Abstract
We present the case of a 78-year-old man with a complex overlap of inclusion body myositis (IBM) and Sjögren’s disease (SjD), complicated by interstitial lung disease (ILD) and esophageal dysfunction. The patient’s neuromuscular decline was managed with periodic intravenous immunoglobulins (IVIG), while his progressive ILD required a combined immunosuppressive and antifibrotic regimen, including prednisone. This case underscores the diagnostic and therapeutic challenges in managing autoimmune overlap syndromes, particularly the rare coexistence of IBM and SjD-related ILD. We highlight the rationale behind the treatment strategy and the importance of a tailored, multidisciplinary approach in addressing both muscular and pulmonary manifestations.
Introduction
Inclusion body myositis (IBM) is an idiopathic inflammatory myopathy with insidious onset, often presenting with progressive muscle weakness and dysphagia (1).
It is characterized by slowly progressive muscle weakness, frequently accompanied by dysphagia and poor response to immunosuppressive therapies (1).
Although rare, its association with connective tissue diseases such as Sjögren’s disease (SjD) has been reported in the literature (2, 3).
The overlap between IBM and Sjögren’s syndrome (SS) can complicate diagnosis and management, particularly when interstitial lung disease (ILD)—a known extraglandular manifestation of SjD—coexists.
This case report describes a challenging patient with IBM, SjD, and SjD-related ILD. Given the rarity of this specific overlap and the need for a coordinated, multidisciplinary treatment approach, we aimed to shed light on the clinical decision-making process, the rationale for the chosen therapeutic strategy, and the broader implications for managing autoimmune overlaps with pulmonary involvement.
Patient information
We present the case of a 78-year-old man, who was a former heavy smoker (50 cigarettes/day until 1990), with several comorbidities, including hypertension, cardiopathy, hypothyroidism, benign prostatic hyperplasia, osteoporotic L1 fracture, right ankle arthrodesis, suspected transient ischemic attack (TIA), occult hepatitis B virus (HBV) infection, and prior surgery for intestinal obstruction. The patient presented for the first time to the rheumatologic evaluation because of the occurrence of a clinical picture characterized by progressive dysphagia, muscle weakness, and dyspnea.
Clinical findings
Careful diagnostic workup was performed to investigate the diagnosis. In 2017 at the age of 70 years, a diagnosis of IBM was confirmed after a muscle biopsy. At disease onset, the clinical picture was characterized by severely impaired muscle strength; on physical examination, the patient showed a strength deficit of finger flexors, wrist extensors, and knee extensors. He also presented esophageal dysmotility, which was confirmed by double-contrast radiography (RX). The examination showed an altered passage of liquid barium contrast in the patient’s esophagus, indicating a muscular disorder. Examinations revealed a creati kinase of 984 U/L and an aldolase of 20.5 U/L, while myositis antibodies were negative. Electromyography showed signs of acute multiradicular denervation, and a magnetic resonance (MR) imaging also revealed muscle edema and fatty degeneration of the left vastus lateralis muscle and bilateral gastrocnemius muscles. The muscle biopsy revealed endomysial lymphocytic (CD8-positive T lymphocyte) inflammation, atrophic fibers, and multiple myofibers with rimmed vacuoles. No necrotic fibers were detected.
The patient did not report any constitutional or neurological symptoms.
Treatment with intravenous immunoglobulin (0.4 g/kg/day for 5 days) was proposed together with non-pharmacological treatment with cyclic physical rehabilitation. The patient experienced discrete but transient efficacy from the treatment and reported, in particular, a subjective improvement of dysphagia and muscle weakness, despite his need for a walking aid. He subsequently received intravenous immunoglobulin (IVIG) cycles approximately once a year to manage these symptoms. This treatment strategy led to a stabilization of his condition. Physical rehabilitation was also a crucial component of his care.
In June 2023, during a follow-up visit, the patient reported the onset of xerophthalmia, subjective xerostomia, and progressive effort dyspnea. Chest auscultation revealed diminished vesicular breath sounds over the mid-lung fields, absent at the bases, accompanied by bilateral crackles.
Diagnostic assessment
A high-resolution computed tomography (HRCT) was promptly requested, and it revealed the presence of diffuse reticular thickening of the subpleural interstitium extending bilaterally from the apex to the lung bases, particularly in the middle-lower third, with more concentrated areas in the posterior basal segments of both lower lobes, in the lateral segment of the middle lobe, and in the upper segment of the lingula. Global spirometry demonstrated a mild restrictive ventilatory impairment and a moderate reduction in diffusing capacity of the lung for carbon monoxide (DLCO) (forced vital capacity [FVC]: 77%, forced expiratory volume [FEV1]: 81%, single-breath diffusing capacity for carbon monoxide [DLCO-SB]: 41%, and alveolar volume-adjusted diffusing capacity [DLCO-VA]: 72%).
In the suspicion of secondary lung involvement due to connective tissue disease, an extractable nuclear antigen (ENA) blot was performed, which resulted in positive Ro52 antibodies.
This finding, together with the presence of subjective and objective sicca syndrome, raised the suspicion of SjD diagnosis complicated by ILD. Schirmer’s test was performed, revealing hypolacrimia (2 mm in the right eye and 3 mm in the left eye). To complete the diagnosis, a biopsy of the minor salivary glands (grade 2 according to Chisolm and Mason and focus score = 1) was taken. The patient satisfied the 2016 American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) classification criteria for Sjögren’s Disease (4, 5). A diagnosis of SjD with interstitial lung disease (ILD) was confirmed. Local treatment for sicca syndrome was initiated. No immunosuppressive therapy was initiated; instead, we preferred to closely monitor pulmonary involvement through a multidisciplinary evaluation to better investigate the clinical behavior.
In April 2024, the patient reported subjective worsening of dyspnea. An HRCT was repeated and revealed progression of the interstitial lung fibrosis with radiologic classification of probable usual interstitial pneumonia (UIP) pattern, characterized by the appearance of multiple millimetric submantellar bronchiolectasis, predominantly in both lower lobes, in the lingular lobe, and in the middle lobe, concomitant with finer interstitial reticular thickening, which was also present in the upper lobes (Figure 1).
Figure 1

High-resolution computed tomography (HRCT) scans performed in June 2024 show multiple millimetric subpleural bronchiolectases affecting both lower lobes, the lingula, and the middle lobe. There is also a concomitant diffuse fine interstitial reticular thickening, which is also present in the upper lobes.
A pulmonary function test showed a stable restrictive pattern with stable functional parameters (FVC: 84% and FEV1: 86%). However, the carbon monoxide diffusion test revealed a worsening of values (DLCO-SB: 36% and DLCO-SB 61%). An echocardiography excluded indirect signs of pulmonary hypertension.
The INBUILD criteria for the definition of progressive fibrosing ILD (PF-ILD) were satisfied (5).
Therapeutic intervention
Following multidisciplinary assessment and the diagnosis of SjD-associated ILD, the patient was treated with combination therapy aimed at controlling both the inflammatory and fibrosing aspects of the disease. Systemic corticosteroid therapy was initiated with prednisone at a dose of 50 mg daily (0.75 mg/kg/day), followed by a slow tapering schedule over 6 months. The tapering regimen involved progressive dose reductions every 2–3 weeks, in order to minimize long-term corticosteroid-related adverse effects. Intravenous cyclophosphamide was administered every month for a period of 6 months according to the National Institutes of Health (NIH) protocol. The patient received a dose of 1.2 g per infusion, calculated based on a body surface area-adjusted range of 0.5–1 g/m2. This induction regimen was selected due to its established efficacy in connective tissue disease-associated ILD, particularly in patients with evidence of disease progression or severe inflammatory activity (6). Nintedanib was initially prescribed at the standard dose of 150 mg twice daily, based on current evidence supporting its use in progressive fibrosing ILD (7), including connective tissue disease-associated ILDs.
In accordance with current clinical practice for the management of CTD-ILD and to support further steroid tapering, mycophenolate mofetil at a dose of 2 g/day was prescribed as maintenance therapy.
A few months into therapy, the patient developed gastrointestinal symptoms consistent with dyspepsia, a commonly reported adverse effect of nintedanib.
In response, and prior to clinical consultation, the dosage of nintedanib was reduced to 100 mg bid. This adjustment resulted in partial improvement of symptoms.
Given occult HBV infection (HBsAg− and anti-HBc+), hepatic function was monitored during immunosuppressive therapy according to current guidelines.
Follow-up and outcomes
The patient reported subjective improvement in swallowing and energy levels following IVIG cycles. He expressed a preference for continued clinical-radiological follow-up rather than invasive diagnostic procedures. Furthermore, after immunosuppressive and antifibrotic therapy, the patient experienced a stabilization of dyspnea.
However, these perceived improvements contrasted with objective findings during follow-up.
In November 2024, an HRCT scan showed mild disease progression, with increased traction bronchiolectasis in the anterobasal and laterobasal segments of the left lower lobe, the anterior segment of the upper lobe, and the apical segment of the right lower lobe (Figure 2).
Figure 2
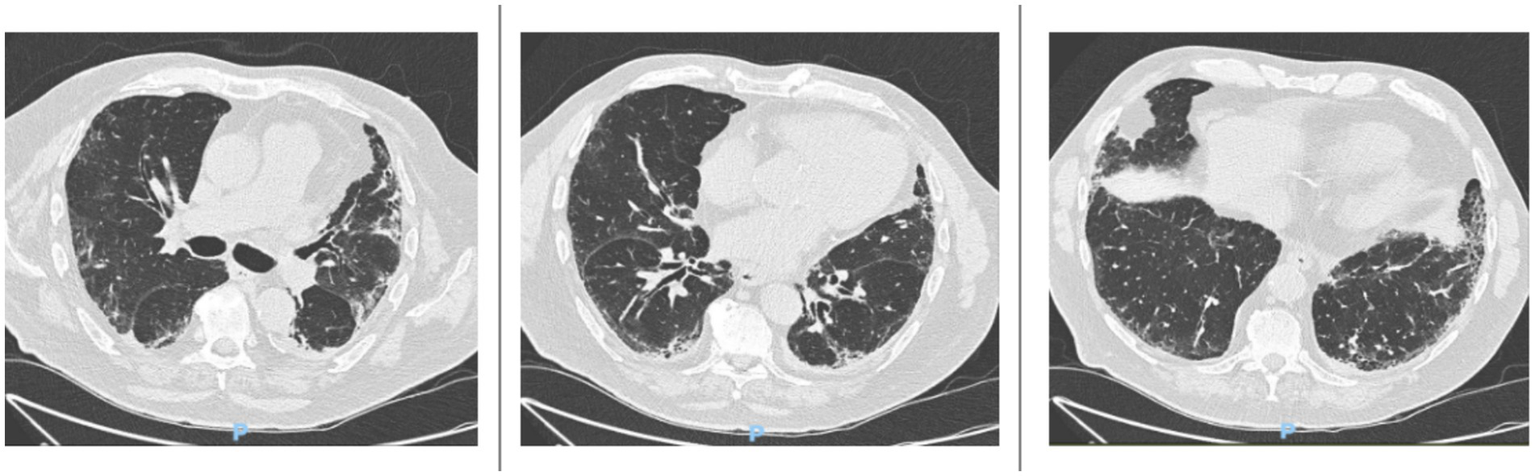
HRCT scans taken in November 2024 reveal a mild progression of interstitial lung disease (ILD). There is a slight increase in traction bronchiolectasis, which is more pronounced in the anterobasal and laterobasal segments of the left lower lobe, the anterior segment of the upper lobe, and the apical segment of the right lower lobe.
A 6-min walk test (6MWT) performed during the same period revealed desaturation to 90% on room air after 160 m. The test was limited by the patient’s reduced physical condition.
This apparent discrepancy between the patient’s subjective improvement and the objective evidence of functional and radiological decline highlights the limitations of relying solely on patient-reported outcomes. The patient’s limited mobility prevented an accurate assessment of dyspnea progression, a critical factor for considering therapeutic escalation or for suggesting functional and radiological evaluations beyond routine monitoring and follow-up.
In agreement with the patient, the nintedanib dose was adjusted to 100 mg twice daily to minimize gastrointestinal side effects while maintaining antifibrotic efficacy (8). As of the most recent follow-up, the patient continues combination therapy.
Multidisciplinary follow-up is ongoing.
Discussion
This case highlights the diagnostic and therapeutic challenges arising from the coexistence of IBM and SjD, particularly when further complicated by ILD.
Although IBM is classified among idiopathic inflammatory myopathies (IIM), it presents with a distinct clinical and histopathological profile. It typically follows an indolent but progressive course, with characteristic involvement of both proximal and distal muscle groups, especially the quadriceps and finger flexors, and is resistant to glucocorticoid therapy.
Dysphagia, as observed in our patient, is a common and often disabling manifestation, resulting from both oropharyngeal and esophageal muscle involvement.
The esophageal dysmotility observed in our patient, confirmed by double-contrast studies, is a hallmark of IBM. Unlike other IIMs, conventional anti-inflammatory and immunosuppressive drugs are often ineffective in IBM; however, intravenous immunoglobulin (IVIG) therapy has demonstrated subjective improvements in muscular weakness and dysphagia in some patients (9). In our case, despite its limited and transient effect, IVIG yielded similar benefits.
SjD is an autoimmune disease primarily affecting exocrine glands such as the salivary and lacrimal glands, typically following an indolent course. However, its systemic involvement is well-recognized, leading to severe manifestations such as ILD, pericarditis, neuropathies, renal insufficiency, and vasculitis. Muscle involvement can also occur, and an association with IIMs has been described. Among patients with both myositis and SjD, IBM is the most common subtype of IIM, with a reported prevalence ranging from 0.5 to 22% (10, 11). Conversely, SjD is also the most frequent connective tissue disease associated with IBM, occurring in 10% of IBM patients (12).
Emerging literature suggests possible shared pathogenic mechanisms between these two diseases, including a common human leukocyte antigen (HLA) background (e.g., HLA A1-B8-DR3-DQ2), the presence of anti-cN1A antibodies (frequently positive in both conditions), and a role for cytotoxic T cells, as supported by the observed association of both diseases with T-cell leukemia and lymphoma (13).
The patient presented with a UIP-like pattern of ILD on high-resolution computed tomography (HRCT), a radiological finding associated with a more aggressive disease course and poorer prognosis in SjD-related ILD (14). Although the literature indicates that UIP-pattern ILD in SjD typically leads to progressive functional decline (15), our patient showed subjective improvement in dyspnea and stabilization of respiratory symptoms following combined antifibrotic and immunosuppressive therapy. However, this symptomatic relief occurred in parallel with mild radiological and functional deterioration, including increased traction bronchiolectasis and decreased exercise tolerance on the 6-min walk test. This apparent contradiction highlights the difficulty in evaluating the patient’s subjective symptoms due to his physical limitations. On the other hand, the mild progression of the interstitial lung disease (ILD) is not in opposition to the treatment’s effectiveness. Given the terrible prognosis of UIP pattern-ILD, the primary goal of therapy is to slow its progression. Therefore, a close follow-up is necessary to accurately study the disease’s course and the patient’s response to treatment.
The coexistence of IBM and ILD also complicates treatment decisions. In our case, the choice of cyclophosphamide for induction therapy was guided by its relatively rapid onset of action and efficacy in progressive connective tissue disease-associated ILD. Although rituximab is also an option, its slower therapeutic onset and less clear benefit for pulmonary involvement influenced our decision. Cyclophosphamide was administered according to the NIH protocol and followed by maintenance therapy with mycophenolate mofetil, which is supported by data from systemic sclerosis-ILD and other CTD-ILDs for maintaining lung function and reducing corticosteroid exposure (16).
Antifibrotic therapy with nintedanib was introduced early due to radiological evidence of PF-ILD. Nintedanib has demonstrated efficacy in slowing FVC decline in patients with progressive fibrosing ILDs, including those associated with autoimmune diseases (17).
Due to gastrointestinal intolerance, the dose was reduced to 100 mg twice daily—a common adjustment in clinical practice that balances tolerability with therapeutic effect.
The patient’s limited mobility precluded an accurate assessment of dyspnea progression, which is a critical factor when considering therapeutic escalation.
For instance, reduced physical capacity due to muscle involvement can hinder the accurate assessment of respiratory function and limit rehabilitation potential. Additionally, dysphagia due to IBM increases the risk of aspiration, which may mimic or exacerbate pulmonary symptoms, potentially confounding ILD monitoring and management.
This case highlights the importance of a multidisciplinary approach involving rheumatologists, pulmonologists, and radiologists in both diagnostic and therapeutic decision-making. Although invasive diagnostic procedures such as bronchoalveolar lavage or lung biopsy could have provided additional diagnostic precision, these were not performed due to patient preference and clinical risk assessment. Nevertheless, longitudinal HRCT imaging and functional assessments allowed us to monitor disease evolution and guide therapy.
Finally, it is important to underline how the combination of immunosuppressive and antifibrotic therapy can change the outcome of these complex patients, leading to the stabilization of the clinical, functional, and radiological picture of interstitial lung involvement. With the emergence of new therapies (such as nerandomilast) (18), we hope to improve the patient’s condition if therapeutic escalation is needed.
Patient perspective
The patient reported subjective improvement in swallowing and energy levels following IVIG cycles and expressed a preference for continued clinical-radiological follow-up over invasive diagnostic procedures. Furthermore, after immunosuppressive and antifibrotic therapy, the patient experienced a stabilization of dyspnea.
Statements
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Comitato etico Area Vasta Emilia Nord. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
EC: Writing – review & editing, Writing – original draft. FC: Writing – review & editing, Investigation. IC: Investigation, Writing – review & editing, Validation, Project administration. FF: Investigation, Writing – review & editing, Supervision, Project administration, Data curation. GB: Project administration, Writing – review & editing, Funding acquisition, Investigation. CS: Validation, Supervision, Writing – review & editing. AM: Supervision, Writing – review & editing, Validation, Investigation.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer EB declared a past co-authorship with author CS.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1.
Dimachkie MM Barohn RJ . Inclusion body myositis. Curr Neurol Neurosci Rep. (2013) 13:321. doi: 10.1007/s11910-012-0321-4
2.
Giannini M Felten R Gottenberg JE Geny B Meyer A . Inclusion body myositis and Sjögren’s syndrome: the association works both ways. Acta Neuropathol Commun. (2022) 10:152. doi: 10.1186/s40478-022-01443-3
3.
Misterska-Skóra M Sebastian A Dzięgiel P Sebastian M Wiland P . Inclusion body myositis associated with Sjögren’s syndrome. Rheumatol Int. (2013) 33:3083–6. doi: 10.1007/s00296-012-2556-4
4.
Shiboski CH Shiboski SC Seror R Criswell LA Labetoulle M Lietman TM et al . 2016 American college of rheumatology/european league against rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Arthritis Rheumatol. (2017) 69:35–45. doi: 10.1002/art.39859
5.
Wells AU Flaherty KR Brown KK Inoue Y Devaraj A Richeldi L et al . Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. (2020) 8:453–60. doi: 10.1016/S2213-2600(20)30036-9
6.
Sullivan KM McSweeney PA Nash RA . Cyclophosphamide in scleroderma lung disease. N Engl J Med. (2006) 355:1173–4. doi: 10.1056/NEJMc061920
7.
Flaherty KR Wells AU Cottin V Devaraj A Walsh SLF Inoue Y et al . Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. (2019) 381:1718–27. doi: 10.1056/NEJMoa1908681
8.
Ruaro B Salotti A Reccardini N Kette S Da Re B Nicolosi S et al . Functional progression after dose suspension or discontinuation of Nintedanib in idiopathic pulmonary fibrosis: a real-life multicentre study. Pharmaceuticals (Basel). (2024) 17. doi: 10.3390/ph17010119
9.
Needham M Mastaglia FL . Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol. (2007) 6:620–31. doi: 10.1016/S1474-4422(07)70171-0
10.
Astouati Q Machet T Houssais C Noury JB Allenbach Y Gallay L et al . Inclusion-body myositis associated with Sjögren’s disease: clinical characteristics and comparison with other Sjögren-associated myositis. Rheumatology (Oxford). (2025) 64:1431–6. doi: 10.1093/rheumatology/keae129
11.
Felten R Giannini M Nespola B Lannes B Levy D Seror R et al . Refining myositis associated with primary Sjögren’s syndrome: data from the prospective cohort ASSESS. Rheumatology (Oxford). (2021) 60:675–81. doi: 10.1093/rheumatology/keaa257
12.
Benveniste O Guiguet M Freebody J Dubourg O Squier W Maisonobe T et al . Long-term observational study of sporadic inclusion body myositis. Brain. (2011) 134:3176–84. doi: 10.1093/brain/awr213
13.
Zeng L Chen K Xiao F Zhu CY Bai JY Tan S et al . Potential common molecular mechanisms between Sjögren syndrome and inclusion body myositis: a bioinformatic analysis and in vivo validation. Front Immunol. (2023) 14:1161476. doi: 10.3389/fimmu.2023.1161476
14.
Luppi F Sebastiani M Silva M Sverzellati N Cavazza A Salvarani C et al . Interstitial lung disease in Sjögren’s syndrome: a clinical review. Clin Exp Rheumatol. (2021) 38:S291–300.
15.
Manfredi A Sambataro G Rai A Cerri S Sambataro D Vacchi C et al . Prevalence of progressive fibrosing interstitial lung disease in patients with primary Sjogren syndrome. J Pers Med. (2024) 14:708. doi: 10.3390/jpm14070708
16.
Volkmann ER Tashkin DP Li N Roth MD Khanna D Hoffmann-Vold AM et al . Mycophenolate mofetil versus placebo for systemic sclerosis-related interstitial lung disease: an analysis of scleroderma lung studies I and II. Arthritis Rheumatol. (2017) 69:1451–60. doi: 10.1002/art.40114
17.
Johnson SR Bernstein EJ Bolster MB Chung JH Danoff SK George MD et al . 2023 American College of Rheumatology (ACR)/American College of Chest Physicians (CHEST) Guideline for the Treatment of Interstitial Lung Disease in People with Systemic Autoimmune Rheumatic Diseases. Arthritis Care Res (Hoboken). (2024) 76:1051–69. doi: 10.1002/acr.25348
18.
Maher TM Assassi S Azuma A Cottin V Hoffmann-Vold AM Kreuter M et al . Nerandomilast in patients with progressive pulmonary fibrosis. N Engl J Med. (2025) 392:2203–14. doi: 10.1056/NEJMoa2503643
Summary
Keywords
inclusion body myositis, Sjögren’s disease, interstitial lung disease, nintedanib, dysphagia
Citation
Cocchiara E, Cozzini F, Chiapparoli I, Falco F, Bajocchi G, Salvarani C and Manfredi A (2025) Case Report: Progressive interstitial lung disease secondary to Sjögren’s disease in a patient with inclusion body myositis complicated by dysphagia—a multidisciplinary approach and therapeutic challenges. Front. Med. 12:1683606. doi: 10.3389/fmed.2025.1683606
Received
11 August 2025
Accepted
22 October 2025
Published
11 November 2025
Volume
12 - 2025
Edited by
Ilias C. Papanikolaou, General Hospital of Corfu, Greece
Reviewed by
Mansoor C. Abdulla, Sultan Qaboos Hospital, Oman
Emanuele Bizzi, Vita-Salute San Raffaele University, Italy
Updates
Copyright
© 2025 Cocchiara, Cozzini, Chiapparoli, Falco, Bajocchi, Salvarani and Manfredi.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emanuele Cocchiara, Emanuele.cocchiara@ausl.re.it
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.