 Yanyun Sun1,2
Yanyun Sun1,2 Xi Chen3Xinyu Zhang1
Xi Chen3Xinyu Zhang1 Xianzhi Shen1Mengwei Wang1Xiaona Wang1,2Wen-Cao Liu4
Xianzhi Shen1Mengwei Wang1Xiaona Wang1,2Wen-Cao Liu4 Chun-Feng Liu1Jie Liu5
Chun-Feng Liu1Jie Liu5 Wenlan Liu6
Wenlan Liu6 Xinchun Jin1,2*
Xinchun Jin1,2*- 1Jiangsu Key Laboratory of Translational Research and Therapy for Neuro-Psycho-Diseases and Institute of Neuroscience, Department of Neurology, The Second Affiliated Hospital of Soochow University, Suzhou, China
- 2School of Pharmacy, Key Laboratory of Molecular Pharmacology and Drug Evaluation, Yantai University, Ministry of Education, Yantai, China
- 3The People’s Hospital of Baoan Shenzhen, Shenzhen, China
- 4Department of Emergency, Shanxi Provincial People’s Hospital, Taiyuan, China
- 5Translational Center for Stem Cell Research, Tongji Hospital, Stem Cell Research Center, Tongji University School of Medicine, Shanghai, China
- 6The Central Laboratory, Shenzhen Second People’s Hospital, Stem Cell Research Center, The First Affiliated Hospital of Shenzhen University, Shenzhen, China
Disruption of the blood brain barrier (BBB) within the thrombolytic time window is an antecedent event to intracerebral hemorrhage in ischemic stroke. Our recent studies showed that 2-h cerebral ischemia induced BBB damage in non-infarcted area and secreted matrix metalloproteinase-2 (MMP-2) accounted for this disruption. However, the factors that affect MMP-2 secretion and regulate BBB damage remains unknown. Since hypoxia-inducible factor-1 alpha (HIF-1α) was discovered as a mater regulator in hypoxia, we sought to investigate the roles of HIF-1α in BBB damage as well as the factors regulating HIF-1α expression in the ischemic brain. in vivo rat middle cerebral artery occlusion (MCAO) and in vitro oxygen glucose deprivation (OGD) models were used to mimic ischemia. Pretreatment with HIF-1α inhibitor YC-1 significantly inhibited 2-h MCAO-induced BBB damage, which was accompanied by suppressed occludin degradation and vascular endothelial growth factor (VEGF) mRNA upregulation. Interestingly, β2-adrenergic receptor (β2-AR) antagonist ICI 118551 attenuated ischemia-induced BBB damage by regulating HIF-1α expression. Double immunostaining showed that HIF-1α was upregulated in ischemic neurons but not in astrocytes andendothelial cells. Of note, HIF-1α inhibition with inhibitor YC-1 or siRNA significantly prevented OGD-induced VEGF upregulation as well as the secretion of VEGF and MMP-2 in neurons. More importantly, blocking β2-AR with ICI 118551 suppressedHIF-1α upregulation in ischemic neurons and attenuated occludin degradation induced by the conditioned media of OGD-treatedneurons. Taken together, blockade of β2-AR-mediated HIF-1α upregulation mediates BBB damage during acute cerebral ischemia. These findings provide new mechanistic understanding of early BBB damage in ischemic stroke and may help reduce thrombolysis-related hemorrhagic complications.
Introduction
Hemorrhagic transformation (HT), the most serious complication in thrombolytic therapy for acute ischemic stroke (Jin et al., 2015; Wang et al., 2015; Liu H. et al., 2016; Ji et al., 2017), occurs as a result of failure of the blood brain barrier (BBB) integrity (Simard et al., 2007). In the past decade, reperfusion-induced BBB injury has been an important focus probably because most of the damaging consequences of BBB injury (hemorrhage and edema) won’t be presented until the blood flow to the ischemic brain is restored (Simard et al., 2007; Hafez et al., 2014; Shi et al., 2016). Accumulating evidence showed that the brain regions with initial BBB damage often undergo HT during thrombolytic reperfusion (Kastrup et al., 2008; Sun et al., 2010; Leigh et al., 2014) and the importance of early ischemic BBB damage is increasingly recognized and is now emerging as pretreatment predictor as well as a promising target for clinical intervention to reduce thrombolysis-associated HT (Jin et al., 2014; Shi et al., 2016). However, ischemia-induced BBB damage, particularly at the early damage that occurs within the 4.5 h thrombolytic time window (Hacke et al., 2008), remains a much less-well studied topic.
Our recent studies showed that 2 h of ischemia caused brain tissue damage in cortex and BBB damage in non-infarcted ventromedial striatum and preoptic area (Jin et al., 2012; Wang et al., 2016). Ischemia-induced matrix metalloproteinase-2 (MMP-2) secretion lead to disrupted BBB and degradation of occludin (Jin et al., 2012; Liu et al., 2012; Wang et al., 2016), which is a key tight junction protein to seal the gaps between adjacent endothelial cells and thus restrict paracellular permeability of the BBB (Wolburg and Lippoldt, 2002). Occludin degradation is frequently seen in the compromised BBB following cerebral ischemia and reperfusion (Yang et al., 2007; Liu et al., 2012). However, we do not know the factors that affect MMP-2 secretion and regulate BBB damage. Hypoxia-inducible factor-1 alpha (HIF-1α) is constitutively transcribed and translated in most cell types, but the protein has only a half-life of less than 5 min under normoxic conditions (Shi, 2009; Semenza, 2014). It is induced in the brain under hypoxia/ischemia conditions and was discovered as a mater regulator in hypoxia (Bernaudin et al., 2002; Zhang et al., 2014). HIF-1α and downstream vascular endothelial growth factor (VEGF) have been shown to play important roles in ischemia-reperfusion induced BBB damage (Chen et al., 2009, 2010). For example, HIF-1α up-regulates VEGF and leads to BBB disruption (Semenza, 2014), while suppression of HIF-1α and VEGF reduces acute hyperglycemia-induced HT in the ischemic brain (Chen et al., 2010; Zhang Z. et al., 2016). Of note, VEGF secreted by hypoxic Müller cells induced MMP-2 activity in endothelial cells (Rodrigues et al., 2013). However, the role of HIF-1α in BBB damage within the first several hours after stroke onset is not known. More important, factors regulating HIF-1α under ischemia remain to be elucidated.
Activation of β2-adrenergic receptor (β2-AR) has been shown to upregulate HIF-1α in cancer cells (Shan et al., 2013). Ischemia produces a significant increase of norepinephrine (NE) in the striatum (Matsumoto et al., 1993), and NE, in turn, upregulates VEGF and MMP-2 (Yang et al., 2006). Therefore, in this study, we tested the hypothesis on an in vivo model of middle cerebral artery occlusion (MCAO) and an in vitro model of oxygen glucose deprivation (OGD) that cerebral ischemia induces β2-AR activation, and activated β2-AR upregulates HIF-1α to promote MMP-2 secretion and BBB disruption. Our data showed that β2-adrenergic receptor inhibition attenuated HIF-1α upregulation as well as BBB damage within the first several hours of cerebral ischemia.
Materials and Methods
Animal Model of Focal Cerebral Ischemia
Sprague-Dawley rats were purchased from SLAC Company (Shanghai, China). They were housed 2–3 per cage under constant temperature (23 ± 1°C) and light-controlled vivarium (12-h light/12-h dark cycle). Rats housed in the same cage underwent the same manipulations. Food and water were available ad libitum. All animal procedures were approved by the University Committee on Animal Care of Soochow University and performed according to the NIH Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and to reduce the number of animals. Rats weighing 270–290 g were subjected to 2-h MCAO using the intraluminal suture occlusion model, as we described previously (Liu et al., 2017). Prior to reperfusion, all rats included in this study displayed typical neurologic deficit of MCAO, circling to the left (non-ischemic side). Detailed animal usage for each experiment was listed in figure legends. Successful MCAO was further confirmed by 2,3,5-triphenyltetrazolium chloride (TTC) staining.
YC-1 Administration
To establish a causal role of HIF-1α in early ischemic BBB damage, the HIF-1α inhibitor YC-1 (Cayman Chemical Company, Ann Arbor, MI, USA dissolved in a solution of 1% dimethyl sulfoxide, DMSO) or vehicle was administered at 2 mg/kg body weight through femoral vein at 24 h and 30 min prior to the onset of ischemia. To inhibit HIF-1α, researchers have used various dosages of YC-1 ranging from 1 mg/kg to 30 mg/kg body weight (Hsiao et al., 2004; Yeh et al., 2007). We chose the dose based on previous publications (Fischer et al., 2002; Schoch et al., 2002; Yan et al., 2011).
ICI 118551 Administration
To block interaction of NE with β2-AR, β2-AR antagonist ICI 118551 (30 nmol/μL, 0.5 mL, Sigma) or vehicle was injected into the ventral striatum of the ischemia hemisphere with a Hamilton syringe at a rate of 0.25 μL/min 10 min before MCAO. The coordinates for ventral striatum are taken from the atlas of Paxinos and Watson (1986) (AP, −1.0 mm; ML, −2.0 mm; DV, −7.0 mm).
Evan’s Blue Leakage Detection
Immediately after 2-h MCAO, Evan’s blue dye (EB; Sigma, St. Louis, MO, USA, 2% wt/vol in PBS) was intravenously administered (3 mL/kg) via the left femoral vein. All rats were reperfused for 10 min to allow sufficient circulation of EB to the ischemic brain, but minimize the impact of reperfusion on BBB integrity. At the end of reperfusion, the rat was transcardially perfused with ice-cold PBS and then the brain was quickly taken out.
1. EB leakage was used to check the spatial distribution of BBB damage. Ten consecutive 1-mm-thick coronary slices were sectioned from a 10-mm-thick brain region which was 3 mm away from the tip of the frontal lobe. Besides characterizing the topographic distribution of EB leakage, the mean leakage area was calculated as averaged area proportion of the sections measured.
2. EB leakage was used to detect the co-localization of BBB damage and occludin. Twenty-micrometer-thick cryosection was cut from the 8-mm-thick brain region as we described (Jin et al., 2012) and mounted for fluorescence microscopy observation. BBB disruption was visualized as leakage of EB, which appeared as red fluorescence on brain sections. Brain sections were subjected to immunostain analysis for occludin expression. The experimental procedures for immunostain were described in details below.
3. EB was infused via femoral vein and its content in ischemic hemisphere compared to contralateral hemisphere to quantify BBB disruption as previously reported (Liu et al., 2017). Briefly, rat brain was quickly removed after transcardial perfusion with PBS. Non-ischemic and ischemic hemispheric tissues were harvested as described above and homogenized in 50% wt/vol trichloroacetic acid (Sigma, St. Louis, MO, USA). After centrifugation, the supernatant was diluted four folds with ethanol, and fluorescence intensity (μg/mL) was measured on a microplate fluorescence reader (Infinite M200 Pro; TECAN, Austria). The total EB content (μg) in each sample was derived from concentrations of external standards (1–20 μg/mL). The difference of dye content between ischemic and nonischemic (NI) hemispheric tissue was calculated as EB leakage and expressed as per gram of brain tissue (μg/g).
Immunostaining
Immediately after 2-h MCAO and 10 min reperfusion, the rat was perfused with ice-cold PBS followed by 4% PFA. The 20-μm-thick cryosection was used for immunostaining analysis for occludin, HIF-1α and VEGF as we described previously (Wang et al., 2017). In brief, non-specific binding sites were blocked by pre-incubating tissue for 1 h at room temperature in PBS containing 0.1% Triton X-100, 1% BSA, and 5% goat serum. Sections were then incubated overnight with anti-HIF-1α (1:200 dilution, Novus), anti-VEGF antibody (1:200 dilution, Abcam), anti-NeuN (1:200 dilution, Millipore), anti-GFAP (1:2000 dilution, Millipore), anti-RECA-1 (1:100 dilution, Abcam) or anti-occludin (1:150 dilution, Invitrogen) primary antibody at 4°C. The latter was followed by incubation with Cy3 conjugated secondary antibody (anti-mouse, 1:800 dilution) or 488-conjugated secondary antibody (anti-rabbit, 1:800 dilution) for 2 h at room temperature. Immunostain was visualized under LSM 700 confocal laser-scanning microscope (Zeiss), and images were taken from the ischemic region and the mirrored region on the NI hemisphere.
Cell Culture
Mouse brain microvascular endothelial cells bEND3 (American Type Culture Collection) were grown as a monolayer in DMEM with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/mL streptomycin at 37°C in a humidified incubator with 5% CO2 and 95% room air. The cells were subcultured into 60 mm dishes coated with type I collagen (Nakamuta et al., 2005) and allowed to grow to confluence before exposure to OGD for 2 h. After OGD treatment (3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium, MTT; Sigma) assay was used to assess the cytotoxicity.
SH-SY5Y cells (neurons) were purchased from American Type Culture and was cultured in DMEM/1640 (1:1; Gibico, Life technologies) containing 10% FBS, 100U/mL penicillin and 100 μg/mL streptomycin, respectively, at 37°C in a humidified incubators with 5% CO2 and 95% room air. The cells were subcultured into 6-well cell culture cluster and allowed to grow to 80%–90% confluence before exposure to OGD for 2 h.
Primary astrocyte cultures were prepared from 2-day-old neonatal C57BL/6J mice. In brief, dissociated cortical cells were suspended in DMEM/1640 (Gibico, Life Technology) containing 25 mM glucose, 4 mM glutamine, 1 mM sodium pyruvate, and 10% FBS and plated on uncoated 25 cm2 flasks at a density of 6 × 105 cells cm−2. Monolayers of type 1 astrocytes were obtained 12–14 days after plating. Non-astrocytic cells were detached from the flasks by shaking and removed by changing the medium. Astrocytes were dissociated by trypsinization and then reseeded on uncoated T75 flasks. These cells were allowed to grow to 80%–90% confluence before exposure to 2-h OGD treatment.
OGD Treatment
To mimic acute ischemia-like conditions in vitro, SH-SY5Y cells were exposed to OGD for 2 h as we described previously (Liu J. et al., 2016). In brief, confluent SH-SY5Y cells were subjected to an ischemic injury by transferring cultures to glucose free medium (DMEM/1640 without glucose) pre-equilibrated with 95% N2 and 5% CO2. Cells were then incubated in a humidified airtight chamber (MIC-101, Billups-Rothberg Inc., Del Mar, CA, USA) equipped with an air lock and flushed with 5% CO2/95% N2 for 15 min. The chamber was then sealed and kept at 37°C for another 105 min. Control cultures were incubated with normal DMEM/1640 medium without FBS for 2 h at 37°C in 5% CO2/95% air. Immediately after OGD treatment, the conditioned media (CM) and cells were collected separately for further analyses.
YC-1 and ICI 118551 Treatment for In Vitro Study
Cells grown on six-well plates at 80%–90% confluence were incubated with the corresponding medium containing HIF-1α inhibitor YC-1 (10 μmol/L) at 2 h before OGD or β2-AR antagonist ICI 118551 (1 μmol/L) at 10 min before OGD.
Occludin Degradation by Conditioned Media from OGD-Neurons
At 24 h after seeding, the media were replaced with conditioned media collected from OGD-treated neurons (OGD-neuron CM) or media from control neurons without OGD treatment (Neuron media). Endothelial cells that were maintained in regular endothelial cell media (Media) served as controls. To investigate whether the β2-AR antagonist ICI 118551 suppressed occludin degradation, endothelial cells exposed to OGD-neuron CM were co-treated with vehicle or β2-AR antagonist ICI118 551 at the concentrations of 1 μmol/L.
siRNA Transfection
SH-SY5Y cells at 60%–70% confluence were transfected with 6 μL HIF-1α siRNA (Santa Cruz, sc-35561) or control siRNA-A (Santa Cruz, sc-37007) which was diluted with the same volume of transfection reagent (Santa Cruz, sc-29528) according to manufacturer’s instruction. Specific silencing was confirmed by western blot.
Gel Gelatin Zymography
Tissue were homogenized in MMP lysis buffer (50 mM Tris-HCl PH 7.6, 150 mM NaCl, 5 mM CaCl2•2H2O, 0.05% Brij-35, 0.02% NaN3, 1% Triton X-100) and MMP-2/9 levels in homogenates were assessed by gel gelatin zymography as we described previously (Shu et al., 2015).
Western Blot Analysis for Occludin, HIF-1α and VEGF
Homogenate aliquots (30 μg of total protein) were boiled and then electrophoresed in 10% SDS-PAGE acrylamide gels, transferred onto nitrocellulose membranes (Bio-Rad) and incubated for 1 h in Tris-buffered saline and 0.1% Tween 20 (TBS-T) containing 5% nonfat milk. Membranes were then incubated overnight at 4°C with primary antibodies against occludin (1:300, Invitrogen), HIF-1α (1:300, Novus), VEGF (1:500, Abcam), washed in TBS-T, and then incubated for 2 h at room temperature with corresponding HRP-conjugated anti-rabbit or anti-mouse antibodies (1:3000, Boster). The membranes were developed with the SuperSignal West Pico HRP substrate kit (Pierce) and photographed on a Gel DOCTM XR+ image station (Bio-rad). Protein band intensities were quantitated after normalization to β-actin or total protein stained by Ponceau S.
Real-Time RT-PCR
Total cellular RNA was isolated using Trizol reagents (Invitrogen) according to manufacturer’s protocol as we described previously (Jin et al., 2015). RNA (0.5 μg) was reverse-transcribed (RT) with random primers in a 20 μL final reaction volume using TaqMan® Reverse Transcription Kits (Applied Biosystems). 0.5 μL RT products were amplified with the 7900HT Fast Real-Time PCR System (Applied Biosystems) in a 10 μL final reaction volume using SYBR® Green PCR Master Mix (Applied Biosystems) under the following conditions: 2 min at 50°C and 10 min at 95°C, followed by a total of 40 cycles of two temperature cycles (15 s at 95°C and 1 min at 60°C). Primers (Integrated DNA Technologies) for VEGF and glyceraldehydes 3-phosphate dehydrogenase (GAPDH) were designed against known mouse sequences: VEGF forward: 5′-AGAAAGCCCATGAAGT GGTG-3′, reverse: 5′-ACTCCAGGGCTTCATCATTG-3′; β-actin forward: 5′-ACTATCGGCAATGAGCGGTTCC-3′, reverse: 5′-AGCACTGTGTTGGCATAGAGGTC-3′. The fluorescence threshold value (Ct value) was calculated using the SDS Enterprise Database software (Applied Biosystems). The relative value of mRNA expression was calculated by the comparative ΔΔCt method described in our previous publication (Liu et al., 2007). In brief, mean Ct values were normalized to the internal control GAPDH and the difference was defined as ΔΔDCt. The difference between the mean ΔCt values of treated and untreated cells was calculated and defined as ΔΔDCt. The comparative mRNA expression level was expressed as 2−ΔΔDCt.
ELISA Assay for VEGF
VEGF contents in cell culture supernatants were assessed using an ELISA kit from Boster (Wuhan, China). Aliquots of 100 μL of cell conditioned media were added to the appropriate microtiter wells provided by the kit. For the standard curve, 100 μL of different concentration VEGF standards and the blank standard were added to the appropriate microtiter wells. The plate was then incubated for 1.5 h at 37°C. After discarding the liquid of each well (no wash), 100 μL of biotinylated anti-human VEGF antibody solution was added into each well except the chromogen blanks and incubated for 1 h at 37°C. After washing three times with the wash buffer, 100 μL avidin-peroxidase complex working solution was added to each well, except the chromogen blanks, and incubated for 30 min at 37°C. Lastly, 100 μL of stabilized chromogen was added to each well, and incubated for 30 min at 37°C in the dark before the addition of 100 μL of stop solution. The absorbance at 450 nm was recorded on a plate reader. A standard curve was generated and the concentrations for unknown samples were obtained from the standard curve.
Statistical Analysis
The data are presented as mean ± SEM. Statistical analysis was carried out using ANOVA (SPSS software, version 17.0). Significant effects were probed using Newman-Keuls post hoc comparison. A value of P < 0.05 was considered statistically significant.
Results
HIF-1α and VEGF Changes after 2-h MCAO
HIF-1α is a key mediator of the adaptive cellular response to hypoxia and VEGF has been implicated in BBB permeability increase (Schoch et al., 2002; Yan et al., 2011). To determine the roles of HIF-1α and VEGF in BBB damage after 2-h MCAO, western blot was used to compare HIF-1α and VEGF levels in the regions of interest (ROI 1, tissue damage area; ROI 2, BBB damage area; Jin et al., 2012). As shown in Figure 1, 2-h MCAO induced a nearly 1.5-fold increase of HIF-1α protein in ROI 2 (P < 0.05), but not ROI 1 (P > 0.05, Figure 1C). Pretreatment with HIF-1α inhibitor YC-1 at a dose of 2 mg/kg body weight significantly prevented this increase (P < 0.05, Figure 1C). Surprisingly, 2-h MCAO did not change the protein levels of VEGF (Figure 1D), but significantly increased VEGF mRNA expression, and YC-1 significantly inhibited VEGF upregulation (Figure 1E). The results suggested that 2-h MCAO induced VEGF mRNA expression in a HIF-1α-dependent manner.
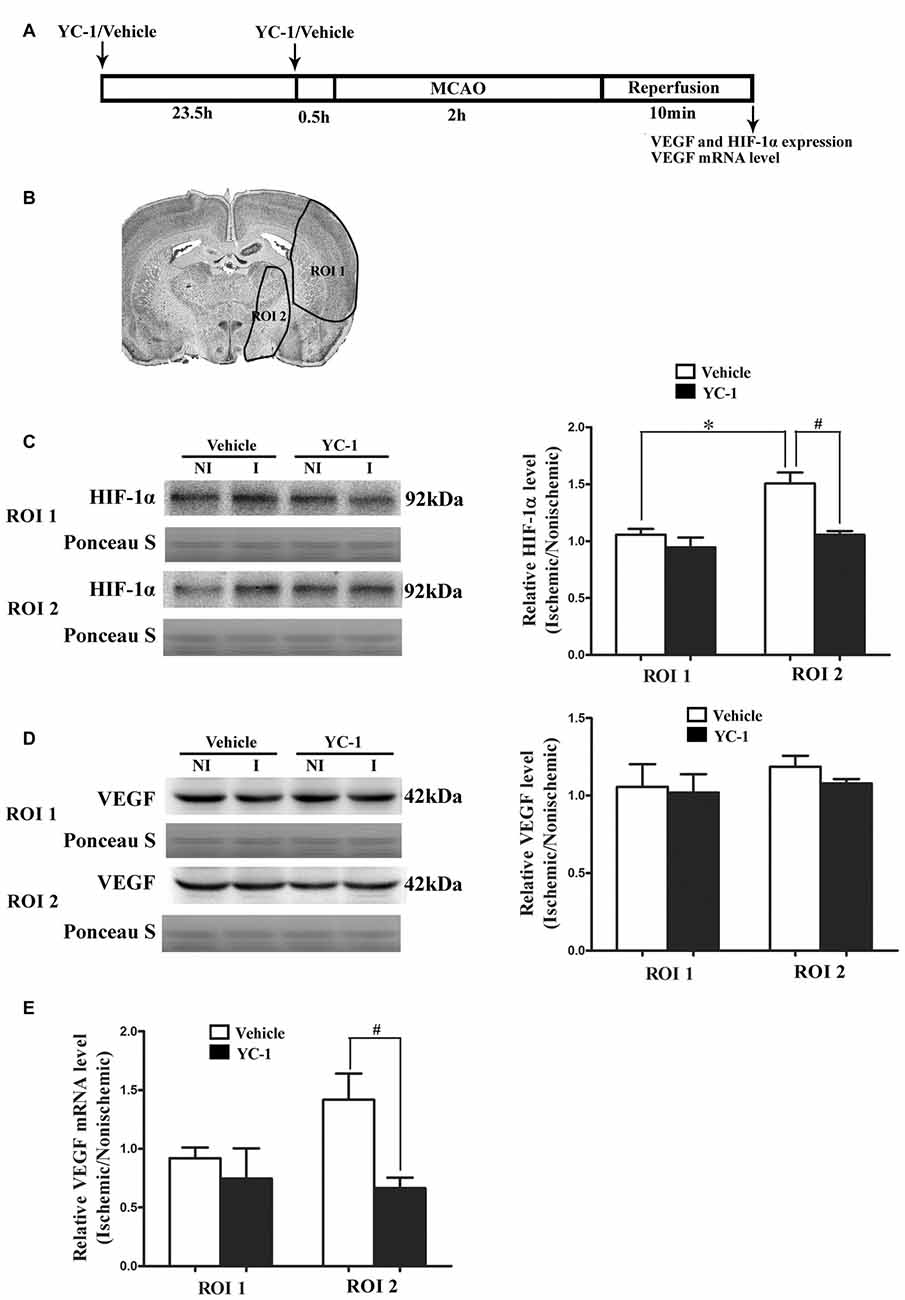
Figure 1. Effect of 2-h ischemia on hypoxia-inducible factor-1 alpha (HIF-1α) and vascular endothelial growth factor (VEGF) expression. (A) Diagram of the experimental procedure. (B) The black outlines showed the two regions of interest (ROI): 1, parietal and insular cortex and dorsal striatum; 2, ventral striatum and preoptic area. (C) Representative western blot images of HIF-1α protein in ROI 1 and 2 in ischemic (I) and nonischemic (NI) hemisphere (left panel). Ratios of HIF-1α (I/NI) were quantitated (right panel). (D) Representative western blot images of VEGF protein in region of ROI 1 and 2 in I and NI hemisphere (left panel). Ratios of VEGF (I/NI) were quantitated (right panel). After 2-h middle cerebral artery occlusion (MCAO), there was a significant increase of HIF-1α protein (C) but not VEGF protein (D) in ROI 2 but not ROI 1 and pretreatment with HIF-1α inhibitor YC-1 significantly prevented this increase. #P < 0.05 vs. Vehicle group in ROI 2, *P < 0.05 vs. Vehicle group in ROI 1. (E) Ratio of VEGF mRNA (I/NI) was quantitated. Two-hour MCAO induced significant upregulation of VEGF mRNA and pretreatment with YC-1 significantly prevented this increase (#P < 0.05 vs. Vehicle). Data were expressed as mean ± SEM, n = 4/group.
HIF-1α Inhibitor YC-1 Inhibited 2-h MCAO-Induced BBB Disruption and Occludin Degradation
EB was used as a marker to evaluate BBB integrity (Wang et al., 2017). We next examined the effect of HIF-1α inhibitor YC-1 treatment on 2-h MCAO-induced BBB disruption and occludin degradation (Figure 2A). Representative images of EB dye in wet brain tissue was shown in Figure 2B. Obvious EB leakage was observed in the ipsilateral hemisphere of brain from rats subjected to 2-h MCAO (Figure 2B). HIF-1α inhibitor YC-1 treatment dramatically reduced the area of EB leakage (Figure 2C) and quantification data showed that YC-1 decreased ~78% of EB leakage (Figure 2D). These results indicated that HIF-1α inhibition was effective in reducing BBB damage within the first 2 h after ischemia onset.
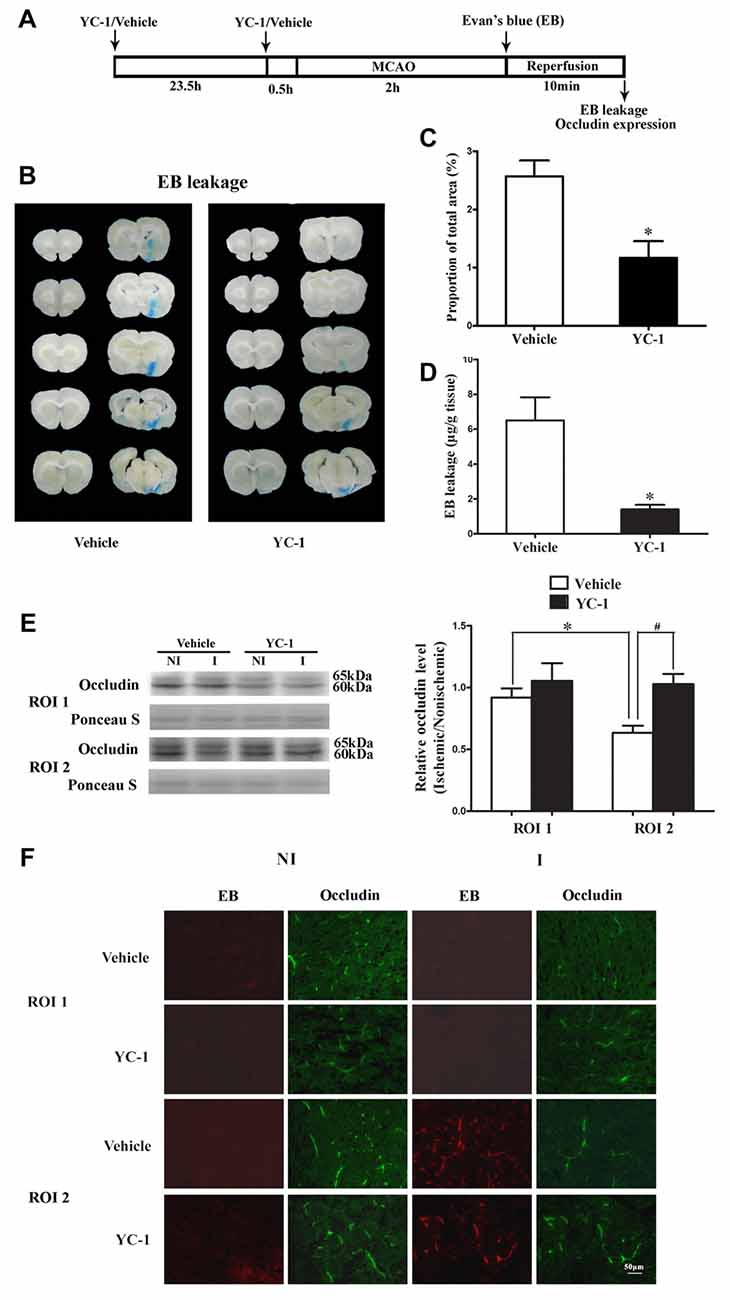
Figure 2. Effect of YC-1 treatment on 2-h ischemia-induced Evan’s blue (EB) leakage and occludin expression. (A) Diagram of the experimental procedure. Rats received YC-1 at 24 h and 30 min before ischemia onset. (B) Ten consecutive sections showed EB leakage from vehicle or YC-1 treated rats. (C) EB leakage was quantitated and expressed as average area proportion of section measured (%). #P < 0.05 vs. Vehicle group. n = 6/group. (D) EB leakage in the brain tissue was quantitated according to the external EB standard curve. EB leakage was expressed as per gram of brain tissue (μg/g). #P < 0.05 vs. Vehicle group. Data were expressed as mean ± SEM, n = 5/group. (E) Western blot was conducted to detect occludin protein levels in the NI and ischemic (I) hemispheric tissue. A representative western blot revealed occludin protein expression in ROI 1 and ROI 2 treated with YC-1 or vehicle (left panel). The band intensity of occludin was quantitated after normalization to the Ponceau S (right panel). Two-hour MCAO induced a significant reduction in occludin protein level in ROI 2 (E, right panel. *P < 0.05 vs. Vehicle group in ROI 1), but not in ROI 1 (P > 0.05). Pretreatment with YC-1 attenuated occludin degradation (#P < 0.05 vs. Vehicle group). Data were expressed as mean ± SEM, n = 5/group. (F) EB leakage (red) was seen in the ischemic ROI 2 after 2-h ischemia and IHC showed occludin degradation in the area where EB leakage occurred. Pretreatment with YC-1 significantly attenuated occludin degradation as well as EB leakage. n = 3/group. Data were expressed as mean ± SEM.
Our recent study showed that loss of occludin contributed to BBB damage after 2-h ischemia (Wang et al., 2016). To determine whether ischemia-induced HIF-1α upregulation contributed to the rapid disruption of occludin, IHC and western blot were conducted to detect occludin protein levels in ROI with BBB leakage. Our data showed that pretreatment with YC-1 significantly inhibited ischemia-induced occludin degradation in ROI 2 (EB leakage area, Figure 2F). Western blot was done to confirm the IHC results and found that 2-h MCAO induced a significant reduction in occludin protein levels in ROI 2, but not ROI 1 (Figure 2E). These results indicated that HIF-1α inhibition significantly attenuated occludin degradation within the first 2 h after ischemia onset (Figure 2E).
Effect of β2-AR Antagonist ICI 118551 on Ischemia-Induced HIF-1α Upregulation and BBB Disruption
β2-AR has been shown to regulate HIF-1α expression in cancer cells (Shan et al., 2013). To investigate the effect of blocking β2-AR on ischemia-induced HIF-1α upregulation and BBB disruption, rats received ICI118551 (30 nmol/mL, 0.5 mL; Qu et al., 2008) 10 min before MCAO (Figure 3A). Our results showed that blocking β2-AR significantly inhibited 2-h ischemia-induced HIF-1α upregulation (Figure 3B, P < 0.05 vs. Vehicle), occludin degradation (Figure 3C, P < 0.05 vs. Vehicle) and BBB damage (Figures 3D–F, P < 0.05 vs. Vehicle), suggesting that blocking β2-AR inhibited BBB damage through regulating HIF-1α.
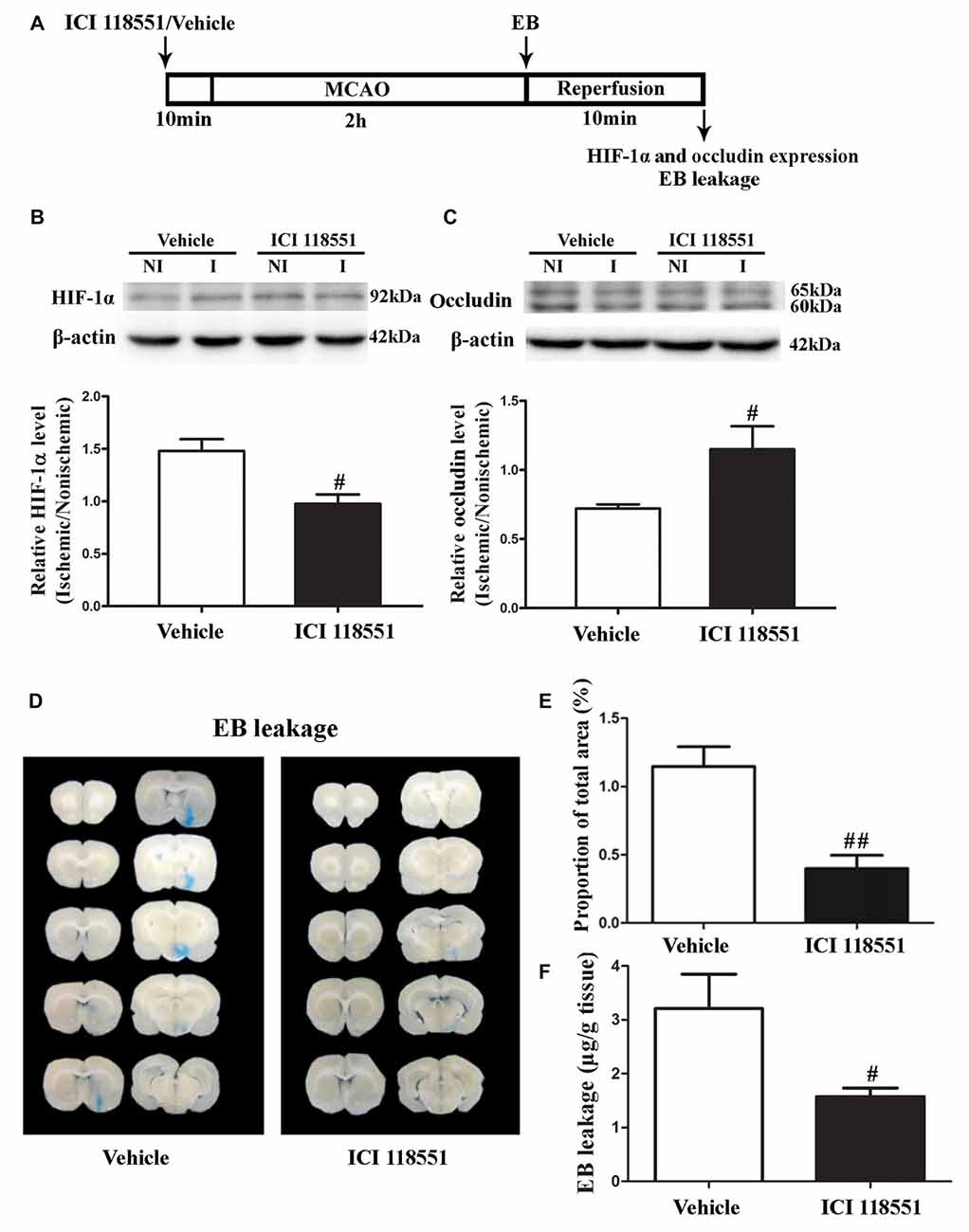
Figure 3. Effect of β2 adrenergic receptor (β2-AR) antagonist ICI118551 on 2-h ischemia-induced HIF-1α expression and blood brain barrier (BBB) disruption. (A) Diagram of the experimental procedure. Rats received ICI118551 10 min before the onset of ischemia. (B,C) Western blot was conducted to detect HIF-1α (B) and occludin (C) protein levels in the NI and ischemic (I) hemispheric tissue. A representative western blot revealed HIF-1α (B) and occludin (C) protein expression treated with YC-1 or vehicle. The band intensity of HIF-1α and occludin was quantitated after normalization to the β-actin. Two-h MCAO induced a significant increase of HIF-1α (B) and reduction of occludin (C). Pretreatment with ICI118551 inhibited HIF-1α upregulation and attenuated occludin degradation (#P < 0.05 vs. Vehicle group). Data were expressed as mean ± SEM, n = 5/group. (D) Ten consecutive sections showed EB leakage from vehicle or YC-1 treated rats. (E) EB leakage was quantitated and expressed as average area proportion of section measured (%). ##P < 0.01 vs. Vehicle group, n = 6/group. (F) EB leakage in the brain tissue was quantitated according to the external EB standard curve. EB leakage was expressed as per gram of brain tissue (μg/g). #P < 0.05 vs. Vehicle group. Data were expressed as mean ± SEM, n = 5/group.
Colocalization of HIF-1α with Endothelial Cells, Neurons and Astrocytes
Cell type-specific action for HIF-1α has been defined in astrocytes and neurons in a rat neonatal stroke model (Mu et al., 2003; Vangeison et al., 2008). However, the cellular pattern of HIF-1α expression was not known in the ischemic brain at 2 h after MCAO onset. Double-immunofluorescence labeling was used to determine the cellular localization of HIF-1α, and DAPI staining was conducted to verify the subcellular localization of HIF-1α. As shown in Figure 4, HIF-1α was mainly expressed in neurons (Figure 4B, upper panel), but not in astrocytes (Figure 4B, middle panel) or endothelial cells (Figure 4B, bottom panel) in the ROI 2 at 2 h after MCAO onset. YC-1 significantly decreased the number of HIF-1α-positive neurons, but had no effect on HIF-1α expression in astrocytes or endothelial cells (Figure 4C). These results indicated that the neurons might be major cellular source of HIF-1α early after cerebral ischemia onset.
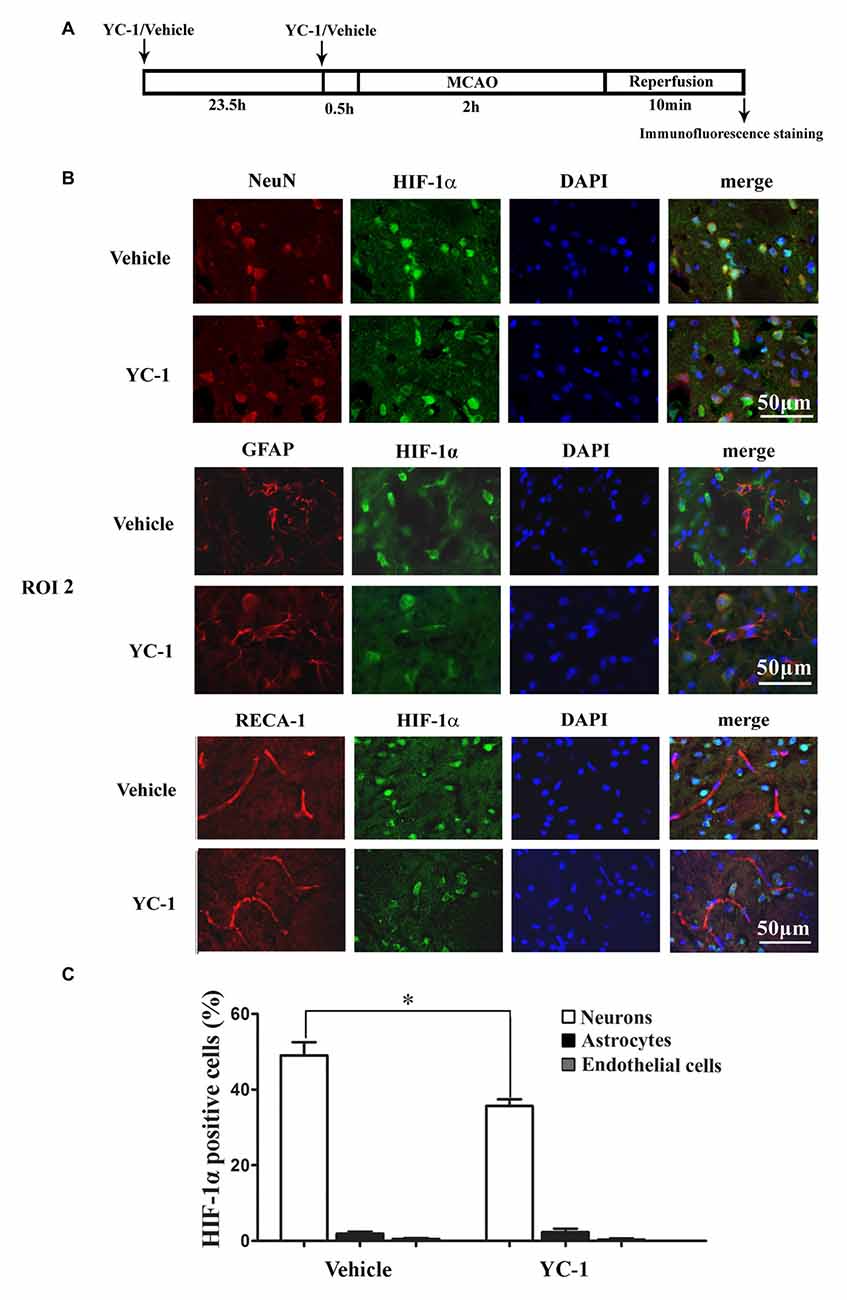
Figure 4. Effect of YC-1 on 2-h ischemia-induced HIF-1α expression in endothelial cells, neurons and astrocytes. (A) Diagram of the experimental procedure. Rats received YC-1 at 24 h and 30 min before ischemia onset. The cellular pattern of HIF-1α expression was analyzed by double-immunofluorescence labeling. Double immunostain of HIF-1α (green) and NeuN (marker of neurons, red) showed a good colocalization of HIF-1α and neurons (B, upper panel). Double immunostain of HIF-1α (green) and GFAP (marker of astrocytes, red) showed no co-localization of HIF-1α and astrocytes (B, middle panel). Double immunostain of HIF-1α (green) and RECA-1 (marker of endothelial cells, red) showed no co-localization of HIF-1α and endothelial cells (B, bottom panel). n = 3/group. Scale bar = 50 μm. (C) YC-1 significantly decreased the proportion of HIF-1α-positive neurons but not astrocytes or endothelial cells *P < 0.05 vs. Vehicle group. n = 3/group.
The cellular distribution and expression of VEGF protein was determined by immunofluorescence with a polyclonal VEGF antibody. VEGF-positive cells were identified to be neurons (colocalization with NeuN but not with GFAP, data not shown).
Effect of HIF-1α Inhibition on VEGF mRNA Transcription and Protein Secretion
Hypoxia rapidly increased the HIF-1α as well as the mRNA levels of VEGF (Bernaudin et al., 2002), but VEGF protein increase was much later, which occurred at 4 h or 8 h during the phase of reperfusion (Mu et al., 2003). We applied oxygen-glucose deprivation (OGD) to mimic ischemia in vitro (Zhang Y. et al., 2016). Human dopaminergic neurons (SH-SY5Y cells) were subjected to 2-h OGD. YC-1 or HIF-1α siRNA was applied to inhibit HIF-1α level and non-targeting siRNA was used as a control siRNA (Ctrl siR). ELISA and western blot were recruited to detect VEGF level in cultured media (CM) and cellular extracts (CE), respectively.
Two-hour OGD induced a significant increase of VEGF mRNA expression in neurons (P < 0.05 vs. Ctrl), and pretreatment with YC-1 (Figure 5A, left panel) or HIF-1α siR (Figure 5A, right panel) significantly inhibited this effect (P < 0.05 vs. OGD). However, 2-h OGD had no effect on the protein levels of VEGF in the CE (Figure 5B), but significantly increased its levels in the CM (Figure 5C, P < 0.05 vs. Ctrl). Pretreatment with YC-1 (Figure 5C, left panel) or HIF-1α siR (Figure 5C, right panel) significantly inhibited VEGF increase in the CM (P < 0.05 vs. OGD) suggesting that VEGF secretion was mediated by HIF-1α.
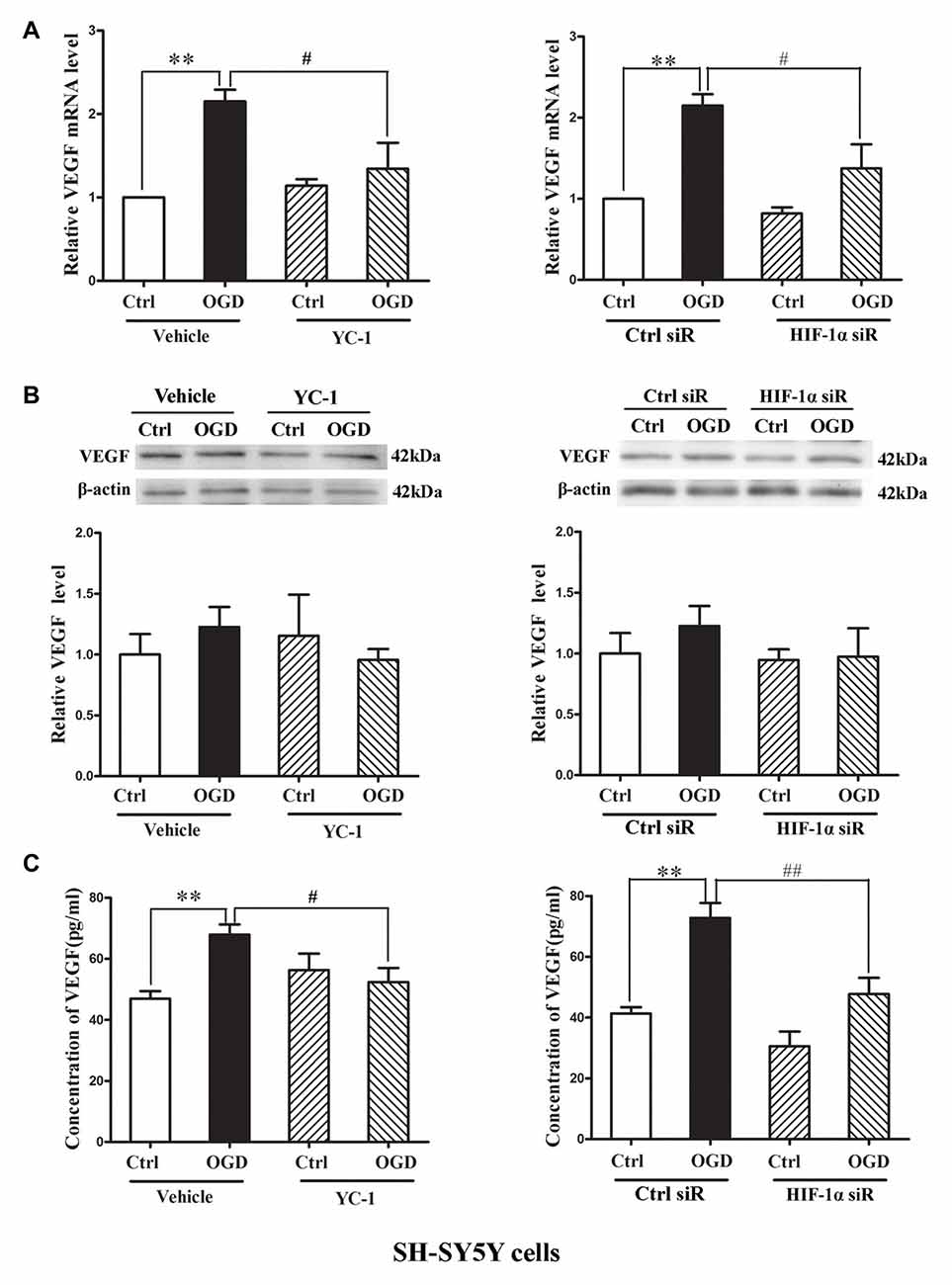
Figure 5. Effect of HIF-1α inhibition by YC-1 or HIF-1α siRNA on 2-h oxygen glucose deprivation (OGD)-induced VEGF secretion in neurons. The neurons were exposed to OGD for 2 h and VEGF secretion was assessed by measuring its content in cellular extracts (CE) and conditioned media (CM) using western blot and ELISA respectively. Two-hours OGD induced a significant increase in VEGF mRNA expression in neurons and pretreatment with YC-1 (A, left panel) or HIF-1α siR (A, right panel) significantly inhibited this effect. **P < 0.01 vs. control (Ctrl), #P < 0.05 vs. OGD, n = 4/group. (B) Representative blots for VEGF and β-actin (upper panel). The relative VEGF protein level was calculated after normalization to β-actin (bottom panel). Two-hours OGD had no effect on the VEGF protein levels in neurons but induced a significant increase of VEGF in cultured media and pretreatment with HIF-1α inhibitor YC-1 (C, left panel) or HIF-1α siR (C, right panel) attenuated this VEGF secretion induced by 2-h OGD. **P < 0.01 vs. Ctrl. #P < 0.05 vs. OGD. ##P < 0.01 vs. OGD. HIF-1α siR represents HIF-1α siRNA. Data are expressed as mean ± SEM, n = 3/group.
Effect of HIF-1α Inhibition on MMP-2 Secretion in Neurons and Endothelial Cells
We applied in vitro cultured brain microvascular endothelial monolayer to mimic the BBB as bEND3 cell line has the ability to maintain BBB characteristics over many passages, is easy to grow and can form functional barriers (Camós and Mallolas, 2010). We have shown that OGD induces MMP-2 secretion in neurons, astrocytes and endothelial cells and the secreted MMP-2 disrupts BBB by degrading occludin (Liu et al., 2012), here we explored the effect of YC-1 on MMP-2 secretion in neurons and endothelial cells. After 2-h OGD, MMP-2 levels in CM and CE were detected by gel gelatin zymography. As shown in Figure 6, OGD-induced MMP-2 increase in the conditioned media accompanied by a decrease in the CE, indicating an OGD-induced MMP-2 secretion in neurons. Pretreatment with YC-1 significantly inhibited MMP-2 secretion in neurons (Figures 6A,B, P < 0.05 vs. OGD), but not in endothelial cells (Figures 6C,D). Of note, YC-1 did not reverse OGD-induced occludin degradation in endothelial cells (Figure 6E). These results suggested that HIF-1α mediated OGD-induced MMP-2 secretion in neurons but not in endothelial cells, and further studies are warranted to define the mechanisms underlying MMP-2 secretion in endothelial cells under OGD conditions.
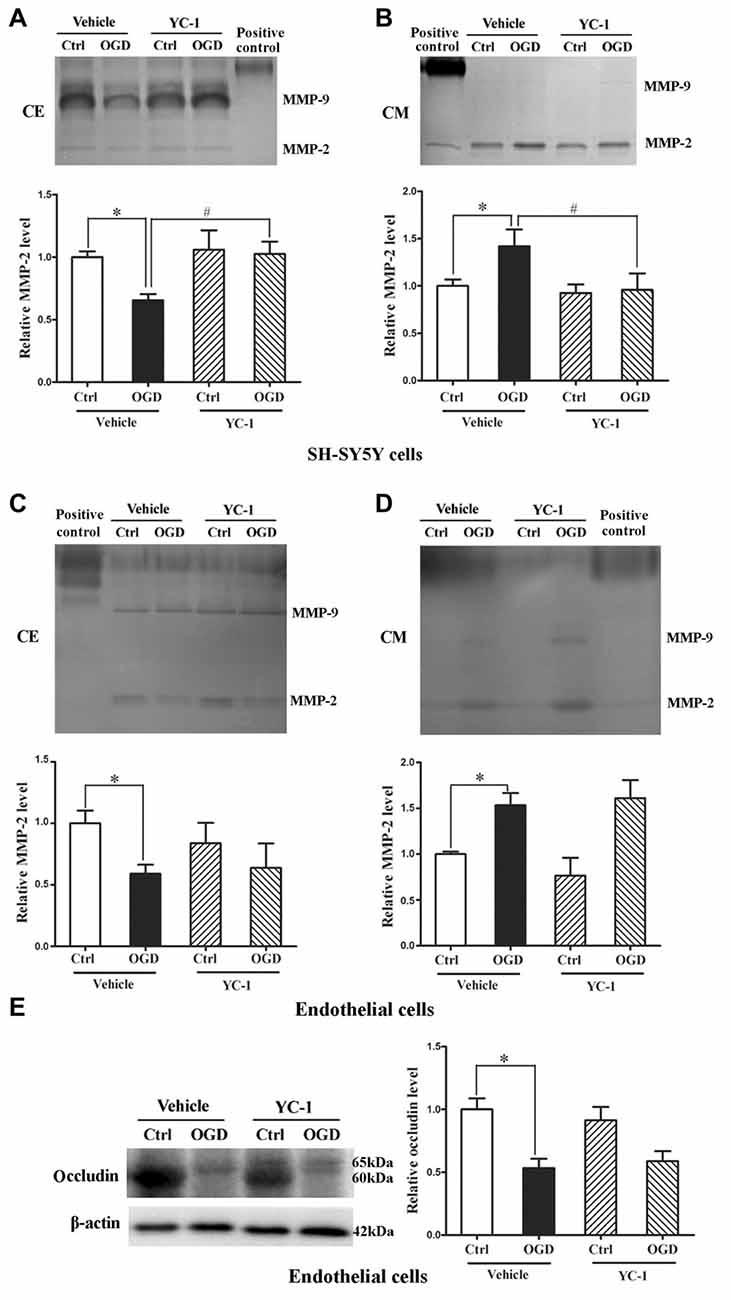
Figure 6. Effect of YC-1 on ischemia-induced matrix metalloproteinase-2 (MMP-2) secretion in neurons and endothelial cells. Gelatin zymography results showed that 2-h OGD elevated MMP-2 levels in conditioned media (CM), which was accompanied by a significant reduction in their levels in whole cellular extracts (CE). (A) Representative gelatin zymogram (upper panel) showing that OGD decreased MMP-2 levels in CE of SH-SY5Y cells and YC-1 treatment prevents this decrease. The relative levels of MMP-2 were quantitated (bottom panel). (B) Representative gelatin zymogram (upper panel) showing that OGD increased MMP-2 levels in CM of SH-SY5Y cells and YC-1 treatment prevents this increase. The relative levels of MMP-2 were quantitated (bottom panel). (C) Representative gelatin zymogram (upper panel) showing that YC-1 treatment did not significantly inhibited OGD-induced MMP-2 decrease in CE of endothelial cells. The relative levels of MMP-2 were quantitated (bottom panel). (D) Representative gelatin zymogram (upper panel) showing that YC-1 treatment did not inhibit OGD-induced MMP-2 increase in CM of endothelial cells. The relative levels of MMP-2 were quantitated (bottom panel). (E) Pretreatment with ICI118551 did not prevent 2-h OGD-induced occludin degradation in endothelial cells. *P < 0.05 vs. control (Ctrl), #P < 0.05 vs. OGD. Data are expressed as mean ± SEM, n = 6/group.
Effect of β2-AR Antagonist ICI 118551 on Ischemia-Induced HIF-1α Upregulation in Neurons and Endothelial Cells
We next examined the effect of blocking β2-AR on HIF-1α expression in neurons and endothelial cells subjected to 2-h OGD. Neurons or endothelial cells were incubated with ICI 118551 for 10 min before exposing to 2-h OGD. As shown in Figure 7, 2-h OGD significantly upregulated HIF-1α in neurons (Figure 7A), but not in endothelial cells (Figure 7B, upper and middle panel) and astrocytes (Figure 7C, upper and middle panel). Of note, 2-h OGD did not induce VEGF secretion in astrocytes (Figure 7C, bottom panel). Although pretreatment with ICI 118551 did not inhibit OGD-induced occludin degradation in endothelial cells (Figure 7B, upper and bottom panel), it significantly inhibited OGD-induced HIF-1α upregulation in neurons (Figure 7A, P < 0.05 vs. OGD). These results suggested that blocking β2-AR inhibited OGD-induced HIF-1α upregulation in neurons.
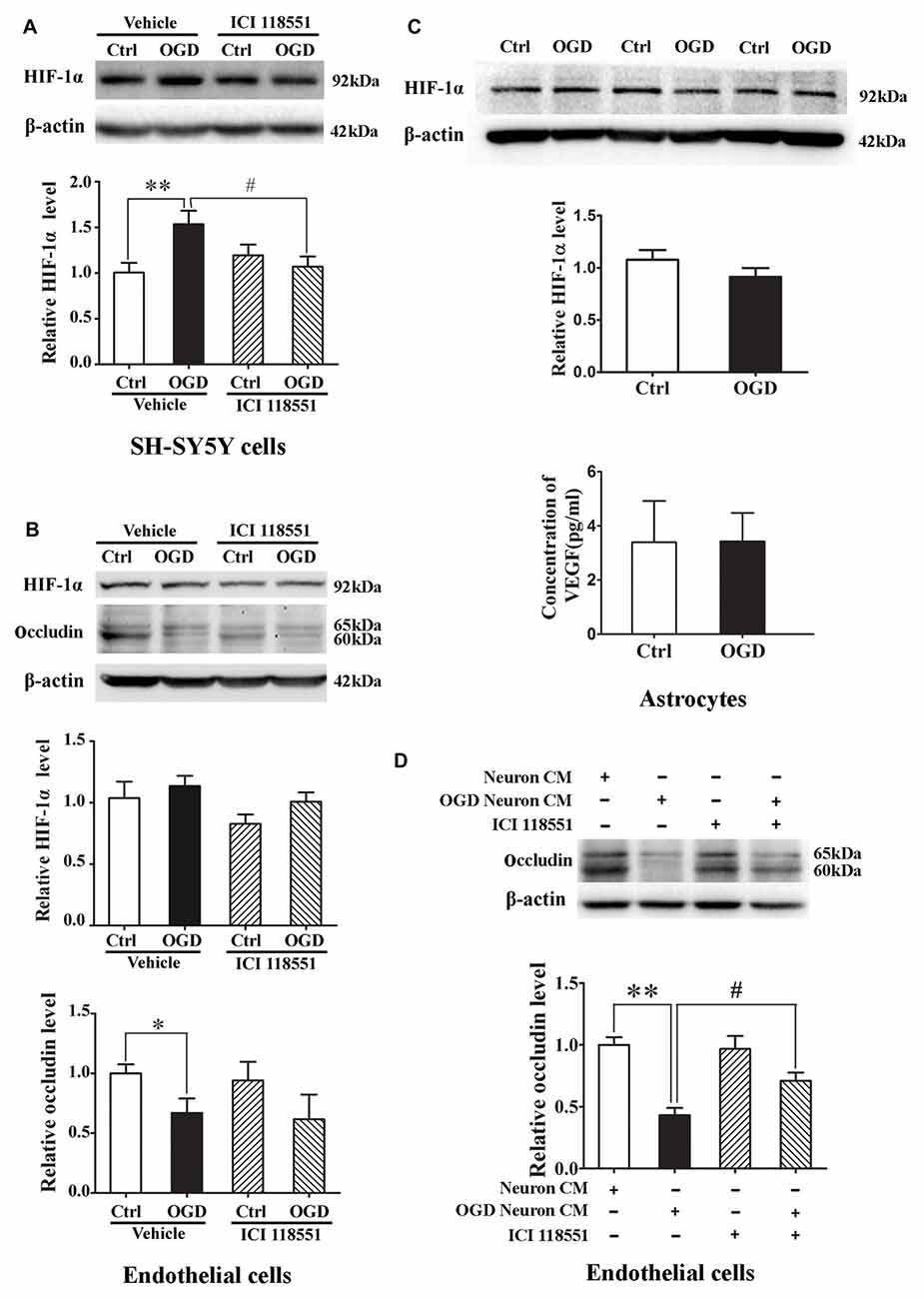
Figure 7. Effect of ICI118551 on ischemia-induced HIF-1α expression and occludin degradation induced by the conditioned media of OGD-treated neurons. HIF-1α expression in neurons, endothelial cells and astrocytes were detected after 2-h OGD. Two-hour OGD induced a significant HIF-1α upregulation in neurons (A) but not endothelial cells (B) or astrocytes (C). Pretreatment with ICI118551 inhibited OGD-induced HIF-1α upregulation in neurons (A), but did not attenuated OGD-induced occludin degradation in endothelial cells (B) *P < 0.05 vs. control (Ctrl), **P < 0.01 vs. control (Ctrl), #P < 0.05 vs. OGD. (D) Western blot analysis showed that conditioned media (CM) from OGD-treated neurons induced occludin degradation in endothelial cells and pretreatment neurons with ICI 118551 attenuated this degradation. **P < 0.01 vs. neuron CM (Ctrl), #P < 0.05 vs. neuron CM.
Effect of Blocking β2-AR on Occludin Degradation Induced by the Conditioned Media of OGD-Treated Neurons
The above results have demonstrated that OGD induced the secretion of MMP-2 and VEGF into extracellular medium through activating β2-AR-HIF-1α pathway, which mediated occludin degradation in endothelial cells. To further clarify the interaction between neurons and endothelial cells, we exposed endothelial cells to the conditioned media from OGD-treated neurons for 2 h. Western blot analysis showed that the OGD-neuron conditioned media induced occludin degradation in endothelial cells and pretreatment neurons with ICI 118551 inhibited the degradation (Figure 7D).
Discussion
Tightness of the BBB at the time of thrombolytic intervention is an important factor determining the efficacy of thrombolysis with tPA (Benchenane et al., 2005; Leigh et al., 2014, 2016) and ischemic BBB damage within thrombolytic time window is a promising target (Jin et al., 2014; Liu J. et al., 2016) for clinical intervention to reduce intracranial hemorrhage in patients with intravenous tPA (Leigh et al., 2014) or post-endovascular treatment (Leigh et al., 2016). In our current study, we found that: (1) 2-h ischemia induced BBB damage in non-infarcted ventral striatum accompanied by HIF-1α protein and VEGF mRNA upregulation; (2) HIF-1α inhibition prevented BBB damage by preventing VEGF and MMP-2 secretion in neuron and occludin degradation in endothelial cells; and (3) blocking β2-AR alleviated ischemia-induced BBB damage by reducing HIF-1α level. Taken together, our results provide strong evidence that BBB damage is reduced by blockade of β2-adrenergic receptor-mediated HIF-1α upregulation during acute cerebral ischemia.
Our current results show that 2-h MCAO induces HIF-1α upregulation, which mainly occurs in the non-infarcted ventral striatum and preoptic area, where the brain tissue has been shown to experience moderate cerebral blood flow (CBF) reduction during MCAO (Jin et al., 2012). This finding is consistent with a previous report that HIF-1α was found to be mostly induced in the salvageable tissue in the ischemic brain, a moderate but not prolonged decrease in CBF, induced HIF-1α in the penumbra (Bergeron et al., 1999). We speculate that moderately-reduced CBF may trigger reperfusion-like damaging events such as increased reactive oxygen species generations in hypoperfused tissue to disrupt BBB (Qutub and Popel, 2008). Future studies are warranted to test this hypothesis. It is worth of note, HIF-1α expression in neuron is cell-type-specific in response to hypoxic/ischemic insults, interneurons express a significantly higher level of HIF-1α protein than pyramidal neurons (Ramamoorthy and Shi, 2014) and there are more interneurons than pyramidal neurons in the striatum.
Our data that 2-h ischemia induces HIF-1α upregulation and increased the mRNA of VEGF. This is consistent with a previous study that hypoxia, rapidly increased the nuclear contents of HIF-1α as well as the mRNA levels of VEGF (Bernaudin et al., 2002). Here, we did not observe an increase in VEGF protein levels, which is different from previous reports that VEGF protein is elevated in adult brain in response to stroke (Yeh et al., 2007; Chen et al., 2010; Yan et al., 2011). Lack of prolonged reperfusion in our experimental design may explain this discrepancy, because it has been reported that the protein levels of VEGF after ischemia (without reperfusion) are comparable to the untreated control but are increased two and three folds after 4 h or 8 h of reperfusion, respectively (Mu et al., 2003), indicating that VEGF protein is produced in the reperfusion stage and the ischemia duration may only stimulate the HIF-1α-mediated VEGF mRNA expression, while may not be long enough to induce VEGF protein synthesis. These results are further confirmed by our in vitro experiments showing that 2-h OGD induces VEGF mRNA but not protein increase in neurons and pretreatment with either YC-1 or HIF-1α siRNA significantly inhibits this increase. Our in vitro data show that there is a significant increase in VEGF protein in the conditioned medium and but does not change VEGF protein contents in the cellular extracts. Our explanation for this finding is that being a secretory protein, the new synthesized VEGF in OGD-treated cells may be secreted into the conditioned media. Therefore, the VEGF protein levels in the cellular extracts are not significantly changed. Our in vivo data show that MCAO did not induce VEGF increase in ROI 2 by western blot that measures the total level of VEGF in ischemic brain tissue. Under this experimental condition, we could not accurately separate the extracellular VEGF from intracellular VEGF, as a substantial portion of the extracellular fluid and the soluble proteins would be inevitably lost during the process of protein extraction, and in addition, the amount of extracellular fluid protein in the total amount of tissue protein is very small, or even negligible. From this point of view, our in vitro and in vivo results of VEGF are consistent.
Our data that HIF-1α is colocalized with the neurons but not astrocytes or endothelial cells indicate that HIF-1α is mainly upregulated in neurons under our experimental conditions. In the brain, the neurons are the most sensitive cell type and robustly respond to hypoxic/ischemic stimulus to upregulate HIF-1α. Unlike many other cells, astrocytes are very resistant to hypoxic stress and severe oxygen deprivation is required to induce HIF-1α signaling and modulate subsequent survival and proliferation in astrocytes (Schmid-Brunclik et al., 2008). Two-hour ischemia may not be strong enough for astrocytes to produce HIF-1α. If the duration of ischemia or reperfusion is extended, astrocytes could then act as an important player for BBB damage. As an example, Li et al. (2014) have reported that 3-h OGD promotes neurons to activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. In addition, cell specific temporal-spatial modulation of HIF-1α signaling pathways has been shown to be a key determinant of functional outcome (Ogunshola and Al-Ahmad, 2012). Understanding cell-type dependent effects of HIF-1α will undoubtedly shed new lights on its role in cerebral ischemia and thus provides potential approaches to augment its beneficial effect and reduce its detrimental function (Engelhardt et al., 2014).
Our previous finding showed that BBB damage is mediated by secreted but not new synthesized MMP-2 at a very stage of ischemia (Liu et al., 2012) has led us to investigate the factors that induces MMP-2 secretion. HIF-1α is well implicated in the control of MMPs that critically contribute to vascular permeability increase under various conditions (Liu and Rosenberg, 2005; Yang et al., 2007; Jin et al., 2013). Here, our data show that OGD induces HIF-1α-dependent MMP-2 secretion in neurons and HIF-1α-independent MMP-2 secretion in endothelial cells. Therefore, when endothelial cells are exposed to OGD, the increased MMP-2 secretion can lead to occludin degradation, and this process is independently of HIF-1α or neurons. However, during MCAO, affected by surrounding cells and the extracellular matrix, endothelial cells may behave differently in the ischemic brain compared to the OGD model. Owing to its high sensitivity to OGD or ischemia, the neurons may be the first cell type responding to ischemic stress and secreting MMP-2 and VEGF in a HIF-1α dependent manner. The secreted MMP-2 and VEGF may then affect the activity of endothelial cells to secret more MMP-2 and VEGF. Therefore, in the MCAO model, we can’t exclude the possibility that the involvement of endothelial cells in BBB disruption may also be dependent on HIF-1α or neurons.
It has been shown that β2-AR-HIF-1α axis played an important role for stress-induced pancreatic tumor growth and angiogenesis (Shan et al., 2013) as well as in NNK-induced pancreatic cancer progression (Zhang et al., 2015). In addition, β2-AR antagonist propranolol has been shown to regress infantile hemangiomas in a HIF-1α-dependent manner. More important, MMP-2 has been shown to mediate β-AR-induced apoptosis in ventricular myocytes (Menon et al., 2005) and cardiac hypertrophy in rats (Miura et al., 2003). Here our data showed that blocking β2-AR significantly inhibits ischemia-induced BBB damage through suppressing HIF-1α expression and occludin degradation, indicating that β2-AR-HIF-1α signaling may represent promising therapeutic targets for preventing ischemia-induced BBB damage.
In summary, BBB damage is reduced by blockade of β2-adrenergic receptor-mediated HIF-1α upregulation during acute cerebral ischemia. The findings may provide new ideas to protect the BBB against ischemic damage and to extend the time window of thrombolysis and reduce cerebral hemorrhage.
Author Contributions
The work was performed and accomplished by all authors. YS, XC, XZ, XS, MW and XW contributed to the execution of the entire project and the statistical analyses. W-CL, JL, WL and C-FL provided advice on experimental design and interpretation, and comments on the manuscript. YS, XC and XJ wrote the manuscript. All authors have read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81571149, 81671145), by Natural Science Foundation of Jiangsu Province of China (L221506415), by Doctoral Fund of Ministry of Education of China (K521507713), by Suzhou Science and Technology Plan (N321500715) and by Grants from Shenzhen Science and Technology Innovation Commission (JCYJ20150402152005623, GCZX2015050411225563, GJHZ20160301163900284), Guangdong Province Natural Science Foundation (2016A030313027). This work was also partly supported by Priority Academic Program Development of Jiangsu Higher Education Institutions of China.
Abbreviations
BBB, blood brain barrier; β2-AR, β2-adrenergic receptor; CBF, cerebral blood flow; CE, cellular extract; CM, conditioned media; EB, Evan’s blue dye; HIF-1α, hypoxia-inducible factor-1 alpha; HT, hemorrhagic transformation; MCAO, middle cerebral artery occlusion; MMP, matrix metalloproteinase; NE, norepinephrine; OGD, oxygen glucose deprivation; ROI, region of interest; TTC, 2,3,5-triphenyltetrazolium chloride; VEGF, vascular endothelial growth factor.
References
Benchenane, K., Berezowski, V., Fernández-Monreal, M., Brillault, J., Valable, S., Dehouck, M. P., et al. (2005). Oxygen glucose deprivation switches the transport of tPA across the blood-brain barrier from an LRP-dependent to an increased LRP-independent process. Stroke 36, 1065–1070. doi: 10.1161/01.STR.0000163050.39122.4f
Bergeron, M., Yu, A. Y., Solway, K. E., Semenza, G. L., and Sharp, F. R. (1999). Induction of hypoxia-inducible factor-1 (HIF-1) and its target genes following focal ischaemia in rat brain. Eur. J. Neurosci. 11, 4159–4170. doi: 10.1046/j.1460-9568.1999.00845.x
Bernaudin, M., Nedelec, A. S., Divoux, D., MacKenzie, E. T., Petit, E., and Schumann-Bard, P. (2002). Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J. Cereb. Blood Flow Metab. 22, 393–403. doi: 10.1097/00004647-200204000-00003
Camós, S., and Mallolas, J. (2010). Experimental models for assaying microvascular endothelial cell pathophysiology in stroke. Molecules 15, 9104–9134. doi: 10.3390/molecules15129104
Chen, C., Hu, Q., Yan, J., Yang, X., Shi, X., Lei, J., et al. (2009). Early inhibition of HIF-1α with small interfering RNA reduces ischemic-reperfused brain injury in rats. Neurobiol. Dis. 33, 509–517. doi: 10.1016/j.nbd.2008.12.010
Chen, C., Ostrowski, R. P., Zhou, C., Tang, J., and Zhang, J. H. (2010). Suppression of hypoxia-inducible factor-1α and its downstream genes reduces acute hyperglycemia-enhanced hemorrhagic transformation in a rat model of cerebral ischemia. J. Neurosci. Res. 88, 2046–2055. doi: 10.1002/jnr.22361
Engelhardt, S., Patkar, S., and Ogunshola, O. O. (2014). Cell-specific blood-brain barrier regulation in health and disease: a focus on hypoxia. Br. J. Pharmacol. 171, 1210–1230. doi: 10.1111/bph.12489
Fischer, S., Wobben, M., Marti, H. H., Renz, D., and Schaper, W. (2002). Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc. Res. 63, 70–80. doi: 10.1006/mvre.2001.2367
Hacke, W., Kaste, M., Bluhmki, E., Brozman, M., Dávalos, A., Guidetti, D., et al. (2008). Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 359, 1317–1329. doi: 10.1056/NEJMoa0804656
Hafez, S., Coucha, M., Bruno, A., Fagan, S. C., and Ergul, A. (2014). Hyperglycemia, acute ischemic stroke, and thrombolytic therapy. Transl. Stroke Res. 5, 442–453. doi: 10.1007/s12975-014-0336-z
Hsiao, G., Huang, H. Y., Fong, T. H., Shen, M. Y., Lin, C. H., Teng, C. M., et al. (2004). Inhibitory mechanisms of YC-1 and PMC in the induction of iNOS expression by lipoteichoic acid in RAW 264.7 macrophages. Biochem. Pharmacol. 67, 1411–1419. doi: 10.1016/j.bcp.2003.12.010
Ji, B., Zhou, F., Han, L., Yang, J., Fan, H., Li, S., et al. (2017). Sodium tanshinone IIA sulfonate enhances effectiveness Rt-PA treatment in acute ischemic stroke patients associated with ameliorating blood-brain barrier damage. Transl. Stroke Res. 8, 334–340. doi: 10.1007/s12975-017-0526-6
Jin, X., Liu, J., and Liu, W. (2014). Early ischemic blood brain barrier damage: a potential indicator for hemorrhagic transformation following tissue plasminogen activator (tPA) thrombolysis? Curr. Neurovasc. Res. 11, 254–262. doi: 10.2174/1567202611666140530145643
Jin, X., Liu, J., Liu, K. J., Rosenberg, G. A., Yang, Y., and Liu, W. (2013). Normobaric hyperoxia combined with minocycline provides greater neuroprotection than either alone in transient focal cerebral ischemia. Exp. Neurol. 240, 9–16. doi: 10.1016/j.expneurol.2012.11.018
Jin, X., Liu, J., Yang, Y., Liu, K. J., and Liu, W. (2012). Spatiotemporal evolution of blood brain barrier damage and tissue infarction within the first 3 h after ischemia onset. Neurobiol. Dis. 48, 309–316. doi: 10.1016/j.nbd.2012.07.007
Jin, X., Sun, Y., Xu, J., and Liu, W. (2015). Caveolin-1 mediates tissue plasminogen activator-induced MMP-9 up-regulation in cultured brain microvascular endothelial cells. J. Neurochem. 132, 724–730. doi: 10.1111/jnc.13065
Kastrup, A., Gröschel, K., Ringer, T. M., Redecker, C., Cordesmeyer, R., Witte, O. W., et al. (2008). Early disruption of the blood-brain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke 39, 2385–2387. doi: 10.1161/STROKEAHA.107.505420
Leigh, R., Christensen, S., Campbell, B. C., Marks, M. P., Albers, G. W., and Lansberg, M. G. (2016). Pretreatment blood-brain barrier disruption and post-endovascular intracranial hemorrhage. Neurology 87, 263–269. doi: 10.1212/WNL.0000000000002862
Leigh, R., Jen, S. S., Hillis, A. E., Krakauer, J. W., and Barker, P. B. (2014). Pretreatment blood-brain barrier damage and post-treatment intracranial hemorrhage in patients receiving intravenous tissue-type plasminogen activator. Stroke 45, 2030–2035. doi: 10.1161/STROKEAHA.114.005249
Li, Y. N., Pan, R., Qin, X. J., Yang, W. L., Qi, Z., Liu, W., et al. (2014). Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 129, 120–129. doi: 10.1111/jnc.12611
Liu, W., Furuichi, T., Miyake, M., Rosenberg, G. A., and Liu, K. J. (2007). Differential expression of tissue inhibitor of metalloproteinases-3 in cultured astrocytes and neurons regulates the activation of matrix metalloproteinase-2. J. Neurosci. Res. 85, 829–836. doi: 10.1002/jnr.21179
Liu, J., Jin, X., Liu, K. J., and Liu, W. (2012). Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J. Neurosci. 32, 3044–3057. doi: 10.1523/JNEUROSCI.6409-11.2012
Liu, Y., Liu, W. C., Sun, Y., Shen, X., Wang, X., Shu, H., et al. (2017). Normobaric hyperoxia extends neuro- and vaso-protection of N-acetylcysteine in transient focal ischemia. Mol. Neurobiol. 54, 3418–3427. doi: 10.1007/s12035-016-9932-0
Liu, K. J., and Rosenberg, G. A. (2005). Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radic Biol. Med. 39, 71–80. doi: 10.1016/j.freeradbiomed.2005.03.033
Liu, H., Wang, Y., Xiao, Y., Hua, Z., Cheng, J., and Jia, J. (2016). Hydrogen sulfide attenuates tissue plasminogen activator-induced cerebral hemorrhage following experimental stroke. Transl. Stroke Res. 7, 209–219. doi: 10.1007/s12975-016-0459-5
Liu, J., Weaver, J., Jin, X., Zhang, Y., Xu, J., Liu, K. J., et al. (2016). Nitric oxide interacts with caveolin-1 to facilitate autophagy-lysosome-mediated claudin-5 degradation in oxygen-glucose deprivation-treated endothelial cells. Mol. Neurobiol. 53, 5935–5947. doi: 10.1007/s12035-015-9504-8
Matsumoto, M., Zornow, M. H., Rabin, B. C., and Maze, M. (1993). The α 2 adrenergic agonist, dexmedetomidine, selectively attenuates ischemia-induced increases in striatal norepinephrine concentrations. Brain Res. 627, 325–329. doi: 10.1016/0006-8993(93)90337-m
Menon, B., Singh, M., and Singh, K. (2005). Matrix metalloproteinases mediate β-adrenergic receptor-stimulated apoptosis in adult rat ventricular myocytes. Am. J. Physiol. Cell Physiol. 289, C168–C176. doi: 10.1152/ajpcell.00606.2004
Miura, S., Ohno, I., Suzuki, J., Suzuki, K., Okada, S., Okuyama, A., et al. (2003). Inhibition of matrix metalloproteinases prevents cardiac hypertrophy induced by β-adrenergic stimulation in rats. J. Cardiovasc. Pharmacol. 42, 174–181. doi: 10.1097/00005344-200308000-00004
Mu, D., Jiang, X., Sheldon, R. A., Fox, C. K., Hamrick, S. E., Vexler, Z. S., et al. (2003). Regulation of hypoxia-inducible factor 1α and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol. Dis. 14, 524–534. doi: 10.1016/j.nbd.2003.08.020
Nakamuta, M., Kotoh, K., Enjoji, M., and Nawata, H. (2005). Effects of fibril- or fixed-collagen on matrix metalloproteinase-1 and tissue inhibitor of matrix metalloproteinase-1 production in the human hepatocyte cell line HLE. World J. Gastroenterol. 11, 2264–2268. doi: 10.3748/wjg.v11.i15.2264
Ogunshola, O. O., and Al-Ahmad, A. (2012). HIF-1 at the blood-brain barrier: a mediator of permeability? High Alt. Med. Biol. 13, 153–161. doi: 10.1007/978-1-4899-1054-7_5
Paxinos, G., and Watson, C. (1986). The Rat Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press.
Qu, L. L., Guo, N. N., and Li, B. M. (2008). β1- and β2-adrenoceptors in basolateral nucleus of amygdala and their roles in consolidation of fear memory in rats. Hippocampus 18, 1131–1139. doi: 10.1002/hipo.20478
Qutub, A. A., and Popel, A. S. (2008). Reactive oxygen species regulate hypoxia-inducible factor 1α differentially in cancer and ischemia. Mol. Cell. Biol. 28, 5106–5119. doi: 10.1128/mcb.00060-08
Ramamoorthy, P., and Shi, H. (2014). Ischemia induces different levels of hypoxia inducible factor-1α protein expression in interneurons and pyramidal neurons. Acta Neuropathol. Commun. 2:51. doi: 10.1186/2051-5960-2-51
Rodrigues, M., Xin, X., Jee, K., Babapoor-Farrokhran, S., Kashiwabuchi, F., Ma, T., et al. (2013). VEGF secreted by hypoxic Muller cells induces MMP-2 expression and activity in endothelial cells to promote retinal neovascularization in proliferative diabetic retinopathy. Diabetes 62, 3863–7383. doi: 10.2337/db13-0014
Schmid-Brunclik, N., Burgi-Taboada, C., Antoniou, X., Gassmann, M., and Ogunshola, O. O. (2008). Astrocyte responses to injury: VEGF simultaneously modulates cell death and proliferation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R864–R873. doi: 10.1152/ajpregu.00536.2007
Schoch, H. J., Fischer, S., and Marti, H. H. (2002). Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain 125, 2549–2557. doi: 10.1093/brain/awf257
Semenza, G. L. (2014). Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 9, 47–71. doi: 10.1146/annurev-pathol-012513-104720
Shan, T., Ma, J., Ma, Q., Guo, K., Guo, J., Li, X., et al. (2013). β2-AR-HIF-1α: a novel regulatory axis for stress-induced pancreatic tumor growth and angiogenesis. Curr. Mol. Med. 13, 1023–1034. doi: 10.2174/15665240113139990055
Shi, H. (2009). Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr. Med. Chem. 16, 4593–4600. doi: 10.2174/092986709789760779
Shi, Y., Leak, R. K., Keep, R. F., and Chen, J. (2016). Translational stroke research on blood-brain barrier damage: challenges, perspectives, and goals. Transl. Stroke Res. 7, 89–92. doi: 10.1007/s12975-016-0447-9
Shu, H., Zheng, G. Q., Wang, X., Sun, Y., Liu, Y., Weaver, J. M., et al. (2015). Activation of matrix metalloproteinase in dorsal hippocampus drives improvement in spatial working memory after intra-VTA nicotine infusion in rats. J. Neurochem. 135, 357–367. doi: 10.1111/jnc.13283
Simard, J. M., Kent, T. A., Chen, M., Tarasov, K. V., and Gerzanich, V. (2007). Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 6, 258–268. doi: 10.1016/s1474-4422(07)70055-8
Sun, L., Zhou, W., Mueller, C., Sommer, C., Heiland, S., Bauer, A. T., et al. (2010). Oxygen therapy reduces secondary hemorrhage after thrombolysis in thromboembolic cerebral ischemia. J. Cereb. Blood Flow Metab. 30, 1651–1660. doi: 10.1038/jcbfm.2010.50
Vangeison, G., Carr, D., Federoff, H. J., and Rempe, D. A. (2008). The good, the bad and the cell type-specific roles of hypoxia inducible factor-1α in neurons and astrocytes. J. Neurosci. 28, 1988–1993. doi: 10.1523/JNEUROSCI.5323-07.2008
Wang, W., Li, M., Chen, Q., and Wang, J. (2015). Hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke: mechanisms, models and biomarkers. Mol. Neurobiol. 52, 1572–1579. doi: 10.1007/s12035-014-8952-x
Wang, X., Liu, Y., Sun, Y., Liu, W., and Jin, X. (2016). Blood brain barrier breakdown was found in non-infarcted area after 2-h MCAO. J. Neurol. Sci. 363, 63–68. doi: 10.1016/j.jns.2016.02.035
Wang, X., Xue, G. X., Liu, W. C., Shu, H., Wang, M., Sun, Y., et al. (2017). Melatonin alleviates lipopolysaccharide-compromised integrity of blood-brain barrier through activating AMP-activated protein kinase in old mice. Aging Cell 16, 414–421. doi: 10.1111/acel.12572
Wolburg, H., and Lippoldt, A. (2002). Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul. Pharmacol. 38, 323–337. doi: 10.1016/S1537-1891(02)00200-8
Yan, J., Zhou, B., Taheri, S., and Shi, H. (2011). Differential effects of HIF-1 inhibition by YC-1 on the overall outcome and blood-brain barrier damage in a rat model of ischemic stroke. PLoS One 6:e27798. doi: 10.1371/journal.pone.0027798
Yang, E. V., Sood, A. K., Chen, M., Li, Y., Eubank, T. D., Marsh, C. B., et al. (2006). Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2 and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 66, 10357–10564. doi: 10.1158/0008-5472.can-06-2496
Yang, Y., Estrada, E. Y., Thompson, J. F., Liu, W., and Rosenberg, G. A. (2007). Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 27, 697–709. doi: 10.1038/sj.jcbfm.9600375
Yeh, W. L., Lu, D. Y., Lin, C. J., Liou, H. C., and Fu, W. M. (2007). Inhibition of hypoxia-induced increase of blood-brain barrier permeability by YC-1 through the antagonism of HIF-1α accumulation and VEGF expression. Mol. Pharmacol. 72, 440–449. doi: 10.1124/mol.107.036418
Zhang, D., Lei, J., Ma, J., Chen, X., Sheng, L., Jiang, Z., et al. (2015). β2-adrenogenic signaling regulates NNK-induced pancreatic cancer progression via upregulation of HIF-1α. Oncotarget 7, 17760–17772. doi: 10.18632/oncotarget.5677
Zhang, Y., Wang, T., Yang, K., Xu, J., Ren, L., Li, W., et al. (2016). Cerebral microvascular endothelial cell apoptosis after ischemia: role of enolase-phosphatase 1 activation and aci-reductone dioxygenase 1 translocation. Front. Mol. Neurosci. 9:79. doi: 10.3389/fnmol.2016.00079
Zhang, Z., Yan, J., and Shi, H. (2016). Role of hypoxia inducible factor 1 in hyperglycemia-exacerbated blood-brain barrier disruption in ischemic stroke. Neurobiol. Dis. 95, 82–92. doi: 10.1016/j.nbd.2016.07.012
Keywords: cerebral ischemia, HIF-1α, β2-AR, blood brain barrier, tight junction proteins, matrix metalloproteinase
Citation: Sun Y, Chen X, Zhang X, Shen X, Wang M, Wang X, Liu W-C, Liu C-F, Liu J, Liu W and Jin X (2017) β2-Adrenergic Receptor-Mediated HIF-1α Upregulation Mediates Blood Brain Barrier Damage in Acute Cerebral Ischemia. Front. Mol. Neurosci. 10:257. doi: 10.3389/fnmol.2017.00257
Received: 20 February 2017; Accepted: 31 July 2017;
Published: 14 August 2017.
Edited by:
Detlev Boison, Legacy Health, United StatesReviewed by:
Nikki Katherine Lytle, University of California, San Diego, United StatesGiuseppe Pignataro, University of Naples Federico II, Italy
Sookyong Koh, Emory University School of Medicine, United States
Copyright © 2017 Sun, Chen, Zhang, Shen, Wang, Wang, Liu, Liu, Liu, Liu and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinchun Jin, eGluY2h1bmppbkBnbWFpbC5jb20=