 Takuto Hideyama1,2 and Shin Kwak1,2*
Takuto Hideyama1,2 and Shin Kwak1,2*- 1 Core Research for Evolutional Science and Technology, Japan Science Technology Agency, Kawaguchi-shi, Saitama, Japan
- 2 Department of Neurology, Graduate School of Medicine, University of Tokyo, Bunkyo-ku, Tokyo, Japan
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease. More than 90% of ALS cases are sporadic, and the majority of sporadic ALS patients do not carry mutations in genes causative of familial ALS; therefore, investigation specifically targeting sporadic ALS is needed to discover the pathogenesis. The motor neurons of sporadic ALS patients express unedited GluA2 mRNA at the Q/R site in a disease-specific and motor neuron-selective manner. GluA2 is a subunit of the AMPA receptor, and it has a regulatory role in the Ca2+-permeability of the AMPA receptor after the genomic Q codon is replaced with the R codon in mRNA by adenosine–inosine conversion, which is mediated by adenosine deaminase acting on RNA 2 (ADAR2). Therefore, ADAR2 activity may not be sufficient to edit all GluA2 mRNA expressed in the motor neurons of ALS patients. To investigate whether deficient ADAR2 activity plays pathogenic roles in sporadic ALS, we generated genetically modified mice (AR2) in which the ADAR2 gene was conditionally knocked out in the motor neurons. AR2 mice showed an ALS-like phenotype with the death of ADAR2-lacking motor neurons. Notably, the motor neurons deficient in ADAR2 survived when they expressed only edited GluA2 in AR2/GluR-BR/R (AR2res) mice, in which the endogenous GluA2 alleles were replaced by the GluR-BR allele that encoded edited GluA2. In heterozygous AR2 mice with only one ADAR2 allele, approximately 20% of the spinal motor neurons expressed unedited GluA2 and underwent degeneration, indicating that half-normal ADAR2 activity is not sufficient to edit all GluA2 expressed in motor neurons. It is likely therefore that the expression of unedited GluA2 causes the death of motor neurons in sporadic ALS. We hypothesize that a progressive downregulation of ADAR2 activity plays a critical role in the pathogenesis of sporadic ALS and that the pathological process commences when motor neurons express unedited GluA2.
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease. ALS is characterized by progressive paralysis with muscle wasting due to a selective loss of upper and lower motor neurons. More than 90% of ALS cases are sporadic, whereas the remaining ALS cases have more than one other affected family member (familial ALS) (Table 1). The majority of sporadic ALS cases do not carry mutations in the genes that are known to cause familial ALS, including Cu/Zn superoxide dismutase (SOD1; Rosen et al., 1993; Jackson et al., 1997) FUS/TLS (Kwiatkowski et al., 2009; Vance et al., 2009) and TARDBP (TDP-43; Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008). In contrast, TDP-43 pathology in the spinal cord motor neurons is considered to be a neuropathological hallmark of sporadic ALS and is observed in most sporadic ALS cases (Arai et al., 2006; Neumann et al., 2006; Hasegawa et al., 2008) but not in the majority of familial ALS cases (Mackenzie et al., 2007; Tan et al., 2007; Kabashi et al., 2008; Sreedharan et al., 2008; Van Deerlin et al., 2008; Yokoseki et al., 2008; Kwiatkowski et al., 2009; Vance et al., 2009; Deng et al., 2010). These lines of evidence suggest that there is a common pathogenic mechanism of sporadic ALS, which is not among the mutations in the genes that cause ALS phenotype in familial ALS that have been identified to date. Therefore, an investigation of the molecular abnormalities that occur specifically in the pathological tissues of patients with sporadic ALS is required to elucidate the disease pathogenesis. Because molecular abnormalities found in the patients’ pathological tissues include both the cause and the consequence of pathological changes, it is necessary to demonstrate that the molecular changes of interest induce the ALS phenotype in animals.
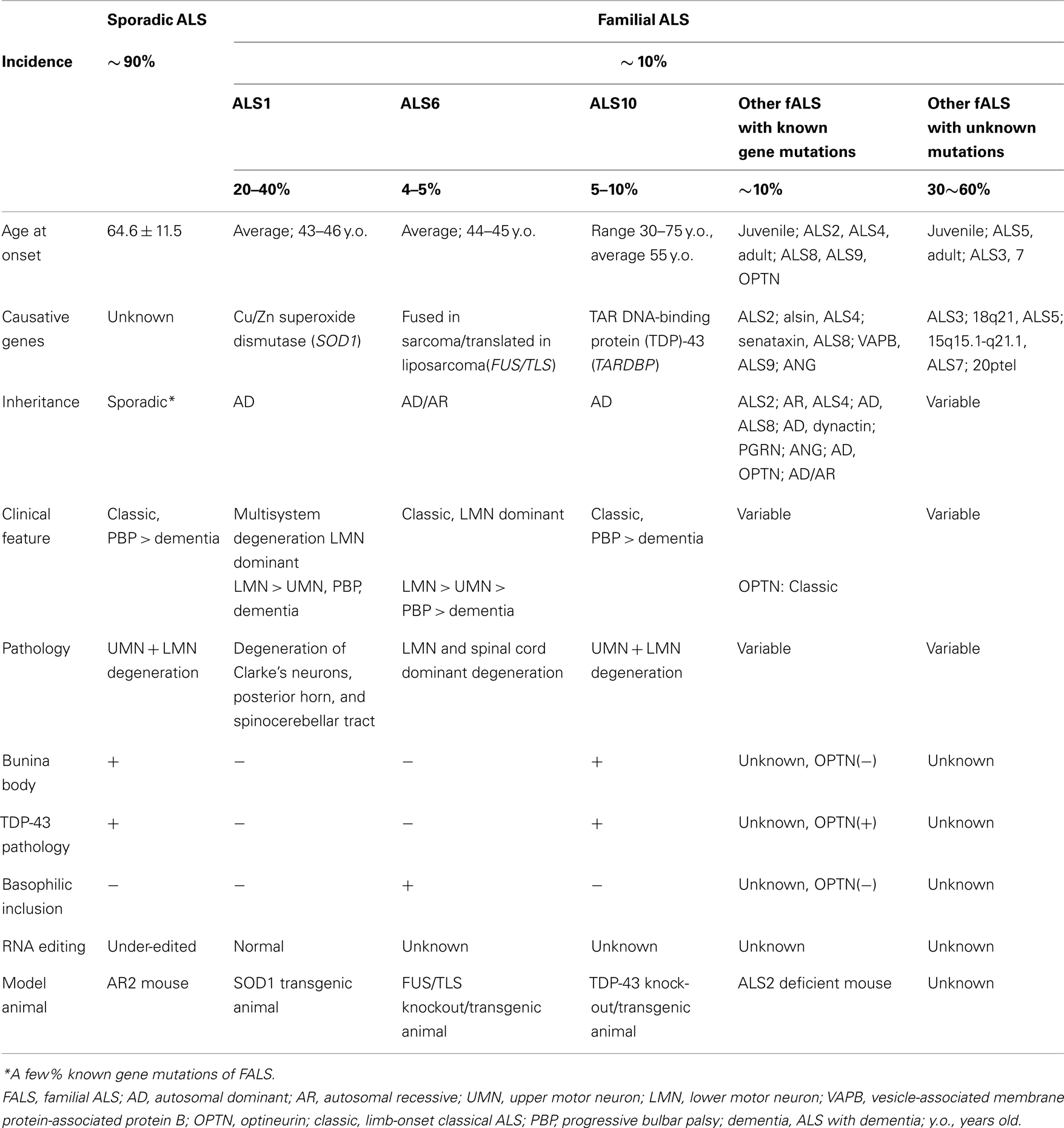
Table 1. Uniform sporadic ALS and multiple different familial ALS.
We have demonstrated that the RNA editing of GluA2, a subunit of the L-α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor, at the glutamine/arginine (Q/R) site is inefficient in the motor neurons of sporadic ALS patients in a disease-specific and motor neuron-selective manner (Takuma et al., 1999; Kawahara et al., 2004). This is in marked contrast to the fact that all GluA2 mRNA was edited in the motor neurons of control subjects (Takuma et al., 1999; Kawahara et al., 2004), in patients with motor neuron diseases other than sporadic ALS (Kawahara et al., 2006), and in dying neurons in other neurodegenerative diseases, including the Purkinje cells of patients with spinocerebellar degeneration (Paschen et al., 1994; Akbarian et al., 1995; Suzuki et al., 2003; Kawahara et al., 2004). The high disease specificity warrants an investigation of how inefficient GluA2 RNA editing leads to neuronal death.
Functional AMPA receptors are tetramers with various combinations of GluA1, GluA2, GluA3, and GluA4 that are produced in a non-stochastic fashion. All the GluA subunits are expressed in the human and rat spinal motor neurons (Tölle et al., 1993; Kawahara et al., 2003; Sun et al., 2005). In mammalian neurons, adenosine in the Q codon (CAG) is converted to inosine (A-to-I conversion) in the Q/R site of virtually all GluA2 mRNA (Figure 1A). This conversion results in the expression of the GluA2 protein with R in the Q/R site because the CIG codon is read as the CGG codon (R) during translation. Because the A-to-I conversion at the Q/R site occurs only in the GluA2 subunit, and because the AMPA receptor subunit with R at the Q/R site critically regulates the Ca2+ permeability of AMPA receptors, AMPA receptors can be impermeable to Ca2+ only when they have Q/R site-edited GluA2 (Sommer et al., 1991; Nutt and Kamboj, 1994; Geiger et al., 1995). This evidence indicates that ALS motor neurons express abundant Ca2+-permeable AMPA receptors with Q/R site-unedited GluA2 (Kwak and Kawahara, 2005) (Figure 1B). Because trafficking of unedited GluA2 is more efficient (Greger et al., 2002), a considerably high proportion of Ca2+-permeable functional AMPA receptors will be expressed in the ALS motor neurons even with small amounts of unedited GluA2 expression.
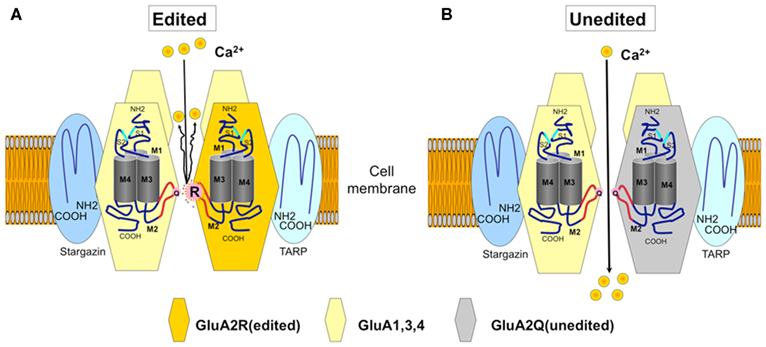
Figure 1. The AMPA receptor and Ca2+-permeability. The GluA2 subunit has four membrane domains (M1–M4). The glutamine/arginine (Q/R) site is located in the M2 domain, and this editing site is the major determinant of the Ca2+-permeability of the AMPA receptor. (A) An AMPA receptor with the GluA2 subunit edited at the Q/R site (orange) in M2 (curved line in red). An arginine (R) residue at the Q/R site prevents Ca2+ (yellow circle) from passing through the channel pore. The AMPA receptors expressed on neurons contain Q/R site-edited GluA2 in their assembly. (B) Ca2+-permeable AMPA receptors do not contain the Q/R site-edited GluA2 subunit. Naturally occurring Ca2+-permeable AMPA receptors consist of GluA1, GluA3, and GluA4 (yellow). In sporadic ALS, Ca2+-permeable AMPA receptors containing unedited GluA2 (gray) are expressed in motor neurons.
RNA editing at the GluA2 Q/R site is catalyzed by adenosine deaminase acting on RNA 2 (ADAR2), a member of the ADAR family (Figure 2; Higuchi et al., 2000). Therefore, investigating the following questions would help to reveal a neuronal death-causing mechanism in sporadic ALS: whether ADAR2-lacking motor neurons die, whether deficient ADAR2 is a direct cause of neuronal death, and whether failure of the A-to-I conversion at the GluA2 Q/R site plays a critical role in the death of ADAR2-lacking motor neurons. To address these aims, we developed mutant mice in which the ADAR2 gene is targeted selectively to motor neurons using the Cre-loxP system.
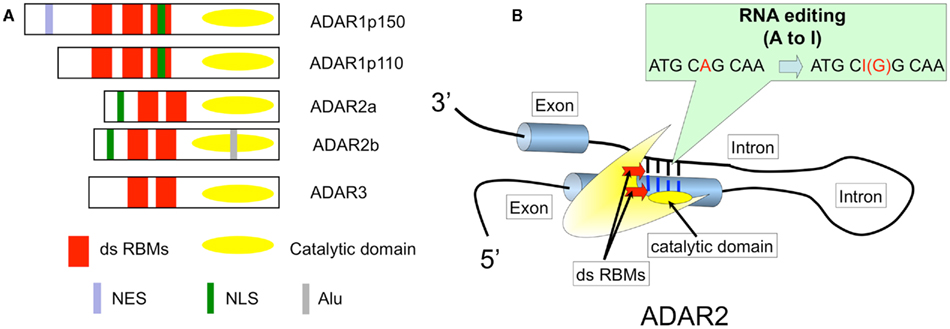
Figure 2. ADAR structure and RNA editing. (A) Three members of the ADAR family. ADAR2 specifically catalyzes the A-to-I conversion at the GluA2 Q/R site, and two isoforms, ADAR2a and more abundantly ADAR2b, are expressed in human brains and spinal cords (Aizawa et al., 2010). RBM, RNA binding motif; NES, A nuclear export signal; NLS, A nuclear localization signal; Alu, Alu sequences; (B). ADAR2 catalyzed adenosine to inosine conversion (A-to-I RNA editing) in the double-stranded RNA structure in both coding and non-coding regions of various transcripts expressed in mammalian brains. When this type of editing occurs in the reading frame, the resulting changes in the amino acid residues alter the biological function of the translated molecules, including 5-HT2c receptors, GluAs, and GluKs. A-to-I RNA editing at the GluA2 Q/R site is specifically catalyzed by ADAR2, a member of the adenosine deaminase acting on RNA (ADAR) family. ADARs act on double-stranded RNA, which is formed with the distal end of exon 11 that includes the coding sequence for the Q/R site and the exon complementary sequence in intron 11 of GluA2 pre-mRNA (Higuchi et al., 1993).
ADAR2 Conditional Knockout Mice (AR2)
It is not clear whether neuronal death occurs in neurons lacking GluA2 Q/R site editing or in those lacking ADAR2 because both systemic ADAR2-null mice (Higuchi et al., 2000) and genetically engineered mice that cannot edit the GluA2 Q/R site (Brusa et al., 1995) die young from status epilepticus. We crossed ADAR2flox/flox mice with VAChT-Cre.Fast mice that displayed restricted Cre expression under the control of the vesicular acetylcholine transporter gene promoter in a subset of cholinergic neurons, including the spinal motor neurons (Misawa et al., 2003). By intercrossing the resulting ADAR2+/flox/VAChT-Cre.Fast mice, we obtained ADAR2flox/flox/VAChT-Cre.Fast (AR2) mice (Figure 3A; Hideyama et al., 2010). The AR2 mice displayed a slow, progressive motor dysfunction (Figures 3B,C) with a low rotarod performance (Figure 4A) and grip strength (Figure 4B). AR2 mice had slightly shorter life spans than the control mice (Figure 4C). We investigated the AR2 mice for morphological changes in the brain, spinal cord, motor nerves, and muscles, as well as for functional changes of the neuromuscular units at various postnatal periods. There were several degenerating large neurons in the anterior horn (AHCs) with cytoplasmic vacuoles (Figure 5A). To evaluate the progression of motor neuron death, we counted the number of motor neurons (SMI32-positive AHCs) with and without ADAR2 immunoreactivity. The number of SMI32-positive AHCs in AR2 mice markedly decreased between 1 and 2 months of age and slowly decreased beyond 1 year of age (Figure 5B). The motor neuron reduction is attributable to the loss of ADAR2-lacking AHCs. The number of ADAR2-positive AHCs remained unchanged after 2 months of age. There were darkly stained, degenerating axons with a decreased number of myelinated axons in the ventral roots (Figure 5C); these were the consequence of AHC degeneration. Skeletal muscles exhibited morphological characteristics of denervation, including muscle fiber atrophy, centrally placed nuclei, and pyknotic nuclear clumps. Electromyographic examination demonstrated fibrillation and fasciculation potentials, which are commonly observed in the denervated and reinnervated muscle fibers of ALS patients (Hideyama et al., 2010). Some neuromuscular junctions (NMJs) were abnormally innervated in AR2 mice; these NMJs were either lacking innervation (denervated NMJs), or they were innervated by ramified axons that innervated more than one NMJ (reinnervated NMJs; Figure 5D). The proportion of denervated NMJs was higher in AR2 mice at 4 months of age than at 12 months, whereas the proportion of reinnervated NMJs was higher in AR2 mice at 12 months of age than at 4 months of age (Figure 5D). These results indicate that degeneration of ADAR2-lacking AHCs led to the degeneration of their axons with a resultant denervation of NMJs, which were reinnervated by collaterally sprouted axons of the ADAR2-expressing normal AHCs. With the degeneration of AHCs, a marked proliferation of glial fibrillary acidic protein (GFAP)-positive astrocytes and MAC2-positive microglial cells was detected in the anterior horns of AR2 mice (Figures 6A,B).

Figure 3. Generation of conditional ADAR2 knockout mice (AR2). (A) One loxP sequence (filled triangle) was inserted into intron 6 and another into intron 9, which covers the majority of the catalytic domain encoded in the ADAR2 gene between the two loxP sites. (B) An AR2 mouse at 12 months of age (left panel) exhibits a low grip strength score with an abnormal hind limb and tail posture compared to a control littermate (right panel). (C) Locomotion and walking distances for an interval of 1 min were traced using a video capture system. An AR2 mouse at 12 months of age (red line) showed low locomotion levels compared to a control littermate (blue line).
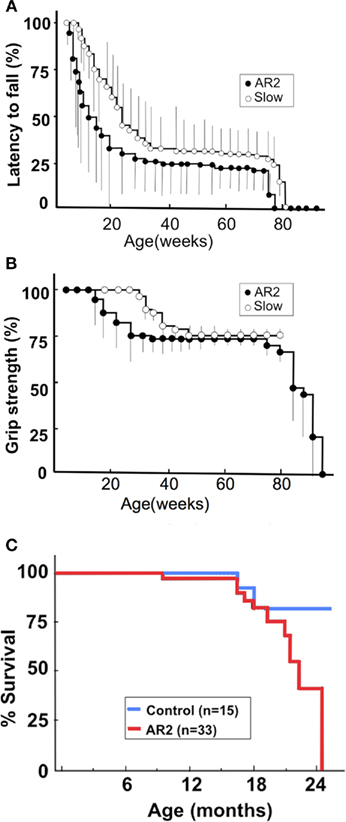
Figure 4. The phenotype of conditional ADAR2 knockout mice (AR2) and slow mice. We generated two lines of conditional ADAR2 knockout mice with different Cre expression patterns: AR2 (ADAR2flox/flox/VAChT-Cre.Fast) mice express Cre in progressively larger proportions of motor neurons, reaching ~50% of the maximum by postnatal week 5. Slow (ADAR2flox/flox/VAChT-Cre.Slow) mice express Cre more slowly, reaching ~50% of motor neurons by 8 months of age. (A) Rotarod performance of AR2 mice (filled circle; n = 28) and Slow mice (open circle; n = 33). Using a mouse-specific rotarod (SN-445, Neuroscience Corp., Tokyo Japan), the maximal time before falling at 10 rpm during a 180-s period among three trials was used for the analysis. The scores are indicated as a percentage of the performance by control mice (n = 15), which exhibited full performance until approximately 12 months of age, followed by a slightly lower performance (greater than 164.5 ± 6.4 s) until 24 months. (B) Grip power measured with a dynamometer (NS-TRM-M, Neuroscience, Japan). The score is indicated as a percentage of the performance by control mice. (C) AR2 mice had long life spans, but the rate of death increased after month 18 compared to control mice.
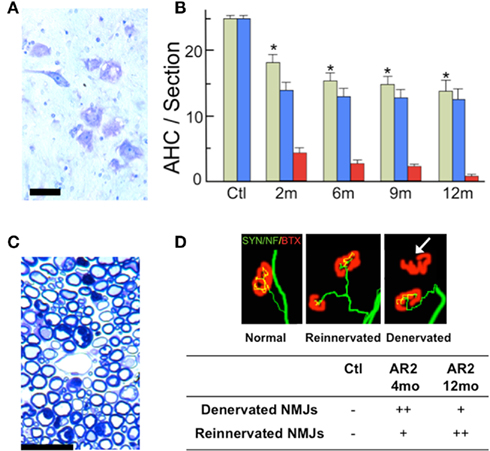
Figure 5. Loss of ADAR2-deficient motor neurons. (A) Degenerating large motor neurons in the anterior horn (AHC) of AR2 mice at 2 months of age (Nissl staining). Scale bar, 50 μm. (B) The numbers (mean ± SEM) of total AHCs (green columns), ADAR2-positive AHCs (blue columns), and ADAR2-negative AHCs (red columns) in AR2 mice at different ages (2, 6, 9, 12 months). The number of AHCs significantly decreased in AR2 mice after 2 months of age due to the reduction of ADAR2-lacking AHCs (*p < 0.01, repeated ANOVA). (C) The ventral root (L5) of AR2 mice at 4 months of age showing degenerating dark axons. Scale bar, 20 μm. (D) Neuromuscular junctions (NMJs) and distal motor nerve axons. The quadriceps muscles from an AR2 mouse (Reinnervated, Denervated) at 12 months of age and an age-matched, wild-type mouse (Ctl; normal) are stained with tetramethylrhodamine-bungarotoxin (BTX; red) and immunostained concomitantly with anti-synaptophysin and neurofilament antibodies (SYN/NF; green). The endplates (red) were counted as “innervated” if they were merged with axon terminals (yellow). A thick axon terminal in the Ctl mouse innervates each endplate, whereas in AR2 mice, some NMJs are innervated by axons that simultaneously innervate more than one NMJ (reinnervated NMJ), and other NMJs are devoid of axon terminals (denervated NMJs; white arrow).
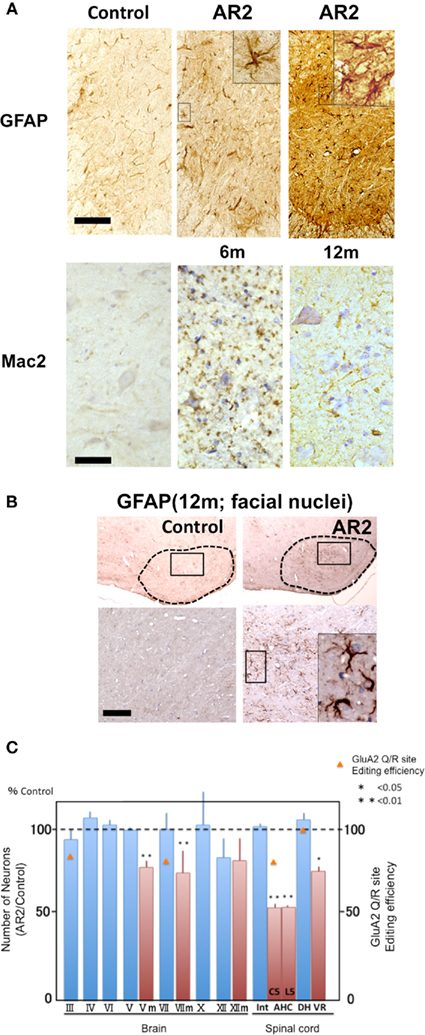
Figure 6. Reactive glial cells in the spinal anterior horns and cranial motor nerve nuclei of AR2 mice (A,B). Immunohistochemistry in the anterior horn (C5) and motor trigeminal nuclei (Vm). There was an increase in GFAP- and MAC2-immunoreactivity in the spinal anterior horn of the AR2 mice, indicating the proliferation of activated astrocytes and microglia in association with motor neuron degeneration. m: months of age, inset; activated astroglia. Scale bars, 100 μm (GFAP) and 50 μm (insets and Mac2). (C) In AR2 mice at 12 months of age, the number of large neurons in the cranial nuclei of Vm, VII, and XII were lower than those in age-matched control mice, whereas those in the cranial nuclei III, IV, and VI were not. The GluA2 Q/R site editing efficiency was decreased in III, VII, and AH, indicating that the cranial motor neurons that innervate extraocular muscles are exceptionally resistant to deficient-ADAR2-mediated neuronal death.
Other than the motor neurons in the spinal cord, large neurons in the facial (VII), and hypoglossal nerve nuclei (XII) in AR2 mice at 12 months of age were significantly decreased in number. In contrast, the number of neurons in the nuclei of the extraocular motor nerves (III, IV, VI) was not decreased (Figure 6C). Conversely, GluA2 Q/R site editing was significantly decreased in the oculomotor nerve nuclei (89.7% of control mice, p = 0.0048) and the facial nerve nuclei (83.3% of control mice, p = 0.0017) of AR2 mice at 12 months of age (Figure 6C). These results indicate that subsets of motor neurons, including those in the oculomotor nerve nucleus, are relatively resistant to cell death mediated by deficient ADAR2. Notably, the selective sparing of motor neurons that innervate the extraocular muscles as compared to those that innervate the bulbar and limb muscles is characteristically seen in ALS patients. Motor neurons in the nuclei of the oculomotor nerves are much less vulnerable in ALS patients. Notably, the expression of Ca2+-binding proteins, particularly parvalbumin, is high in oculomotor neurons and low in the facial and spinal motor neurons (Ince et al., 1993). It has been shown that over-expression of parvalbumin attenuated kainate-induced Ca2+ transients and protected spinal motor neurons from the resultant neurotoxicity in parvalbumin transgenic mice (Van Den Bosch et al., 2002). In AR2 mice and in patients with sporadic ALS, it is likely that neurons with abundant parvalbumin, such as ocular motor neurons, are more resistant to Ca2+ overload from Ca2+-permeable AMPA receptors than those with low parvalbumin levels, such as spinal motor neurons.
The Crucial Role of GluA2 RNA Editing in the Death of ADAR2-Lacking Motor Neurons in AR2 Mice
ADAR2 predominantly catalyzes the RNA editing at the Q/R site of GluA2 both in vivo and in vitro (Melcher et al., 1996; Higuchi et al., 2000; Wang et al., 2000), but there are numerous A-to-I positions in mammalian brains, some of which are specifically catalyzed by ADAR2. Among the A-to-I positions specifically mediated by ADAR2 (Nishimoto et al., 2008), we found a significant reduction in the editing efficiency at the GluK2 (GluR6) Q/R site (AR2 mice vs. control mice, 15.3 vs. 31.8%, p = 0.04416) and at the K/E site of cytoplasmic fragile X mental retardation interacting protein 2 (CYFIP2) mRNA (Figure 7). These results indicate that RNA editing at the ADAR2-mediated A-to-I positions is universally defective in AR2 motor neurons.
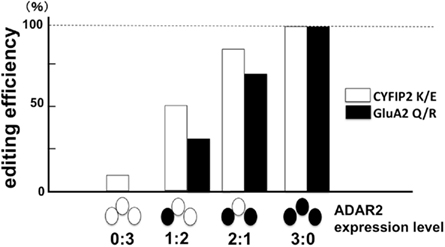
Figure 7. The failure of CYFIP2 K/E site editing and GluA2 Q/R site editing in ADAR2-lacking motor neurons. When the extent of RNA editing was measured in the pooled lysates of three motor neurons obtained from AR2 mice, the proportions of CYFIP2 mRNA edited at the lysine/glutamic acid (K/E) site and those of Q/R site-edited GluA2 mRNA were lower in the lysates containing ADAR2-lacking motor neurons. This finding indicates that ADAR2 specifically mediates A-to-I conversion at the K/E site of CYFIP2 pre-mRNA. Ratios in abscissa indicate the number of ADAR2-expressing motor neurons (filled circle) and ADAR2-lacking motor neurons (open circle) in the three motor neuron lysates of AR2 mice. (see Figure 10).
To examine the possible role of defective RNA editing at A-to-I positions other than the GluA2 Q/R site in motor neuron death, we investigated the effects of edited GluA2 expression in ADAR2-lacking motor neurons (Figure 8). We exchanged the endogenous GluA2 alleles that encoded Q at the Q/R site in AR2 mice with the GluR-BR alleles (Kask et al., 1998), which encode R at the Q/R site of GluA2. This exchange circumvented the need for ADAR2-mediated RNA editing in the expression of edited GluA2. We intercrossed ADAR2flox/+/VAChT-Cre.Fast/GluR-BR/+ mice to generate AR2/GluR-BR/R mice. AR2/GluR-BR/R mice (AR2res) were phenotypically normal and had full motor function until 6 months of age. The AHCs, including those lacking ADAR2 due to Cre-mediated recombination, were viable in AR2res mice at 6 months of age, and the total number of AHCs was the same as in age-matched control mice (Figure 9A). Consistent with a lack of AHC loss, there was no detectable increase in GFAP- or MAC2-immunoreactivity in the anterior horns (Hideyama et al., 2010). These results demonstrate that an ADAR2 deficiency induces the slow death of motor neurons specifically via the GluA2 Q/R site editing failure (Figure 9B).
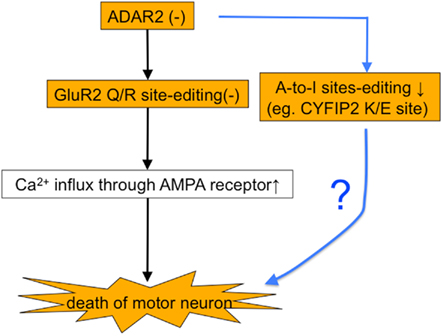
Figure 8. Do other ADAR2 substrates play a role in the death of ADAR2-lacking motor neurons? There is a possibility that the death of motor neurons in AR2 mice is mediated by a failure of RNA editing at ADAR2-mediated A-to-I positions other than the GluA2 Q/R site.
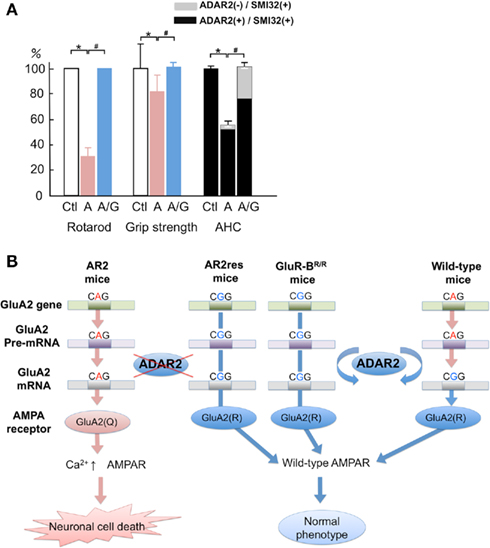
Figure 9. The crucial role of GluA2 Q/R site editing in the death of ADAR2-deficient motor neurons. (A) AR2/GluR-BR/R mice (AR2res) are phenotypically normal with a preservation of ADAR2-lacking AHCs that account for 30% of the total AHCs at 6 months of age. (B) A scheme illustrating that the death of ADAR2-lacking motor neurons due to failure to edit the GluA2 Q/R site in the AR2 mice is rescued in the AR2res mice. The exonic Q codon (CAG) for the Q/R site of GluA2 is substituted by an R codon (CGG) in the endogenous GluA2 alleles, and only edited GluA2 is expressed in the absence of ADAR2 activity.
Death of Motor Neurons in Heterozygous AR2 (HeteroAR2) Mice
The results of our experiments with the AR2 mice indicate that deficient-ADAR2-mediated death of motor neurons in the spinal cord and the cranial motor nerve nuclei is specifically mediated by the failure to edit the GluA2 Q/R site. In the ALS spinal cord, some motor neurons express unedited GluA2, but others express only edited GluA2. The majority of motor neurons expressing unedited GluA2 also express edited GluA2 (Kawahara et al., 2004). Furthermore, a recent immunohistochemical study demonstrated that both ADAR2-positive and ADAR2-negative motor neurons coexist in patients with sporadic ALS, whereas all motor neurons are ADAR2-positive in control subjects (Aizawa et al., 2010). However, we do not know the expression level of ADAR2 that is required to edit all GluA2 mRNA or the proportion of unedited GluA2 that is not harmful to motor neurons.
To answer these questions, we investigated the extent of GluA2 Q/R site editing in motor neurons lacking one ADAR2 allele in the heterozygous ADAR2flox/+/VAChT-Cre (HeteroAR2) mice. Additionally, we investigated whether motor neurons lacking one ADAR2 allele can survive in HeteroAR2 mice compared to AR2 and control mice. In HeteroAR2 mice, the proportion of motor neurons that express Cre is the same as in AR2 mice; the Cre-expressing motor neurons express only one ADAR2 allele. Therefore, if the expression level of ADAR2 in normal motor neurons is sufficiently above the requirement to edit the Q/R site of all GluA2 mRNAs expressed (i.e., threefold or more), all motor neurons would express only edited GluA2. However, if normal motor neurons express ADAR2 at a level that is only sufficient to edit GluA2 (i.e., less than twofold), motor neurons with one ADAR2 allele would express abundant unedited GluA2 and die. Furthermore, if motor neurons expressing one ADAR2 allele express both edited and unedited GluA2 and undergo degeneration in HeteroAR2 mice, we would expect to find the proportion of unedited GluA2 that is toxic to motor neurons.
When the extent of GluA2 Q/R site editing was examined in the lysates of three laser-captured motor neurons of 2-month-old HeteroAR2 mice, GluA2 Q/R site editing was incomplete in approximately 20% of the lysates (Figure 10). The proportion of edited GluA2 was above 70% in all the lysates; however, in the AR2 mice of the same age, unedited GluA2 was detected in more than 60% of the lysates of three motor neurons, and the editing efficiency was 0 in 7% of the lysates examined (Hideyama et al., 2010). Therefore, it is likely that motor neurons exhibit considerable editing activity in the expression of only one ADAR2 allele; however, this is not sufficient to edit the Q/R site of all GluA2 mRNA in HeteroAR2 mice.
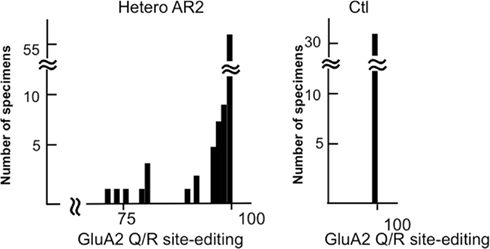
Figure 10. Some of the motor neurons from HeteroAR2 mice express Q/R site-unedited GluA2 mRNA. A frequency histogram of editing efficiency at the GluA2 Q/R site in the lysates of three motor neurons obtained from heterozygous ADAR2flox/+/VAChT-Cre.Fast (HeteroAR2) mice and control mice (Ctl). Motor neurons (252 neurons) were dissected from 14-μm-thick frozen sections of spinal cord obtained with a laser microdissector from HeteroAR2 mice at 2 months of age (n = 4). The lysates containing three motor neurons (84 specimens) were subjected to a measurement of the editing efficiency of GluA2 mRNA at the Q/R site. This histogram shows that approximately a quarter of the lysates, or 15–20% of AHCs, express unedited GluA2. Notably, the extent of GluA2 editing is above 70%, which is in contrast with the AR2 mice, in which approximately 8% of the lysates expressed only unedited GluA2. All 96 motor neurons dissected from control mice (Ctl; ADAR2flox/flox and VAChT-Cre mice) expressed only Q/R site-edited GluA2 mRNA.
We also examined whether there was a loss of AHCs in HeteroAR2 mice. The extent of AHC loss in HeteroAR2 mice was approximately half (26%) of that observed in AR2 mice (46%) at 12 months of age (Figure 11A). A moderate increase in GFAP- and MAC2-immunoreactivity was detected in the anterior horns of HeteroAR2 mice at 12 months of age (Figure 11B). HeteroAR2 mice did not exhibit significant behavioral changes until 12 months of age, indicating that mild loss of motor neurons would not affect motor function at least until 1 year of age.
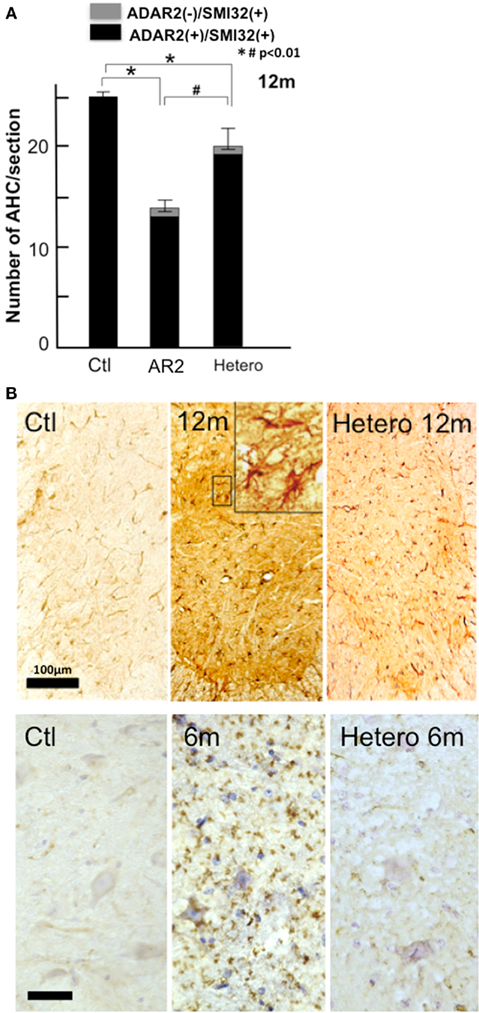
Figure 11. HeteroAR2 mice display a loss of AHCs. (A) The numbers (mean ± SEM) of ADAR2-positive AHCs (black columns) and those of ADAR2-lacking AHCs (gray columns) in HeteroAR2 mice at 12 months of age compared to age-matched AR2 and control mice. There was a loss of AHCs in the HeteroAR2 mice (Hetero; *p < 0.01) but to a lesser extent than in the AR2 mice (#p < 0.01). (B) Immunohistochemistry in the anterior horn of the spinal cord (C5). There was an increase in GFAP-immunoreactivity but to a lesser extent than in the AR2 mice (upper panel) and a slight increase of MAC2-immunoreactivity (lower panel) in the HeteroAR2 mice. Scale bars: 100 μm (upper panel) and 50 μm (insets and lower panel).
These results show that one ADAR2 gene allele is sufficient to edit all GluA2 mRNA in half of the motor neurons but is insufficient in the other half of motor neurons. Because the editing efficiency was above 70% in the motor neuron lysates of HeteroAR2 mice, it is likely that the minimal expression level of ADAR2 required for complete GluA2 editing is slightly higher than half of the normal level in mouse motor neurons.
More than a quarter of the motor neurons in HeteroAR2 mice underwent degeneration by the age of 1 year. Furthermore, from the results of our experiments with AR2 and AR2res (Hideyama et al., 2010), it is likely that only the motor neurons expressing unedited GluA2 have undergone degeneration. The proportions of unedited GluA2 are 30% at the maximum and less than 10% in the majority of motor neurons in HeteroAR2 mice, indicating that expression of unedited GluA2, even in a small proportion, is not favorable for the survival of motor neurons in mice.
The ADAR2 Hypothesis for Sporadic ALS
Progression of ALS is rather slow, taking several years to progress from the onset of the initial symptoms to death, which results from failure of the respiratory muscles. From our findings in the AR2 mice, we learned that some ADAR2-lacking motor neurons died within 1 month of ADAR2 knockout, whereas others could survive more than 1 year even with only unedited GluA2 expression (Hideyama et al., 2010). Therefore, it is likely that the timing of motor neuron death may be a stochastic phenomenon that depends on environmental factors and the level of the compensatory activity in the individual neurons, including the firing frequency of the motor neurons, the strength of the Ca2+ buffering system, and the density of functional Ca2+-permeable AMPA receptors. Furthermore, the results from the HeteroAR2 mice indicate that motor neurons expressing unedited GluA2, regardless of the proportion, are destined to die in sporadic ALS patients.
The progressive downregulation of ADAR2 activity increases the number of motor neurons expressing unedited GluA2 in sporadic ALS. The mechanism underlying the reduction of ADAR2 activity in ALS motor neurons is not clear; however, considering that inefficient GluA2 RNA editing was found only in sporadic cases (Kawahara et al., 2006), undefined postnatal factors regulating the ADAR2 activity should not be neglected. Because ADAR2 SNPs are associated with longevity syndrome (Sebastiani et al., 2009) and the age-dependent downregulation of ADAR2 activity has been shown in human brains (Nicholas et al., 2011), the acceleration of age-related neuronal dysfunction may have a role in the progressive reduction of ADAR2 activity in ALS motor neurons.
We recently reported that TDP-43 pathology, which is a hallmark of ALS, appeared only in the motor neurons lacking ADAR2 immunoreactivity in patients with sporadic ALS (Aizawa et al., 2010). It is likely that motor neurons lacking ADAR2 immunoreactivity represent those expressing unedited GluA2, and motor neurons expressing ADAR2 immunoreactivity with normal TDP-43 immunoreactivity represent those expressing only edited GluA2. Because a reduction of ADAR2 likely begins before motor neurons express unedited GluA2 in ALS motor neurons, TDP-43 pathology may be induced by the expression of unedited GluA2 rather than TDP-43 pathology causes reduced ADAR2 activity. We do not know whether the reduction of ADAR2 immunoreactivity in the ALS motor neurons results from a reduced gene expression or accelerated ADAR2 protein degradation catalyzed by Ca2+-activated proteinase as demonstrated in ischemic rat brains (Mahajan et al., 2011).
Based on this evidence, we propose a hypothesis for the pathogenesis of sporadic ALS. ALS motor neurons express progressively lower ADAR2 activity before manifesting an ALS phenotype, and the pathological process commences when motor neurons begin to express unedited GluA2. Motor neurons expressing unedited GluA2 do not function normally but do not immediately die. The timing of the entry of these motor neurons into the death cascade may be regulated in a stochastic manner. With a sequential progression of these events, the pool of normally functioning motor neurons expressing only edited GluA2 decreases, which ultimately induces the ALS phenotype in patients (Figure 12). Because mutations in the coding molecules of the genes involved in RNA regulation, including TDP-43 and FUS/TLS, were recently found in patients with familial ALS (Arai et al., 2006; Neumann et al., 2006; Kwiatkowski et al., 2009; Vance et al., 2009), dysregulation of RNA metabolism in ALS pathogenesis is now attracting the interest of researchers (Lemmens et al., 2010). Because ADAR2 is an RNA regulatory molecule, future studies are needed to elucidate the molecular link between abnormalities of these ALS-linked RNA regulatory molecules with the ADAR2 downregulation in ALS motor neurons. Forcing motor neurons to express only edited GluA2 may be a future therapy for sporadic ALS.
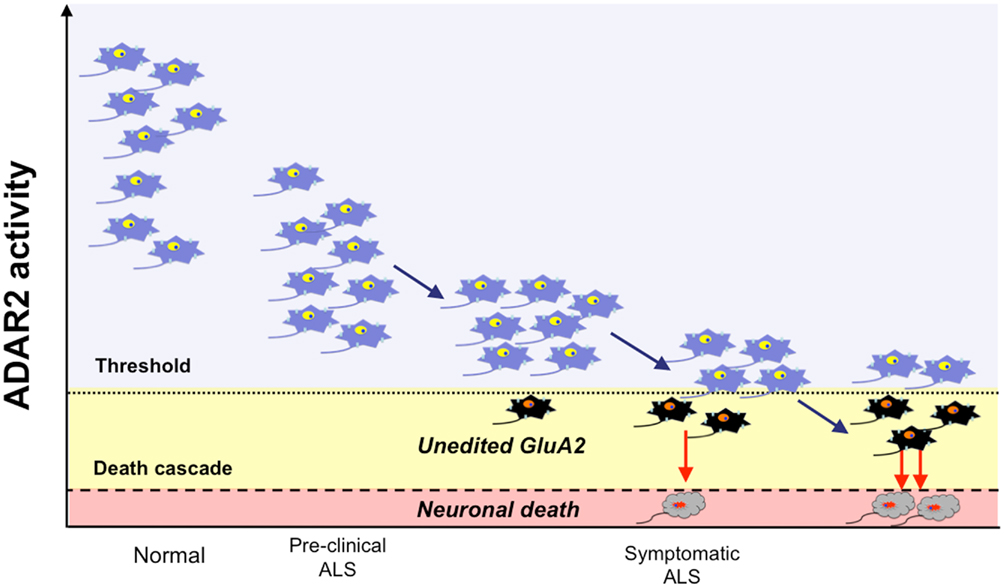
Figure 12. The ADAR2-GluA2 hypothesis for ALS pathogenesis. ADAR2 activity is preserved well above the level that enables motor neurons to edit the Q/R site of all GluA2 mRNA expressed in healthy individuals (normal; neurons in blue). In the motor neurons of sporadic ALS patients, ADAR2 activity progressively decreases, and the ADAR2 activity in all motor neurons is initially low but is within a level that is sufficient to edit the Q/R site of all GluA2 mRNA; thus, motor neurons function normally (pre-clinical ALS). With a decline of ADAR2 activity, some motor neurons express ADAR2 below a threshold level for GluA2 RNA editing (dotted line) and express unedited GluA2 (neurons in black). These motor neurons can survive for some period of time, but they do not function normally and ultimately enter into the death cascade in a stochastic manner. With the progressive decline of ADAR2 activity, the number of motor neurons expressing unedited GluA2, and therefore those destined to die, increases. Patients and neurologists recognize ALS only after a considerable proportion of motor neurons has already malfunctioned (neurons in black) or been lost (neurons in gray).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aizawa, H., Sawada, J., Hideyama, T., Yamashita, T., Katayama, T., Hasebe, N., Kimura, T., Yahara, O., and Kwak, S. (2010). TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 120, 75–84.
Akbarian, S., Smith, M., and Jones, E. (1995). Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer’s disease, Huntington’s disease and schizophrenia. Brain Res. 699, 297–304.
Arai, T., Hasegawa, M., Akiyama, H., Ikeda, K., Nonaka, T., Mori, H., Mann, D., Tsuchiya, K., Yoshida, M., Hashizume, Y., and Oda, T. (2006). TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611.
Brusa, R., Zimmermann, F., Koh, D., Feldmeyer, D., Gass, P., Seeburg, P., and Sprengel, R. (1995). Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science 270, 1677–1680.
Deng, H. X., Zhai, H., Bigio, E. H., Yan, J., Fecto, F., Ajroud, K., Mishra, M., Ajroud-Driss, S., Heller, S., Sufit, R., Siddique, N., Mugnaini, E., and Siddique, T. (2010). FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 67, 739–748.
Geiger, J. R., Melcher, T., Koh, D. S., Sakmann, B., Seeburg, P. H., Jonas, P., and Monyer, H. (1995). Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron 15, 193–204.
Greger, I. H., Khatri, L., and Ziff, E. B. (2002). RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 34, 759–772.
Hasegawa, M., Arai, T., Nonaka, T., Kametani, F., Yoshida, M., Hashizume, Y., Beach, T. G., Buratti, E., Baralle, F., Morita, M., Nakano, I., Oda, T., Tsuchiya, K., and Akiyama, H. (2008). Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70.
Hideyama, T., Yamashita, T., Suzuki, T., Tsuji, S., Higuchi, M., Seeburg, P. H., Takahashi, R., Misawa, H., and Kwak, S. (2010). Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J. Neurosci. 30, 11917–11925.
Higuchi, M., Maas, S., Single, F. N., Hartner, J., Rozov, A., Burnashev, N., Feldmeyer, D., Sprengel, R., and Seeburg, P. H. (2000). Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406, 78–81.
Higuchi, M., Single, F., Kohler, M., Sommer, B., Sprengel, R., and Seeburg, P. (1993). RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell 75, 1361–1370.
Ince, P., Stout, N., Shaw, P., Slade, J., Hunziker, W., Heizmann, C. W., and Baimbridge, K. G. (1993). Parvalbumin and calbindin D-28k in the human motor system and in motor neuron disease. Neuropathol. Appl. Neurobiol. 19, 291–299.
Jackson, M., Al-Chalabi, A., Enayat, Z. E., Chioza, B., Leigh, P. N., and Morrison, K. E. (1997). Copper/zinc superoxide dismutase 1 and sporadic amyotrophic lateral sclerosis: analysis of 155 cases and identification of a novel insertion mutation. Ann. Neurol. 42, 803–807.
Kabashi, E., Valdmanis, P. N., Dion, P., Spiegelman, D., McConkey, B. J., Vande Velde, C., Bouchard, J. P., Lacomblez, L., Pochigaeva, K., Salachas, F., Pradat, P. F., Camu, W., Meininger, V., Dupre, N., and Rouleau, G. A. (2008). TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574.
Kask, K., Zamanillo, D., Rozov, A., Burnashev, N., Sprengel, R., and Seeburg, P. H. (1998). The AMPA receptor subunit GluR-B in its Q/R site-unedited form is not essential for brain development and function. Proc. Natl. Acad. Sci. U.S.A. 95, 13777–13782.
Kawahara, Y., Ito, K., Sun, H., Aizawa, H., Kanazawa, I., and Kwak, S. (2004). Glutamate receptors: RNA editing and death of motor neurons. Nature 427, 801.
Kawahara, Y., Kwak, S., Sun, H., Ito, K., Hashida, H., Aizawa, H., Jeong, S. Y., and Kanazawa, I. (2003). Human spinal motoneurons express low relative abundance of GluR2 mRNA: an implication for excitotoxicity in ALS. J. Neurochem. 85, 680–689.
Kawahara, Y., Sun, H., Ito, K., Hideyama, T., Aoki, M., Sobue, G., Tsuji, S., and Kwak, S. (2006). Underediting of GluR2 mRNA, a neuronal death inducing molecular change in sporadic ALS, does not occur in motor neurons in ALS1 or SBMA. Neurosci. Res. 54, 11–14.
Kwak, S., and Kawahara, Y. (2005). Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J. Mol. Med. 83, 110–120.
Kwiatkowski, T. J. Jr., Bosco, D. A., Leclerc, A. L., Tamrazian, E., Vanderburg, C. R., Russ, C., Davis, A., Gilchrist, J., Kasarskis, E. J., Munsat, T., Valdmanis, P., Rouleau, G. A., Hosler, B. A., Cortelli, P., De Jong, P. J., Yoshinaga, Y., Haines, J. L., Pericak-Vance, M. A., Yan, J., Ticozzi, N., Siddique, T., Mckenna-Yasek, D., Sapp, P. C., Horvitz, H. R., Landers, J. E., and Brown, R. H. Jr. (2009). Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208.
Lemmens, R., Moore, M. J., Al-Chalabi, A., Brown, R. H. Jr., and Robberecht, W. (2010). RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci. 33, 249–258.
Mackenzie, I. R., Bigio, E. H., Ince, P. G., Geser, F., Neumann, M., Cairns, N. J., Kwong, L. K., Forman, M. S., Ravits, J., Stewart, H., Eisen, A., Mcclusky, L., Kretzschmar, H. A., Monoranu, C. M., Highley, J. R., Kirby, J., Siddique, T., Shaw, P. J., Lee, V. M., and Trojanowski, J. Q. (2007). Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 61, 427–434.
Mahajan, S. S., Thai, K. H., Chen, K., and Ziff, E. (2011). Exposure of neurons to excitotoxic levels of glutamate induces cleavage of the RNA editing enzyme, adenosine deaminase acting on RNA 2, and loss of GLUR2 editing. Neuroscience 189, 305–315.
Melcher, T., Maas, S., Herb, A., Sprengel, R., Seeburg, P., and Higuchi, M. (1996). A mammalian RNA editing enzyme. Nature 379, 460–464.
Misawa, H., Nakata, K., Toda, K., Matsuura, J., Oda, Y., Inoue, H., Tateno, M., and Takahashi, R. (2003). VAChT-Cre. Fast and VAChT-Cre.Slow: postnatal expression of Cre recombinase in somatomotor neurons with different onset. Genesis 37, 44–50.
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., Bruce, J., Schuck, T., Grossman, M., Clark, C. M., Mccluskey, L. F., Miller, B. L., Masliah, E., Mackenzie, I. R., Feldman, H., Feiden, W., Kretzschmar, H. A., Trojanowski, J. Q., and Lee, V. M. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Nicholas, A., De Magalhaes, J. P., Kraytsberg, Y., Richfield, E. K., Levanon, E. Y., and Khrapko, K. (2011). Age-related gene-specific changes of A-to-I mRNA editing in the human brain. Mech. Ageing Dev. 131, 445–447.
Nishimoto, Y., Yamashita, T., Hideyama, T., Tsuji, S., Suzuki, N., and Kwak, S. (2008). Determination of editors at the novel A-to-I editing positions. Neurosci. Res. 61, 201–206.
Nutt, S., and Kamboj, R. (1994). Differential RNA editing efficiency of AMPA receptor subunit GluR-2 in human brain. Neuroreport 5, 1679–1683.
Paschen, W., Hedreen, J., and Ross, C. (1994). RNA editing of the glutamate receptor subunits GluR2 and GluR6 in human brain tissue. J. Neurochem. 63, 1596–1602.
Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O’regan, J. P., Deng, H. X., Rahmani, Z., Krizus, A., McKenna-Yasek, D., Cayabyab, A., Gaston, S. M., Berger, R., Tanzi, R. E., Halperin, J. J., Herzfeldt, B., Van den Bergh, R., Hung, W.-Y., Bird, T., Deng, G., Mulder, D. W., Smyth, C., Laing, N. G., Soriano, E., Pericak–Vance, M. A., Haines, J., Rouleau, G. A., Gusella, J. S., Horvitz, H. R., and Brown, R. H. Jr. (1993). Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62.
Sebastiani, P., Montano, M., Puca, A., Solovieff, N., Kojima, T., Wang, M. C., Melista, E., Meltzer, M., Fischer, S. E., Andersen, S., Hartley, S. H., Sedgewick, A., Arai, Y., Bergman, A., Barzilai, N., Terry, D. F., Riva, A., Anselmi, C. V., Malovini, A., Kitamoto, A., Sawabe, M., Arai, T., Gondo, Y., Steinberg, M. H., Hirose, N., Atzmon, G., Ruvkun, G., Baldwin, C. T., and Perls, T. T. (2009). RNA editing genes associated with extreme old age in humans and with lifespan in C. elegans. PLoS ONE 4, e8210. doi:10.1371/journal.pone.0008210
Sommer, B., Kohler, M., Sprengel, R., and Seeburg, P. H. (1991). RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67, 11–19.
Sreedharan, J., Blair, I. P., Tripathi, V. B., Hu, X., Vance, C., Rogelj, B., Ackerley, S., Durnall, J. C., Williams, K. L., Buratti, E., Baralle, F., de Belleroche, J., Mitchell, J. D., Leigh, P. N., Al-Chalabi, A., Miller, C. C., Nicholson, G., and Shaw, C. E. (2008). TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672.
Sun, H., Kawahara, Y., Ito, K., Kanazawa, I., and Kwak, S. (2005). Expression profile of AMPA receptor subunit mRNA in single adult rat brain and spinal cord neurons in situ. Neurosci. Res. 52, 28–34.
Suzuki, T., Tsuzuki, K., Kameyama, K., and Kwak, S. (2003). Recent advances in the study of AMPA receptors. Folia Pharmacol. Jpn. 122, 515–526.
Takuma, H., Kwak, S., Yoshizawa, T., and Kanazawa, I. (1999). Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann. Neurol. 46, 806–815.
Tan, C. F., Eguchi, H., Tagawa, A., Onodera, O., Iwasaki, T., Tsujino, A., Nishizawa, M., Kakita, A., and Takahashi, H. (2007). TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol. 113, 535–542.
Tölle, T. R., Berthele, A., Zieglgänsberger, W., Seeburg, P. H., and Wisden, W. (1993). The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and periaqueductal grey. J. Neurosci. 13, 5009–5028.
Van Deerlin, V. M., Leverenz, J. B., Bekris, L. M., Bird, T. D., Yuan, W., Elman, L. B., Clay, D., Wood, E. M., Chen-Plotkin, A. S., Martinez-Lage, M., Steinbart, E., McCluskey, L., Grossman, M., Neumann, M., Wu, I. L., Yang, W. S., Kalb, R., Galasko, D. R., Montine, T. J., Trojanowski, J. Q., Lee, V. M., Schellenberg, G. D., and Yu, C. E. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416.
Van Den Bosch, L., Schwaller, B., Vleminckx, V., Meijers, B., Stork, S., Ruehlicke, T., Van Houtte, E., Klaassen, H., Celio, M. R., Missiaen, L., Robberecht, W., and Berchtold, M. W. (2002). Protective effect of parvalbumin on excitotoxic motor neuron death. Exp. Neurol. 174, 150–161.
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., Hu, X., Smith, B., Ruddy, D., Wright, P., Ganesalingam, J., Williams, K. L., Tripathi, V., Al-Saraj, S., Al-Chalabi, A., Leigh, P. N., Blair, I. P., Nicholson, G., De Belleroche, J., Gallo, J. M., Miller, C. C., and Shaw, C. E. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211.
Wang, Q., Khillan, J., Gadue, P., and Nishikura, K. (2000). Requirement of the RNA editing deaminase ADAR1 gene for embryonic erythropoiesis. Science 290, 1765–1768.
Keywords: ADAR2, RNA editing, GluA2, Q/R site, ALS, neuronal death, AMPA
Citation: Hideyama T and Kwak S (2011) When does ALS start? ADAR2–GluA2 hypothesis for the etiology of sporadic ALS. Front. Mol. Neurosci. 4:33. doi: 10.3389/fnmol.2011.00033
Received: 02 September 2011;
Paper pending published: 29 September 2011;
Accepted: 03 October 2011;
Published online: 02 November 2011.
Edited by:
William Wisden, Imperial College, UKReviewed by:
Peter H. Seeburg, Max Planck Institute for Medical Research, GermanyWilliam Wisden, Imperial College, UK
Copyright: © 2011 Hideyama and Kwak. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Shin Kwak, Department of Neurology, Graduate School of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan. e-mail:a3dhay10a3lAdW1pbi5hYy5qcA==