 Miao Zhang1,2
Miao Zhang1,2 Chendan Wang1,2,3*
Chendan Wang1,2,3*- 1The Fifth Clinical Medical College of Shanxi Medical University, Taiyuan, China
- 2Shanxi Provincial Key Laboratory of Kidney Disease, Taiyuan, China
- 3Medicinal Basic Research Innovation Center of Chronic Kidney Disease, Ministry of Education, Shanxi Medical University, Taiyuan, China
Fabry disease (FD), as an X-linked lysosomal storage disorder (LSD), has seen significantly improved in patient prognosis since enzyme replacement therapy (ERT) was applied clinically in 2001. However, ERT has drawbacks such as immunogenicity, individual efficacy variability, and long-term treatment burdens. With deeper insights into the multidimensional pathological mechanisms of FD and breakthroughs in new delivery systems (such as mRNA therapy, engineered adeno-associated virus vectors), various emerging therapies have gradually developed. Nevertheless, each therapeutic strategy still faces areas needing improvement due to high disease heterogeneity, cytotoxicity, insufficient targeted tissue delivery efficiency, and a lack of long-term safety data. This article systematically reviews the development of different treatment strategies for FD, outlines the evolution from ERT clinical application to emerging technologies like novel vector delivery, analyzes the technical breakthroughs and clinical limitations of therapies at each stage, reveals the intrinsic connection between deepened understanding of pathological mechanisms and innovative treatments, and explores potential optimization directions for future treatment strategies based on global accessibility data.
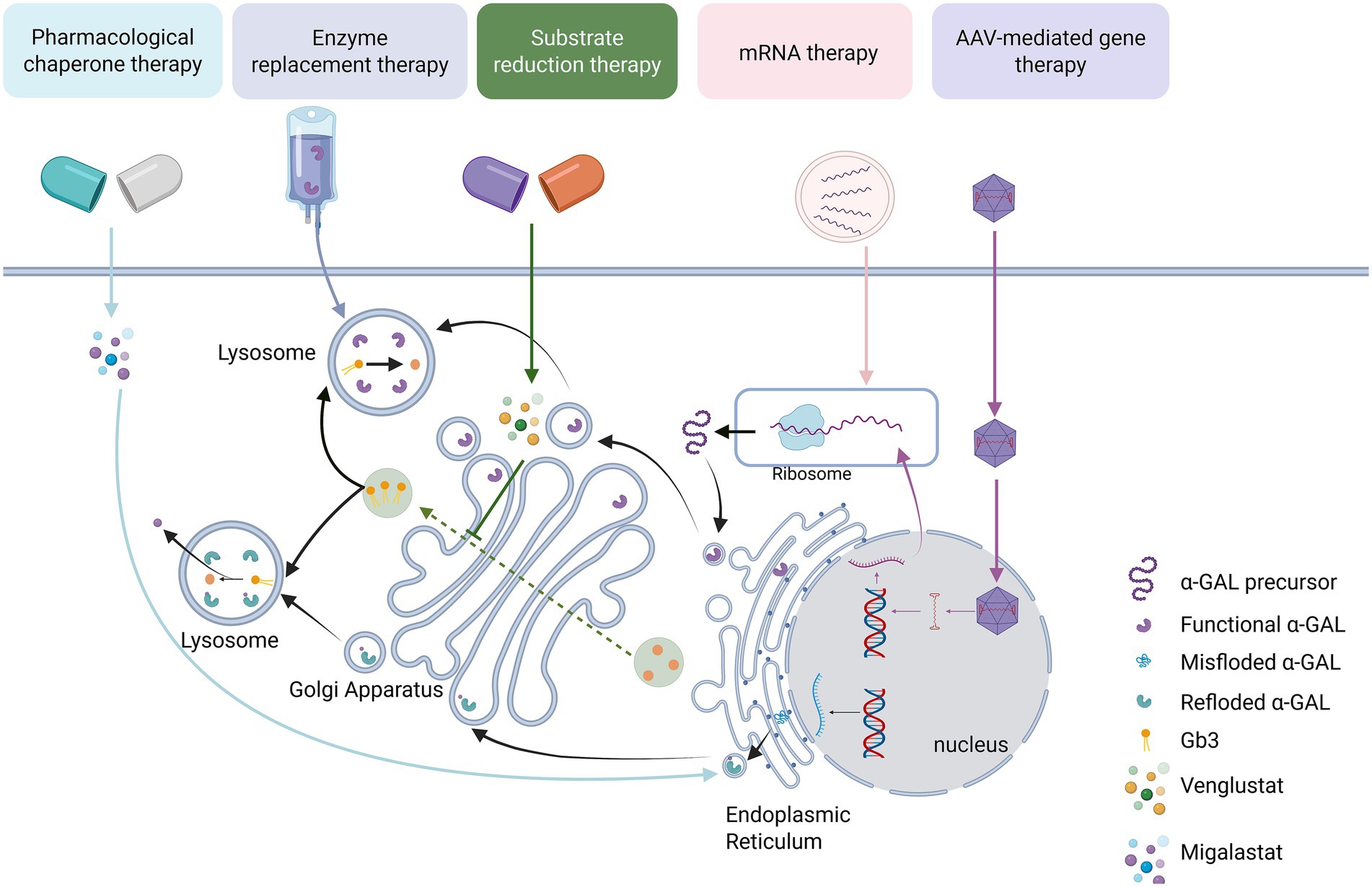
Graphical Abstract.
1 Introduction
Fabry disease (FD) is a rare X-linked lysosomal storage disorder (LSD) with an estimated birth prevalence of 1 in 6,264 based on a large-scale Chinese newborn-screening cohort (1). FD is caused by pathogenic variants in the GLA gene that reduce or absent the activity of the encoded lysosomal enzyme α-galactosidase A (α-Gal A). Consequently, the metabolic substrates globotriaosylceramide (Gb3) and its deacylated derivative globotriaosylsphingosine (lyso-Gb3) accumulate in multiple cell types—including endothelial cells, podocytes, smooth-muscle cells, cardiomyocytes, and neurons (2, 3). Progressive accumulation underlies the multi-systemic pathology of FD. GLA variants encompass missense, nonsense, frameshift, splice-site, and large copy-number alterations. Missense mutations are the most common, and over 1,000 different genotypes have been identified (4). FD can be classified into classic and non-classic/late-onset forms based on clinical presentation, with a clear distinction between the two types in males. The classic form is typically characterized by absent or significantly decreased α-Gal A enzyme activity, with symptoms usually beginning in childhood or adolescence and progressing to renal, cardiac, and central nervous system pathology in adulthood. In contrast, late-onset patients, retaining higher residual enzyme activity, experience milder phenotypes and a delayed disease course (Table 1). Plasma Lyso-Gb3 levels help to differentiate between the classic and late-onset forms of FD, with classic patients typically exhibiting significantly higher Lyso-Gb3 levels than late-onset patients (5). Due to the stochastic and dynamic nature of X-chromosome inactivation (XCI), and mechanisms such as skewed XCI, female patients exhibit marked heterogeneity in disease severity, ranging from asymptomatic to severe classic manifestations (6). Nevertheless, XCI pattern alone fails to predict individual phenotype. In a single-centre, cross-tissue study of 95 female GLA-variant carriers, skewed XCI (≥75:25) was detected in 35% of blood, 33% of skin fibroblast and only 7% of oral epithelial samples. Irrespective of tissue source or XCI pattern (skewed vs. random, mutant-allele-active vs. inactive), no significant association emerged between XCI skewing and clinical severity, organ involvement, leukocyte α-Gal A activity, or plasma Gb3 (p > 0.05). Thus, even in blood—the tissue most prone to skewing—XCI status does not stratify symptom severity (7).
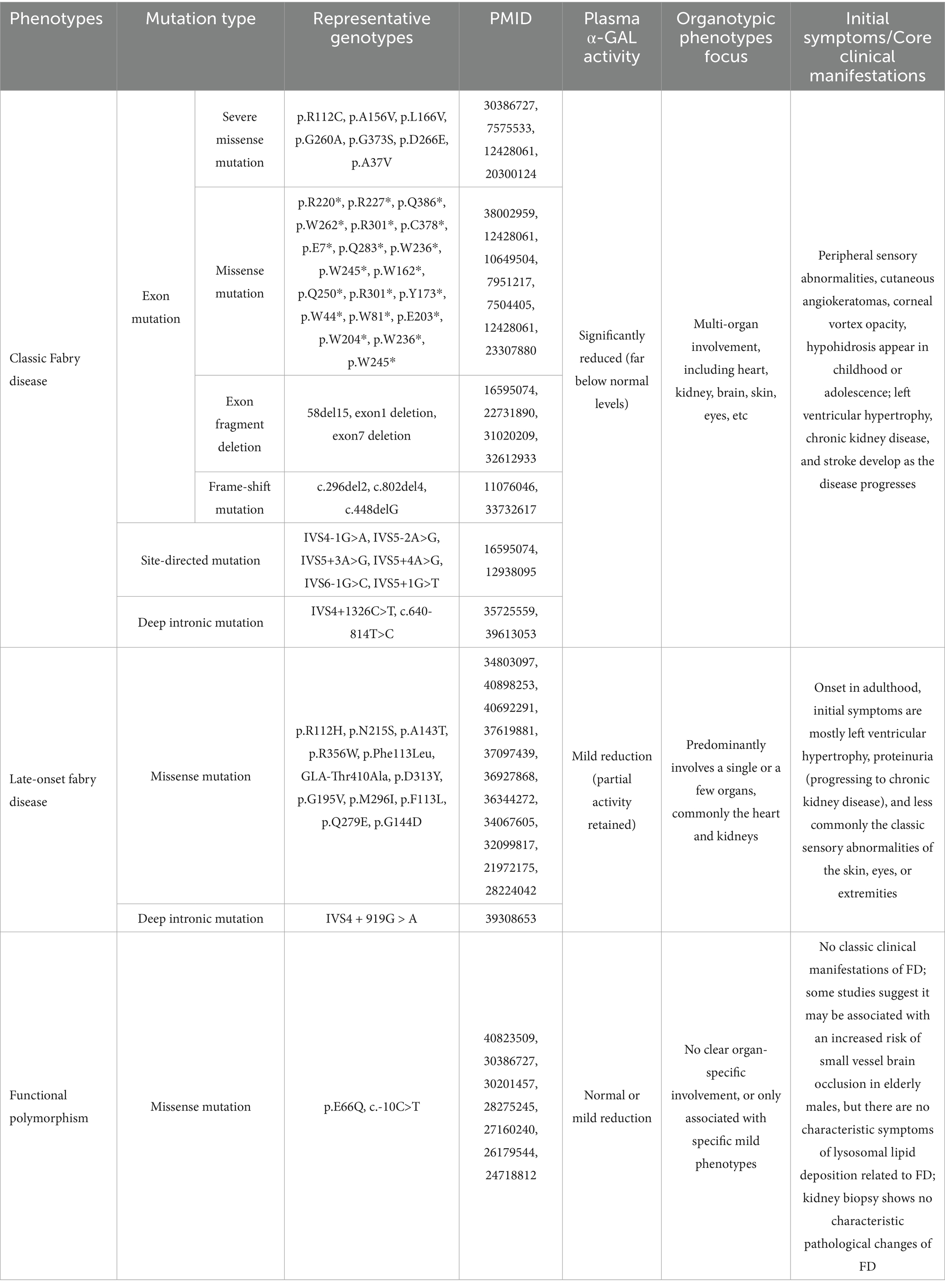
Table 1. Fabry disease genotype–phenotype spectrum and clinical characteristics overview.
The disease is characterized by insidious early symptoms and multisystem involvement, making early diagnosis challenging. The median delay from first symptom to confirmed diagnosis is 15 years (8). Renal biopsies from nine paediatric patients with only trace proteinuria already showed widespread Gb3 deposition, independent of overt nephropathy. Structural changes in the kidneys, including glomerulosclerosis and interstitial fibrosis, were observed in nearly 80% of patients. These findings suggest that severe pathological damage to the target organs may already be present early in the disease course (9). Patients who initiated enzyme replacement therapy (ERT) in childhood or early adulthood exhibited a slower progression of renal and cardiac disease compared to those who began ERT in late adulthood (10, 11). The introduction of dried-blood-spot (DBS) screening has now streamlined newborn and high-risk detection, enabling a genuine “early-diagnosis, early-treatment” paradigm.
Gb3 accumulates in glomerular endothelial, mesangial and podocyte compartments as well as peritubular capillaries (12). This accumulation can cause cellular dysfunction, which in turn leads to impaired renal filtration. As the disease progresses, the kidneys can develop fibrosis and scar formation, eventually leading to end-stage renal disease (13). Proteinuria is a common manifestation of renal involvement in FD, often presenting as microproteinuria in the early stages. The degree of proteinuria increases progressively with disease advancement. Massive proteinuria is not only a marker of renal injury but also exacerbates the renal burden, accelerating the deterioration of renal function (14, 15).
Gb3 and its metabolites accumulate extensively in cardiac tissues, interfering with normal cellular metabolic processes. This accumulation leads to dysfunction and injury of cardiomyocytes and cardiac vascular endothelial cells (16). Damage to the endothelial cells of cardiac blood vessels results in vessel wall thickening and luminal narrowing, thereby affecting myocardial blood supply (17). Left ventricular hypertrophy (LVH), characterized by increased left ventricular wall thickness, is a common manifestation of cardiac involvement in FD (18). This thickening can impair both diastolic and systolic cardiac function. As the disease progresses and the heart undergoes chronic damage, myocardial contractile and diastolic functions are progressively compromised, potentially leading to heart failure (19).
In the nervous system, patients with FD experience a variety of neurologic symptoms, including neuropathic pain, impaired sweating, and hearing deficit. Gb3 and its metabolites are primarily deposited in Schwann cells, dorsal root ganglia, and neurons within the central nervous system. This deposition interferes with the normal metabolic processes of nerve cells, leading to nerve fiber damage and disruption of nerve signaling (20). As the disease progresses, nerve cells may undergo pathological changes such as degeneration and apoptosis, leading to neurological dysfunction. Pain, a common symptom of neurological involvement, is typically manifested as episodes of severe pain, including tingling, burning, or electric shock-like sensations, often localized to the extremities, particularly the hands and feet (21). Additionally, the accumulation of Gb3 triggers inflammatory responses and oxidative stress, which further exacerbate target organ damage (22).
As research deepens and clinical experience accumulates, the clinical presentation spectrum of FD has been greatly expanded. As a disease affecting multiple systems, its manifestations are highly heterogeneous and camouflaged. More and more atypical or easily overlooked symptoms, such as decreased lung function (23), gastrointestinal manifestations (abdominal pain, diarrhea, vomiting) (24, 25), and azoospermia (26, 27), have been identified, sometimes even as the initial or only symptom, greatly challenging traditional diagnostic approaches. Since ERT was introduced into clinical treatment, emerging therapies that are continuously iterated are gradually reshaping its treatment landscape. From the initial ERT “alternative therapy” aimed at supplementing exogenous α-Gal A, to the “functional repair therapies” represented by small-molecule pharmacological chaperones and substrate reduction therapy, and then to the “root cause correction therapies” centered on gene therapy, mRNA delivery, and gene editing, the treatment philosophy has undergone three generational leaps. Registered clinical studies regarding the emerging therapies of FD are summarized in Table 2.
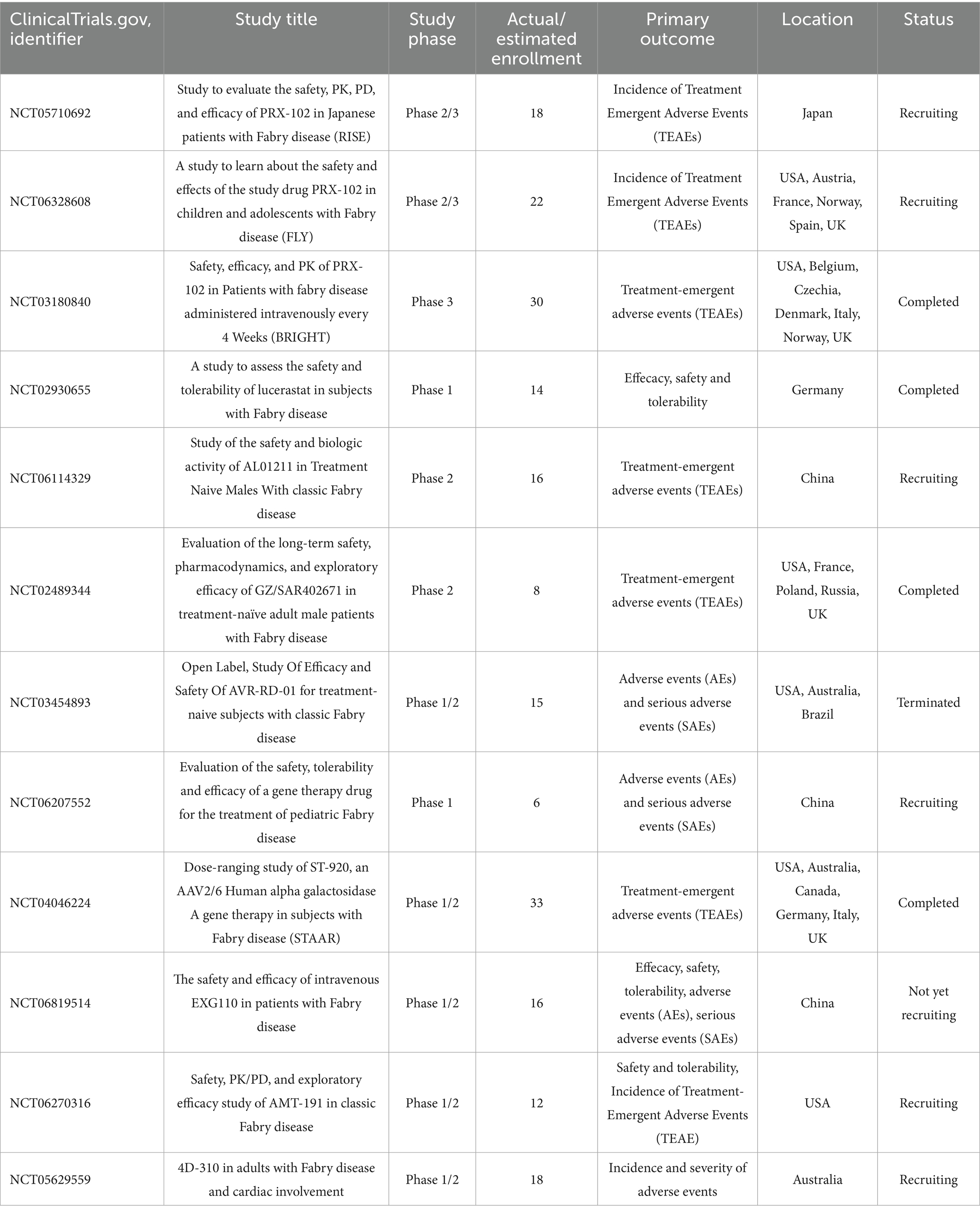
Table 2. Registered clinical studies regarding the emerging therapies of FD.
2 Enzyme replacement therapy
In 2001, ERT was introduced into clinical practice, based on the principle of promoting the degradation of Gb3 and Lyso-Gb3 through intravenous infusion of recombinant α-Gal A (Figure 1). Currently, three ERT drugs have been approved for clinical use worldwide: agalsidase alfa (Replagal®; Takeda), agalsidase beta (Fabrazyme®; Sanofi), and pegunigalsidase alfa (Elfabrio®; Chiesi) (28, 29).
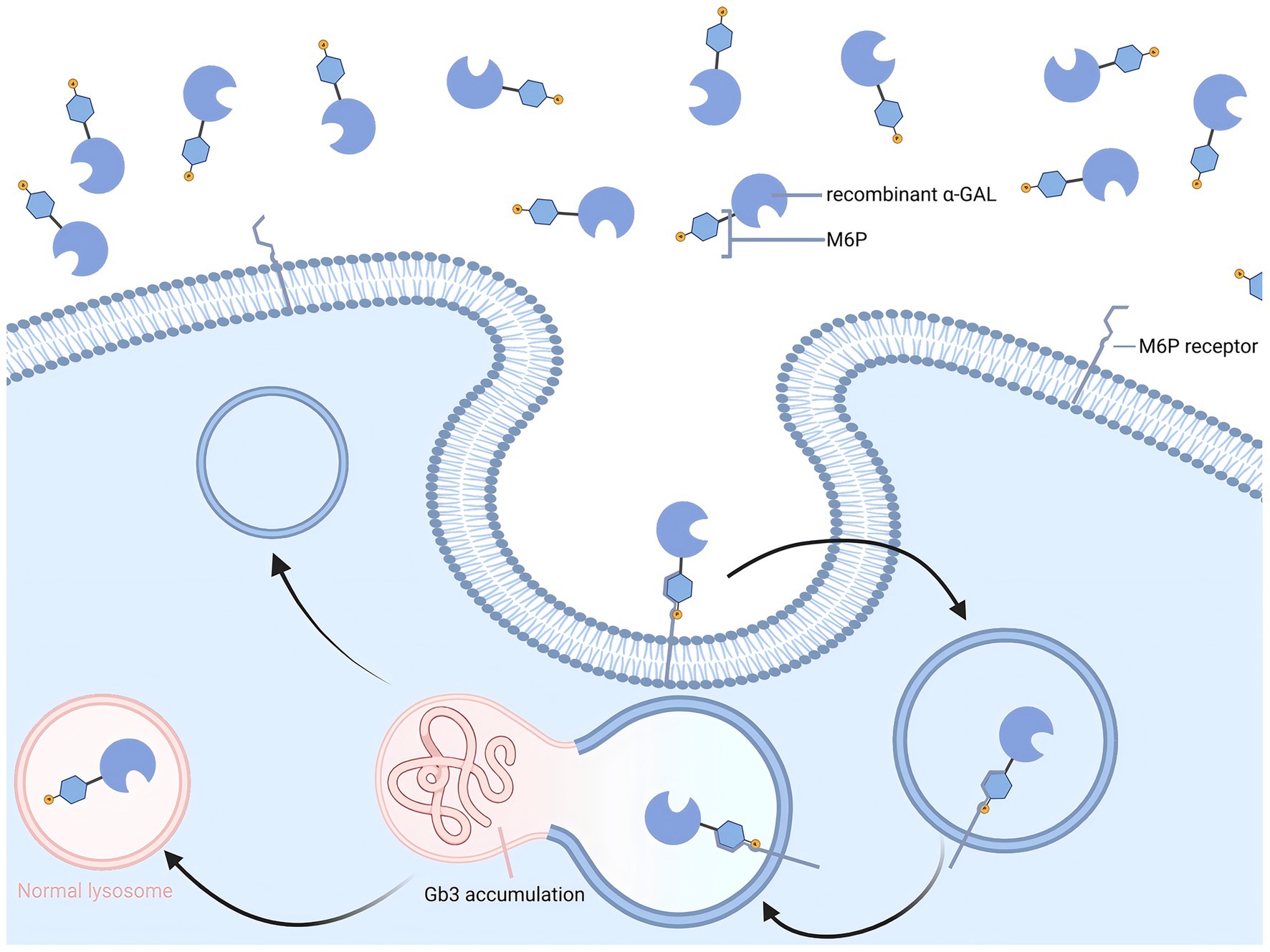
Figure 1. Enzyme replacement therapy for Fabry disease.
In a 6-month randomized, double-blind, placebo-controlled trial, recombinant α-Gal A markedly reduced renal Gb3 deposition: mesangial widening fell by 12.5 ± 5.0% in the ERT group but rose by 16.5 ± 7.7% with placebo, yielding a 29% absolute difference (95% CI 8–50%, p = 0.01). Concurrently, mean creatinine clearance increased by 2.1 mL min−1 1.73 m−2 with ERT and declined by 16.1 mL min−1 1.73 m−2 with placebo, giving a between-group difference of 18.2 mL min−1 1.73 m−2 (95% CI 2.6–33.8, p = 0.02) (30). In a 20-year observational study based on Fabry Outcome Survey (FOS), adult patients receiving ERT were indirectly compared with an untreated cohort. The results showed that the annual decline rate of eGFR in the treatment group was significantly lower than that in the untreated group; among the eGFR subgroups: for males with baseline eGFR ≥60 mL min−1 1.73 m−2, the annual decline rate in the treatment group was −1.68 ± 0.10 mL min−1 1.73 m−2 yr.−1, and in the untreated group was −3.0 ± 0.1 mL min−1 1.73 m−2 yr.−1 (p < 0.0001); for males with baseline eGFR <60 mL min−1 1.73 m−2, the annual decline rate in the treatment group was −1.60 ± 0.34 mL min−1 1.73 m−2 yr.−1, and in the untreated group was −6.8 ± 1.5 mL min−1 1.73 m−2 yr.−1; stratified by baseline proteinuria, regardless of the patient’s 24-h urine protein concentration being ≥1.0 g, 0.1- < 1.0 g, or <0.1 g, the annual decline rate of eGFR in the treatment group was significantly lower than that in the corresponding untreated subgroup. Therefore, ERT treatment can significantly slow down the progression of renal function deterioration in FD patients; even when further subdivided by treatment duration, the decline rate of eGFR in patients with more than 10 years of ERT still shows an advantage, and the overall decline rate in the treatment group has always been lower than that in the untreated cohort. Among adult treated patients, those with treatment duration less than 10 years had an annual decline rate of eGFR of −1.42 ± 0.06 mL min−1 1.73 m−2 yr.−1, and those with treatment duration over 10 years had an annual decline rate of −1.10 ± 0.08 mL min−1 1.73 m−2 yr.−1, both significantly lower than the corresponding levels in the untreated group (31). In a retrospective analysis of 21 years of clinical data for male patients receiving agalsidase alfa 0.2 mg/kg intravenous infusion every other week in the FOS cohort, those with baseline proteinuria ≤0.5 g/24 h (n = 114) had an average annual eGFR loss of −1.61 mL min−1 1.73 m−2; those with proteinuria >0.5 g/24 h (n = 51) had an average annual decline of −3.62 mL min−1 1.73 m−2, with a difference of 2.01 mL min−1 1.73 m−2 yr.−1 (95% CI 1.20–2.82, p < 0.0001), suggesting that early initiation of ERT in the early stage of renal damage can significantly slow down subsequent renal function decline (32).
A retrospective FOS analysis (NCT03289065) enrolled 560 males on agalsidase alfa who started ERT at ≤18 years (n = 151); baseline LVMI averaged 36.7 ± 7.9 g/m2 and only 6.8% exceeded the 50 g/m2 pathological threshold. Median follow-up was 6.3 ± 4.3 years. Long-term follow-up results showed that the annual change slope of LVMI in Cohort 1 was −0.17 g/m2 (95% CI: −0.66 ~ 0.31), although the statistical test p-value was 0.490, the numerical trend and clinical significance indicated that LVMI not only did not increase but showed a continuous slight decrease, and the mean LVMI remained stable at 33.6 g/m2 (95% CI: 29.5 ~ 37.7) during the follow-up period, significantly lower than the baseline level (33). A decade-long FOS extension showed that baseline-non-LVH patients (females ≤48 g/m2, males ≤50 g/m2) exhibited minimal LVMI increases: +0.52 g/m2 yr.−1 (95% CI: −0.13 ~ +1.17) and +0.57 g/m2 yr.−1 (95% CI: +0.02 ~ +1.13) for females and males, respectively, indicating absence of significant progression. At the same time, the annual change slope of posterior wall thickness at end-diastole (PWTd) in different baseline renal function subgroups was controlled at +0.01 ~ +0.11 mm yr.−1, remaining within the normal range, further confirming the long-term stable effect of ERT on cardiac structure (34). Nevertheless, sudden cardiac death and other major adverse cardiac events (MACE) continue to occur on ERT (35), indicating that decelerated progression does not equate to full arrhythmic or microvascular risk elimination.
ERT can effectively alleviate pain symptoms in FD patients and improve their autonomic function to some extent. Systematic reviews of relevant studies have demonstrated significant improvement in neuropathic pain and benefit for stroke prevention among patients treated with ERT (28, 36, 37). Several clinical trials have well documented significant reductions in pain scores, as well as in the frequency and severity of pain episodes, in patients treated with agalsidase alfa. ERT treatment increases the threshold for detecting hot and cold sensations in the hands and feet and improves sweating and heat tolerance (38, 39). Despite these positive results, ERT does not fully normalize the function of the peripheral nervous system (20).
The majority of patients currently follow a conventional dosing regimen. However, available studies continue to yield divergent conclusions regarding the correlation between dose and efficacy. A 53-week, III/IV phase, multicenter study (NCT01124643) indicates that in initially treated FD adults with left ventricular hypertrophy (LVMI >50 g/m2 for males, >47 g/m2 for females) at baseline, increasing the dosing frequency of agalsidase alfa from the approved 0.2 mg/kg every two weeks (EOW) to 0.2 mg/kg weekly, with LVMI as the primary endpoint—monitored at 53 weeks, EOW group +3.2 g/m2, weekly group +0.5 g/m2; LSM difference −2.20 g/m2 (95.005% CI –12.26 ~ 7.85, p = 0.6585), there was no statistically significant difference in changes between the two groups (40). However, for Fabry patients who have been regularly receiving 0.2 mg/kg agalsidase alfa infusion every two weeks and whose renal function continues to decline, switching to the same dose administered weekly significantly slowed the average decline rate of eGFR within 24 months; multivariate analysis shows that the weekly dosing frequency is the strongest independent predictor of improved eGFR slope (β = 0.6486, p = 0.0008) (41). A 14-year follow-up study of 20 classic FD patients found that the dose of ERT had a significant impact on efficacy. Although there was no significant difference in baseline renal function and proteinuria levels between the two groups before treatment, patients receiving higher doses showed more significant clearance of podocyte Gb3 deposits (average decrease of 23.16 points, p = 0.002), which was significantly higher than the low-dose group. In addition, the reduction of podocyte Gb3 was positively correlated with cumulative enzyme dose (r = 0.69, p = 0.001), and the high-dose group also showed a more optimal effect in the clearance of Gb3 in the intima of arteries/arterioles (p = 0.03). The residual plasma lysoGb3 levels were significantly lower in the high-dose group (10.4 vs. 20.1 nmol/L, p = 0.04) and were significantly associated with the cumulative dose in male patients (r = 0.71, p = 0.01) (42). Therefore, higher doses of agalsidase treatment can more effectively clear kidney Gb3 deposits, delay tissue damage, and support a clear dose-effect relationship between ERT dosing and the efficacy of FD. However, another 3-year follow-up observation systematically evaluated the efficacy changes after switching from agalsidase beta (1.0 mg/kg) to agalsidase alfa (0.2 mg/kg). The results showed that after the switch, patients’ cardiac structural indicators continued to significantly improve: the mean LVMI decreased from 58.1 g/m2 to 50.7 g/m2 (p = 0.045), while renal function remained stable, and plasma Gb3 and lyso-Gb3 levels did not significantly increase, and pain scores and quality of life also did not show significant changes. The above data indicate that even with a significant reduction in dose, efficacy was not weakened, and no evidence of additional efficacy improvement with increased dose was observed (43). The differences in the above conclusions mainly arise from the different observation time windows of the endpoint. Given that irreversible complications such as end-stage renal failure, stroke, and heart failure are concentrated at the end stage of the disease, existing switch or down-titration studies were mostly triggered by agalsidase-beta shortages and are limited to short-term surrogate endpoints. Consequently, “non-inferior” surrogate stability after dose reduction cannot yet be extrapolated to long-term hard-end-point risk. Therefore, current data are insufficient to inform long-term dosing strategies, and prospective, comparative trials with extended follow-up are urgently warranted.
Agalsidase alfa and agalsidase beta, the primary ERT agents for FD, exhibit differences in clinical efficacy and safety despite both being derived from recombinant α-Gal A. Sirrs et al. followed patients treated with agalsidase alfa and agalsidase beta for 10 years. The results showed that in male patients, the incidence of renal events (including renal replacement therapy, doubling of serum creatinine, and proteinuria >3.5 g/day) was lower in the agalsidase beta group than in the agalsidase alfa group. Additionally, the rate of decline in eGFR was slower in male patients treated with agalsidase beta (44). Krämer et al. conducted a study involving 112 patients with FD who received agalsidase beta for more than 1 year. In a subgroup analysis, patients were divided into two groups: the conversion group, which switched from agalsidase beta to agalsidase alfa, and the reconversion group, which switched back from agalsidase alfa to agalsidase beta. The results showed that the conversion group’s eGFR decreased by 4.6 ± 9.1 mL min−1 1.73 m−2. In contrast, the reconversion group exhibited a significantly lesser decrease in eGFR after switching back to agalsidase beta. These findings suggest that agalsidase beta may be more advantageous than agalsidase alfa in maintaining stable renal function (45). In terms of safety, multiple studies suggest that the immunogenicity of α-galactosidase alfa is lower than that of α-galactosidase beta. Vedder et al. provided quantitative data from a prospective cohort study of 52 patients with FD: after 6 months of treatment, the positive rate of anti-α-Gal A antibodies in male patients increased with dose, with the α-galactosidase alfa 0.2 mg/kg group at 43% (4/10), the α-galactosidase beta 0.2 mg/kg group reaching 83% (6/8), and the α-galactosidase beta 1.0 mg/kg group even higher at 89% (8/10); overall comparisons showed that the antibody incidence in the beta 1.0 mg/kg group was significantly higher than that in the alfa 0.2 mg/kg group (p = 0.005). Further functional experiments showed that 83% of male patients’ serum could neutralize standard enzyme activity, and the antibody titer was highly correlated with ELISA results (r = 0.80, p < 0.001). Notably, no antibodies were detected in all female patients (n = 24), suggesting that gender is also an important covariate for immune response (46).
Pegunigalsidase alfa achieves molecular structure upgrade through dual modification of “chemical cross-linking-PEGylation”: Covalently bridging two molecules of α-Gal A with a 2000 Da branched PEG forms a homodimeric network, which has an 11 °C increase in thermal stability compared to the unmodified enzyme; in human plasma, the activity of the unmodified enzyme is almost completely lost after 30 min, while Pegunigalsidase alfa retains 30–40% activity after 30 min; under lysosome-like conditions, the activity of the unmodified enzyme remains only 10% after 10 days, while the modified Pegunigalsidase alfa maintains 80% activity, meeting the demand for administration every 2 weeks or even monthly; at the same time, the antibody binding curve slope of Pegunigalsidase alfa is smoother, proving that the PEG chain effectively shields the immunogenic epitopes, reduces antibody recognition, and lowers the risk of neutralizing antibody production. (47–49).
III phase cluster data (BALANCE, BRIGHT, BRIDGE, and 6-year extension) show that the 2-year eGFR slope is not worse than high-dose agalsidase beta (49); following switch from agalsidase alfa 0.2 mg/kg, annual eGFR slope improved from −5.9 to −1.19 mL min−1 1.73 m−2 yr.−1 (50); plasma lyso-Gb3 maintained low levels, and renal capillary Gb3 inclusion bodies decreased by 84% (48); no new fibrosis occurred in the left ventricular mass index at 60 months (51). Across BALANCE, BRIDGE and real-world cohorts, exposure-adjusted infusion-reaction rates were 3.6- to 7.8-fold lower than with agalsidase beta; no de-novo neutralizing antibodies; 75% of the titers decreased in previous ADA-positive individuals after crossover; 97% of adverse events were mild to moderate, severe events <3%, and there were no deaths or III-IV grade hypersensitivity. In summary, pegunigalsidase alfa, with a chemical cross-linking-PEGylation strategy, simultaneously improves stability, extends half-life, and reduces immunogenicity (49–51). On the basis of efficacy non-inferior to high-dose beta preparations, it significantly improves safety and ease of administration, providing a better choice for ERT in FD.
3 Pharmacological chaperone therapy
Approximately 35 to 50% of FD patients exhibit structural instability of the enzyme but retain some catalytic activity due to specific missense mutations in the GLA gene, such as p. N215S and p. F113L. Migalastat is a small molecule drug that is structurally similar to the substrate of α-Gal A, the Gb3-terminal galactose. Consequently, it can specifically and reversibly bind to the active site of α-Gal A mutants generated by GLA gene mutations, effectively providing an “auxiliary scaffold” for the mutated enzyme (Figure 2). This binding stabilizes the three-dimensional structure of the enzyme, preventing its recognition and degradation by the endoplasmic reticulum’s quality control system, thereby enhancing the stability of the mutant enzyme (52, 53). In vitro assays demonstrated that mutants were classified as therapeutic if they exhibited an absolute increase (≥ 3% of wild-type enzyme activity) and a relative increase (≥ 1.2-fold) in α-GAL activity after incubation with migalastat.
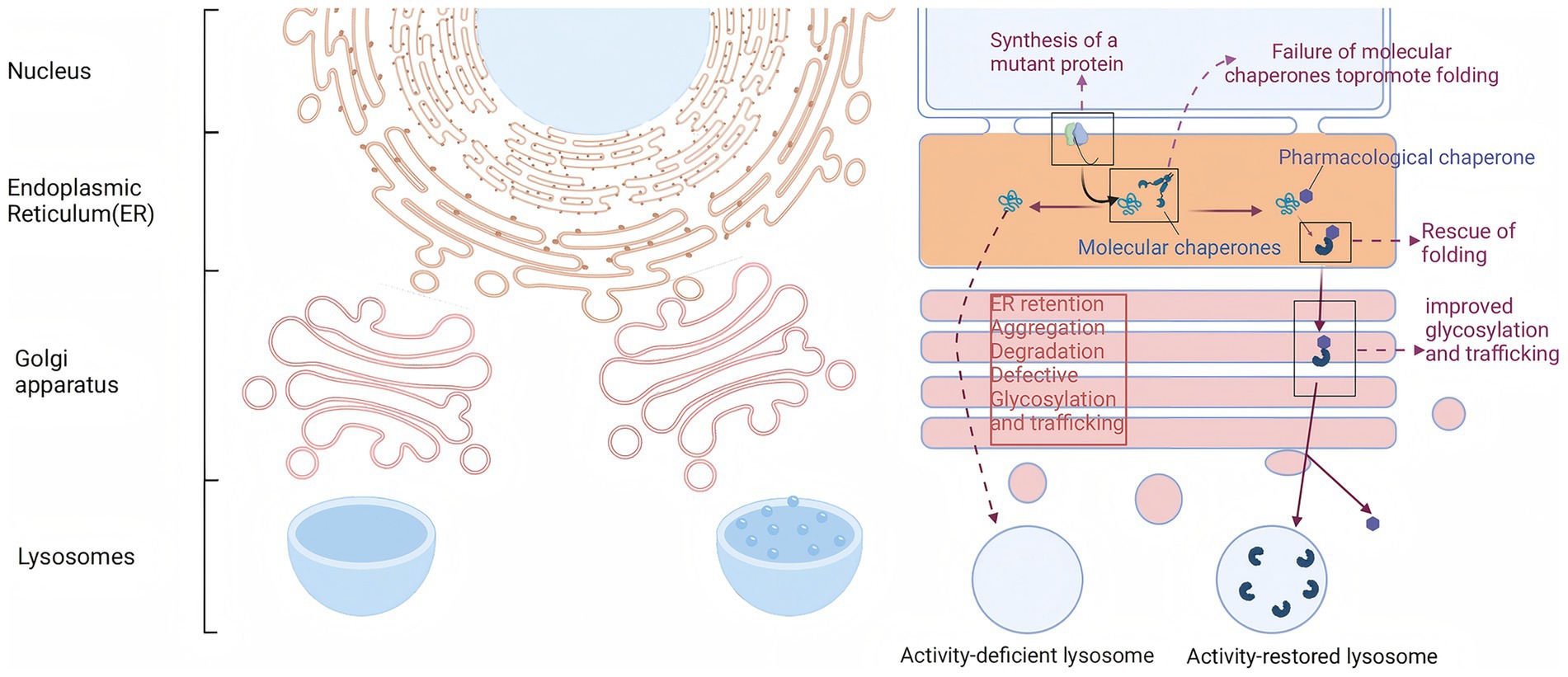
Figure 2. Migalastat stabilizes mutated α-Gal A enzymes to enhance enzymatic activity.
Long-term follow-up data show that for both naive and treated ERT patients, the oral migalastat 123 mg every other day regimen can significantly slow the decline in renal function. Summary of the FACETS, ATTRACT, and its open-label extension studies (median follow-up 5.9 years, longest 8.6 years, n = 78): after adjusting for gender, age, and baseline renal function with a random coefficients model, the mean annualized eGFR change in the naive ERT group was only −0.1 mL/min/1.73 m2, and in the treated ERT group was +0.1 mL/min/1.73 m2, both superior to the historical untreated cohort. Subgroup analysis showed that the mean annualized decline in classic male naive patients was −0.5 mL/min/1.73 m2, significantly lower than the −2.2 mL/min/1.73 m2 decline in the agalsidase beta 54-month cohort; those with baseline eGFR >90 mL/min had a decline of −0.2 mL/min/1.73 m2, and those with proteinuria <300 mg/24 h had a decline of −0.1 mL/min/1.73 m2 (54). Another paired renal biopsy study performed quantitative electron microscopy morphometry on 8 adult classic male patients carrying a potentially inducible GLA mutation, showing that after 6 months of oral migalastat 150 mg every other day treatment, the mean total Gb3 inclusion body volume in podocytes decreased from 4,200 ± 1,600 μm3 to 2,500 ± 1,100 μm3, with an average reduction of 41% (p = 0.02); at the same time, the mean podocyte volume decreased synchronously by 37% (p = 0.004), and the reduction in podocyte width was highly correlated with the decrease in Gb3 volume (r = 0.82, p = 0.02). Plasma lyso-Gb3 levels were also significantly associated with changes in podocyte Gb3 content (r = 0.79, p = 0.02) (55).
In a 18-month randomized, open-label, active-controlled phase III clinical trial (ATTRACT study, NCT00925301), a total of 57 adult patients with FD who had previously received ERT for ≥12 months were enrolled and randomly assigned in a 1.5:1 ratio to the oral drug migalastat group (n = 36, 150 mg once every other day) or to continue the original ERT regimen group (n = 21, agalsidase alfa 0.2 mg/kg or agalsidase beta 1.0 mg/kg, once every two weeks). After 18 months of treatment, the LVMI in the migalastat group significantly decreased from baseline by −6.6 g/m2 (95% CI −11.0 to −2.2, p < 0.01), while the ERT group showed only a non-significant change of −2.0 g/m2 (95% CI −11.0 to 7.0). Subgroup analysis showed that the migalastat recipients with pre-existing left ventricular hypertrophy (n = 13) had a more significant decrease, reaching −8.4 g/m2. This result was moderately correlated with the change in interventricular septum thickness (IVSWT) (r2 = 0.26, p = 0.0028) (56). In an 8.6-year observational study of migalastat treatment, it was found that although some patients still experienced cardiac-related events, overall cardiac function remained relatively stable (57). These findings suggest that migalastat has a protective and stabilizing effect on cardiac function by maintaining enzyme activity and reducing substrate buildup in the heart, potentially reducing the risk of heart disease progression.
Migalastat, the first oral agent for the treatment of FD, is now widely used in clinical practice. Compared to ERT, which requires frequent intravenous infusions, migalastat greatly improves patient convenience and compliance. However, the mechanism of action of migalastat limits its effectiveness to only some missense mutations (58). Recent studies have found that the in vitro treatability of mutations does not necessarily correspond to the in vivo treatability of migalastat-treated patients. This suggests that mutations shown to respond favorably to migalastat in in vitro assays may not have the desired effect in actual patient treatment (59). For example, in the national FD cohort in Switzerland, some patients carrying “combinable mutations” still had less effective treatment than ERT after receiving migalastat. A classic male patient (mutation p. S276N) switched from ERT to migalastat, and although the peripheral blood leukocyte α-Gal A activity increased from 3.0 to 9.9%, the plasma lyso-Gb3 level continued to rise, indicating insufficient substrate clearance, further deterioration of renal function, and ultimately, ERT was restarted due to “ineffective treatment” (58). Therefore, it is important to monitor patients’ clinical symptoms and laboratory markers during treatment to comprehensively assess the actual therapeutic efficacy of migalastat.
Migalastat is overall safe and well-tolerated in patients with FD. In the randomized controlled Phase III trial and its open-label extension study, patients treated with migalastat for up to 8.6 years did not show any new safety signals, serious adverse events were rare, and no patient discontinued due to drug-related toxicity (57). The Phase IIIb ASPIRE study in adolescents (n = 21, median age 14.7 years) also showed that migalastat’s tolerability was consistent with adults, and no unexpected adverse reactions specific to children were found (60). Additionally, the German multicenter MALTA-FABRY prospective cohort (n = 40, followed up for 24 months) reported that 92.5% of patients maintained high adherence, with only one case rated as low adherence due to gastrointestinal discomfort, and significant improvements in pain and physical function limitations (61). Notably, renal clearance removes approximately 74% of the dose during dialysis; pharmacokinetic study indicates that 123 mg every other day maintains therapeutic exposure in ESRD without adjustment or accumulation (62).
4 Substrate reduction therapy
Venglustat is an oral glucose ceramide synthase (GCS) inhibitor that acts by inhibiting the enzymatic conversion of ceramides to glucosylceramides, thereby reducing the available substrates for the synthesis of more complex glycosphingolipids (Figure 3).
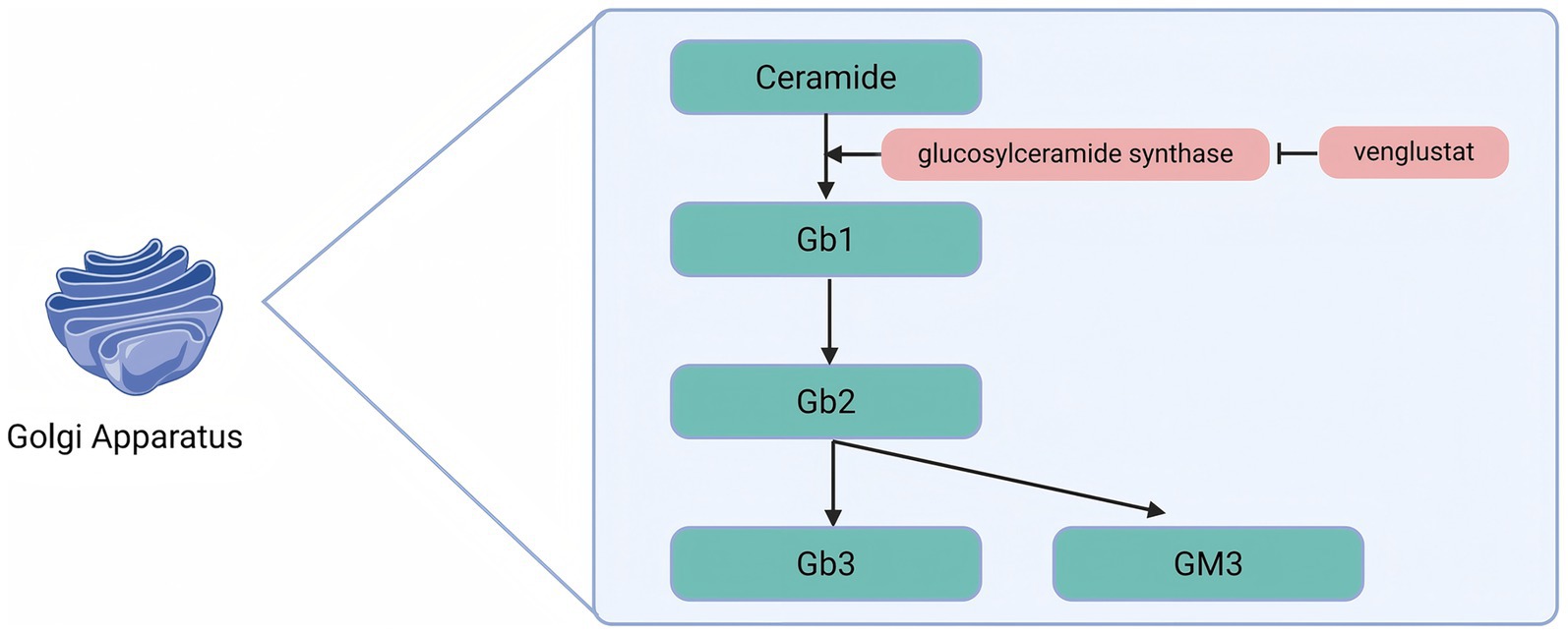
Figure 3. Venglustat inhibits glucose ceramide synthase to reduce Gb3 production.
Patients with FD suffer from α-Gal A deficiency, which leads to the progressive accumulation of Gb3 in the lysosomes of various cell types. By inhibiting GCS, Venglustat reduces the production of Gb1, which in turn decreases the synthesis of complex glycosphingolipids such as Gb3. This mechanism alleviates substrate accumulation at its source and mitigates the pathological process of FD. An international, open-label, single-arm, Phase 2a uncontrolled 26-week clinical trial (NCT02228460) and its 130-week extension study (NCT02489344) evaluated the safety, pharmacodynamics, pharmacokinetics, and exploratory efficacy of Venglustat in untreated adult male patients with classic FD. Patients received 15 mg of Venglustat orally once daily. Of the 11 patients enrolled, 9 completed the 26-week study, and 7 completed the extension study. At week 26, a decrease in Gb3 scores of the superficial skin capillary endothelium (SSCE) was observed in some patients. By the end of the extension study, Gb3 scores had decreased in 5 of 6 patients. The Gb3 inclusion body volume fraction in the cytoplasm of the SSCE, as measured by electron microscopy, was significantly lower at both week 26 and week 156 compared with baseline. In addition, Venglustat reduced the levels of markers associated with the major glycosphingolipid synthesis and degradation pathways, with a rapid decrease in proximal markers and a gradual decrease in distal markers. After 3 years of follow-up, no progression of FD was detected, confirming the target engagement and pharmacodynamic effects of Venglustat in this patient population (63).
As an oral drug, Venglustat offers the advantage of easy administration compared to traditional ERT, which can improve patient compliance. Due to its distinct pharmacological properties, Venglustat is able to cross the blood–brain barrier and reduce the levels of glycosphingolipids accumulated in the brain, an advantage that α-Gal A lacks (64). Therefore, the combination of Venglustat and ERT may provide complementary and additive therapeutic benefits.
5 mRNA therapy
The application of mRNA therapy in FD treatment is primarily reflected in its ability to effectively increase α-Gal A levels and improve related symptoms (65, 66) (Figure 4). A preclinical study showed that intravenous injection of mRNA encoding human α-Gal A into wild-type mice, FD mouse models, and wild-type non-human primates resulted in significantly higher α-Gal A activity in the liver and heart of the 0.5 mg/kg dose group compared to wild-type mice after 72 h, with kidney activity reaching 50% of wild-type levels, and plasma activity being more than 500 times higher than wild-type after 6 h. It also reduced lyso-Gb3 levels in the liver and spleen by 80%, and in the kidneys and heart by more than 50%. After a single dose, the reduction effect on tissue substrates lasted for at least 6 weeks, and in the plasma for about 4 weeks. These results suggest that mRNA therapy is capable of sustained in vivo production of α-Gal A and effective reduction of substrate levels, indicating its potential applicability to the treatment of FD (67).
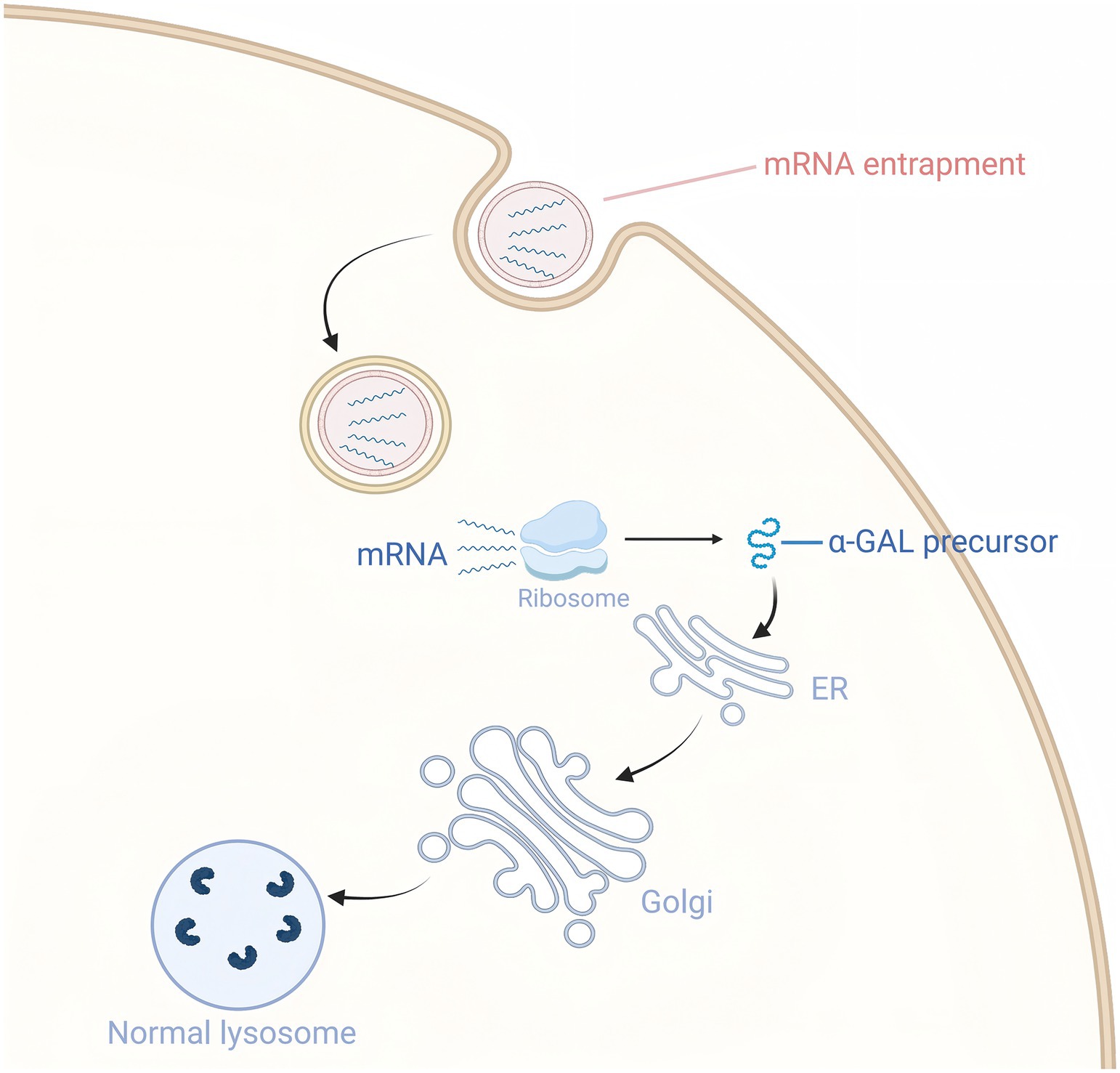
Figure 4. Exogenous mRNA delivery enhances α-GalA expression.
Based on evidence from multiple preclinical studies, repeated administration of LNP-encapsulated α-Gal A mRNA achieves sustained pharmacodynamic benefits and overall safety for Fabry disease. The Moderna team systematically evaluated the multi-dose regimen in α-Gal A knockout mice: the experimental group received intravenous injections of 0.2 mg/kg or 0.5 mg/kg α-Gal A mRNA every two weeks (EOW), or 0.5 mg/kg monthly, for 3 months; the control group was given an equivalent amount of eGFP mRNA. Organs were collected 7 days after the last dose, and liver IHC showed a dose-dependent increase in α-Gal A protein (0.5 mg/kg EOW > 0.5 mg/kg monthly > 0.2 mg/kg EOW), consistent with cardiac, renal, and liver enzyme activity. The 0.5 mg/kg EOW group showed the best efficacy, with liver and heart α-Gal A activity at 8 and 4 times that of the wild type, respectively, and the kidney maintained 25%. After 3 or 5 repeat doses, there was no increase in anti-α-Gal A antibodies, and enzyme activity accumulation was significantly better than with a single dose, with further accumulation of α-Gal A activity in the multi-dose group indicating no neutralizing anti-α-Gal A antibodies were produced (67). In vitro experiments showed that HEK293 cells transfected with α-Gal A mRNA had significantly higher mRNA and protein expression levels of α-Gal A and significantly increased enzyme activity in the cell lysate compared to the negative control; in vivo experiments, after mRNA-LNP injection in Fabry disease mice, α-Gal A activity in plasma significantly increased within 6 h and maintained a high level from 6 to 24 h, with significantly higher enzyme activity in the heart, liver, and kidneys than the control group. Immunohistochemistry showed that at 24 h, α-Gal A protein expression was enhanced in these tissues, and the effect was more significant in the high-dose group. At the same time, the levels of NF-κB, IL-6, and TNF-α in the liver of high-dose mRNA group mice were significantly reduced, and metabolomics identified 20 significantly different metabolites (VIP > 1, p < 0.05), with increased arachidonic acid, 5-HETE, and phosphatidylinositol 16/17/18, and decreased adenosine 5′-diphosphate, tryptophan, and glycolysis intermediates, mainly enriched in arachidonic acid, amino acids, and glycolysis/gluconeogenesis pathways; PCA/OPLS-DA showed dose-dependent metabolic profile separation, with significant changes in the high-dose group, suggesting that α-Gal A mRNA corrects the enzyme defect while reshaping the global metabolic homeostasis (66).
In terms of application, mRNA therapy can theoretically be used in various types of FD patients, particularly those with poor response to conventional ERT or the presence of anti-drug antibodies. Although mRNA therapeutics show promise in FD treatment, several practical challenges remain. These include optimizing the delivery system of mRNA, improving its targeting and stability, and determining the optimal dosage and frequency of administration.
6 AAV-mediated gene therapy
Viruses possess the natural ability to invade cells and can break through cellular barriers to deliver their genetic material into the cell interior, achieving stable gene expression in host cells. Among various viral vectors, the adeno-associated virus (AAV) vector is one of the preferred vectors in gene therapy due to its unique advantages (68). AAV is characterized by its small size, stable gene expression, low immunogenicity, and adjustable targeting, making it widely applicable in gene therapy. Wild-type AAV consists of single-stranded DNA that carries the Rep and Cap genes. The Rep gene encodes four proteins that control viral replication, packaging, and genome integration, while the Cap gene encodes the three major subunits that constitute the viral capsid: VP1, VP2, and VP3 (69). At the ends of the AAV genome, inverted terminal repeat sequences (ITRs) are present. These sequences are essential for the replication and packaging of the viral DNA (70).
Recombinant AAV (rAAV) is an AAV vector modified through genetic engineering technology. During the construction of rAAV, all gene sequences encoding the Rep and Cap proteins in the AAV genome are removed, retaining only the ITRs at both ends of the genome. These ITRs act as packaging signals to ensure that exogenous genes are correctly packaged into the viral particle (68, 71). With these modifications, rAAV vectors no longer contain the genes required for viral replication, thereby reducing the likelihood of replication in host cells and enhancing their safety. Upon entering the body, the rAAV vector is capable of specifically infecting target cells and delivering the therapeutic genes it carries into the cell interior (72). Within the cell, these therapeutic genes are expressed, and the corresponding proteins are produced, thereby compensating for proteins that are missing or functioning abnormally in the patient’s body due to genetic defects and exerting a therapeutic effect (Figure 5).
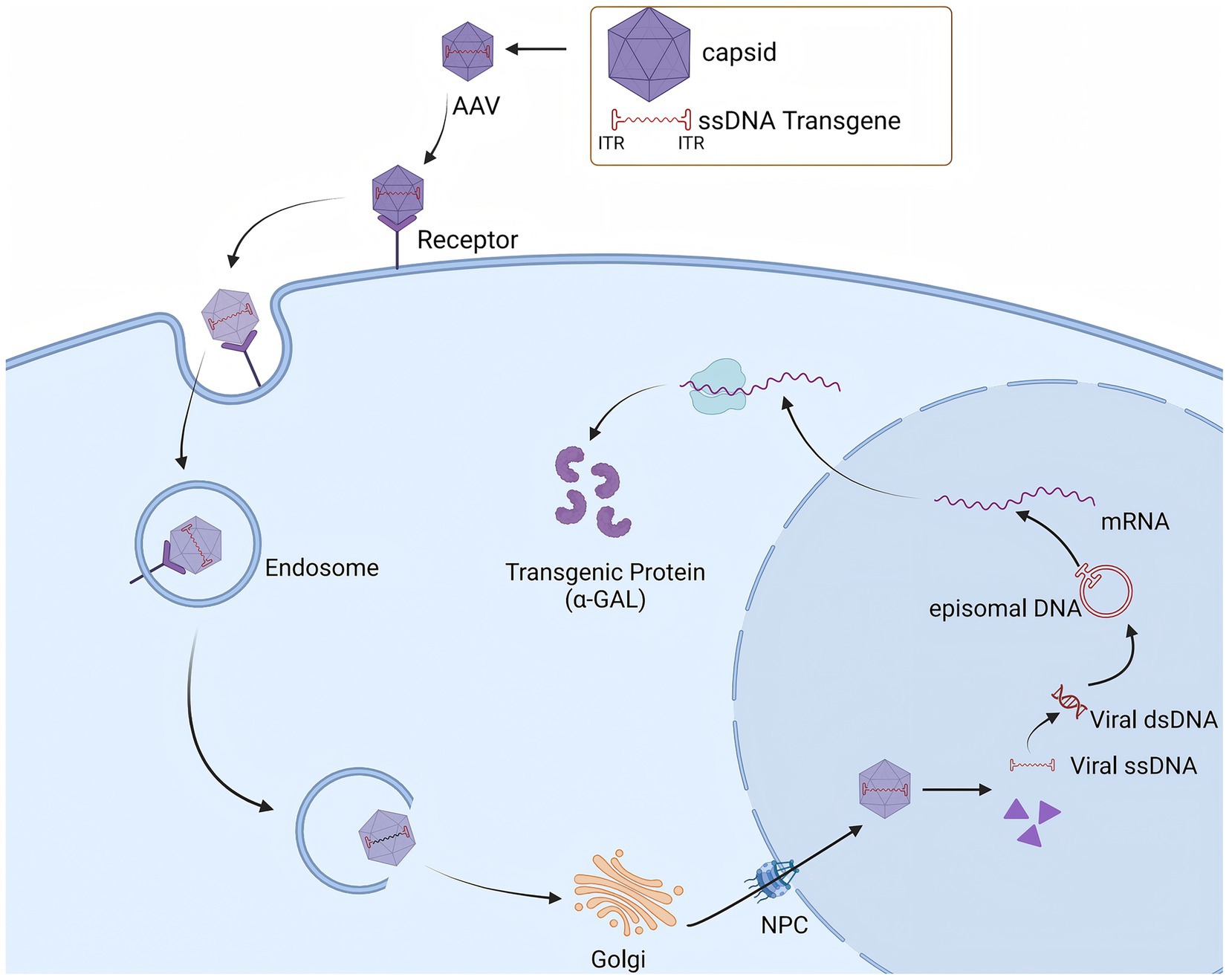
Figure 5. AAV vectors mediate specific target cell infection to facilitate GLA Gene delivery and α-GAL Expression.
AAV has multiple serotypes, and the capsid of each serotype determines its affinity for different tissues and cells (Figure 6). These natural differences in tropism provide the basis for gene therapy targeting specific tissues or organs (72–74). Capsid engineering optimizes the properties of rAAV by modifying the rAAV capsid protein, offering a more effective tool for gene therapy and other biomedical applications. The modification strategies mainly include adjusting the tissue tropism and reducing immunogenicity, thereby enabling precise delivery of therapeutic genes to target cells. This approach significantly improves the utilization of therapeutic genes and therapeutic efficacy while reducing infection of non-target cells and tissues, and minimizing side effects caused by off-target effects (75, 76).
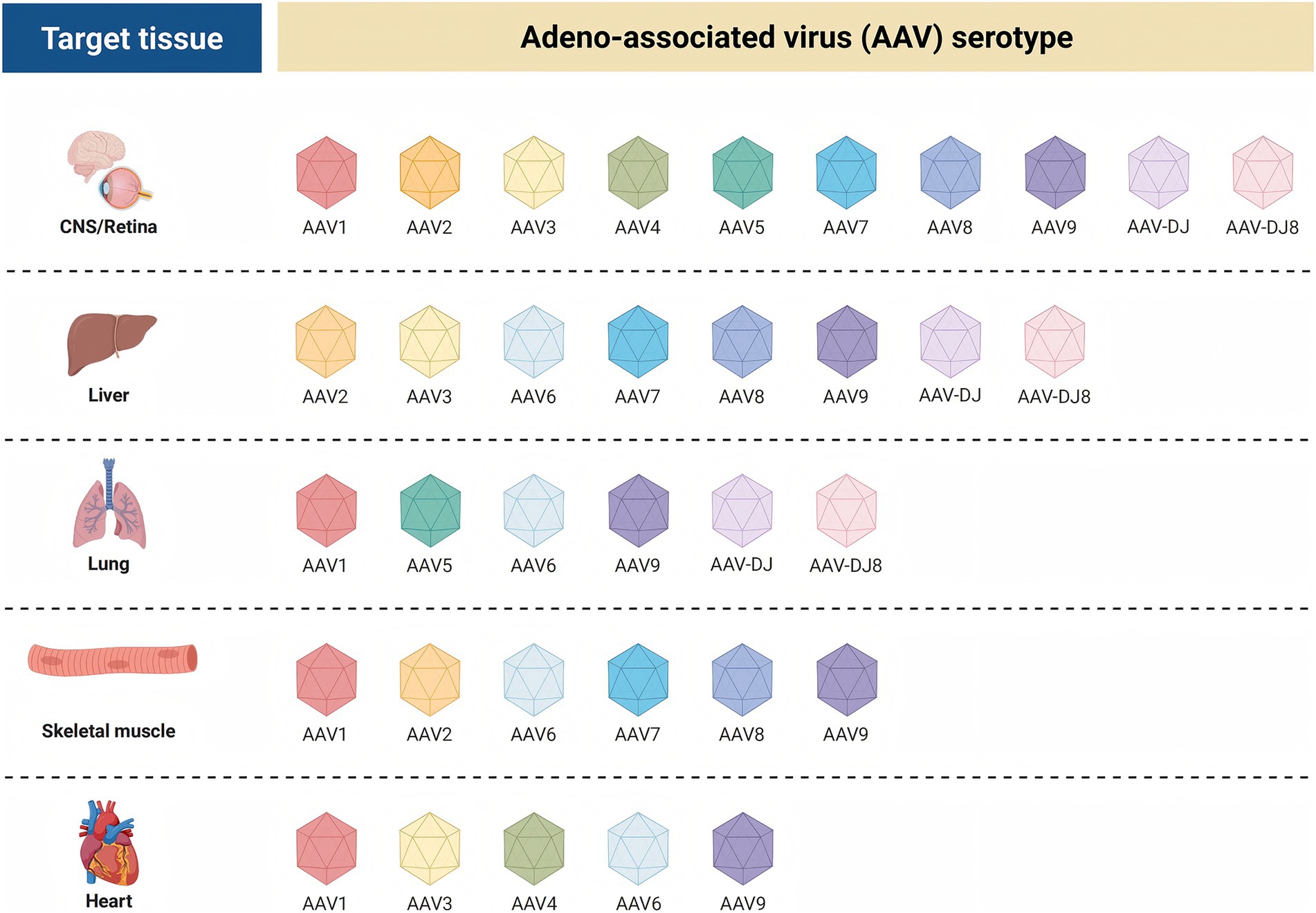
Figure 6. Tissue-specific affinities of adeno-associated virus AAV serotypes for different target tissue.
In mouse studies, the genome was pseudotyped with AAV8 for efficient transduction. AAV vectors exhibit favorable biosafety and tissue-targeting profiles, enabling precise delivery of the human GLA gene into liver cells. The liver-specific promoter FRE1 ensures that the human GLA gene is predominantly expressed in the liver, thereby avoiding non-specific expression in other tissues and enhancing therapeutic precision and safety. Pseudotyping with AAV8 further enhances the infectivity of the vector to target cells, enabling more liver cells to take up and express the human GLA gene (77).
ST-920 utilizes the AAV2/6 vector, which is capable of delivering the human GLA gene from the liver. This vector carries human GLA cDNA driven by a liver-specific expression cassette into patients with FD. The liver-specific expression cassette ensures that human GLA gene expression is initiated primarily in liver cells, thereby avoiding non-specific expression in other tissues and improving therapeutic precision. The human GLA gene that enters the nucleus of liver cells expresses functional α-Gal A under the action of intracellular transcriptional and translational mechanisms. In experiments using the Fabry mouse model, AAV2/6 vector-mediated gene therapy significantly increased α-Gal A activity in plasma and tissues, and the levels of Gb3 and Lyso-Gb3 in key pathological sites were essentially restored to normal. These findings indicate that α-Gal A successfully decomposed substrates, reducing their accumulation in the body, thereby alleviating damage to the kidneys, heart, and other organs caused by substrate accumulation and improving the pathological process of the disease (78).
While AAV-mediated gene therapy holds promise, immunogenicity and safety remain key considerations. Although rAAV typically does not integrate into the host genome, rare integration events can occur. These events are somewhat stochastic and may insert into key gene loci, thereby interfering with normal gene expression and regulation and potentially affecting the normal physiological functions of cells. Upon entering hepatocytes, AAV vectors may integrate their genomes into the host cell genome, thereby triggering hepatocyte injury (79). The vector dose is closely related to hepatotoxicity, with higher doses of AAV vectors being more likely to induce hepatotoxicity. When a large amount of vector enters the liver, it may exceed the processing capacity of hepatocytes, leading to metabolic overload and triggering cellular injury. In studies involving high-dose intravenous injection of AAV vectors in non-human primates and pigs, some animals exhibited marked hepatotoxicity, characterized by significant elevation of aminotransferases, hepatocellular damage, and even acute liver failure (80). The immune system’s recognition and response to AAV vectors are major contributors to hepatotoxicity. AAV vectors are recognized as foreign substances by the immune system, which triggers an immune response. Immune cells, such as macrophages and dendritic cells, recognize AAV vectors, activate relevant signaling pathways, and release pro-inflammatory cytokines, including TNF-α and IL-6. These cytokines induce an inflammatory response in the liver, leading to hepatocyte damage. The adaptive immune response is also involved. When T cells and B cells are activated, T cells attack AAV-infected hepatocytes. Antibodies produced by B cells may bind to antigens on the surface of AAV vectors or infected cells, forming immune complexes that are deposited in the liver and further exacerbate liver damage (81–83). Neurotoxic manifestations are also significant. Several studies have demonstrated that AAV vectors administered via different routes (e.g., intravenous, intrathecal injection, etc.) may lead to dorsal root ganglion toxicity, characterized by neuronal degeneration and necrosis, nerve fiber degeneration, and monocyte infiltration. In the central nervous system, motor dysfunction was observed in rats after intrathalamic injection of AAV-Cre. Similarly, in AAV-mediated gene therapy for FD mouse models, high-dose AAV injection was found to increase the expression of CXCL9 and CXCL10, recruit lymphocyte infiltration, and affect central nervous system function (84).
Theoretically, AAV therapy is expected to achieve the goal of “once-and-for-all, long-term benefit.” In a study involving 18 male patients with hemophilia A treated with an AAV vector (SPK-8011), factor VIII expression was maintained in 16 patients, with 12 followed for more than 2 years. No significant decrease in factor VIII activity was observed at weeks 26–52 and beyond, and the annualized bleeding rate was reduced by 91.5%. However, not all patients achieved such desirable outcomes, and some patients lost factor expression due to cellular immune responses against the AAV capsid. This suggests that there are individual differences in the durability of AAV therapy with immune responses being one of the key factors influencing durability (85). In a study for the treatment of MPS IIIA, healthy Beagles were administered an cerebrospinal fluid (CSF) AAV9 vector carrying the canine sulfonamidase gene. Results showed consistent detection of sulfonamidase activity in the CSF throughout the 7-year study period, with widespread sulfonamidase expression and activity in brain tissue, spinal cord, and dorsal root ganglia, and no obvious histologic abnormalities (86). In addition, the durability of AAV therapy is also closely related to vector design, disease progression, and other factors. For example, in the case of Leber’s congenital amaurosis (LCA) caused by RPE65 deficiency, early studies using AAV vector therapy improved visual function in some patients, but the duration of efficacy was limited by the progression of retinal degeneration. However, through continuous optimization of vector design, such as the development of new AAV2/5-OPTIRPE65 vectors, stronger effects than those of the original vectors were demonstrated in mouse experiments (87).
In terms of vector immunogenicity, preclinical studies consistently show that humoral responses are mild and do not affect efficacy. Chang et al. used GLA−/y mice as a model, administering a single moderate dose (1 × 1011 vg/mouse) of AAV8-GLA or AAV9-GLA via the tail vein, and monitoring serum anti-GLA-IgG titers at baseline and weeks 2, 4, 6, and 8 after administration: all were below the threshold (mean + 3 SD) during the first 4 weeks, with only a slight increase at week 8, indicating an extremely limited humoral immune response to the transgene (88). Takeda’s PET tracing study further confirmed that [18F]AGAL heart signals persisted for 26 weeks and were highly correlated with in vitro enzyme levels (r > 0.9), indicating that anti-GLA antibodies did not significantly neutralize the expressed enzyme (89). Additionally, using the enzyme-stabilized mutant Eng-C, the vector dose can be reduced to ≤5 × 1011 vg/kg while maintaining equivalent efficacy, further lowering the capsid protein burden and potential immune activation risk (90).
7 Symptomatic treatments
7.1 Protective role of ACEIs/ARBs in FD kidneys
Angiotensin-converting enzyme inhibitors (ACEIs) reduce the production of angiotensin II by inhibiting angiotensin-converting enzyme. Angiotensin receptor blockers (ARBs) selectively block the binding of angiotensin II to receptor 1 (AT1), effectively inhibiting its vasoconstrictor effect. This action reduces the elevated capillary pressure in the glomeruli and lowers the intraglomerular pressure. This makes it more difficult for proteins to pass through the glomerular filtration membrane into the urine when blood flows through the glomeruli, thereby reducing urine protein levels (91). In a prospective observational study of 24 adult FD patients undergoing ERT, the effect of anti-proteinuria therapy with ACEIs/ARBs on renal function was evaluated, and the results showed that 75% of patients achieved the urine protein-to-creatinine ratio (UPCR) target with ACEIs/ARBs therapy, and ACEIs/ARBs treatment reduced proteinuria and slowed the decline in eGFR even when baseline eGFR was low. Although some patients have decreased renal function due to severe baseline renal impairment, ACEIs/ARBs remain a key means of controlling proteinuria and delaying the progression of kidney disease (92). The overall therapeutic goal of ACEIs/ARBs in combination with ERT is to reduce urinary protein excretion to less than 500 mg/day and stabilize the decline in renal function to −1 mL/min/1.73 m2/year. In the absence of ACEIs/ARBs, ERT alone does not reduce proteinuria in Fabry patients (93). With the increasing clinical use of sodium-glucose cotransporter-2 (SGLT2) inhibitors and mineralocorticoid receptor antagonists (MRAs), these drugs may confer renal benefits in FD patients.
7.2 Cardiovascular system
A long-term follow-up study of adult FD patients found no significant differences in cardiac symptoms and arrhythmias in patients treated with ERT compared with untreated patients (19). This may be due to the fact that the mechanism of arrhythmogenesis is more complex and is associated with abnormal ion channel function, inflammatory response, and fibrosis, in addition to metabolic substrate accumulation (94, 95). Anticoagulants, such as warfarin and rivaroxaban, are among the important therapeutic agents that can effectively reduce the risk of thromboembolic events (96). β-blockers can be used to control the ventricular rate in patients and alleviate symptoms such as palpitations. Antiarrhythmic drugs, such as amiodarone, can also be used to restore and maintain sinus rhythm (97). However, their use may affect other bodily functions, such as thyroid function, lysosomal pH and enzyme activity, and thus requires close monitoring in patients with FD. Catheter ablation, including radiofrequency ablation, is an effective therapeutic option that restores normal cardiac rhythm by disrupting abnormal electrical conduction pathways. For some patients with severe disease who do not respond well to medications, it may be necessary to consider implanting a pacemaker or implantable cardioverter defibrillator (ICD). Implanted pacemakers can be used to treat severe bradycardia or heart block, while ICD can prevent sudden death by administering a shock defibrillator in the event of a life-threatening arrhythmia (94, 98, 99).
7.3 Neuralgia
In actual treatment, some patients continue to experience neuropathic pain symptoms despite overall improvement in their condition with ERT. Antiepileptic drugs, such as gabapentin and carbamazepine, relieve neuralgia by modulating neuronal excitability and reducing nerve impulse transmission, and are widely regarded as first-line drugs for treating neuropathic pain in FD (100). A study showed that male FD patients aged 15–45 years treated with gabapentin for 4 weeks experienced a reduction in pain compared to pre-treatment levels, and the treatment was generally well-tolerated (101). Additionally, a case report documented a 7-year-old patient experiencing intermittent, intense “burning” sensations in the hands and feet, which were unresponsive to aspirin, acetaminophen, acetylcodeine, and phenytoin. Carbamazepine and phenytoin sodium reduced the frequency and duration of pain episodes to 3–4 per year, indicating that while antiepileptic drugs significantly decreased the frequency of FD-related neuralgia, they did not provide complete relief. Over 28 consecutive months of observation, 7 crises occurred. During these crises, intravenous morphine was administered at 0.06 mg/kg, followed by a continuous infusion of 0.02 mg/kg/h of the drug and 0.25 mg/kg of amitriptyline at bedtime, which rapidly alleviated the pain symptoms (102). A recent study analyzing the surface proteome of sensory-like neurons derived from induced pluripotent stem cells (IPSCs) in FD patients identified 48 abnormally expressed surface proteins, among which pain-related proteins like CACNA2D3 and GPM6A were significantly dysregulated. Surface protein interaction network analysis revealed enrichment of extracellular matrix proteins (e.g., COL5A1, POSTN) and transmembrane receptors (e.g., ROBO2, SEMA6D), suggesting impaired neuronal network integrity and axon guidance abnormalities. Although there were no significant differences in mRNA levels of related genes, the protein interaction network unveiled potential abnormalities in neuronal plasticity and inflammatory pathways. Combining the core lysosomal dysfunction in FD, it is hypothesized that Gb3 accumulation indirectly affects ion channels and the neurochemical environment through membrane structural changes and impaired secretion, collectively contributing to the development of neuropathic pain (103).
8 Accessibility and economic feasibility of Fabry disease treatment drugs
Orphan drugs for the treatment of FD exhibit a significant “north–south divide” in terms of accessibility and affordability worldwide: High-income countries rely on institutional collaboration and policy innovation to significantly optimize drug access and health insurance coverage, continuously improving patient accessibility; while middle and low-income countries are trapped in a “high clinical value—low actual accessibility” dilemma due to structural barriers such as delayed approval, absent health insurance, and excessive concentration of medical resources. At the same time, although orphan drugs for FD have clear efficacy, they exert a sustained squeeze on health insurance funds due to high pricing and an imbalanced cost-effectiveness ratio, further amplifying global health inequalities.
8.1 Accessibility disparities: well-developed systems in high-income countries vs. multiple barriers in middle and low-income countries
In high-income countries, the approval, supply, and use system for orphan drugs has become mature. In the approval process, the United States, through the Orphan Drug Act, provides 7 years of market exclusivity, 50% tax credit for clinical research and development, free designation, and full technical support, significantly speeding up the time to market; the EU, based on Regulation (EC) No 141/2000, grants 10 years of market exclusivity and fee reductions, and through the “centralized approval” and “mutual recognition” mechanisms, allows drugs to be marketed simultaneously or rapidly sequentially in member states, greatly simplifying the process (104–106). In the payment process, Germany’s statutory health insurance fully reimburses almost all OMPs approved by the EMA, despite the annual drug cost for Fabry disease reaching 264,896 euros (107), the patients’ actual burden is extremely low. In the use process, Germany, Norway, Italy, and other countries incorporate home infusion into routine management (107–109), making home treatment more comfortable, less stressful, and more effective, significantly improving the accessibility of treatment and patients’ compliance.
Compared to high and middle-income countries, there are significant barriers to the accessibility of orphan drugs for Fabry disease in low-income countries. A study in India shows that only 51.8% of 54 patients could receive enzyme replacement therapy, with an average diagnosis delay of 11.7 years (110). Although Turkey has included 43 orphan drugs in its reimbursement list, 22 of them have not been launched domestically and require individual applications for import by the social security agency (111). Among the 242 new orphan drugs approved by the US FDA from 2013 to 2023, as of January 1, 2025, China has only approved 119, with a significant delay: the median lag time for approved varieties is 1,004 days, with the longest delay reaching 3,785 days, and there is still a significant lag compared to Asian countries such as Japan and South Korea. Before being included in the national health insurance in a first-tier city in Northeast China, 15 out of 16 FD orphan drugs exceeded the “catastrophic medical expenditure” threshold for rural residents; even after inclusion in the health insurance, 8 of them still did not meet the standard (112). The National Essential Medicines List of Thailand only includes 22.93% of registered orphan drugs, and five-year procurement data show that only 31.7% of registered varieties were actually supplied to hospitals (113), reflecting the triple accessibility barriers of “diagnosis delay—market lag—reimbursement gap.”
8.2 Economic differences: high drug costs dominate vs. healthcare burden and cost-effectiveness imbalance
Globally, the payment for orphan drugs for FD continues to place a heavy burden on public finances and healthcare systems. Canada once tried a “triple-sharing” model: pharmaceutical companies invested in the University Health Network (UHN) through provincial governments to support Canadian Fabry Disease Initiative (CFDI) research; provincial governments procured drugs with assistance from the federal government, creating a hybrid mechanism where “companies bear the research costs and governments share the procurement costs” to spread the risk of a single entity. However, after about a year, the federal government exited, citing “a lack of a clear endpoint.” The core economic consideration was that the ERT involved in the CFDI was one of the most expensive treatment methods globally at the time, requiring continuous funding to maintain patient registration systems, collect real-world evidence, and with no clear end to the research period. The federal government may have been concerned that such long-term, indefinite funding would crowd out resources in other public health areas, and without a national budget framework for rare disease treatment, it would be difficult to ensure sustained financial support, so it chose to withdraw to avoid long-term financial risks (114). The case in Chengdu, China further highlights the impact of high-priced orphan drugs on local finances. In 2019, the total budget for 98 rare disease patients’ orphan drugs, if fully self-paid, reached 1.79 billion yuan; under six simulated reimbursement scenarios, it still required 0.32–1.56 billion yuan, and the annual funding needs are expected to rise to 2.00–13.03 billion yuan over the next three years (115). India, as a traditional populous country, insufficient coverage exacerbates fiscal fragility. Only 37.2% of the population has any form of health insurance, with significant inter-state and population differences; the National Health Account (NHA) data shows that while government health spending has increased year by year, the proportion paid by individuals remains high, and high-priced orphan drugs are prone to push families into catastrophic spending (115). It is evident that whether in high-income or middle- and low-income countries, how to establish a sustainable financing and risk-sharing mechanism for rare drugs within limited budgets has become a global fiscal governance challenge.
In the cost-effectiveness dimension, although ERT can bring measurable clinical benefits, it is accompanied by the sharp contradiction of “high gain, higher price” in the long term. Agalsidase beta can delay renal function decline, but the incremental health benefits are extremely limited: the Dutch cohort study shows that lifetime Quality-adjusted life years (QALYs) increase by only 1.6 years, and the years free of end-organ damage (YFEOD) are extended by 1.5 years, with slightly higher benefits for males. The corresponding Incremental cost effectiveness ratio (ICER) reaches 6.1 million euros/QALY, the incremental cost of YFEOD is 6.6 million euros/year, about 8 times that of Gaucher’s ERT at the same time; sensitivity analysis suggests that even if the drug price is reduced by 25% (from 200,000 to 150,000 euros/year), the ICER is still 4.55 million euros/QALY, far exceeding the Dutch healthcare threshold (≈50,000 euros/QALY). Drug costs account for 70–90% of the total treatment cost and are the only key variable determining the ICER (116). China reduced the ERT price to the lowest global range through national medical insurance negotiations in 2021, with an ICER of 148,072 yuan/QALY, within the 1–3 times per capita GDP threshold (89,358–268,074 yuan/QALY), theoretically making it economically viable (117). However, this ICER is still based on the “medical insurance payment price,” and if local fiscal support is insufficient, residents in economically underdeveloped areas need to bear out-of-pocket costs as well as hidden costs such as transportation and care, which still limit actual accessibility. Therefore, while maintaining the national negotiation price, it is necessary to increase the central-provincial fiscal subsidy ratio and establish a special co-payment fund for rare diseases to truly transform “cost-effectiveness acceptable” into “budget-burden acceptable” actual accessibility.
9 Conclusion
Significant advancements have been achieved in the pathogenesis, diagnosis, and treatment of FD as research progresses. However, numerous challenges persist. ERT, while effective in alleviating symptoms and delaying disease progression, is not without limitations. For patients with advanced organ damage, ERT provides only limited improvement and cannot reverse severe organ disease. Furthermore, due to the difficulty of ERT in penetrating the blood–brain barrier, it is challenging to improve central nervous system symptoms. Migalastat, a pharmacological chaperone therapy, is only effective for patients with specific missense mutations. Although it can stabilize kidney function and improve heart structure, it requires close monitoring due to the differences in efficacy between in vivo and in vitro effects. Emerging therapies, such as substrate reduction therapy, mRNA therapy, and AAV-mediated gene therapy, have demonstrated potential in the treatment of FD. Among these, AAV-mediated gene therapy, which leverages precise targeting and efficient expression through multiple serotypes of vectors, has significantly ameliorated the pathological process of FD in animal experiments and is theorized to provide long-term benefits to patients. However, it faces safety concerns, including the optimization of dosing regimens, enhancement of delivery systems, and management of immunogenicity (Table 3).
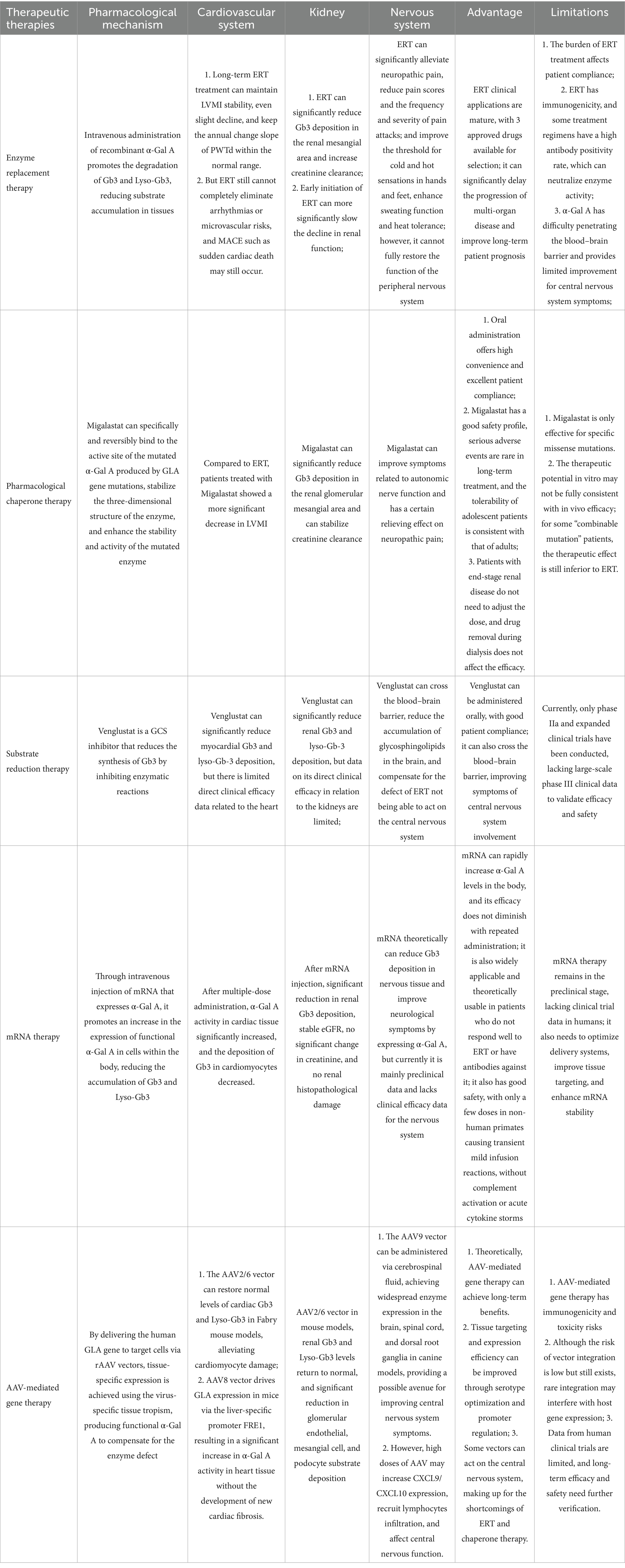
Table 3. Comparison of the pharmacological mechanisms, organ benefits, and limitations of the main treatment strategies for Fabry disease.
The future of FD treatment will be centered on the goal of “precision, efficiency, and cure.” This will be achieved through the optimization of existing therapies, breakthroughs in emerging technologies such as mRNA targeted delivery, AAV capsid modification, and CRISPR gene editing, reliance on precision medicine for early screening and subtyping, and enhancement of patient care quality through interdisciplinary integration and long-term management. Interdisciplinary integration and long-term management will improve the quality of patients’ full-cycle care. With the synergy of policy support and technological innovation, FD is expected to become a model for gene therapy in rare diseases, facilitating the transition from “lifelong replacement therapy” to “gene-level cure.”
Author contributions
MZ: Writing – original draft, Writing – review & editing. CW: Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the ShanXi Natural Science Foundation (No. 202103021224382).
Acknowledgments
We would like to thank Biorender.com for the drawing material for all the figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chang, S, Zhan, X, Liu, Y, Song, H, Gong, Z, Han, L, et al. Newborn screening for 6 lysosomal storage disorders in China. JAMA Netw Open. (2024) 7:e2410754. doi: 10.1001/jamanetworkopen.2024.10754
2. Svarstad, E, and Marti, HP. The changing landscape of Fabry disease. Clin J Am Soc Nephrol. (2020) 15:569–76. doi: 10.2215/CJN.09480819
3. Terryn, W, Cochat, P, Froissart, R, Ortiz, A, Pirson, Y, Poppe, B, et al. Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European renal best practice. Nephrol Dial Transplant. (2013) 28:505–17. doi: 10.1093/ndt/gfs526
4. Klein, A, Klug, K, Breyer, M, Grüner, J, Medala, VK, Nordbeck, P, et al. Genetic variants of unknown significance in alpha-galactosidase a: cellular delineation from Fabry disease. J Inherit Metab Dis. (2024) 47:805–17. doi: 10.1002/jimd.12743
5. Nowak, A, Mechtler, T, Kasper, DC, and Desnick, RJ. Correlation of Lyso-Gb3 levels in dried blood spots and sera from patients with classic and later-onset Fabry disease. Mol Genet Metab. (2017) 121:320–4. doi: 10.1016/j.ymgme.2017.06.006
7. Wagenhäuser, L, Rickert, V, Sommer, C, Wanner, C, Nordbeck, P, Rost, S, et al. X-chromosomal inactivation patterns in women with Fabry disease. Mol Genet Genomic Med. (2022) 10:e2029. doi: 10.1002/mgg3.2029
8. Expert consensus for diagnosis and treatment of Fabry disease in China. Zhonghua Nei Ke Za Zhi. (2021) 60:321–30. doi: 10.3760/cma.j.cn112138-20201218-01028
9. Tøndel, C, Bostad, L, Hirth, A, and Svarstad, E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. (2008) 51:767–76. doi: 10.1053/j.ajkd.2007.12.032
10. Hughes, D, Linhart, A, Gurevich, A, Kalampoki, V, Jazukeviciene, D, and Feriozzi, S. Prompt Agalsidase alfa therapy initiation is associated with improved renal and cardiovascular outcomes in a Fabry outcome survey analysis. Drug Des Devel Ther. (2021) 15:3561–72. doi: 10.2147/DDDT.S313789
11. Arends, M, Wijburg, FA, Wanner, C, Vaz, FM, van Kuilenburg, ABP, Hughes, DA, et al. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol Genet Metab. (2017) 121:157–61. doi: 10.1016/j.ymgme.2017.05.001
12. Basic-Jukic, N, Kes, P, Coric, M, and Basic-Kes, V. Renal complications of Fabry disease. Curr Pharm Des. (2013) 19:6046–50. doi: 10.2174/13816128113199990346
13. Pisani, A, Visciano, B, Imbriaco, M, Di Nuzzi, A, Mancini, A, Marchetiello, C, et al. The kidney in Fabry’s disease. Clin Genet. (2014) 86:301–9. doi: 10.1111/cge.12386
14. Azimpour, K, Tordoff-Gibson, C, Dorling, P, Koulinska, I, Kunduri, S, Laliman-Khara, V, et al. Influence of treatment effect modifiers in Fabry disease: a systematic literature review. Adv Ther. (2025) 42:579–96. doi: 10.1007/s12325-024-03062-x
15. Nowak, A, Koch, G, Huynh-Do, U, Siegenthaler, M, Marti, HP, and Pfister, M. Disease progression modeling to evaluate the effects of enzyme replacement therapy on kidney function in adult patients with the classic phenotype of fabry disease. Kidney Blood Press Res. (2017) 42:1–15. doi: 10.1159/000464312
16. Kertész, AB, and Édes, I. Fabry disease cardiomyopathy: from genes to clinical manifestations. Curr Pharm Biotechnol. (2012) 13:2477–84. doi: 10.2174/138920112804583069
17. Faro, DC, Di Pino, FL, and Monte, IP. Inflammation, oxidative stress, and endothelial dysfunction in the pathogenesis of vascular damage: unraveling novel cardiovascular risk factors in Fabry disease. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25158273
18. Linhart, A, and Elliott, PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. (2007) 93:528–35. doi: 10.1136/hrt.2005.063818
19. Madsen, CV, Bundgaard, H, Rasmussen, ÅK, Sørensen, SS, Petersen, JH, Køber, L, et al. Echocardiographic and clinical findings in patients with Fabry disease during long-term enzyme replacement therapy: a nationwide Danish cohort study. Scand Cardiovasc J. (2017) 51:207–16. doi: 10.1080/14017431.2017.1332383
20. Schiffmann, R. Neuropathy and Fabry disease: pathogenesis and enzyme replacement therapy. Acta Neurol Belg. (2006) 106:61–5.
21. Üçeyler, N, Ganendiran, S, Kramer, D, and Sommer, C. Characterization of pain in fabry disease. Clin J Pain. (2014) 30:915–20. doi: 10.1097/AJP.0000000000000041
22. Ivanova, MM, Dao, J, Friedman, A, Kasaci, N, and Goker-Alpan, O. Sex differences in circulating inflammatory, immune, and tissue growth markers associated with Fabry disease-related cardiomyopathy. Cells. (2025) 14. doi: 10.3390/cells14050322
23. Wang, RY, Abe, JT, Cohen, AH, and Wilcox, WR. Enzyme replacement therapy stabilizes obstructive pulmonary Fabry disease associated with respiratory globotriaosylceramide storage. J Inherit Metab Dis. (2008) 31:S369–74. doi: 10.1007/s10545-008-0930-x
24. Feriozzi, S, Torre, ES, Ranalli, TV, Cardello, P, Morrone, A, and Ancarani, E. A diagnosis of Fabry gastrointestinal disease by chance: a case report. Eur J Gastroenterol Hepatol. (2007) 19:163–5. doi: 10.1097/MEG.0b013e32800fef46
25. Concolino, D, Rapsomaniki, M, Disabella, E, Sestito, S, Pascale, MG, Moricca, MT, et al. Co-existence of phenylketonuria and Fabry disease on a 3 year-old boy: case report. BMC Pediatr. (2010) 10:32. doi: 10.1186/1471-2431-10-32
26. Papaxanthos-Roche, A, Deminière, C, Bauduer, F, Hocké, C, Mayer, G, and Lacombe, D. Azoospermia as a new feature of Fabry disease. Fertil Steril. (2007) 88:e15:212–8. doi: 10.1016/j.fertnstert.2006.11.036
27. Lacombe, D, Germain, DP, and Papaxanthos-Roche, A. Azoospermia as a new feature of Fabry disease. Rev Med Interne. (2010) 31:S214–6. doi: 10.1016/S0248-8663(10)70014-X
28. Germain, DP, Elliott, PM, Falissard, B, Fomin, VV, Hilz, MJ, Jovanovic, A, et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: a systematic literature review by a European panel of experts. Molecular Genetics Metabolism Reports. (2019) 19. doi: 10.1016/j.ymgmr.2019.100454
29. Germain, DP, and Linhart, A. Pegunigalsidase alfa: a novel, pegylated recombinant alpha-galactosidase enzyme for the treatment of Fabry disease. Front Genet. (2024) 15. doi: 10.3389/fgene.2024.1395287
30. Schiffmann, R, Kopp, JB, Austin, HA 3rd, Sabnis, S, Moore, DF, Weibel, T, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. (2001) 285:2743–9. doi: 10.1001/jama.285.21.2743
31. Ramaswami, U, Pintos-Morell, G, Kampmann, C, Nicholls, K, Niu, DM, Reisin, R, et al. Two decades of experience of the Fabry outcome survey provides further confirmation of the long-term effectiveness of agalsidase alfa enzyme replacement therapy. Mol Genet Metab Rep. (2025) 43:101215. doi: 10.1016/j.ymgmr.2025.101215
32. Cybulla, M, Nicholls, K, Feriozzi, S, Linhart, A, Torras, J, Vujkovac, B, et al. Renoprotective effect of Agalsidase alfa: a long-term follow-up of patients with Fabry disease. J Clin Med. (2022) 11. doi: 10.3390/jcm11164810
33. Parini, R, Pintos-Morell, G, Hennermann, JB, Hsu, TR, Karabul, N, Kalampoki, V, et al. Analysis of renal and cardiac outcomes in male participants in the Fabry outcome survey starting Agalsidase alfa enzyme replacement therapy before and after 18 years of age. Drug Des Devel Ther. (2020) 14:2149–58. doi: 10.2147/DDDT.S249433
34. Ramaswami, U, Beck, M, Hughes, D, Kampmann, C, Botha, J, Pintos-Morell, G, et al. Cardio- renal outcomes with long- term Agalsidase alfa enzyme replacement therapy: a 10- year Fabry outcome survey (FOS) analysis. Drug Des Devel Ther. (2019) 13:3705–15. doi: 10.2147/DDDT.S207856
35. Weidemann, F, Niemann, M, Störk, S, Breunig, F, Beer, M, Sommer, C, et al. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. (2013) 274:331–41. doi: 10.1111/joim.12077
36. Alegra, T, Vairo, F, de Souza, MV, Krug, BC, and Schwartz, IV. Enzyme replacement therapy for Fabry disease: a systematic review and meta-analysis. Genet Mol Biol. (2012) 35:947–54. doi: 10.1590/s1415-47572012000600009
37. Sheng, S, Wu, L, Nalleballe, K, Sharma, R, Brown, A, Ranabothu, S, et al. Fabry’s disease and stroke: effectiveness of enzyme replacement therapy (ERT)in stroke prevention, a review with meta-analysis. J Clin Neurosci. (2019) 65:83–6. doi: 10.1016/j.jocn.2019.03.064
38. Schiffmann, R, Floeter, MK, Dambrosia, JM, Gupta, S, Moore, DF, Sharabi, Y, et al. Enzyme replacement therapy improves peripheral nerve and sweat function in Fabry disease. Muscle Nerve. (2003) 28:703–10. doi: 10.1002/mus.10497
39. Whybra, C, Miebach, E, Mengel, E, Gal, A, Baron, K, Beck, M, et al. A 4-year study of the efficacy and tolerability of enzyme replacement therapy with agalsidase alfa in 36 women with Fabry disease. Genet Med. (2009) 11:441–9. doi: 10.1097/GIM.0b013e3181a23bec
40. Goláň, L, Goker-Alpan, O, Holida, M, Kantola, I, Klopotowski, M, Kuusisto, J, et al. Evaluation of the efficacy and safety of three dosing regimens of agalsidase alfa enzyme replacement therapy in adults with Fabry disease. Drug Des Devel Ther. (2015) 9:3435–44. doi: 10.2147/DDDT.S80928
41. Schiffmann, R, Askari, H, Timmons, M, Robinson, C, Benko, W, Brady, RO, et al. Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J Am Soc Nephrol. (2007) 18:1576–83. doi: 10.1681/ASN.2006111263
42. Skrunes, R, Tøndel, C, Leh, S, Larsen, KK, Houge, G, Davidsen, ES, et al. Long-term dose-dependent Agalsidase effects on kidney histology in Fabry disease. Clin J Am Soc Nephrol. (2017) 12:1470–9. doi: 10.2215/CJN.01820217
43. Tsuboi, K, and Yamamoto, H. Clinical course of patients with Fabry disease who were switched from agalsidase-β to agalsidase-α. Genet Med. (2014) 16:766–72. doi: 10.1038/gim.2014.28
44. Sirrs, SM, Bichet, DG, Casey, R, Clarke, JTR, Lemoine, K, Doucette, S, et al. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol Genet Metab. (2014) 111:499–506. doi: 10.1016/j.ymgme.2014.01.014
45. Krämer, J, Lenders, M, Canaan-Kühl, S, Nordbeck, P, Üçeyler, N, Blaschke, D, et al. Fabry disease under enzyme replacement therapy-new insights in efficacy of different dosages. Nephrol Dial Transplant. (2018) 33:1362–72. doi: 10.1093/ndt/gfx319
46. Vedder, AC, Breunig, F, Donker-Koopman, WE, Mills, K, Young, E, Winchester, B, et al. Treatment of Fabry disease with different dosing regimens of agalsidase: effects on antibody formation and GL-3. Mol Genet Metab. (2008) 94:319–25. doi: 10.1016/j.ymgme.2008.03.003
47. Ruderfer, I, Shulman, A, Kizhner, T, Azulay, Y, Nataf, Y, Tekoah, Y, et al. Development and analytical characterization of Pegunigalsidase alfa, a chemically cross-linked plant recombinant human α-galactosidase-a for treatment of Fabry disease. Bioconjug Chem. (2018) 29:1630–9. doi: 10.1021/acs.bioconjchem.8b00133
48. Schiffmann, R, Goker-Alpan, O, Holida, M, Giraldo, P, Barisoni, L, Colvin, RB, et al. Pegunigalsidase alfa, a novel PEGylated enzyme replacement therapy for Fabry disease, provides sustained plasma concentrations and favorable pharmacodynamics: a 1-year phase 1/2 clinical trial. J Inherit Metab Dis. (2019) 42:534–44. doi: 10.1002/jimd.12080
49. Wallace, EL, Goker-Alpan, O, Wilcox, WR, Holida, M, Bernat, J, Longo, N, et al. Head-to-head trial of pegunigalsidase alfa versus agalsidase beta in patients with Fabry disease and deteriorating renal function: results from the 2-year randomised phase III BALANCE study. J Med Genet. (2024) 61:520–30. doi: 10.1136/jmg-2023-109445
50. Linhart, A, Dostálová, G, Nicholls, K, West, ML, Tøndel, C, Jovanovic, A, et al. Safety and efficacy of pegunigalsidase alfa in patients with Fabry disease who were previously treated with agalsidase alfa: results from BRIDGE, a phase 3 open-label study. Orphanet J Rare Dis. (2023) 18:332. doi: 10.1186/s13023-023-02937-6
51. Hughes, D, Gonzalez, D, Maegawa, G, Bernat, JA, Holida, M, Giraldo, P, et al. Long-term safety and efficacy of pegunigalsidase alfa: a multicenter 6-year study in adult patients with Fabry disease. Genet Med. (2023) 25:100968. doi: 10.1016/j.gim.2023.100968
52. Nowicki, M, Bazan-Socha, S, Błażejewska-Hyżorek, B, Kłopotowski, MM, Komar, M, Kusztal, MA, et al. A review and recommendations for oral chaperone therapy in adult patients with Fabry disease. Orphanet J Rare Dis. (2024) 19:16. doi: 10.1186/s13023-024-03028-w
53. Wang, H, Shen, Y, Zhao, L, and Ye, Y. 1-Deoxynojirimycin and its derivatives: a Mini review of the literature. Curr Med Chem. (2021) 28:628–43. doi: 10.2174/0929867327666200114112728
54. Bichet, DG, Torra, R, Wallace, E, Hughes, D, Giugliani, R, Skuban, N, et al. Long-term follow-up of renal function in patients treated with migalastat for Fabry disease. Mol Genet Metab Rep. (2021) 28:100786. doi: 10.1016/j.ymgmr.2021.100786
55. Mauer, M, Sokolovskiy, A, Barth, JA, Castelli, JP, Williams, HN, Benjamin, ER, et al. Reduction of podocyte globotriaosylceramide content in adult male patients with Fabry disease with amenable GLA mutations following 6 months of migalastat treatment. J Med Genet. (2017) 54:781–6. doi: 10.1136/jmedgenet-2017-104826
56. Hughes, DA, Nicholls, K, Shankar, SP, Sunder-Plassmann, G, Koeller, D, Nedd, K, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. (2017) 54:288–96. doi: 10.1136/jmedgenet-2016-104178
57. Hughes, DA, Bichet, DG, Giugliani, R, Hopkin, RJ, Krusinska, E, Nicholls, K, et al. Long-term multisystemic efficacy of migalastat on Fabry-associated clinical events, including renal, cardiac and cerebrovascular outcomes. J Med Genet. (2023) 60:722–31. doi: 10.1136/jmg-2022-108669
58. Nowak, A, Huynh-Do, U, Krayenbuehl, PA, Beuschlein, F, Schiffmann, R, and Barbey, F. Fabry disease genotype, phenotype, and migalastat amenability: insights from a national cohort. J Inherit Metab Dis. (2020) 43:326–33. doi: 10.1002/jimd.12167
59. Lenders, M, Stappers, F, and Brand, E. In vitro and in vivo amenability to Migalastat in Fabry disease. Mol Ther Methods Clin Dev. (2020) 19:24–34. doi: 10.1016/j.omtm.2020.08.012
60. Ramaswami, U, Font-Montgomery, E, Goker-Alpan, O, Ortiz, D, Sanchez-Valle, A, Whitley, CB, et al. Safety and efficacy of migalastat in adolescent patients with Fabry disease: results from ASPIRE, a phase 3b, open-label, single-arm, 12-month clinical trial, and its open-label extension. Mol Genet Metab. (2025) 145:109102. doi: 10.1016/j.ymgme.2025.109102
61. Müntze, J, Lau, K, Cybulla, M, Brand, E, Cairns, T, Lorenz, L, et al. Patient reported quality of life and medication adherence in Fabry disease patients treated with migalastat: a prospective, multicenter study. Mol Genet Metab. (2023) 138:106981. doi: 10.1016/j.ymgme.2022.106981
62. Johnson, FK, Wu, S, Schmith, G, Williams, H, Rutecki, J, Halabi, A, et al. Pharmacokinetic evaluation of single-dose migalastat in non-Fabry disease subjects with ESRD receiving dialysis treatment, and use of modeling to select dose regimens in Fabry disease subjects with ESRD receiving dialysis treatment. PLoS One. (2024) 19:e0314030. doi: 10.1371/journal.pone.0314030
63. Deegan, PB, Goker-Alpan, O, Geberhiwot, T, Hopkin, RJ, Lukina, E, Tylki-Szymanska, A, et al. Venglustat, an orally administered glucosylceramide synthase inhibitor: assessment over 3 years in adult males with classic Fabry disease in an open-label phase 2 study and its extension study. Mol Genet Metab. (2023) 138:106963. doi: 10.1016/j.ymgme.2022.11.002
64. Ashe, KM, Budman, E, Bangari, DS, Siegel, CS, Nietupski, JB, Wang, B, et al. Efficacy of enzyme and substrate reduction therapy with a novel antagonist of glucosylceramide synthase for Fabry disease. Mol Med. (2015) 21:389–99. doi: 10.2119/molmed.2015.00088
65. DeRosa, F, Smith, L, Shen, Y, Huang, Y, Pan, J, Xie, H, et al. Improved efficacy in a Fabry disease model using a systemic mRNA liver depot system as compared to enzyme replacement therapy. Mol Ther. (2019) 27:878–89. doi: 10.1016/j.ymthe.2019.03.001
66. Zhang, Z, Liu, Q, Deng, Z, Liu, J, Li, S, Hong, M, et al. Evaluating the metabolic basis of α-Gal a mRNA therapy for Fabry disease. Biology (Basel). (2024) 13. doi: 10.3390/biology13020106
67. Zhu, X, Yin, L, Theisen, M, Zhuo, J, Siddiqui, S, Levy, B, et al. Systemic mRNA therapy for the treatment of Fabry disease: preclinical studies in wild-type mice, Fabry mouse model, and wild-type non-human Primates. Am J Hum Genet. (2019) 104:625–37. doi: 10.1016/j.ajhg.2019.02.003
68. Wang, JH, Gessler, DJ, Zhan, W, Gallagher, TL, and Gao, G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct Target Ther. (2024) 9:78. doi: 10.1038/s41392-024-01780-w
69. Wörner, TP, Bennett, A, Habka, S, Snijder, J, Friese, O, Powers, T, et al. Adeno-associated virus capsid assembly is divergent and stochastic. Nat Commun. (2021) 12:1642. doi: 10.1038/s41467-021-21935-5
70. Taylor, NK, Guggenbiller, MJ, Mistry, PP, King, OD, and Harper, SQ. A self-complementary AAV proviral plasmid that reduces cross-packaging and ITR promoter activity in AAV vector preparations. Mol Ther Methods Clin Dev. (2024) 32:101295. doi: 10.1016/j.omtm.2024.101295
71. Wang, D, Tai, PWL, and Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. (2019) 18:358–78. doi: 10.1038/s41573-019-0012-9
72. Gonzalez, TJ, Mitchell-Dick, A, Blondel, LO, Fanous, MM, Hull, JA, Oh, DK, et al. Structure-guided AAV capsid evolution strategies for enhanced CNS gene delivery. Nat Protoc. (2023) 18:3413–59. doi: 10.1038/s41596-023-00875-y
73. Wang, JH, Cui, M, Liu, H, Guo, P, McGowan, J, Cheng, SY, et al. Cell-penetrating peptide-grafted AAV2 capsids for improved retinal delivery via intravitreal injection. Mol Ther Methods Clin Dev. (2025) 33:101426. doi: 10.1016/j.omtm.2025.101426
74. Biferi, MG, Cohen-Tannoudji, M, García-Silva, A, Souto-Rodríguez, O, Viéitez-González, I, San-Millán-Tejado, B, et al. Systemic treatment of Fabry disease using a novel AAV9 vector expressing α-galactosidase a. Mol Ther Methods Clin Dev. (2021) 20:1–17. doi: 10.1016/j.omtm.2020.10.016
75. Biber, J, Gandor, C, Becirovic, E, and Michalakis, S. Retina-directed gene therapy: achievements and remaining challenges. Pharmacol Ther. (2025) 271:108862. doi: 10.1016/j.pharmthera.2025.108862
76. Noriega, HA, Wang, Q, Yu, D, and Wang, XS. Structural studies of Parvoviridae capsid assembly and evolution: implications for novel AAV vector design. Front Artif Intell. (2025) 8:1559461. doi: 10.3389/frai.2025.1559461
77. Jeyakumar, JM, Kia, A, Tam, LCS, McIntosh, J, Spiewak, J, Mills, K, et al. Preclinical evaluation of FLT190, a liver-directed AAV gene therapy for Fabry disease. Gene Ther. (2023) 30:487–502. doi: 10.1038/s41434-022-00381-y
78. Yasuda, M, Huston, MW, Pagant, S, Gan, L, St Martin, S, Sproul, S, et al. AAV2/6 gene therapy in a murine model of Fabry disease results in supraphysiological enzyme activity and effective substrate reduction. Mol Ther. (2020) 18:607–19. doi: 10.1016/j.omtm.2020.07.002
79. Sabatino, DE, Bushman, FD, Chandler, RJ, Crystal, RG, Davidson, BL, Dolmetsch, R, et al. Evaluating the state of the science for adeno-associated virus integration: an integrated perspective. Mol Ther. (2022) 30:2646–63. doi: 10.1016/j.ymthe.2022.06.004
80. Hinderer, C, Katz, N, Buza, EL, Dyer, C, Goode, T, Bell, P, et al. Severe toxicity in nonhuman Primates and piglets following High-dose intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther. (2018) 29:285–98. doi: 10.1089/hum.2018.015
81. Ertl, HCJ. Immunogenicity and toxicity of AAV gene therapy. Front Immunol. (2022) 13:975803. doi: 10.3389/fimmu.2022.975803
82. Shao, W, Huang, W, Wang, Y, Sima, H, Ma, K, Chen, R, et al. Exosome-modified AAV gene therapy attenuates autoimmune hepatitis via enhanced regulatory T cell targeting and immune modulation. Microorganisms. (2025) 13. doi: 10.3390/microorganisms13040823
83. Sun, CP, Wu, TH, Chen, CC, Wu, PY, Shih, YM, Tsuneyama, K, et al. Studies of efficacy and liver toxicity related to adeno-associated virus-mediated RNA interference. Hum Gene Ther. (2013) 24:739–50. doi: 10.1089/hum.2012.239
84. Panzer, E, Boch, L, Cosquer, B, Grgurina, I, Boutillier, AL, de Vasconcelos, AP, et al. Disconnecting prefrontal cortical neurons from the ventral midline thalamus: loss of specificity due to progressive neural toxicity of an AAV-Cre in the rat thalamus. J Neurosci Methods. (2024) 405:110080. doi: 10.1016/j.jneumeth.2024.110080
85. George, LA, Monahan, PE, Eyster, ME, Sullivan, SK, Ragni, MV, Croteau, SE, et al. Multiyear factor VIII expression after AAV gene transfer for hemophilia a. N Engl J Med. (2021) 385:1961–73. doi: 10.1056/NEJMoa2104205
86. Marcó, S, Haurigot, V, Jaén, ML, Ribera, A, Sánchez, V, Molas, M, et al. Seven-year follow-up of durability and safety of AAV CNS gene therapy for a lysosomal storage disorder in a large animal. Mol Ther Methods Clin Dev. (2021) 23:370–89. doi: 10.1016/j.omtm.2021.09.017
87. Georgiadis, A, Duran, Y, Ribeiro, J, Abelleira-Hervas, L, Robbie, SJ, Sünkel-Laing, B, et al. Development of an optimized AAV2/5 gene therapy vector for Leber congenital amaurosis owing to defects in RPE65. Gene Ther. (2016) 23:857–62. doi: 10.1038/gt.2016.66
88. Chang, FP, Lee, YT, Liu, PH, Chen, PS, Chen, YR, and Niu, DM. Comparative evaluation of AAV8 and AAV9 gene therapy in Fabry knockout (Gla(−/y)) and symptomatic (G3S(Tg/+)Gla(−/y)) murine models. Genes (Basel). (2025) 16. doi: 10.3390/genes16070766
89. Kaittanis, C, Teceno, T, Knight, A, Petibon, Y, Sandoval, P, Cohen, L, et al. Longitudinal imaging of therapeutic enzyme expression after gene therapy for Fabry disease using positron emission tomography and the radiotracer [(18)F]AGAL. Mol Ther. (2025) 33:4381–93. doi: 10.1016/j.ymthe.2024.11.021
90. Ruangsiriluk, W, Deshpande, M, Boukharov, N, Rajarshi, G, Mukherji, S, Yuan, S, et al. Reversing pathology in an aggravated Fabry mouse model using low-dose engineered human alpha-galactosidase a AAV gene therapy. Biomedicine. (2025) 13. doi: 10.3390/biomedicines13030577
91. Furumatsu, Y, Nagasawa, Y, Tomida, K, Mikami, S, Kaneko, T, Okada, N, et al. Effect of renin-angiotensin-aldosterone system triple blockade on non-diabetic renal disease: addition of an aldosterone blocker, spironolactone, to combination treatment with an angiotensin-converting enzyme inhibitor and angiotensin II receptor blocker. Hypertens Res. (2008) 31:59–67. doi: 10.1291/hypres.31.59
92. Warnock, DG, Thomas, CP, Vujkovac, B, Campbell, RC, Charrow, J, Laney, DA, et al. Antiproteinuric therapy and Fabry nephropathy: factors associated with preserved kidney function during agalsidase-beta therapy. J Med Genet. (2015) 52:860–6. doi: 10.1136/jmedgenet-2015-103471
93. Jain, G, and Warnock, DG. Blood pressure, proteinuria and nephropathy in Fabry disease. Nephron Clin Pract. (2011) 118:c43–8. doi: 10.1159/000320903
94. Piccolo, S, Casal, M, Rossi, V, Ferrigni, F, Piccoli, A, Bolzan, B, et al. Ventricular arrhythmias and primary prevention of sudden cardiac death in Anderson-Fabry disease. Int J Cardiol. (2024) 415:132444. doi: 10.1016/j.ijcard.2024.132444
95. Roy, A, Cumberland, MJ, O’Shea, C, Holmes, A, Kalla, M, Gehmlich, K, et al. Arrhythmogenesis in Fabry disease. Curr Cardiol Rep. (2024) 26:545–60. doi: 10.1007/s11886-024-02053-2
96. Elliott, PM, Anastasakis, A, Borger, MA, Borggrefe, M, Cecchi, F, Charron, P, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. (2014) 35:2733–79. doi: 10.1093/eurheartj/ehu284
97. Tognola, C, Ruzzenenti, G, Maloberti, A, Varrenti, M, Mazzone, P, Giannattasio, C, et al. Anderson-Fabry disease: an overview of current diagnosis, arrhythmic risk stratification, and therapeutic. Strategies Diagnostics. (2025) 15. doi: 10.3390/diagnostics15020139
98. Vijapurapu, R, Bradlow, W, Leyva, F, Moon, JC, Zegard, A, Lewis, N, et al. Cardiac device implantation and device usage in Fabry and hypertrophic cardiomyopathy. Orphanet J Rare Dis. (2022) 17:6. doi: 10.1186/s13023-021-02133-4
99. Sené, T, Lidove, O, Sebbah, J, Darondel, JM, Picard, H, Aaron, L, et al. Cardiac device implantation in Fabry disease: a retrospective monocentric study. Medicine (Baltimore). (2016) 95:e4996. doi: 10.1097/MD.0000000000004996
100. Stepien, KM, Broomfield, A, Cole, D, Deegan, PB, Forshaw-Hulme, S, Hughes, D, et al. Management of pain in Fabry disease in the UK clinical setting: consensus findings from an expert Delphi panel. Orphanet J Rare Dis. (2023) 18:203. doi: 10.1186/s13023-023-02796-1
101. Ries, M, Mengel, E, Kutschke, G, Kim, KS, Birklein, F, Krummenauer, F, et al. Use of gabapentin to reduce chronic neuropathic pain in Fabry disease. J Inherit Metab Dis. (2003) 26:413–4. doi: 10.1023/A:1025127723729
102. Gordon, KE, Ludman, MD, and Finley, GA. Successful treatment of painful crises of Fabry disease with low dose morphine. Pediatr Neurol. (1995) 12:250–1. doi: 10.1016/0887-8994(95)00007-3
103. Breyer, M, Lamer, S, Schlosser, A, and Üçeyler, N. Human sensory-like neuron surfaceome analysis. PLoS One. (2025) 20:e0320056. doi: 10.1371/journal.pone.0320056
104. Alqahtani, S, Seoane-Vazquez, E, Rodriguez-Monguio, R, and Eguale, T. Priority review drugs approved by the FDA and the EMA: time for international regulatory harmonization of pharmaceuticals? Pharmacoepidemiol Drug Saf. (2015) 24:709–15. doi: 10.1002/pds.3793
105. Giannuzzi, V, Conte, R, Landi, A, Ottomano, SA, Bonifazi, D, Baiardi, P, et al. Orphan medicinal products in Europe and United States to cover needs of patients with rare diseases: an increased common effort is to be foreseen. Orphanet J Rare Dis. (2017) 12:64. doi: 10.1186/s13023-017-0617-1
106. Ebbers, HC, Langedijk, J, Bouvy, JC, Hoekman, J, Boon, WP, de Jong, JP, et al. An analysis of marketing authorisation applications via the mutual recognition and decentralised procedures in Europe. Eur J Clin Pharmacol. (2015) 71:1237–44. doi: 10.1007/s00228-015-1904-1
107. Heinrich, R, Claus, F, and Schoenfelder, T. Health care costs of home care enzyme replacement therapy for patients with lysosomal storage diseases in Germany. Orphanet J Rare Dis. (2024) 19:462. doi: 10.1186/s13023-024-03492-4
108. Guest, JF, Jenssen, T, Houge, G, Aaseboe, W, Tøndel, C, and Svarstad, E. Modelling the resource implications of managing adults with Fabry disease in Norway favours home infusion. Eur J Clin Investig. (2010) 40:1104–12. doi: 10.1111/j.1365-2362.2010.02363.x
109. Guest, JF, Concolino, D, Di Vito, R, Feliciani, C, Parini, R, and Zampetti, A. Modelling the resource implications of managing adults with Fabry disease in Italy. Eur J Clin Investig. (2011) 41:710–8. doi: 10.1111/j.1365-2362.2010.02458.x
110. Nampoothiri, S, Yesodharan, D, Bhattacherjee, A, Ahamed, H, Puri, RD, Gupta, N, et al. Fabry disease in India: a multicenter study of the clinical and mutation spectrum in 54 patients. JIMD Rep. (2020) 56:82–94. doi: 10.1002/jmd2.12156
111. Czech, M, Baran-Kooiker, A, Atikeler, K, Demirtshyan, M, Gaitova, K, Holownia-Voloskova, M, et al. A review of rare disease policies and orphan drug reimbursement systems in 12 Eurasian countries. Front Public Health. (2019) 7:416. doi: 10.3389/fpubh.2019.00416
112. Wang, Y, Zhou, N, Li, B, Lv, Z, Duan, S, Li, X, et al. Utilization and affordability of health insurance coverage for rare disease drugs in a first-tier city in Northeast China from 2018 to 2021: a study based on the health insurance claims database. Int J Equity Health. (2024) 23:151. doi: 10.1186/s12939-024-02225-0
113. Suwattanapreeda, S, Hirunrassamee, S, Sooksriwong, C, Maluangnon, K, Chuachantra, T, Kuchaisit, K, et al. Unlocking access: a comprehensive analysis of medicines accessibility for rare diseases in Thailand. Orphanet J Rare Dis. (2025) 20:258. doi: 10.1186/s13023-025-03754-9
114. Douglas, CMW, and Grunebaum, S. Lessons learned from the Canadian Fabry disease initiative for future risk-sharing and managed access agreements for pharmaceutical and advanced therapies in Canada. Health Policy. (2024) 143:105044. doi: 10.1016/j.healthpol.2024.105044
115. Zhang, X, Zhou, T, Zhou, J, Zhang, D, Yang, Y, and Pan, J. Budget impact analysis of high-priced orphan medicinal products intended for the treatment of rare diseases in China: evidence from a densely populated metropolis of Chengdu. BMC Health Serv Res. (2024) 24:1123. doi: 10.1186/s12913-024-11632-6
116. Katsigianni, EI, and Petrou, P. A systematic review of economic evaluations of enzyme replacement therapy in lysosomal storage diseases. Cost Eff Resour Alloc. (2022) 20:51. doi: 10.1186/s12962-022-00369-w
117. Huang, Y, Yuan, H, and Huang, Z. Cost-effectiveness analysis of enzyme replacement therapy for the treatment of Chinese patients with fabry disease: a Markov model. Front Pharmacol. (2025) 16:1546018. doi: 10.3389/fphar.2025.1546018
Glossary
FD - Fabry Disease
LSD - Lysosomal storage disease
α-GAL - α-galactosidase A
Gb3 - Globotriosylceramide
Lyso-Gb3 - Globotriosylsphingosine
ERT - Enzyme replacement therapy
LVH - Left ventricular hypertrophy
Ccr - Creatinine clearance
eGFR - Estimated glomerular filtration rate
RCT - Randomized controlled trial
GCS - Glucose ceramide synthase
SSCE - Superficial skin capillary endothelium
AAV - Adeno-associated virus
ITRs - Inverted terminal repeat sequences
rAAV - Recombinant AAV
CNS - Central nervous system
CSF - Cerebrospinal fluid
LCA - Leber’s congenital amaurosis
ACEI - Angiotensin-converting enzyme inhibitor
ARB - Angiotensin receptor blocker
AT1 - Angiotensin II to receptor 1
SGLT2 - Sodium-glucose cotransporter-2
MRA - Mineralocorticoid receptor antagonist
ICD - Implantable cardioverter defibrillator
IPSC - Induced pluripotent stem cell
UHN - University Health Network
CFDI - Canadian Fabry Disease Initiative
NHA - National Health Account
QALY - Quality-adjusted life year
YFEOD - years free of end-organ damage
ICER - Incremental cost effectiveness ratio
Keywords: Fabry disease, enzyme replacement therapy, pharmacological chaperone therapy, substrate reduction therapy, mRNA therapy, AAV-mediated gene therapy
Citation: Zhang M and Wang C (2025) Therapeutic landscape of Fabry disease: advances and challenges from classical strategies to emerging therapies. Front. Med. 12:1662867. doi: 10.3389/fmed.2025.1662867
Edited by:
Piergiorgio Messa, University of Milan, ItalyReviewed by:
Yan Ouyang, Shanghai Jiao Tong University, ChinaMichal Nowicki, Medical University of Lodz, Poland
Copyright © 2025 Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chendan Wang, d3AwMDQyNDBAc3htdS5lZHUuY24=