| TJP1 | CTGAGTTGCCCGCGACG | CTGCTCCCGCACGTAACTTC |
| CASP9 | AGTTCCCGGGTGCTGTCTAT | GCCATGGTCTTTCTGCTCAC |
| CASP3 | CCTCAGAGAGACATTCATGG | GCAGTAGTCGCCTCTGAAGA |
| BCL2 | CTCGTCGCTACCGTCGTGACTTCG | CAGATGCCGGTTCAGGTACTCAGTC |
| BCL2L1 | TGGAGTAAACTGGGGGTCGCATCG | AGCCACCGTCATGCCCGTCAGG |
| BAX | AAGCTGAGCGAGTGTCTCCGGCG | GCCACAAAGATGGTCACTGTCTGCC |
| GAPDH | GCCCAATACGACCAAATCC | AGCCACATCGCTCAGACAC |
Primers used in RT-qPCR.
Immunohistochemical and histological analysis
Formalin-fixed, paraffin-embedded (FFPE) tissue specimens were sectioned at 4 μm, mounted on Apex Bond Slides (Leica Biosystems), and subjected to immunohistochemical and histological analyses. Immunohistochemistry was performed using a BOND III automated stainer (Leica Biosystems, Wetzlar, Germany) with the Bond Polymer Refine Detection Kit, encompassing deparaffinization through hematoxylin counterstaining. Sections were incubated for 30 min at room temperature with primary antibodies against Kir6.1 (monoclonal, AB174629, Abcam, Toronto, Canada), SUR2 (monoclonal, AB271996; Abcam, Toronto, Canada), Caspase-3 (polyclonal, 9662; Cell Signaling Technology, Danvers, MA, USA), Occludin (monoclonal, 91131; Cell Signaling Technology), and Claudin-1 (polyclonal, 4933; Cell Signaling Technology). Antigen retrieval was performed using BOND Epitope Retrieval Solution 2 (Leica Biosystems). For histological evaluation, sections were stained with hematoxylin and eosin (H&E) and reviewed by a board-certified pathologist. Inflammatory scoring was based on epithelial integrity, immune cell infiltration, and tissue/muscle remodeling, whereas morphological assessment included crypt architecture, goblet cell preservation, and mucosal organization. Pathological alterations were quantified using standardized criteria to ensure reproducibility. Images were captured using a Nikon Eclipse Ti2 microscope.
Data analysis and statistics
All data were collected and analyzed using Excel (Microsoft Office 2010) and SigmaPlot 10.0 (Systat Software). Results are presented as mean ± SEM unless otherwise specified. The corresponding figure legends detail the number of biological and technical replicates for each experiment. Statistical analyses were conducted using one-way analysis of variance (ANOVA) followed by post hoc multiple comparisons to assess differences among groups, with a significance level of p < 0.05 unless otherwise stated. Student's t-test was applied for pairwise comparisons, with statistical significance defined as p < 0.05. Sample sizes were determined via power analysis (α = 0.05, power = 0.8) based on pilot data. Experimental groups included Kir6.1wt/VM (n = 4), SUR2wt/AV (n = 4), SUR2AV/AV (n = 4), and wild-type (WT) controls (n = 4). These sample sizes were chosen based on previous studies in similar models, which consistently demonstrated robust and reproducible effects using comparable cohorts, particularly for mechanistic and histological endpoints. Investigators were blinded to genotype during data collection and analysis. Lower power of 0.72 is calculated with a sample size of n = 3.
Conclusion
The present study demonstrates that GOF mutations in Kir6.1 and SUR2, characteristic of CS, induce profound alterations in TJ protein expression and epithelial barrier integrity. CS murine models exhibited disrupted mRNA and protein levels of Occludin, Claudin-1, and ZO-1, correlating with structural epithelial defects, increased paracellular permeability, and inflammatory infiltration. Additionally, smooth muscle hypertrophy and fibrosis indicate broader neuromuscular dysfunction in the colon.
Mechanistically, KATP channel hyperactivity likely drives oxidative stress and caspase-3-mediated apoptosis, compromising barrier integrity and sustaining a cycle of inflammation and tissue damage. These findings underscore the essential role of KATP channels in intestinal homeostasis and link their dysregulation to epithelial and neuromuscular pathology in CS.
Given that SUR1 and SUR2 subunits are well-established drug targets in diabetes and cardiovascular diseases, the high expression of KATP channel subunits in the colon suggests their potential for pharmacological modulation of intestinal motility and barrier function. Targeting oxidative stress or KATP channel activity may offer novel therapeutic strategies to restore barrier function and mitigate intestinal complications in CS and related disorders.
Statements
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee (IACUC) of Washington University School of Medicine, as well as by the Italian Ministry of Health and the University of Bari's OPBA (Organization for Animal Health) (protocol 8515-X/10, January 30, 2019). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
FM: Funding acquisition, Visualization, Resources, Formal analysis, Project administration, Validation, Supervision, Data curation, Investigation, Writing – review & editing, Writing – original draft, Methodology, Conceptualization, Software. DM: Writing – review & editing, Investigation, Methodology, Formal analysis. AO: Writing – review & editing, Data curation, Visualization. DT: Writing – review & editing, Data curation. CN: Formal analysis, Writing – review & editing. MA: Formal analysis, Writing – review & editing, Methodology, Data curation, Investigation. GB: Formal analysis, Writing – review & editing, Investigation. RA: Investigation, Data curation, Formal analysis, Writing – review & editing, Methodology. IG: Writing – review & editing, Investigation, Methodology. AV: Writing – review & editing. FR: Investigation, Validation, Conceptualization, Supervision, Writing – review & editing, Software, Visualization, Writing – original draft, Formal analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Italian Ministry of Health, Ricerca Corrente 2024–2025 program, at the IRCCS “Saverio de Bellis,” (RC2024_Maqoud). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer EM declared a past co-authorship with the author CN to the handling editor.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Van Bon BWM Gilissen C Grange DK Hennekam RCM Kayserili H Engels H et al . Cantú syndrome is caused by mutations in ABCC9. Am J Hum Genet. (2012) 90:1094–101. 10.1016/j.ajhg.2012.04.014
2.
Cooper PE McClenaghan C Chen X Stary-Weinzinger A Nichols CG . Conserved functional consequences of disease-associated mutations in the slide helix of Kir6.1 and Kir6.2 subunits of the ATP-sensitive potassium channel. J Biol Chem. (2017) 292:17387–98. 10.1074/jbc.M117.804971
3.
Cooper PE Sala-Rabanal M Lee SJ Nichols CG . Differential mechanisms of Cantú syndrome-associated gain of function mutations in the ABCC9 (SUR2) subunit of the KATP channel. J Gen Physiol. (2015) 146:527–40. 10.1085/jgp.201511495
4.
Carvalho AA Ferraz LDA Martelli DRB Machado RA Junior HM . Craniofacial findings in syndromes associated with cafe-Au-lait spots: a literature review. Rev Assoc Med Bras. (2023) 69:195–202. 10.1590/1806-9282.20220866
5.
Cantú JM García-Cruz D Sánchez-Corona J Hernández A Nazará Z . A distinct osteochondrodysplasia with hypertrichosis- Individualization of a probable autosomal recessive entity. Hum Genet. (1982) 60:36–41. 10.1007/BF00281261
6.
Nichols CG Singh GK Grange DK . KATP channels and cardiovascular disease: suddenly a syndrome. Circ Res. (2013) 112:1059–72. 10.1161/CIRCRESAHA.112.300514
7.
Scurr I Wilson L Lees M Robertson S Kirk E Turner A et al . Cantú syndrome: report of nine new cases and expansion of the clinical phenotype. Am J Med Genet A. (2011) 155A:508–18. 10.1002/ajmg.a.33885
8.
Huang Y McClenaghan C Harter TM Hinman K Halabi CM Matkovich SJ et al . Cardiovascular consequences of KATP overactivity in cantu syndrome. JCI Insight. (2018) 3:e121153. 10.1172/jci.insight.121153
9.
Mcclenaghan C Huang Y Matkovich SJ Kovacs A Weinheimer CJ Perez R et al . The mechanism of high-output cardiac hypertrophy arising from potassium channel gain-of-function in cantú syndrome. Function. (2020) 1:zqaa004. 10.1093/function/zqaa004
10.
Aziz Q Li Y Anderson N Ojake L Tsisanova E Tinker A et al . Molecular and functional characterization of the endothelial ATP-sensitive potassium channel. J Biol Chem. (2017) 292:17587–97. 10.1074/jbc.M117.810325
11.
Baukrowitz T Fakler B . K(ATP) channels gated by intracellular nucleotides and phospholipids. Eur J Biochem. (2000) 267:5842–8. 10.1046/j.1432-1327.2000.01672.x
12.
Tricarico D Mele A Lundquist AL Desai RR George AL Camerino DC et al . Hybrid assemblies of ATP-sensitive K channels determine their muscle-type-dependent biophysical and pharmacological properties. (2006) 103:1118–23. 10.1073/pnas.0505974103
13.
Tinker A Aziz Q Li Y Specterman M . ATP-Sensitive potassium channels and their physiological and pathophysiological roles. Compr Physiol. (2018) 8:1463–511. 10.1002/cphy.c170048
14.
Colin GN . KATP channels as molecular sensors of cellular metabolism. Nat. (2006) 440:470–6. 10.1038/nature04711
15.
Li Y Aziz Q Anderson N Ojake L Tinker A . Endothelial ATP-sensitive potassium channel protects against the development of hypertension and atherosclerosis. Hypertension. (2020) 76:776–84. 10.1161/HYPERTENSIONAHA.120.15355
16.
Ashcroft FM . KATP Channels and the metabolic regulation of insulin secretion in health and disease: the 2022 banting medal for scientific achievement award lecture. Diabetes. (2023) 72:693–702. 10.2337/dbi22-0030
17.
Rodríguez-Rivera NS Barrera-Oviedo D . Exploring the pathophysiology of ATP-dependent potassium channels in insulin resistance. Int J Mol Sci. (2024) 25:4079. 10.3390/ijms25074079
18.
Priyamvada S Gomes R Gill RK Saksena S Alrefai WA Dudeja PK et al . Mechanisms underlying dysregulation of electrolyte absorption in inflammatory bowel disease-associated diarrhea. Inflamm Bowel Dis. (2015) 21:2926–35. 10.1097/MIB.0000000000000504
19.
Tomuschat C O'Donnell AM Coyle D Dreher N Kelly D Puri P et al . Altered expression of ATP-sensitive K+ channels in hirschsprung's disease. J Pediatr Surg. (2016) 51:948–52. 10.1016/j.jpedsurg.2016.02.060
20.
York NW Parker H Xie Z Tyus D Waheed MA Yan Z et al . Kir6.1- And SUR2-dependent KATP overactivity disrupts intestinal motility in murine models of Cantú syndrome. JCI Insight. (2020) 5:141443. 10.1172/jci.insight.141443
21.
Han J Lee SH Giebisch G Wang T . Potassium channelopathies and gastrointestinal ulceration. Gut Liver. (2016) 10:881–9. 10.5009/gnl15414
22.
Anderson KJ Cormier RT Scott PM . Role of ion channels in gastrointestinal cancer. World J Gastroenterol. (2019) 25:5732–72. 10.3748/wjg.v25.i38.5732
23.
Sharkey KA Mawe GM . The enteric nervous system. Physiol Rev. (2023) 103:1487–564. 10.1152/physrev.00018.2022
24.
Horii K Suzuki Y Shiina T Saito S Onouchi S Horii Y et al . ATP-dependent potassium channels contribute to motor regulation of esophageal striated muscle in rats. J Vet Med Sci. (2019) 81:1266–72. 10.1292/jvms.19-0197
25.
Cosme D Estevinho MM Rieder F Magro F . Potassium channels in intestinal epithelial cells and their pharmacological modulation: a systematic review. Am J Physiol Cell Physiol. (2020) 320:C520–46. 10.1152/ajpcell.00393.2020
26.
Gade AR Kang M Akbarali HI . Hydrogen sulfide as an allosteric modulator of ATP-sensitive potassium channels in colonic inflammation. Mol Pharmacol. (2013) 83:294–306. 10.1124/mol.112.081596
27.
Radulovic M Anand P Korsten MA Gong B . Targeting ion channels: an important therapeutic implication in gastrointestinal dysmotility in patients with spinal cord injury. J Neurogastroenterol Motil. (2015) 21:494–502. 10.5056/jnm15061
28.
Davis MJ Kim HJ Zawieja SD Castorena-Gonzalez JA Gui P Li M et al . Kir6.1-dependent KATP channels in lymphatic smooth muscle and vessel dysfunction in mice with Kir6.1 gain-of-function. J. Physiol. (2020) 598:3107–27. 10.1113/JP279612
29.
Furness JB . The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. (2012) 9:286–94. 10.1038/nrgastro.2012.32
30.
Spencer NJ Hu H . Enteric nervous system: sensory transduction, neural circuits and gastrointestinal motility. Nat Rev Gastroenterol Hepatol. (2020) 17:338–51. 10.1038/s41575-020-0271-2
31.
Kenton Sanders CM Sanders KM Ward SM . Nitric oxide and its role as a non-adrenergic, non-cholinergic inhibitory neurotransmitter in the gastrointestinal tract. Br J Pharmacol. (2019) 176:212–27. 10.1111/bph.14459
32.
Koh SD Ward SM Sanders KM . Ionic conductances regulating the excitability of colonic smooth muscles. J Neurogastroenterol Motil. (2012) 24:705–18. 10.1111/j.1365-2982.2012.01956.x
33.
Nakayama S Ohya S Liu HN Watanabe T Furuzono S Wang J et al . Sulphonylurea receptors differently modulate ICC pacemaker Ca2+ activity and smooth muscle contractility. J Cell Sci. (2005) 118:4163–73. 10.1242/jcs.02540
34.
Ahn SW Kim SH Kim JH Seok C Yeum CH Wie HW et al . Phentolamine inhibits the pacemaker activity of mouse interstitial cells of dis by activating ATP-sensitive K+ channels. Arch Pharm Res. (2010) 33:479–89. 10.1007/s12272-010-0319-x
35.
Sanders KM Koh SD Ward SM . Interstitial cells of cajal as pacemakers in the gastrointestinal tract. Annu Rev Physiol. (2006) 68:307–43. 10.1146/annurev.physiol.68.040504.094718
36.
Kito Y Ward SM Sanders KM . Pacemaker potentials generated by interstitial cells of cajal in the murine intestine. Am J Physiol Cell Physiol. (2005) 288:710–20. 10.1152/ajpcell.00361.2004
37.
Distrutti E Sediari L Mencarelli A Renga B Orlandi S Antonelli E et al . Evidence that hydrogen sulfide exerts antinociceptive effects in the gastrointestinal tract by activating KATP channels. J Pharmacol Exp Ther. (2006) 316:325–35. 10.1124/jpet.105.091595
38.
Lee B Moon KM Kim CY . Tight junction in the intestinal epithelium: Its association with diseases and regulation by phytochemicals. J Immunol Res Vol. (2018) 2018:2645465. 10.1155/2018/2645465
39.
Zihni C Mills C Matter K Balda MS . Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol. (2016) 17:564–80. 10.1038/nrm.2016.80
40.
Singh AB Uppada SB Dhawan P . Claudin proteins, outside-in signaling, and carcinogenesis. Pflugers Arch. (2017) 469:69–75. 10.1007/s00424-016-1919-1
41.
Günzel D Yu AS . Claudins and the modulation of tight junction permeability. Physiol Rev. (2013) 93:525–69. 10.1152/physrev.00019.2012
42.
Umeda K Ikenouchi J Katahira-Tayama S Furuse K Sasaki H Nakayama M et al . ZO-1 and ZO-2 independently determine where claudins are polymerized in tight-junction strand formation. Cell. (2006) 126:741–54. 10.1016/j.cell.2006.06.043
43.
Scala R Maqoud F Zizzo N Mele A Camerino GM Zito FA et al . Pathophysiological consequences of KATP channel overactivity and pharmacological response to glibenclamide in skeletal muscle of a murine model of cantù syndrome. Front Pharmacol. (2020) 11:604885. 10.3389/fphar.2020.604885
44.
Scala R Maqoud F Zizzo N Passantino G Mele A Camerino GM et al . Consequences of sur2(a478v) mutation in skeletal muscle of murine model of cantu syndrome. Cells. (2021) 10:1791. 10.3390/cells10071791
45.
Bartoszewski R Matalon S Collawn JF . Ion channels of the lung and their role in disease pathogenesis. REVIEW ion channels and transporters in lung function and disease. Am J Physiol Lung Cell Mol Physiol. (2017) 313:859–72. 10.1152/ajplung.00285.2017
46.
Farhadi A Keshavarzian A Ranjbaran Z Fields JZ Banan A . The role of protein kinase C isoforms in modulating injury and repair of the intestinal barrier. J Pharmacol Exp Ther. (2006) 316:1–7. 10.1124/jpet.105.085449
47.
Rao R . Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci. (2008) 13:7210–26. 10.2741/3223
48.
Rao RK Basuroy S Rao VU Karnaky KJJr Gupta A . Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J. (2002) 368:471–81. 10.1042/bj20011804
49.
Lin PY Stern A Peng HH Chen JH Yang HC . Redox and metabolic regulation of intestinal barrier function and associated disorders. Int J Mol Sci. (2022) 23:14463. 10.3390/ijms232214463
Summary
Keywords
Cantú syndrome, murine model, ion channels, KATP channels, gain-of-function mutation, tight junctions, colonic epithelium, barrier dysfunction
Citation
Maqoud F, Mallardi D, Orlando A, Tricarico D, Nichols CG, Antonacci M, Bianco G, Armentano R, Grassi I, Valentini AM and Russo F (2025) Gain-of-function mutations in KATP channel subunits compromise colonic tight junction integrity and epithelial homeostasis in murine models of Cantú syndrome. Front. Med. 12:1656718. doi: 10.3389/fmed.2025.1656718
Received
30 June 2025
Accepted
26 August 2025
Published
11 September 2025
Volume
12 - 2025
Edited by
Mohamed Taha Moutaoufik, Mohammed VI Polytechnic University, Morocco
Reviewed by
Volodymyr Tsvilovskyy, Heidelberg University, Germany
Elsayed Metwally, Suez Canal University, Egypt
Updates
Copyright
© 2025 Maqoud, Mallardi, Orlando, Tricarico, Nichols, Antonacci, Bianco, Armentano, Grassi, Valentini and Russo.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Russo francesco.russo@irccsdebellis.it
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.