


- 1 Department of Biochemistry I – Receptor Biochemistry, Ruhr University Bochum, Bochum, Germany
- 2 Development and Plasticity of the Nervous System, DFG Graduate School 736, Ruhr University Bochum, Bochum, Germany
- 3 Ruhr University Research School, Ruhr University Bochum, Bochum, Germany
Ionotropic glutamate receptors are major players in synaptic transmission and are critically involved in many cognitive events. Although receptors of different subfamilies serve different functions, they all show a conserved domain topology. For most of these domains, structure–function relationships have been established and are well understood. However, up to date the role of the transmembrane domain C in receptor function has been investigated only poorly. We have constructed a series of receptor chimeras and point mutants designed to shed light on the structural and/or functional importance of this domain. We here present evidence that the role of transmembrane domain C exceeds that of a mere scaffolding domain and that several amino acid residues located within the domain are crucial for receptor gating and desensitization. Furthermore, our data suggest that the domain may be involved in receptor interaction with transmembrane AMPA receptor regulatory proteins.
Introduction
Glutamate is the most abundant excitatory neurotransmitter in the vertebrate CNS and plays key roles in synaptoplastic events like long-term potentiation and depression. The superfamily of ionotropic glutamate receptors (iGluRs) in mammals consists of 18 members, and this diversity is even increased by alternative splicing, RNA editing, and posttranslational modifications (Sommer et al., 1990; Hollmann and Heinemann, 1994; Rosenmund et al., 1998; Seeburg et al., 1998; Hollmann, 1999). The iGluRs are classified into four subfamilies distinguishable by function, sequence identity, and pharmacological properties: The α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptors GluA1 to GluA4 are the main mediators of fast synaptic transmission, whereas the N-methyl-D-aspartate (NMDA) receptors (GluN1, GluN2A through GluN2D, and GluN3A plus GluN3B) are silent under resting conditions; they become activated when the membrane is depolarized beyond −20 mV, e.g., by AMPA receptor activity. The kainate receptors GluK1 to GluK5 are located presynaptically as well as postsynaptically, and have been reported to operate in dual fashion as both ionotropic and metabotropic receptors (Lerma, 2003; Rozas et al., 2003). The fourth subfamily of glutamate receptors comprises the “orphan” receptors GluD1 and GluD2; up to now, neither their physiological function nor their ligand(s) have been identified unequivocally. Please note that the new glutamate receptor nomenclature recently recommended by IUPHAR (Collingridge et al., 2009) is used throughout.
All iGluRs share a common domain topology and quaternary structure (Figure 1A); in these characteristics, the iGluR superfamily differs greatly from other ligand-gated ion channels (LGICs) such as acetylcholine (ACh), γ-aminobutyric acid (GABA), or glycine receptors. While these receptor complexes are comprised of five subunits with four transmembrane domains each, functional iGluR complexes are tetramers of subunits that possess three true transmembrane domains (TMDs A to C) and a pore loop between TMDs A and B that does not traverse the membrane. Receptors are tetrameric complexes with the subunit stoichiometry depending on the subfamily: while some AMPAR subunits are capable of forming homomeric as well as heteromeric receptors that are functional when expressed heterologously, NMDARs are imperatively heteromeric (McBain and Mayer, 1994; Wenthold et al., 1996).
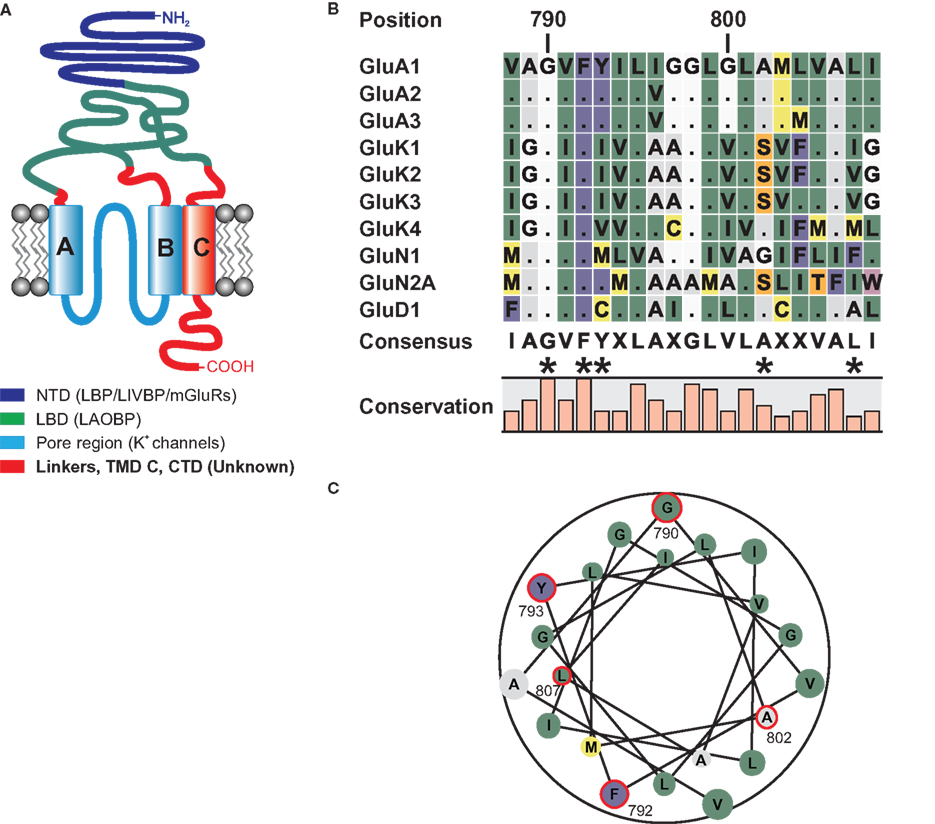
Figure 1. (A) Domain topology of ionotropic glutamate receptors. Other proteins homologous to the domains are given in brackets. The NTD shares homologies with the bacterial leucine binding protein (LBP) and leucine–isoleucine–valine binding protein (LIVBP), as well as the LBD of metabotropic glutamate receptors (O’Hara et al., 1993). The LBD of iGluRs shares sequence similarities with the bacterial leucine–arginine–ornithine binding protein (LAOBP) (Kuryatov et al., 1994), and the pore region shows structural homology to the pore of potassium channels with an inverted orientation (Kuner et al., 2003). For the linker regions, the TMD C, and the CTD no homologous sequences have been identified. (B) Local sequence alignment of the TMD C regions of all iGluR subunits that were used as TMD C donors in domain transplantation constructs. Two residues, G790 and F792 are conserved among all 18 iGluR subunits. Residues that were altered in this study are indicated by asterisks. (C) Helical wheel representation of the TMD C of GluA1. Residues investigated in this study are encircled in red. Numbering refers to the mature GluA1 sequence, without the signal peptide.
The domain composition of iGluRs is modular and strictly conserved among the four subfamilies. For nearly all domains, homologies to other proteins have been found (Figure 1A). The extracellular portion of the protein consists of the N-terminal domain (NTD) and the ligand binding domain (LBD). The latter is not contiguous in sequence, its two segments S1 and S2 are interrupted by the pore region that is comprised of TMDs A and B and the pore loop between them. The LBD binds the agonist and undergoes a conformational change that is relayed to the pore region, ultimately leading to channel opening. The third TMD, TMD C, is linked to the intracellularly located C-terminal domain (CTD). The CTD is highly variable among the glutamate receptors, both in size and in sequence, and is subject to alternative splicing. It has a pivotal role in trafficking, membrane localization, and interaction with cytoskeletal proteins (Sheng and Lee, 2001; Collingridge et al., 2004). In contrast to the CTD, the role and function of the TMD C remain unclear. In GluN2 subunits, several amino acids residing in TMD C appear to be involved in receptor desensitization and in receptor modulation by ethanol (Ren et al., 2003a,b). Additionally, an ER retention signal in GluN2B is masked by the TMD C of GluN1 to enable ER export (Horak et al., 2008). The functional role of the TMD C in AMPA receptors, however, has not been investigated up to now. We therefore conducted a series of experiments designed to elucidate the function of this domain as a whole as well as of single amino acids residing within it. In doing so, we exploited the modular design of iGluRs that allows for homologous domains to be interchanged between subunits from different subfamilies, conveying the properties of the donor domain to the accepting receptor. The feasibility of this domain-swapping approach has been shown before for other domains such as the pore region (Strutz-Seebohm et al., 2003; Hoffmann et al., 2006a,b; Tapken and Hollmann, 2008; Villmann et al., 2008) or the LBD (Schmid et al., 2009).
Two basic roles are conceivable for the TMD C in AMPA receptors: On the one hand, it could constitute a simple membrane anchor. Since the CTD is extremely important for correct trafficking and intracellular localization of the receptor (Hayashi et al., 2000), it needs to be located on the cytoplasmic side of the plasma membrane, a position established and maintained by the TMD C. If membrane anchoring was indeed the key function of TMD C, the sequence of the domain should be of minor importance, as long as it forms a membrane-spanning alpha helix. On the other hand, it is easily imaginable that the domain is actively involved in receptor gating and desensitization, as has been proposed for NMDA receptors (Ren et al., 2003a,b; Jang et al., 2004). In this case, the transplantation of the TMD C from another iGluR subfamily into an AMPA receptor should alter the functional properties of the receptor. Furthermore, the TMD might also be involved in interactions with the transmembrane AMPA receptor regulatory protein (TARP) family of proteins, though no evidence for this has been reported to date. As depicted in Figure 1B, the amino acid sequence of the TMD C is highly conserved within the subfamilies, whereas it is only moderately conserved between them. There are, however, two residues, G790 and F792, which are conserved among all 18 iGluR subunits. The helical wheel representation (Figure 1C) shows that these two residues are located on opposing sides of the helix. Please note that numbering throughout the article refers to the amino acid positions in the sequence of putatively mature GluA1, without the signal peptide.
Materials and Methods
Molecular Biology
All constructs were generated by PCR-directed mutagenesis using oligonucleotides bearing the desired mutations (Microsynth, Balgach, Switzerland). Domain borders were defined based on the Swiss-Prot entries for the respective subunits from Rattus norvegicus (Accession numbers: GluA1: P19490; GluA2: P19491; GluA3: P19492; GluK2: P42460; GluN1: P35439; GluN2A: Q00959; GluD1: Q62640). All chimeras and point mutants were inserted into the Xenopus oocyte expression vector pSGEM.
cRNA Synthesis
cDNA was transcribed using the mMESSAGE mMACHINE Kit (Ambion, Austin, TX, USA) according to the protocol provided. RNA integrity was checked via denaturing agarose gel electrophoresis. RNA concentration was determined photometrically using a NanoPhotometer (Implen GmbH, Munich, Germany).
Heterologous Expression in Xenopus Oocytes
Stage V or VI oocytes were surgically removed from the ovaries of anesthetized Xenopus laevis and prepared as described previously (Schmidt et al., 2006). For homomeric expression of mutants, 8 ng (40 nl) of cRNA were injected within 24 h after surgery. For coexpression with the TARP γ2, 8 ng (40 nl) receptor cRNA and 0.8 ng (40 nl) TARP cRNA were mixed and coinjected. Electrophysiological recordings were carried out 4–5 days after injection. Two electrode voltage clamping was performed using a TurboTec-10CX amplifier (NPI electronic, Tamm, Germany) controlled by Pulse software (HEKA Elektronik, Lambrecht, Germany). Borosilicate glass capillaries were pulled to resistances of 1–3 MΩ and filled with 3 M KCl (potential electrode) or 3 M CsCl (current electrode). Oocytes were clamped at −70 mV and continuously perfused with calcium-free Ringer’s solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM MgCl2, 10 mM HEPES–NaOH, pH 7.2) to avoid artifacts evoked by endogenous Ca2+-gated chloride channels. The agonists glutamate (Sigma, Taufkirchen, Germany) and kainate (Ascent Scientific, Bristol, UK) and glutamate in combination with trichlormethiazide (TCM) (Sigma) were applied for 20 s.
Labeling of Cell-Surface Protein, Western Blotting, and Quantification
Oocytes were used for plasma membrane-resident protein analysis 5 days after cRNA injection following a previously described protocol (Hollmann et al., 1994). Isolation of surface membrane proteins was achieved by labeling with biotinylated concanavalin A followed by streptavidin–agarose-mediated precipitation of glycosylated surface proteins. Crude protein mixtures were separated by SDS–PAGE and blotted onto HyBond ECL-nitrocellulose membranes (GE Healthcare, Chalfont St. Giles, UK) as described previously (Villmann et al., 1999). The detection of proteins was performed using a primary antibody (diluted 1:1000 in TBS) directed against the C-terminus of GluA1 (kind gift from R.L. Huganir, Johns Hopkins University of Medicine, Baltimore, MD, USA). Horseradish peroxidase-labeled mouse anti-rabbit IgG (Pierce poly-HRP, Fisher Thermo Scientific, Waltham, MA, USA) was used as secondary antibody (dilution 1:20,000 in TBS). Immunoreactive bands were visualized using the Pierce SuperSignal pico or SuperSignal femto ECL kits (Fisher Thermo Scientific, Waltham, MA, USA).
Western blots were quantified using the software ImageJ, intensity values were normalized to the wild type band in each blot.
Statistical Analyses
To ensure comparability between different oocyte batches, all recorded currents were normalized batch-wise. Agonist-induced currents of GluA1 wild type were averaged for each batch; subsequently, all recorded currents from that batch were normalized to this average. Significances were calculated using either an unpaired, two-tailed t-test or a one-way ANOVA with Dunnett’s post test. Statistical analyses were performed using Prism 5.0 (GraphPad Software, San Diego, CA, USA).
Structure Analysis
Analysis of the GluA2 crystal structure (PDB# 3KG2, Sobolevsky et al., 2009) was performed using PyMOL (DeLano Scientific, San Francisco, CA, USA).
Results
The Sequence of the TMD C of GluA1 is Crucial for Receptor Function, but not for Assembly and Membrane Integration
The initial question was whether the TMD C constitutes a solely structural domain that simply serves a scaffolding function, or whether its amino acids are involved in specific aspects of receptor function. To test this, constructs were generated in which the TMD C of GluA1 was replaced by the sequence of TMD A from the same receptor, resulting in an A-B-A TMD topology.
When heterologously expressed in Xenopus oocytes, the construct did not yield functional ion channels. Upon application of 300 μM glutamate or 150 μM kainate, no currents could be detected (data not shown). This outcome was unaffected by the coexpression of the TARP γ2, which increases both glutamate and kainate-evoked currents of wild type GluA1 by at least one order of magnitude. Western blot analysis of plasma membrane surface proteins, however, revealed that GluA1(A-B-A) was inserted into the membrane in the absence and presence of γ2 (Figure 2). Replacement of the TMD C by the TMD A sequence resulted in an altered electrophoretic mobility of the protein. A shortening of the protein as a result of the domain exchange can be ruled out, since the antibody used for detection binds to the CTD of the receptor. A chain termination in the TMD C region hence would have caused the signal to disappear. It has long been known that the apparent molecular weight of a peptide can vary from the theoretical weight (Dunker and Rueckert, 1969) and that the exchange of single amino acid residues can cause shifts in mobility up to several kDa (Fasano et al., 1984; Armstrong and Roman, 1992).
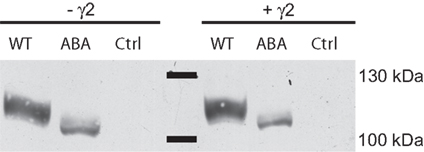
Figure 2. Membrane expression analysis of GluA1 wild type (WT) and GluA1(TMD)ABA in the absence and presence of γ2. Control lanes show preparations of oocytes injected with water or γ2.
The TMD C of GluA1 can be Replaced by the Homologous Domains from Other iGluR Families Without Losing Functionality
The next set of constructs was made to clarify whether domain transplantation between iGluR subunits is possible for the TMD C as it is for other domains. To this end, chimeric receptors were generated in which the TMD C of GluA1 was replaced by that of either GluA2, GluA3, GluK1, GluK2, GluK3, GluK4, GluN1, GluN2A, or GluD1. The chimeras were heterologously expressed in Xenopus oocytes, and agonist-evoked currents were recorded. Since GluA1 readily forms homomeric receptors in oocytes, the chimeric constructs could be expressed without additional subunits.
When expressed in the absence of a TARP, the chimeras GluA1-(TMDC)GluA2, GluA1-(TMDC)GluA3, and GluA1-(TMDC)GluK1 yielded currents that were significantly larger in amplitude than background currents seen in uninjected control oocytes (Figure 3A). GluA1-(TMDC)GluK2 produced kainate-evoked currents corresponding to 25% of the wild type, which correlates with reduced protein expression (Figure 3B). When the TARP γ2 was coexpressed in order to boost GluR expression, nearly all constructs produced distinct current responses significantly different from controls upon application of kainate (Figure 3A, Table 1). Exceptions were the chimeras GluA1-(TMDC)GluN1, GluA1-(TMDC)GluN2A, and GluA1-(TMDC)GluD1. Although the latter did not reach significance levels, kainate-induced currents still amounted to 23% of wild type currents. In contrast, the replacement of the TMD C of GluA1 by that of GluN1 or GluN2A caused a nearly complete loss of function. Western blot analysis revealed that the GluN2A chimera was properly delivered to the membrane, as was the GluD1 chimera, whereas GluA1-(TMDC)GluN1 exhibited a broadened band on the Western blot (Figure 3B). However, densitometric quantification (Figure 3C) showed that the total amount of protein in the lane equaled that of the wild type.
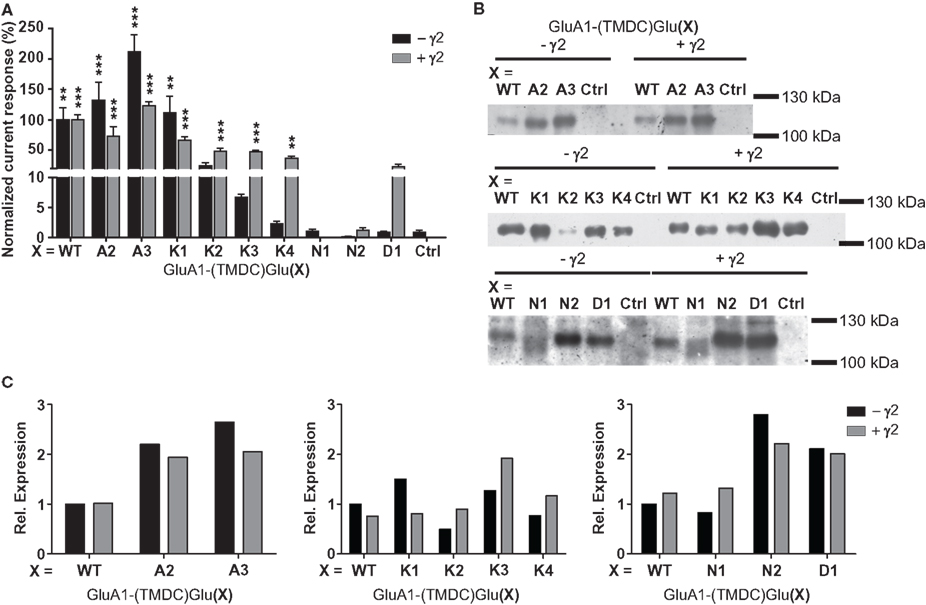
Figure 3. (A) Kainate-induced current amplitudes of TMD C chimeras in the absence and presence of γ2. Currents were normalized to the wild type; error bars show the SEM. Asterisks represent significant differences to controls (uninjected or γ2-injected oocytes, respectively). **p > 0.01, ***p > 0.001, one-way ANOVA with Dunnett’s post test. (B) Western blot analysis of expression of the TMD C chimeras. All constructs are detectable in the membrane fraction, independently of TARP expression. However, GluA1(TMDC)GluK2 is only weakly present in the absence of γ2, and GluA1(TMDC)GluN1 exhibits band broadening. Ctrl, controls injected with water. (C) Densitometric quantification of the Western blots shown in (B). Since the membrane protein preparations were performed in three different batches of oocytes, band intensities were normalized to GluA1 wild type in absence of γ2 for each batch.
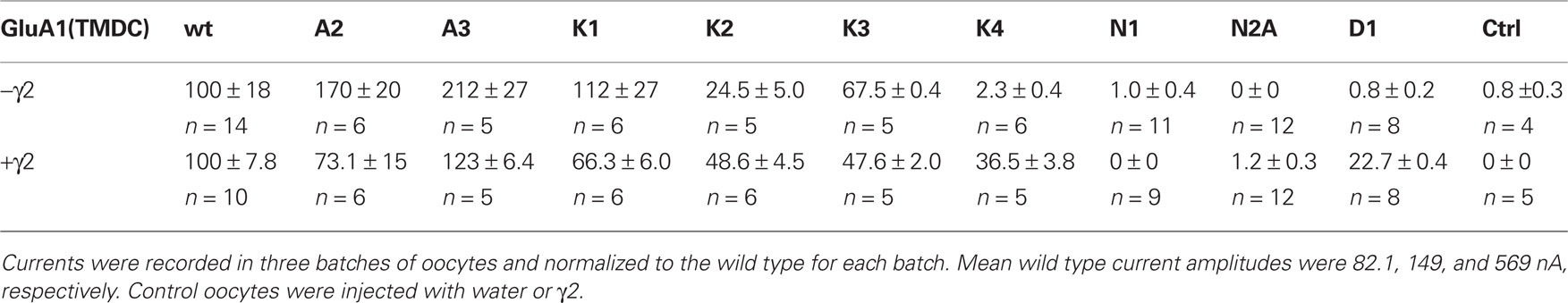
Table 1. Normalized kainate-induced currents of all generated chimeras in presence and absence of the TARP γ2, all values in % ± SEM.
A Single Amino Acid Exchange is Sufficient to Restore Receptor Functionality in a Non-Functional TMD C Chimera
Sequence comparison reveals that the TMD C of GluN1 differs in 14 out of 21 amino acid residues from that of GluA1. However, only four of these residues are exclusive to GluN1 and are not found at the corresponding positions in the TMD C of any other glutamate receptor subunit. Hence, three constructs were made in which the residues M793, G802, and F807 in the chimera GluA1-(TMDC)GluN1 were replaced by the respective residues found in GluA1. Analysis of the fourth residue, L794, was omitted since its mutation would have constituted a leucine to isoleucine exchange that was considered, in all likelihood, to be of minor impact. When expressed in oocytes, all constructs were delivered to the membrane at levels much higher than the initial chimera GluA1-(TMDC)GluN1, as shown by Western blot analysis (Figure 4D).
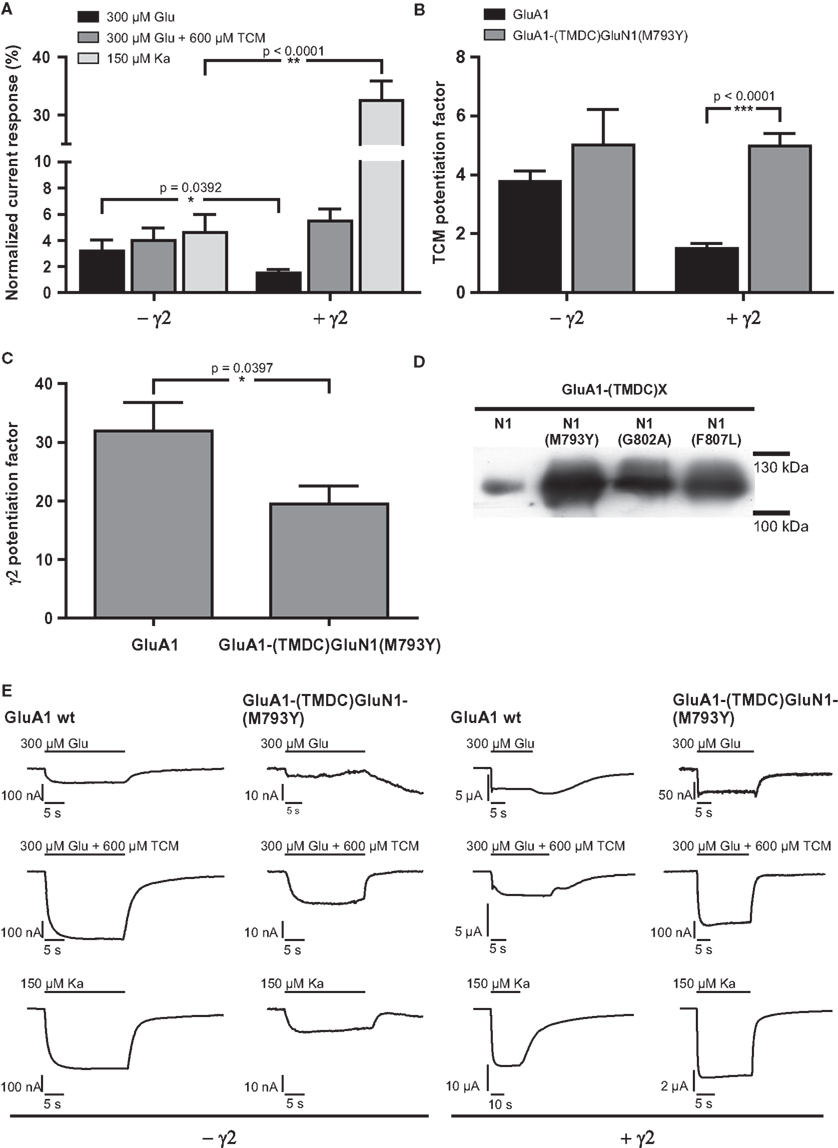
Figure 4. (A) Mean agonist-induced current amplitudes of the construct GluA1-(TMDC)GluN1(M793Y) in absence and presence of γ2. (B) Potentiation of glutamate-evoked currents by TCM, a desensitization inhibitor. (C) Potentiation of glutamate-induced currents by γ2. Levels of significance were calculated by unpaired, two-tailed t-tests. (D) Membrane expression of point mutants of the GluN1 chimera. All mutants are expressed and integrated into the plasma membrane at higher levels than the initial chimera GluA1-(TMDC)GluN1. (E) Exemplary current traces of GluA1 wild type and GluA1-(TMDC)GluN1(M793Y). Glutamate-induced currents are shown in the top row, glutamate-induced currents in presence of TCM are shown in the top row, and kainate-evoked currents in the bottom row; currents recorded in the absence of the TARP γ2 are shown in the left half of the figure, currents observed in its presence in the right half. Please note different scaling and units.
Despite the high expression levels, constructs GluA1-(TMDC-)GluN1(G802A) and GluA1-(TMDC)GluN1(F807L) gave no observable currents upon application of glutamate or kainate (data not shown). The third construct, however, GluA1-(TMDC)GluN1(M793Y), turned out to be a regain-of-function mutation. Upon application of both glutamate and kainate, this construct reproducibly showed currents that were stable, although significantly reduced, reaching 3.2% of mean GluA1 wild type currents in the case of glutamate and 4.6% in the case of kainate (Figures 4A,E, Table 2). The addition of the AMPA receptor-specific desensitization inhibitor TCM led to a slight relative increase in the glutamate-induced current amplitude, corresponding to 4% of GluA1 wild type currents. Yet, the potentiation by TCM was not significantly altered compared to GluA1 wild type (Figure 4B); GluA1-(TMDC)GluN1(M793Y) is potentiated five-fold by TCM, the wild type 3.8-fold.
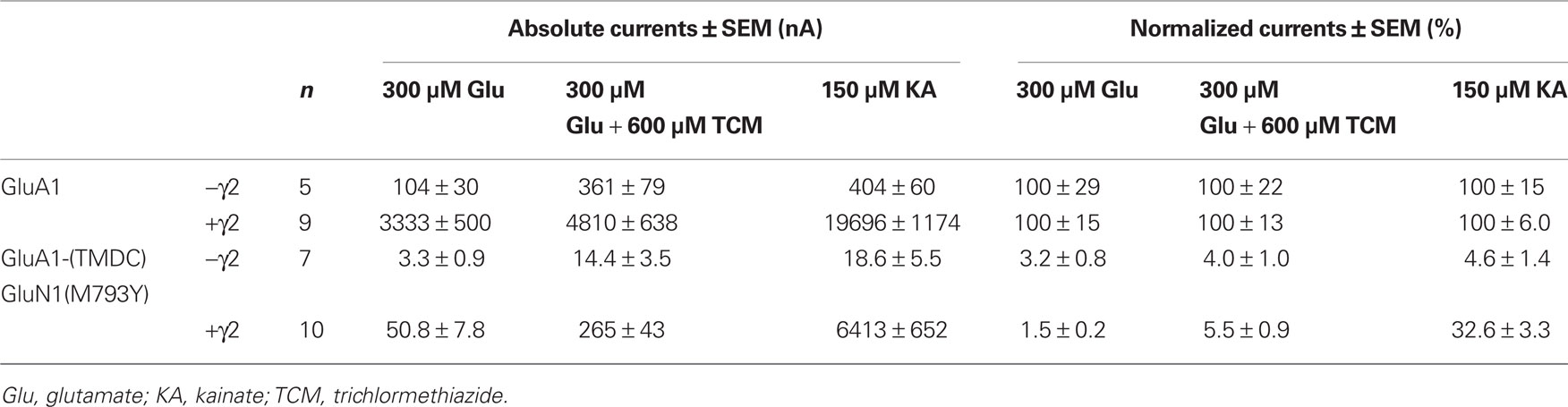
Table 2. Absolute and normalized mean currents of GluA1 wild type and GluA1-(TMDC)GluN1(M793Y), in the absence and presence of the TARP γ2.
The coexpression of the TARP γ2 causes potentiation of agonist-evoked currents by multiple mechanisms; in addition to elevated expression levels, glutamate-induced steady-state currents are increased due to decreased desensitization, whereas kainate-evoked currents are augmented because of an increased kainate efficacy (Priel et al., 2005; Turetsky et al., 2005). For the mutant GluA1-(TMDC)GluN1(M793Y) the latter effect is clearly pronounced, kainate-induced currents reach 32% of the wild type amplitude. Contrary to this, the picture is completely different for glutamate-induced currents: Although absolute amplitudes are still increased compared to expression without γ2, the mutant is only boosted 20-fold, which is significantly lower than the 30-fold potentiation seen in the wild type (Figure 4C). As a result, currents elicited by glutamate only amount to 1.5% of the wild type amplitude in presence of the TARP (Figure 4A).
In wild type GluA1 the effect of γ2 on desensitization outweighs the effect of TCM. Therefore, the TCM potentiation factor is lower for the wild type when the TARP is coexpressed. For GluA1-(TMDC)GluN1(M793Y) this masking effect by γ2 is completely lost. Potentiation by TCM is five-fold in both the absence and presence of γ2, which is a highly significant difference to the wild type (Figure 4B).
Two Highly Conserved Amino Acid Residues in the TMD C of iGluRs are Pivotal for Receptor Functionality
Local sequence alignment of TMD C amino acid residues shows that two residues, G790 and F792, are conserved among all 18 known iGluR subunits (indicated for the subunits investigated in this study in Figure 1B). Since a high degree of conservation often indicates key roles for the respective residues in protein function, these two positions were exchanged for alanine in two independent constructs, GluA1(G790A) and GluA1(F792A). GluA1(G790A) gave stable current responses upon agonist application that corresponded to 56% of mean GluA1 wild type currents for kainate (Figures 5A,E, Table 3). Contrary to that, glutamate-elicited currents exhibited only 12% of the wild type amplitude. In combination with TCM, glutamate-evoked currents of 34% of the wild type amplitude were observed. Coexpression of γ2 caused an increase in all agonist-induced currents. Kainate-evoked currents were potentiated about 10-fold by γ2 in both the wild type and GluA1(G790A); mean currents of the latter reached 54% of the wild type amplitude, which is indistinguishable from the 56% seen in the absence of γ2. However, the picture was completely different for glutamate-induced currents: While responses of the wild type were only boosted roughly 20-fold by the TARP, GluA1(G790A) showed a 40-fold potentiation (Figure 5C). Mean amplitudes of the construct in the presence of a TARP corresponded to 27% of the wild type, which was significantly more than in the absence of γ2. The presence of TCM increased glutamate-elicited currents even stronger to nearly 80% of the wild type currents. Relative potentiation by TCM was increased for GluA1(G790A) in both the presence and the absence of γ2, yet in the latter case this increase was significantly higher, reaching a 10-fold potentiation (Figure 5B) compared to a three-fold potentiation when γ2 is coexpressed. This means that in the absence of γ2 the mutant is potentiated stronger by TCM than is the wild type. In presence of the TARP the effect of TCM is partially masked as explained previously. However, although this masking effect can be seen in the mutant, it is less pronounced than in the wild type. This indicates that the effects on desensitization by TCM and γ2 are mechanistically distinct, and that the influence of the G790A exchange on desensitization kinetics is independent of the open-state stabilization by γ2 and TCM.
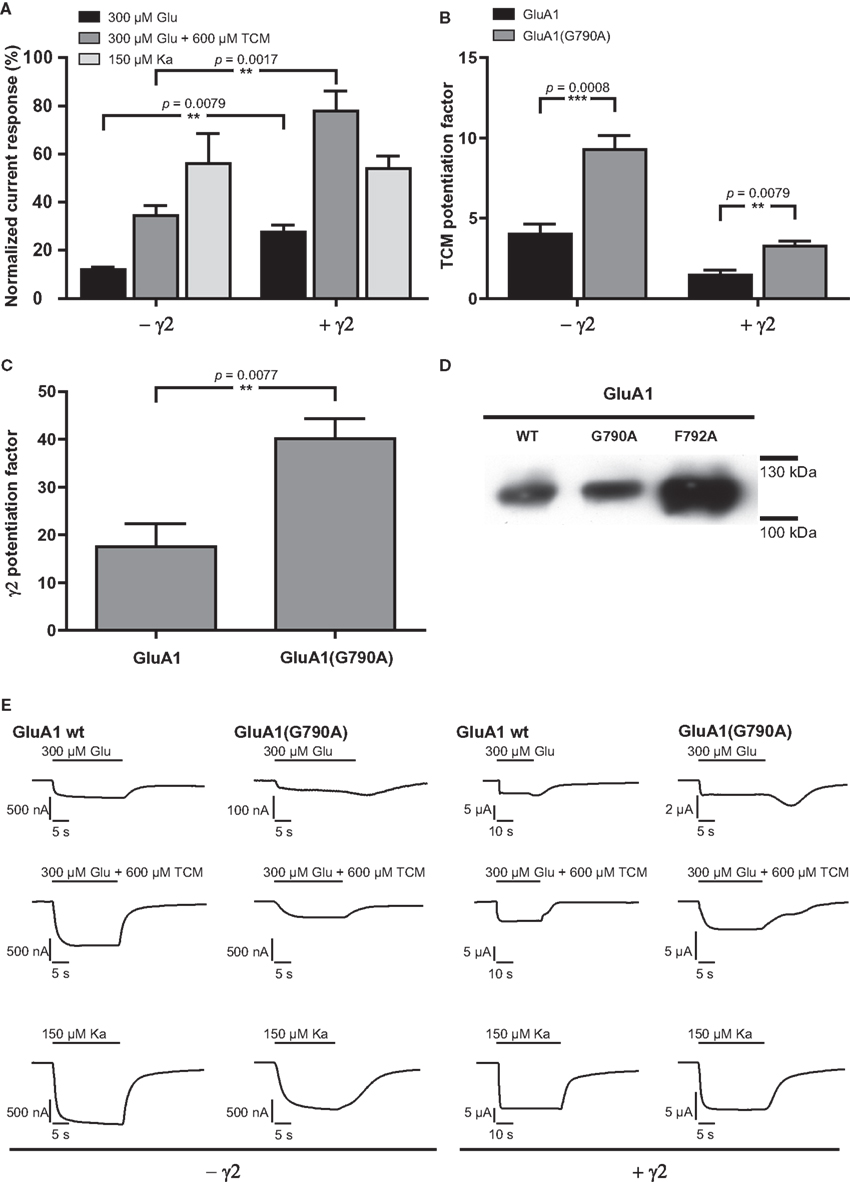
Figure 5. (A) Mean agonist-induced current amplitudes of the point mutant GluA1(G790A) in the absence and presence of γ2. (B) Potentiation of glutamate-evoked currents by TCM, a desensitization inhibitor. (C) Potentiation of glutamate-induced currents by γ2. (D) Membrane expression of point mutants of GluA1 wild type. (E) Exemplary current traces of GluA1 wild type and GluA1(G790A). Glutamate-induced currents are shown in the top row, glutamate-induced currents in presence of TCM are shown in the top row, and kainate-evoked currents in the bottom row; currents recorded in the absence of the TARP γ2 are shown in the left half of the figure, currents observed in its presence in the right half. Please note different scaling and units.
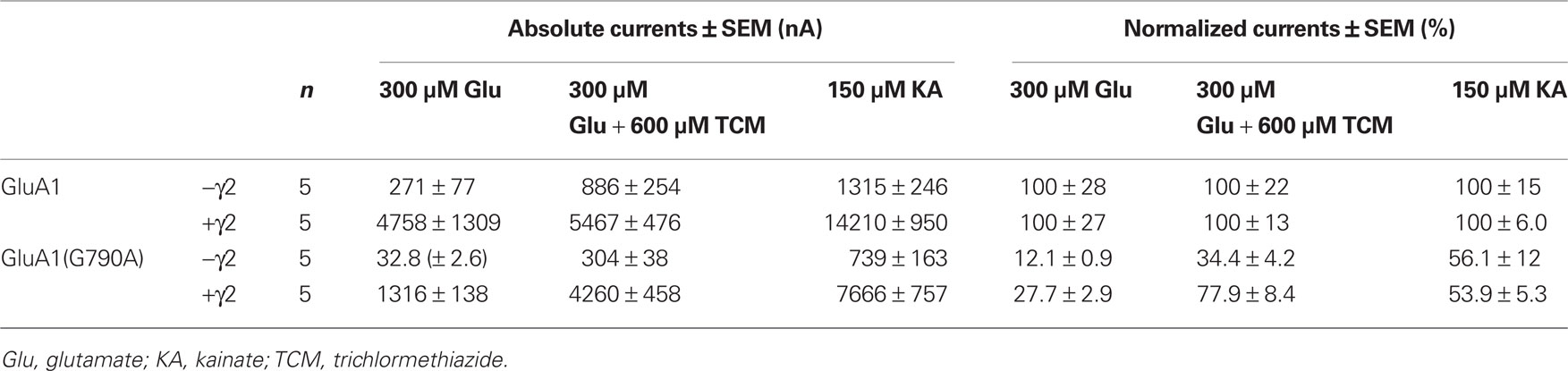
Table 3. Absolute and normalized mean currents of GluA1 wild type and GluA1(G790A), in the absence and presence of the TARP γ2.
The second construct generated, GluA1(F792A), scarcely showed any detectable current responses upon agonist application, irrespective of γ2 expression. Only in the absence of the TARP, kainate-induced currents corresponding to 5.8% of the mean wild type amplitude could be detected. In all other cases the observed relative currents did not exceed 1% of the wild type amplitude (data not shown). However, analysis of surface membrane expression revealed that GluA1(F807L) was present in the plasma membrane at even higher levels than the wild type. This was in sharp contrast to GluA1(G790A), the expression level of which closely resembled those of GluA1 wild type (Figure 5D).
Discussion
The TMD C of Ampa Receptors can be Replaced by Functionally Homologous Domains, but not by Merely Structurally Similar Ones
The initial experiments with an internally exchanged TMD show that the TMD C cannot be replaced by a merely structurally similar TMD without losing receptor function. However, while being non-functional, the A-B-A mutant was expressed in the oocyte plasma membrane, indicating that changes in the TMD C have not much impact on proper membrane integration. However, the A-B-A construct could only be detected when the TARP γ2 was coexpressed. Since TARPs have been shown to act as chaperones (Vandenberghe et al., 2005), it is conceivable that this TARP effect is necessary for the mutant to be inserted into the membrane correctly.
When the TMD C of GluA1 was replaced by a TMD C of other iGluRs, receptor function was retained in most cases, at least partially. Perhaps not surprisingly, any functional impact of TMD C transplantation was least pronounced for constructs carrying the sequence of other AMPA receptors, which only differ in one amino acid in the case of GluA2 and two residues in the cases of GluA3 and GluA4. For the other constructs, the amplitude of currents elicited by kainate was strongly dependent on the coexpression of γ2, which is known to enhance kainate efficacy (Turetsky et al., 2005) and increase membrane expression levels of AMPA receptors (Chen et al., 2000). All kainate receptor chimeras (containing TMD Cs of GluK1–GluK4) as well as the GluD1 TMD C chimera show currents between 23% (GluA1-(TMDC)GluD1) and 66% (GluA1-(TMDC)GluK1) of the wild type amplitude when coexpressed with γ2. Analysis of membrane expression shows that this decrease can partly be attributed to a decrease in receptor protein expression. The chimeras carrying the TMD C of the NMDA receptor subunits GluN1 and GluN2A exhibit a complete loss of function for the GluN1 construct, and show only very small currents for the GluN2A mutant. In the case of GluA1-(TMDC)GluN1 this is in part reflected by a reduction in membrane expression, while for GluA1-(TMDC)GluN2A it is not. This means that receptor function is critically dependent on amino acid residues located in the TMD C.
A Single Amino Acid Exchange Partially Rescues a Loss-of-Function Mutant and Simultaneously Alters Modulation by γ2
Replacement of a methionine residue at position 793 of the chimera GluA1-(TMDC)GluN1 by tyrosine, the amino acid found in wild type GluA1, led to a partial restoration of receptor function. Similar to the chimeras discussed in the preceding section, the amplitude of kainate-induced currents of GluA1-(TMDC)GluN1(M793Y) was dependent on the coexpression of γ2. When the TARP was present, kainate-evoked currents reached more than 30% of the wild type amplitude. Strikingly, the exact opposite was observed for currents elicited by glutamate. Although γ2 still boosted absolute glutamate-induced current amplitudes, the potentiating effect of γ2, which is due to decreased desensitization, was significantly weaker. While glutamate-elicited currents reached 3% of GluA1 wild type amplitudes in the absence of γ2, they only exhibited a mere 1.5% of the wild type current when the TARP was coexpressed.
In wild type GluA1, the interaction with TARPs typically results in up to six-fold reduced desensitization, a mechanism that is independent of the simultaneous alteration of kainate efficacy, with both mechanisms contributing to the observed larger steady state currents (Priel et al., 2005; Turetsky et al., 2005). The data presented here suggest that the reduction of desensitization by γ2 is absent in GluA1-(TMDC)GluN1(M793Y). The residual increase in absolute steady-state currents can be attributed to elevated surface expression facilitated by γ2, an effect that is independent of the pharmacological effects of the TARP. In line with that is a significant change in TCM potentiation of this mutant: For the wild type, the TCM-mediated stabilization of the open state is less pronounced when γ2 is coexpressed, since the TARP itself stabilizes the receptor in the open conformation, thereby partially masking the effect of desensitization inhibitors such as TCM or cyclothiazide (Priel et al., 2005; Kott et al., 2007). For the GluA1-(TMDC)GluN1(M793Y) construct, however, TCM potentiation is unchanged by coexpression of γ2; it is actually indistinguishable from the potentiation seen in GluA1 wild type in the absence of γ2. Since kainate-induced currents are still boosted by γ2, this strongly suggests a specific interaction of γ2 with this region of the TMD C, which thus is potentially involved in the mediation of desensitization-modulating interactions with the TARP. In the recently published crystal structure of GluR2 (Sobolevsky et al., 2009) the TMD C is located on the outer surface of the receptor complex (Figure 6). The tyrosine residue corresponding to Y793 in GluA1 protrudes into the lipid bilayer, thus making an interaction of this domain with a membrane-spanning auxiliary protein conceivable. However, this needs to be proven experimentally.
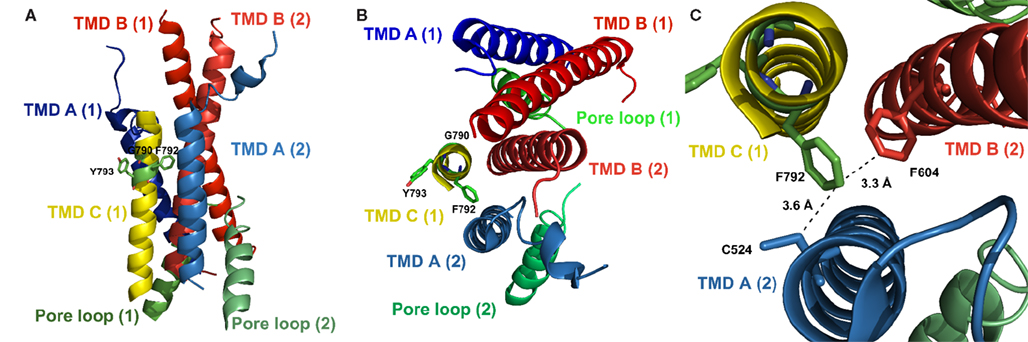
Figure 6. Localization of the residues discussed here in the crystal structure of GluA2 (PDB# 3KG2), with residue numbering, however, referring to the respective positions in GluA1. (A) Side view; (B) Top view; shown are the TMDs and the pore loop of two subunits of the tetramer as indicated by numbers in brackets; the TMD C of the second subunit was omitted for clarity. Side chains of residues investigated in this study are indicated. The tyrosine residue at position 793 is oriented away from the pore, facing the lipid bilayer. (C) Detailed top view; the phenylalanine residue at position 792 is oriented toward the pore and is in close proximity to C524 in TMD A and to F604 in TMD B of the neighboring subunit, suggesting an interaction between those residues and explaining the loss of function evoked by an exchange of F792 to alanine.
A Glycine Residue Conserved Among all iGluRs is Involved in Desensitization of Ampa Receptors
Replacing G790 of GluA1 with alanine causes functional impairment of the receptor, as all agonist-induced current responses are decreased, whereas membrane expression levels are unaltered. Interestingly, currents evoked by different agonists are differentially affected by the point mutation. While currents elicited by kainate in GluA1(G790A) in the absence of γ2 amount to 56% of wild type currents, glutamate evokes much smaller currents of only about 12%. This is counteracted by TCM, which increases glutamate-induced current amplitudes to 34% of the wild type. Coexpression of γ2 does not significantly alter relative kainate-induced currents, but increases glutamate-elicited currents to 27% of the wild type amplitude. Taken together, these data indicate faster desensitization of the G790A mutant. Mechanistically, there are two possible ways of speeding up desensitization that might contribute to the observed effect. On the one hand, it is conceivable that the open state of the receptor is destabilized by the point mutation; this would, however, be rather surprising, since stabilization of the open state usually occurs in the LBD (Sun et al., 2002). Also, if that was the case, currents evoked in the presence of TCM or γ2 should also be clearly reduced, which they are not. On the other hand, it is possible that the reduction of glutamate-induced currents is not the result of destabilization of the open state, but of a faster or more efficient transmission of the desensitization-inducing conformational change from the LBD to the TMDs. A possible explanation for the latter could be the nature of the exchanged amino acid residue; glycine residues are thought to be critically involved in interactions between transmembrane α-helices (Javadpour et al., 1999). Furthermore, glycine adds flexibility to α-helices; hence the G790A mutation would render the TMD C more rigid. Admittedly, the homologous position in GluA2 (G792), as revealed by the crystal structure, shows no residues of other TMDs in close proximity that would support an inter-domain interaction. It should be noted, however, that this crystal structure shows the receptor in the open state, so possible interactions of G792 in the closed state remain speculative.
For the loss-of-function mutant F792A an interpretation based on the crystal structure is more straightforward. The phenylalanine residue is in close proximity to both a cysteine residue in TMD A (3.6 Å) and a phenylalanine residue in TMD B (3.3 Å) of the neighboring subunit (Figure 6C). This suggests an interaction of F792 with at least one of those residues, which is lost when F792 is mutated to alanine. The lack of inter-subunit helix–helix interactions could explain the gating inability of the F792A mutant.
Conclusions
We here for the first time provide experimental evidence that the TMD C of AMPA receptors is involved in channel gating and desensitization. The data presented in this study show that amino acid residues located near the extracellular boundary of the TMD C are crucial for receptor function. The exchange of highly conserved residues either completely abolishes ion channel function (F792A) or greatly impairs it (G790A). For the latter mutant we could show that this particular glycine residue is of importance for receptor desensitization and hypothesize that this may be due to faster or more efficient relay to the membrane spanning domains of the conformational change induced by desensitization. We furthermore found that the mutation of a non-functional chimera, GluA1-(TMDC)GluN1, at a single position in the TMD C can reestablish receptor function to some extent. Strikingly, in this mutant distinct TARP effects are not observed, supporting the possibility that the TMD C is involved in AMPA receptor – TARP interactions, which up to now have been thought to be restricted to the intra- and extracellular domains of these two proteins. Additionally, this could explain, why and how the interaction with TARPs is restricted to AMPA receptors. Amino acid residues in the TMD C could be involved as “recognition sites” for the TARP. However, this remains speculative. In summary, we have established a new role for the TMD C, exceeding that of a mere structural element, but rather being an active participant in AMPA receptor function, modulation, and interaction with auxiliary subunits.
Acknowledgments
The authors would like to thank Stefanie Koll and Björn Peters for expert frog surgery, and Nora C?avara, Sabine Kott, and Daniel Tapken for critical reading of this manuscript. Jan Terhag is supported by a stipend from the DFG graduate school 736 “Development and plasticity of the nervous system: Molecular, synaptic, and cellular mechanisms”.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Armstrong, D. J., and Roman, A. (1992). Mutagenesis of human papillomavirus types 6 and 16 E7 open reading frames alters the electrophoretic mobility of the expressed proteins. J. Gen. Virol. 73(Pt 5), 1275–1279.
Chen, L., Chetkovich, D. M., Petralia, R. S., Sweeney, N. T., Kawasaki, Y., Wenthold, R. J., Bredt, D. S., and Nicoll, R. A. (2000). Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408, 936–943.
Collingridge, G. L., Isaac, J. T. R., and Wang, Y. T. (2004). Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962.
Collingridge, G. L., Olsen, R., Peters, J. A., and Spedding, M. (2009). Ligand gated ion channels. Neuropharmacology 56, 1.
Dunker, A. K., and Rueckert, R. R. (1969). Observations on molecular weight determinations on polyacrylamide gel. J. Biol. Chem. 244, 5074–5080.
Fasano, O., Aldrich, T., Tamanoi, F., Taparowsky, E., Furth, M., and Wigler, M. (1984). Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 81, 4008–4012.
Hayashi, Y., Shi, S. H., Esteban, J. A., Piccini, A., Poncer, J. C., and Malinow, R. (2000). Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287, 2262–2267.
Hoffmann, J., Gorodetskaia, A., and Hollmann, M. (2006a). Ion pore properties of ionotropic glutamate receptors are modulated by a transplanted potassium channel selectivity filter. Mol. Cell Neurosci. 33, 335–343.
Hoffmann, J., Villmann, C., Werner, M., and Hollmann, M. (2006b). Investigation via ion pore transplantation of the putative relationship between glutamate receptors and K+ channels. Mol. Cell Neurosci. 33, 358–370.
Hollmann, M. (1999). “Structure of ionotropic glutamate receptors,” in Handbook of Experimental Pharmacology, Vol. 141, eds P. Jonas and H. Monyer (Berlin: Springer-Verlag), 3–98.
Hollmann, M., Boulter, J., Maron, C., and Heinemann, S. (1994). Molecular biology of glutamate receptors. Potentiation of N-methyl-D-aspartate receptor splice variants by zinc. Ren. Physiol. Biochem. 17, 182–183.
Hollmann, M., and Heinemann, S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17, 31–108.
Horak, M., Chang, K., and Wenthold, R. J. (2008). Masking of the endoplasmic reticulum retention signals during assembly of the NMDA receptor. J. Neurosci. 28, 3500–3509.
Jang, M.-K., Mierke, D. F., Russek, S. J., and Farb, D. H. (2004). A steroid modulatory domain on NR2B controls N-methyl-D-aspartate receptor proton sensitivity. Proc. Natl. Acad. Sci. U.S.A. 101, 8198–8203.
Javadpour, M. M., Eilers, M., Groesbeek, M., and Smith, S. O. (1999). Helix packing in polytopic membrane proteins: role of glycine in transmembrane helix association. Biophys. J. 77, 1609–1618.
Kott, S., Werner, M., Körber, C., and Hollmann, M. (2007). Electrophysiological properties of AMPA receptors are differentially modulated depending on the associated member of the TARP family. J. Neurosci. 27, 3780–3789.
Kuner, T., Seeburg, P. H., and Guy, H. R. (2003). A common architecture for K+ channels and ionotropic glutamate receptors? Trends Neurosci. 26, 27–32.
Kuryatov, A., Laube, B., Betz, H., and Kuhse, J. (1994). Mutational analysis of the glycine-binding site of the NMDA receptor: structural similarity with bacterial amino acid-binding proteins. Neuron 12, 1291–1300.
Lerma, J. (2003). Roles and rules of kainate receptors in synaptic transmission. Nat. Rev. Neurosci. 4, 481–495.
McBain, C. J., and Mayer, M. L. (1994). N-methyl-D-aspartic acid receptor structure and function. Physiol. Rev. 74, 723–760.
O’Hara, P. J., Sheppard, P. O., Thøgersen, H., Venezia, D., Haldeman, B. A., McGrane, V., Houamed, K. M., Thomsen, C., Gilbert, T. L., and Mulvihill, E. R. (1993). The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron 11, 41–52.
Priel, A., Kolleker, A., Ayalon, G., Gillor, M., Osten, P., and Stern-Bach, Y. (2005). Stargazin reduces desensitization and slows deactivation of the AMPA-type glutamate receptors. J. Neurosci. 25, 2682–2686.
Ren, H., Honse, Y., Karp, B. J., Lipsky, R. H., and Peoples, R. W. (2003a). A site in the fourth membrane-associated domain of the N-methyl-D-aspartate receptor regulates desensitization and ion channel gating. J. Biol. Chem. 278, 276–283.
Ren, H., Honse, Y., and Peoples, R. W. (2003b). A site of alcohol action in the fourth membrane-associated domain of the N-methyl-D-aspartate receptor. J. Biol. Chem. 278, 48815–48820.
Rosenmund, C., Stern-Bach, Y., and Stevens, C. F. (1998). The tetrameric structure of a glutamate receptor channel. Science 280, 1596–1599.
Rozas, J. L., Paternain, A. V., and Lerma, J. (2003). Noncanonical signaling by ionotropic kainate receptors. Neuron 39, 543–553.
Schmid, S. M., Kott, S., Sager, C., Huelsken, T., and Hollmann, M. (2009). The glutamate receptor subunit delta2 is capable of gating its intrinsic ion channel as revealed by ligand binding domain transplantation. Proc. Natl. Acad. Sci. U.S.A. 106, 10320–10325.
Schmidt, C., Werner, M., and Hollmann, M. (2006). Revisiting the postulated “unitary glutamate receptor”: electrophysiological and pharmacological analysis in two heterologous expression systems fails to detect evidence for its existence. Mol. Pharmacol. 69, 119–129.
Seeburg, P. H., Higuchi, M., and Sprengel, R. (1998). RNA editing of brain glutamate receptor channels: mechanism and physiology. Brain Res. Brain Res. Rev. 26, 217–229.
Sheng, M., and Lee, S. H. (2001). AMPA receptor trafficking and the control of synaptic transmission. Cell 105, 825–828.
Sobolevsky, A. I., Rosconi, M. P., and Gouaux, E. (2009). X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756.
Sommer, B., Keinänen, K., Verdoorn, T. A., Wisden, W., Burnashev, N., Herb, A., Köhler, M., Takagi, T., Sakmann, B., and Seeburg, P. H. (1990). Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science 249, 1580–1585.
Strutz-Seebohm, N., Werner, M., Madsen, D. M., Seebohm, G., Zheng, Y., Walker, C. S., Maricq, A. V., and Hollmann, M. (2003). Functional analysis of Caenorhabditis elegans glutamate receptor subunits by domain transplantation. J. Biol. Chem. 278, 44691–44701.
Sun, Y., Olson, R., Horning, M., Armstrong, N., Mayer, M., and Gouaux, E. (2002). Mechanism of glutamate receptor desensitization. Nature 417, 245–253.
Tapken, D., and Hollmann, M. (2008). Arabidopsis thaliana glutamate receptor ion channel function demonstrated by ion pore transplantation. J. Mol. Biol. 383, 36–48.
Turetsky, D., Garringer, E., and Patneau, D. K. (2005). Stargazin modulates native AMPA receptor functional properties by two distinct mechanisms. J. Neurosci. 25, 7438–7448.
Vandenberghe, W., Nicoll, R. A., and Bredt, D. S. (2005). Interaction with the unfolded protein response reveals a role for stargazin in biosynthetic AMPA receptor transport. J. Neurosci. 25, 1095–1102.
Villmann, C., Hoffmann, J., Werner, M., Kott, S., Strutz-Seebohm, N., Nilsson, T., and Hollmann, M. (2008). Different structural requirements for functional ion pore transplantation suggest different gating mechanisms of NMDA and kainate receptors. J. Neurochem. 107, 453–465.
Villmann, C., Strutz, N., Morth, T., and Hollmann, M. (1999). Investigation by ion channel domain transplantation of rat glutamate receptor subunits, orphan receptors and a putative NMDA receptor subunit. Eur. J. Neurosci. 11, 1765–1778.
Keywords: AMPA receptor, transmembrane domain, desensitization, gating, TARP, domain transplantation
Citation: Terhag J, Gottschling K and Hollmann M (2010) The transmembrane domain C of AMPA receptors is critically involved in receptor function and modulation. Front. Mol. Neurosci. 3:117. doi: 10.3389/fnmol.2010.00117
Received: 21 May 2010;
Accepted: 30 November 2010;
Published online: 16 December 2010.
Edited by:
Hannah Monyer, The Neurological University Hospital Heidelberg, GermanyReviewed by:
Rolf Sprengel, Max Planck Institute for Medical Research, GermanyValentin Stein, Max Planck Institute of Neurobiology, Germany
Copyright: © 2010 Terhag, Gottschling and Hollmann. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Michael Hollmann, Department of Biochemistry I – Receptor Biochemistry, Building NC 6/171, Ruhr University Bochum, Universitätsstr. 150, D-44780 Bochum, Germany. e-mail:bWljaGFlbC5ob2xsbWFubkBydWIuZGU=