 Marlen Dierich1†
Marlen Dierich1† Michael G. Leitner
Michael G. Leitner- 1Department of Neurophysiology, Institute of Physiology and Pathophysiology, Philipps University of Marburg, Marburg, Germany
- 2Division of Physiology, Department of Physiology and Medical Physics, Innsbruck Medical University, Innsbruck, Austria
The three members of the ether-à-go-go-gene-like (Elk; Kv12.1-Kv12.3) family of voltage-gated K+ channels are predominantly expressed in neurons, but only little information is available on their physiological relevance. It was shown that Kv12.2 channels modulate excitability of hippocampal neurons, but no native current could be attributed to Kv12.1 and Kv12.3 subunits yet. This may appear somewhat surprising, given high expression of their mRNA transcripts in several brain areas. Native Kv12 currents may have been overlooked so far due to limited knowledge on their biophysical properties and lack of specific pharmacology. Except for Kv12.2, appropriate genetically modified mouse models have not been described; therefore, identification of Kv12-mediated currents in native cell types must rely on characterization of unique properties of the channels. We focused on recombinant human Kv12.1 to identify distinct properties of these channels. We found that Kv12.1 channels exhibited significant mode shift of activation, i.e., stabilization of the voltage sensor domain in a “relaxed” open state after prolonged channel activation. This mode shift manifested by a slowing of deactivation and, most prominently, a significant shift of voltage dependence to hyperpolarized potentials. In contrast to related Kv11.1, mode shift was not sensitive to extracellular Na+, which allowed for discrimination between these isoforms. Sensitivity of Kv12.1 and Kv11.1 to the broad-spectrum K+ antagonist 4-aminopyridine was similar. However, 4-AP strongly activated Kv12.1 channels, but it was an inhibitor of Kv11 channels. Interestingly, the agonist of Kv11 channels NS1643 also differentially modulated the activity of these channels, i.e., NS1643 activated Kv11.1, but strongly inhibited Kv12.1 channels. Thus, these closely related channels are distinguished by inverse pharmacological profiles. In summary, we identified unique biophysical and pharmacological properties of Kv12.1 channels and established straightforward experimental protocols to characterize Kv12.1-mediated currents. Identification of currents in native cell types with mode shift that are activated through 4-AP and inhibited by NS1643 can provide strong evidence for contribution of Kv12.1 to whole cell currents.
Introduction
The ether-à-go-go (Eag) superfamily of voltage-gated K+ channels comprises three evolutionary conserved families that share high sequence homology: Ether-à-go-go (Eag; Kv10), ether-à-go-go-related-gene (Erg; Kv11) and ether-à-go-go-gene-like (Elk; Kv12) channels (Bauer and Schwarz, 2001). The best-studied member, Kv11.1 (the human isoform is referred to as HERG channel) mediates rapidly activating K+ current IKr in cardiac myocytes determining heart action potential duration (Sanguinetti et al., 1995). Accordingly, loss of Kv11.1 channel function through mutations or drug treatment causes cardiac arrhythmia and sudden death in humans (Curran et al., 1995; Sanguinetti et al., 1995; Trudeau et al., 1995). Kv11 channels also mediate important K+ currents in neurons of the auditory brainstem (Hardman and Forsythe, 2009), the olfactory bulb (Hirdes et al., 2009) and the midbrain (Ji et al., 2012). Kv10.1 channels regulate cell cycle progression and proliferation (Sanchez et al., 2016; Urrego et al., 2016), and are frequently overexpressed in human cancers with poor prognosis (Pardo and Stuhmer, 2014). Kv10.1 channel mutations cause developmental disorders and epilepsy (Kortum et al., 2015; Simons et al., 2015).
In contrast to Kv10 and Kv11 channels, only little information on physiological relevance is available for the three members of the Kv12 family that are expressed predominantly in neurons (Engeland et al., 1998; Shi et al., 1998; Miyake et al., 1999; Trudeau et al., 1999; Saganich et al., 2001; Zou et al., 2003). Kv12.2 channels regulate excitability in pyramidal neurons of hippocampus in mice (Zhang et al., 2010), but no native current component could be attributed to Kv12.1 and Kv12.3 subunits despite expression of their mRNA transcripts in several brain areas (Shi et al., 1998; Miyake et al., 1999; Saganich et al., 2001; Zou et al., 2003). We consider that Kv12.1/Kv12.3-mediated currents in neurons were overlooked so far due to insufficient knowledge on biophysical properties and lack of specific pharmacological tools.
Recently, it was shown that Kv12.1 channels exhibit mode shift of activation (also termed pre-pulse facilitation or voltage-dependent potentiation) (Li et al., 2015; Dai and Zagotta, 2017). Mode shift denotes time-dependent stabilization of the voltage sensor domain in a “relaxed” open state after prolonged channel activation through depolarized (conditioning) membrane potentials (Bezanilla et al., 1982; Villalba-Galea et al., 2008). It manifests by slowing of deactivation and a shift of voltage dependence to hyperpolarized potentials (Li et al., 2015; Dai and Zagotta, 2017). Accordingly, when measured with routine voltage clamp protocols (e.g., holding potentials of -60 mV) human Kv12.1 channels mediate “conventional” K+ currents that activate similar to many other Kv channels with voltages at half-maximal activation of around -30 mV (c.f. Figure 1) (Li et al., 2015). These currents could easily go unnoticed in cell types expressing different endogenous K+ currents. Taking into account their mode shift, appropriate voltage protocols (e.g., depolarized holding potentials) may uncover Kv12-mediated currents. However, it remains to be explored whether mode shift can be detected in cells expressing various K+ currents and whether it may be employed to identify native Kv12.1 channels. Mode shift is not an exclusive feature of Kv12.1 channels, but has been demonstrated also for voltage-gated Na+ channels (Bezanilla et al., 1982), HCN (Mannikko et al., 2005; Bruening-Wright and Larsson, 2007), Shaker (Olcese et al., 1997; Tilegenova et al., 2017), and Kv11.1 (Erg1) channels (Piper et al., 2003; Tan et al., 2012; Goodchild et al., 2015). Nevertheless, it may constitute a prominent hallmark to distinguish Kv12 channels from other K+ current components in native tissue.
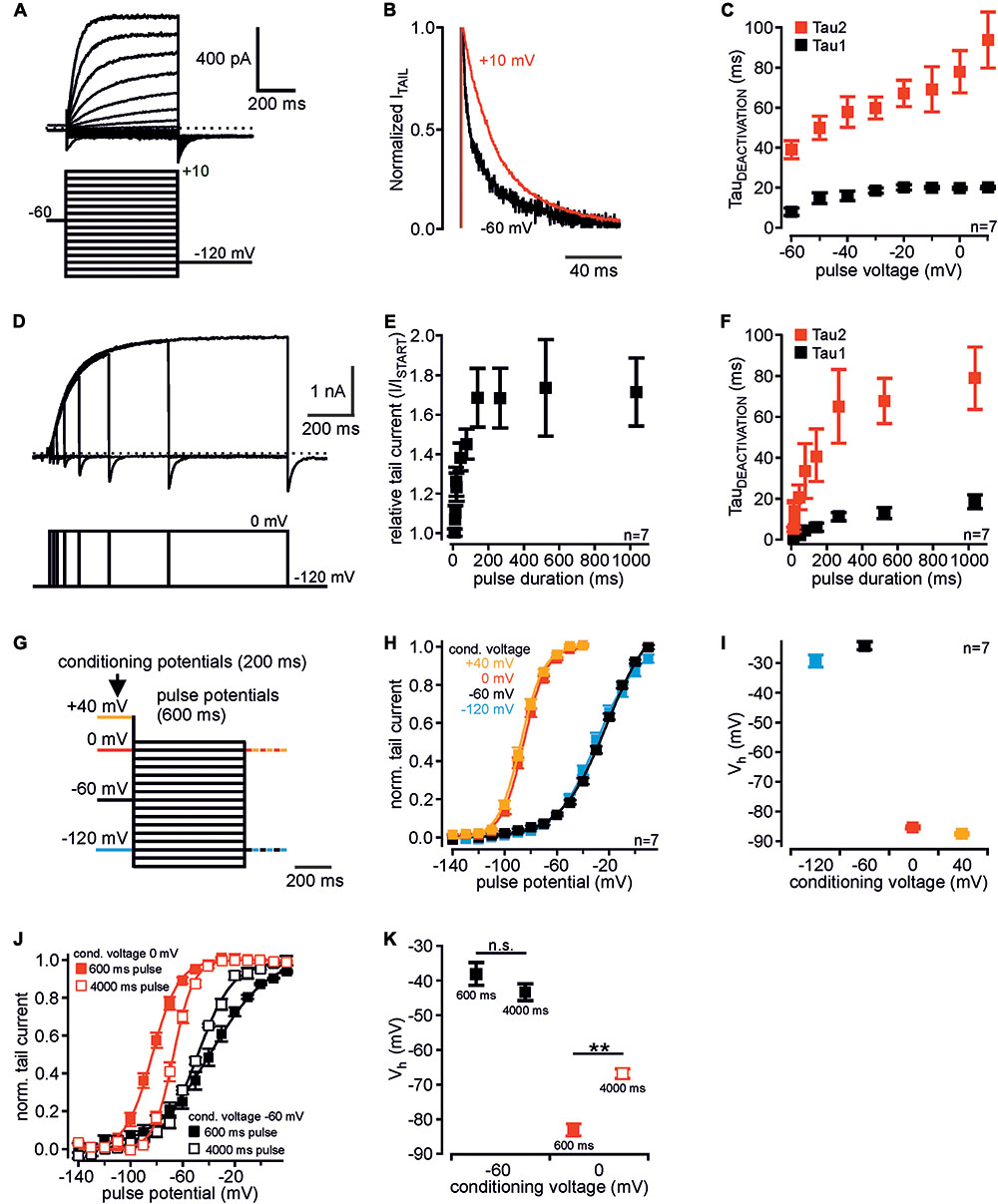
FIGURE 1. Voltage-dependent mode shift of human Kv12.1 channels. (A) Representative patch clamp recording of a CHO cell transiently transfected with human Kv12.1 channels measured with the indicated voltage protocol. (B) Representative tail currents elicited at –120 mV after activating pulses of –60 mV (black) and +10 mV (red). Currents were normalized to maximum amplitude for visualization of deactivation kinetics (currents from recording shown in A). (C) Tail current deactivation was best described by double exponential kinetics: Tau1 and tau2 were derived from double-exponential fits to the decaying phase of tail currents (voltage protocols as in A). Deactivation slowed down with channel activation at more depolarized potentials. (D–F) When Kv12.1 channels were activated at 0 mV for different time intervals, (E) tail current amplitudes increased and (F) deactivation slowed-down with pre-pulse duration. Tail current increase and slowing of deactivation saturated at a pulse duration of about 200 ms and 400–500 ms, respectively (D shows a representative recording). (G–I) Analysis of voltage dependence of recombinant Kv12.1 channels. (G) Voltage protocols consisted of a 200 ms conditioning potential step to –120 mV (blue), –60 mV (black), 0 mV (red), or +40 mV (orange), followed by 600 ms activating pulse potentials from –140 to +10 mV (10 mV increments). In these experiments, tail currents were elicited either at –120 mV or at 0 mV (for representative recordings see Supplementary Figures 1A,C). (H) Summary of voltage dependence of human Kv12.1 channels derived from Boltzmann fits to individual recordings as shown in Supplementary Figures 1A,C (solid line represents a Boltzmann fit to averaged data). Depolarized conditioning potentials of +40 and 0 mV induced a large shift of voltage dependence to hyperpolarized potentials. (I) shows mean Vh of channel activation in dependence of conditioning potentials (derived from fits shown in H). (J,K) Upon extension of the activating pulses from 600 ms (filled black squares) to 4000 ms (open black squares), voltage dependence of recombinant Kv12.1 channels was not significantly changed when measured after a conditioning pulse of –60 mV. However, extension of the activating pulse to 4000 ms significantly shifted voltage dependence of Kv12.1 channels to depolarized potentials after conditioning voltage of 0 mV (P ≤ 0.01; open red squares). For data shown in (J) eight independent recordings were averaged (each cell measured with all four voltage protocols). There was no significant difference between corresponding data in (J,K) and (H,I) (conditioning potential –60 mV, 600 ms).
Here we describe biophysical and pharmacological properties of Kv12.1 channels and demonstrate straightforward experimental protocols that may be employed to identify Kv12 currents in neurons. We show, that these properties allowed detection of Kv12.1-mediated currents in cells expressing a variety of different K+ channels. Our findings may be utilized to identify physiological roles of Kv12.1 channels.
Results
Voltage-Dependent Mode Shift of Human Kv12.1 Channels
Mode shift of human Kv12.1 channels was recently demonstrated (Li et al., 2015), but a detailed characterization is currently not available. In order to identify exclusive properties of human Kv12.1 channels, we thus set out with detailed biophysical analysis of these channels in an overexpression system. In CHO cells, activation of Kv12.1 channels through depolarizing voltage steps produced robust outwardly rectifying currents (Figure 1A) (Zou et al., 2003; Li et al., 2015; Dai and Zagotta, 2017). Channel deactivation at hyperpolarized potentials was best described by double exponential kinetics, and deactivation slowed down with more depolarized pre-potentials (Figures 1B,C). When we activated Kv12.1 channels at 0 mV for different time intervals, tail current amplitudes increased (Figures 1D,E) and deactivation slowed down with the duration of the pre-pulse (Figure 1F). Tail current increase and slowing of deactivation saturated at a pulse duration of about 200 ms and 400–500 ms, respectively.
We then analyzed voltage dependence of human Kv12.1 channels with voltage protocols established previously to study mode shift of related Kv11.1 (Tan et al., 2012). We applied depolarizing holding potentials (conditioning potentials; 200 ms) before a series of activating voltage steps (pulse potentials from -140 mV to +10 mV; 600 ms) (Figure 1G and Supplementary Figures 1A–C). To minimize time intervals at hyperpolarized potentials that may counteract mode shift (c.f. Villalba-Galea, 2017), we at start recorded tail currents at correspondingly depolarized potentials (Figures 1G–I). In these experiments, amplitudes of Kv12.1-mediated outward currents were similar irrespective of conditioning potentials. This indicated that comparable steady-state channel activation was reached with all voltage protocols (Supplementary Figures 1A–C). After a conditioning potential of -60 mV, voltage dependence of Kv12.1 channels also did not change relevantly upon extension of the activating steps to 4000 ms (Figures 1J,K). This additionally demonstrated that steady-state activation of Kv12.1 channels was already reached by activating pulses as short as 600 ms. For 600 ms activating pulses, half-maximal voltages of activation (Vh) were -29.3 ± 2.3 mV and -24.1 ± 1.6 mV for negative conditioning potentials of -120 mV or -60 mV, respectively (n = 7; 600 ms activating pulses; Figures 1H,I). When cells were held at depolarized conditioning pulses of 0 mV or +40 mV, Vh was -85.3 ± 0.9 mV and -87.6 ± 0.8 mV, respectively (Figures 1H,I; 600 ms activating pulses). In these experiments, slope factors derived from Boltzmann fits to the recordings changed from -15.7 ± 0.7 mV (conditioning pulse of -120 mV) and -16.4 ± 0.9 mV (-60 mV) to -8.6 ± 0.1 mV (0 mV) and -8.9 ± 0.6 (+40 mV) (n = 7; Supplementary Figure 1D; 600 ms activating pulses). Accordingly, depolarizing conditioning potentials induced a large shift of voltage dependence by about -60 mV, and the full shift occurred across a potential range of 60 mV (between holding potentials of -60 and 0 mV). In analogous experiments, the same conditioning voltages shifted Vh of related Kv11.1 channels from -6.8 ± 4.4 mV (conditioning voltage -60 mV) to -62.4 ± 1.5 (conditioning voltage +40 mV; n = 7; Figures 2A,B), consistent with a previous report (Tan et al., 2012).
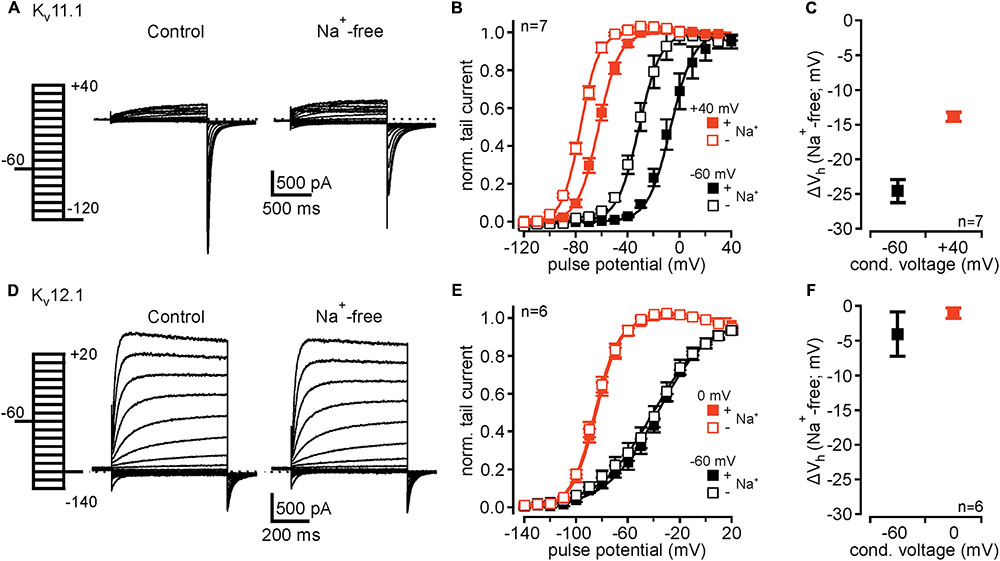
FIGURE 2. Kv12.1 are not sensitive to extracellular Na+. (A) Representative recordings from a CHO cell transiently transfected with Kv11.1 under control conditions (left) and after removal of external Na+ (right). (B) Kv11.1 channels exhibit mode shift of activation, as previously reported. After conditioning potential of +40 mV voltage dependence of Kv11.1 channels shifted to hyperpolarized potentials, compared to conditional potential of –60 mV [solid lines represent a Boltzmann fit to averaged data; from recordings as shown in (A)]. Upon removal of extracellular Na+, voltage dependence of Kv11.1 shifted to hyperpolarized potentials after conditioning potentials of –60 and +40 mV. (C) Shows averaged shifts of Vh (ΔVh) for conditional potentials of –60 and +40 mV after removal of extracellular Na+. (D–F) Recombinant Kv12.1 channels are insensitive to extracellular Na+. (D) Representative recordings of Kv12.1-mediated currents under control conditions (left) and in absence of extracellular Na+ (right). (E) Summary of voltage dependence of Kv12.1 channels derived from experiments presented in (D). Note that neither current amplitudes nor voltage dependence of Kv12.1 channels were affected by removal of extracellular Na+. (F) shows that removal of extracellular Na+ did not shift Vh (ΔVh) of activation of Kv12.1 channels. In these experiments, Na+ in the extracellular solution was replaced by NMDG without changing extracellular K+ concentration.
We next tested whether mode shift was sensitive to the employed voltage protocol. Therefore, we again applied 200 ms depolarized conditioning pulses, but this time we extended the activating pulses to 4000 ms (Figures 1J,K). When in these experiments cells were held at conditioning pulses of -60 and 0 mV, Vh was -43.3 ± 2.5 mV and -66.8 ± 1.5 mV, respectively (Figures 1J,K). Thus, mode shift of Kv12.1 channels was readily induced also when activating the channels for 4000 ms. However, the extent of mode shift was significantly reduced in these experiments compared to experiments with 600 ms activating pulses (Figure 1K; P ≥ 0.01), i.e., increased time intervals at hyperpolarized holding potentials during these protocols presumably counteracted development of mode shift. Similarly, the extent of mode shift was reduced when we activated channels for 600 ms after conditioning pulses of -60 and 0 mV, but recorded tail currents at hyperpolarized holding potentials (-120 mV; Supplementary Figures 1E,F), i.e., we introduced additional hyperpolarizing potentials after every activating pulse. Thus, also in these experiments hyperpolarizing holding potentials between the conditioning pulses reduced the expression of mode shift.
Taken together, human Kv12.1 channels exhibited significant voltage-dependent mode shift that in response to depolarized holding potentials manifested by slowed channel deactivation and by a large hyperpolarizing shift of voltage dependence. This mode shift of Kv12.1 channels can be induced robustly employing different voltage protocols, but the extent of mode shift significantly varies with duration of hyperpolarized holding potentials.
In Contrast to Kv11.1, Kv12.1 Channels Are Insensitive to Extracellular Na+
Inhibition of Kv11 channels by extracellular Na+ is well established (Numaguchi et al., 2000; Sturm et al., 2005) and a hallmark used to identify Kv11-mediated currents in neurons (e.g., Hardman and Forsythe, 2009). In control experiments, replacement of extracellular Na+ with NMDG without altering extracellular K+ concentration slightly increased Kv11.1-mediated outward currents (Figure 2A), as previously reported (Numaguchi et al., 2000). Upon removal of extracellular Na+, activation voltage range of Kv11.1 channels conditioned at -60 and +40 mV shifted to hyperpolarized potentials by -24.4 ± 1.5 mV and -13.9 ± 0.7 mV, respectively (n = 7; Figures 2B,C). Accordingly, overall mode shift of voltage dependence was attenuated from -55.7 ± 4.4 mV under control conditions to -45.1 ± 3.5 mV in absence of extracellular Na+ (n = 7; P ≤ 0.05). We then tested whether related Kv12.1 channels also exhibited such Na+ sensitivity. We found that neither current amplitudes nor voltage dependence of Kv12.1 channels were affected by removal of extracellular Na+ (Figures 2D–F). Consequently, the extent of mode shift was also insensitive to changes of the Na+ concentration. To conclude, despite the high similarity in mode shift behavior, the absence of Na+ sensitivity in Kv12.1 channels distinguishes Kv11.1-mediated currents from Kv12.1.
Kv12.1 Channels Are Not Sensitive to Kv Channel Blockers E-4031, XE991, and TEA
We then evaluated whether Kv12.1 channels were sensitive to channel inhibitors that are widely used to attribute neuronal K+ currents to particular channel families. At concentrations generally applied to inhibit established target channels, human Kv12.1 channels were insensitive to both E-4031 (20 μM), a specific inhibitor of Kv11 channels (Supplementary Figures 2A,B) (Trudeau et al., 1995), and XE991 (10 μM), a specific antagonist of neuronal Kv7 (KCNQ) channels (Supplementary Figures 2D,E) (Wang et al., 1998). Kv12.1-mediated currents were also insensitive to E-4031 and XE991 concentrations up to 100 μM (Supplementary Figures 2C,F). Kv12.1 channels also were not affected by the broad-spectrum K+ channel inhibitor tetraethylammonium (TEA) at a concentration that completely inhibited Kv7.2 channels (5 mM; Supplementary Figures 2G,H) (Hadley et al., 2000). However, Kv12.1 channels were slightly inhibited by 50 and 100 mM TEA (I50mM TEA/IStart = 0.90 ± 0.01; I100mM TEA/IStart = 0.83 ± 0.01; n = 9; Supplementary Figure 2I). Of note, low sensitivity of Kv12.1 channels to E-4031 and TEA has been shown in an early report (Shi et al., 1998). In summary, E-4031, XE991 and TEA cannot be used to inhibit Kv12.1 channels in native tissue, but may be used to isolate Kv12.1 channel activity through inhibition of other K+ channels.
The Broad-Spectrum K+ Channel Antagonist 4-Aminopyridine (4-AP) Activates Kv12.1
We then tested whether human Kv12.1 channels were sensitive to 4-AP, another established antagonist of several Kv families (Gutman et al., 2005). Surprisingly, 4-AP at a concentration well in the range often used to isolate neuronal K+ currents (3 mM; e.g., Marcotti et al., 2003) increased Kv12.1-mediated steady-state and tail currents within seconds (Figures 3A–C). Current potentiation was the same for conditioning pulses of -60 and 0 mV, and 4-AP potentiated currents at all holding potentials positive to -60 mV (Figure 3C). 4-AP-dependent current increase was reversible within 2–3 min after washout of the drug (Figure 3D). 4-AP (3 mM) not only potentiated Kv12.1-mediated currents, but also significantly shifted the voltage dependence of Kv12.1 channels to hyperpolarized potentials (Figure 3E). Compared to control recordings measured before application of the substance, 3 mM 4-AP shifted Vh by -19.7 ± 1.7 mV (n = 7; P ≤ 0.001) and by -10.4 ± 1.5 mV (n = 7; P ≤ 0.001) after condition voltage pulses of -60 and 0 mV, respectively. Thus, the shift of Vh was significantly more pronounced for currents conditioned at -60 mV (P ≤ 0.01; c.f. Figure 3E).
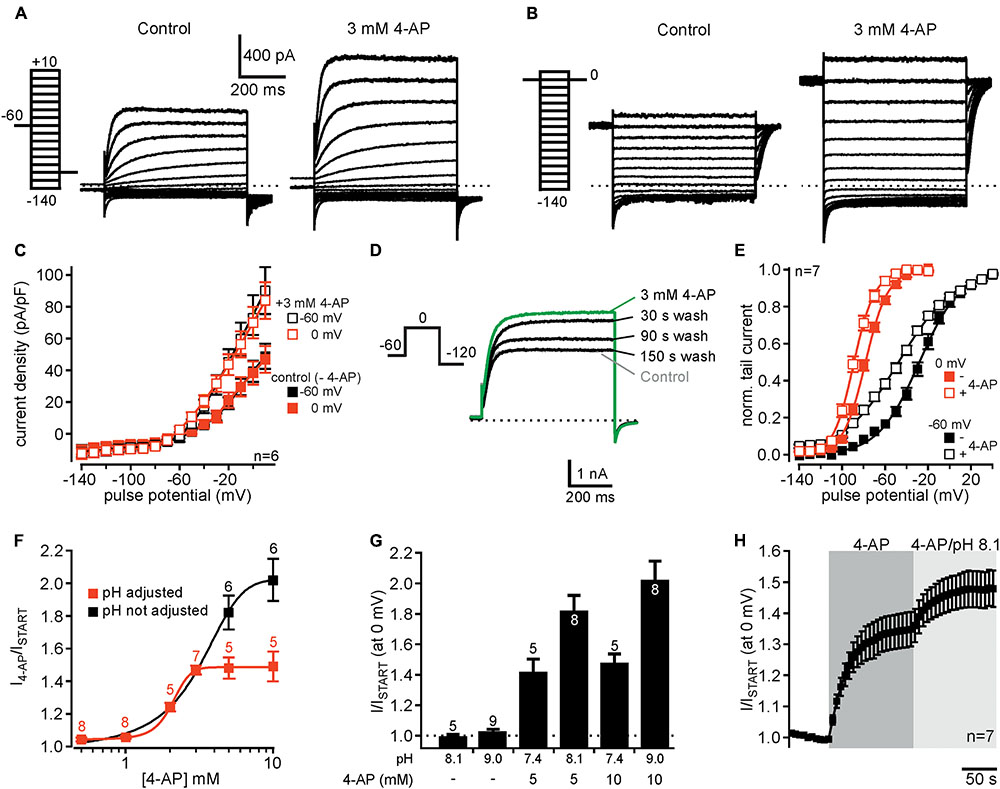
FIGURE 3. 4-AP potentiates currents through human Kv12.1 channels. (A,B) Representative recordings from a CHO cell transiently transfected with Kv12.1 before (left) and after extracellular application of 3 mM 4-AP (right) after conditioning pulses of (A) –60 mV and (B) 0 mV. (C) 4-AP-dependent activation of Kv12.1-mediated steady-state outward currents was independent on employed voltage protocols (summary of recordings as presented in A,B). 4-AP potentiated currents at all holding potentials positive to –60 mV. (D) 4-AP-dependent potentiation of Kv12.1 currents was reversible, as current amplitudes returned to control levels within 2–3 min after removal of the substance [shown are representative recordings under control conditions (gray), after application of 3 mM 4-AP (green) and at different time points (30, 90, and 150 s) after washout of 4-AP]. (E) 3 mM 4-AP shifted the voltage dependence of human Kv12.1 channels to hyperpolarized potentials (open squares) [the panel shows Boltzmann fits to averaged data; data obtained from recordings as shown in (A,B)]. (F) 4-AP activated Kv12.1 channels in a dose-dependent manner. When pH of the extracellular solution was adjusted to 7.4 at higher 4-AP concentrations, the EC50 was about 2.1 mM and the Hill coefficient was about 0.8 (red trace). When pH of the solutions was not adjusted, the EC50 was about 3.1 and Hill coefficient was 0.9 (black trace). Parameters were derived from fits of averaged data to a Hill equation described in Section “Materials and Methods” (solid line represents fit; note that addition of 4-AP to 5 and 10 mM 4-AP increased the pH of the solution to 8.1 and 9.0, respectively). (G) Increasing the pH of the extracellular solution from 7.4 to 8.1 or 9.0 without addition of 4-AP did not increase Kv12.1-mediated steady-state currents. In contrast, application of 5 mM or 10 mM 4-AP in extracellular solution without adjusting pH activated Kv12.1 channels even more strongly than when pH of the solution was adjusted to pH 7.4. (H) Application of 3 mM 4-AP that does not change pH of the extracellular solution strongly potentiated Kv12.1-mediated currents. Current potentiation through 3 mM 4-AP was further increased when the pH of the extracellular solution was increased to pH 8.1.
We then analyzed the dose-response relationship of 4-AP action on Kv12.1 channels. As 5 and 10 mM 4-AP increased pH of the solution to about 8.1 and 9.0, respectively, we adjusted pH of these solutions to 7.4 after addition of 4-AP. Of note, 3 mM 4-AP or lower 4-AP concentrations did not alter the pH of the solution relevantly and thus no adjustment of pH was necessary. Under these experimental conditions, 4-AP potentiated Kv12.1 currents in a concentration-dependent manner with an EC50 of about 2.1 and a Hill coefficient of about 0.8 (Figure 3F, red trace). In contrast, Kv11.1 channels were inhibited by 4-AP with an IC50 of approximately 2.6 mM and a Hill coefficient of about 0.7 (Supplementary Figure 3), consistent with previous reports (e.g., Ridley et al., 2003). Thus, despite inverse effects of 4-AP on Kv12.1 and Kv11.1, the sensitivity of both channels to 4-AP was quite similar. When we applied 5 mM or 10 mM 4-AP without adjusting pH, we found that these concentrations activated Kv12.1 channel even stronger than at physiological pH (Figures 3F,G). Without adjusting pH at higher concentrations, the EC50 was about 3.1 and the Hill coefficient was 0.9 (Figure 3F, black trace). As in line with a previous study (Kazmierczak et al., 2013) increasing pH of the extracellular solution from 7.4 to 8.1 or 9.0 without addition of 4-AP did not potentiate Kv12.1-mediated steady-state currents (Figure 3G), these data suggested that 4-AP activated Kv12.1 channels more efficiently at more alkaline pH. Indeed, increasing pH of the extracellular solution containing 3 mM 4-AP to 8.1 further increased Kv12.1-mediated steady-state outward currents (Figure 3H).
Kv12.1 Channels Are Also Activated by Isomeric Aminopyridines
Several other aminopyridines have been shown to inhibit voltage-gated K+ channels, albeit antagonistic efficiency of these substances was lower than that of 4-AP (Robertson and Nelson, 1994; Sedehizadeh et al., 2012; Strupp et al., 2017). We thus wondered whether we could identify isomeric aminopyridines that activated Kv12.1, at best without affecting other K+ channels. We found that 2-aminopyridine (2-AP) and 3-aminopyridine (3-AP) potentiated Kv12.1-mediated currents at a concentration close to the EC50 of 4-AP (3 mM) by about 20% (P ≤ 0.001) and 7% (P ≤ 0.001), respectively (Figure 4). At the same concentration 3,4-diaminopyridine (3,4-DAP; 3 mM) was ineffective. Hence, 2-AP (P ≤ 0.01) and 3-AP (P ≤ 0.001) activated Kv12.1 channels significantly less than 4-AP mirroring efficacy of inhibition of other K+ channels by these substances (Robertson and Nelson, 1994; Sedehizadeh et al., 2012; Strupp et al., 2017).
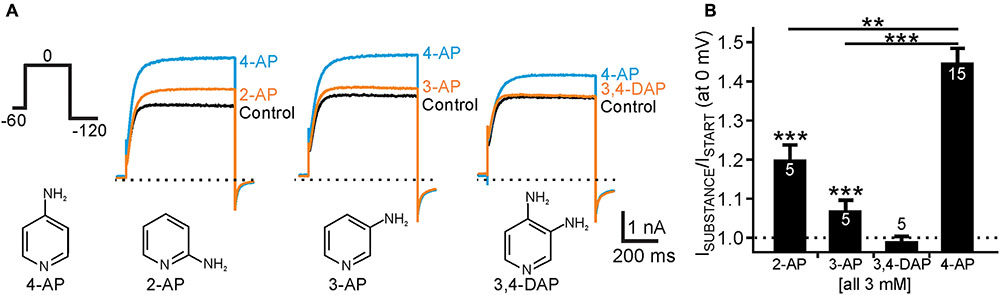
FIGURE 4. Activation of human Kv12.1 channels by isomeric aminopyridines. (A) Representative whole cell currents through human Kv12.1 channels elicited by voltage steps to 0 mV (voltage protocol as indicated). The upper panel shows effects of 2-AP (left), 3-AP (middle), or 3,4-DAP (right) on human Kv12.1 channels. 4-AP was applied at the end of every recording for comparison of current potentiation (concentration of all substances was 3 mM). The lower panel displays chemical structures of 4-AP, 2-AP, 3-AP, and 3,4-DAP. (B) Summary of recordings as presented in A (steady-state outward currents at 0 mV were analyzed at the end of the activating pulse). At the same concentration (all 3 mM), 2-AP and 3-AP significantly potentiated Kv12.1-mediated currents, albeit to a lower extent than 4-AP (∗∗P ≤ 0.01; ∗∗∗P ≤ 0.001).
NS1643, an Activator of Kv11 Channels, Inhibits Kv12.1 Channels
We then turned to NS1643, a partial agonist of the Kv11 channel family (Casis et al., 2006). As shown earlier (c.f. Hansen et al., 2006), NS1643 (30 μM) slowed channel deactivation and potentiated Kv11.1-mediated outward currents (Figures 5A–C). In contrast, the same concentration of NS1643 (30 μM) completely inhibited Kv12.1 channels with a time constant of 16.8 ± 2.6 s (n = 7; Figures 5A–C). When we applied only 10 μM NS1643, a concentration close to the reported EC50 of NS1643 for activation of Kv11.1 channels (Casis et al., 2006), Kv12.1-mediated currents were reduced to 37.1 ± 4.1% of initial current amplitudes. Inhibition of currents by 10 μM was much slower than when 30 μM NS1643 was applied (n = 6; compare Figure 5B and Supplementary Figure 4). We then analyzed voltage dependence of residual Kv12.1 currents in the presence of 10 μM NS1643 (Figures 5D–F): Application of NS1643 (10 μM) shifted Vh by +8.8 ± 2.9 mV (n = 6; P ≤ 0.05) and by +21.7 ± 5.2 mV (n = 6; P ≤ 0.01) after condition voltage pulses of -60 and 0 mV, respectively (Figures 5D–F). At the same time, slope factors changed by +8.0 ± 1.5 mV (conditioning pulse of -60 mV; n = 6; P ≤ 0.05) and -1.8 ± 0.9 mV (conditioning pulse 0 mV; n = 6; P ≤ 0.01). Furthermore, NS1643 (10 μM) significantly accelerated deactivation of Kv12.1 channels at hyperpolarized potentials (Figures 5G,H; P ≤ 0.05; n = 6).
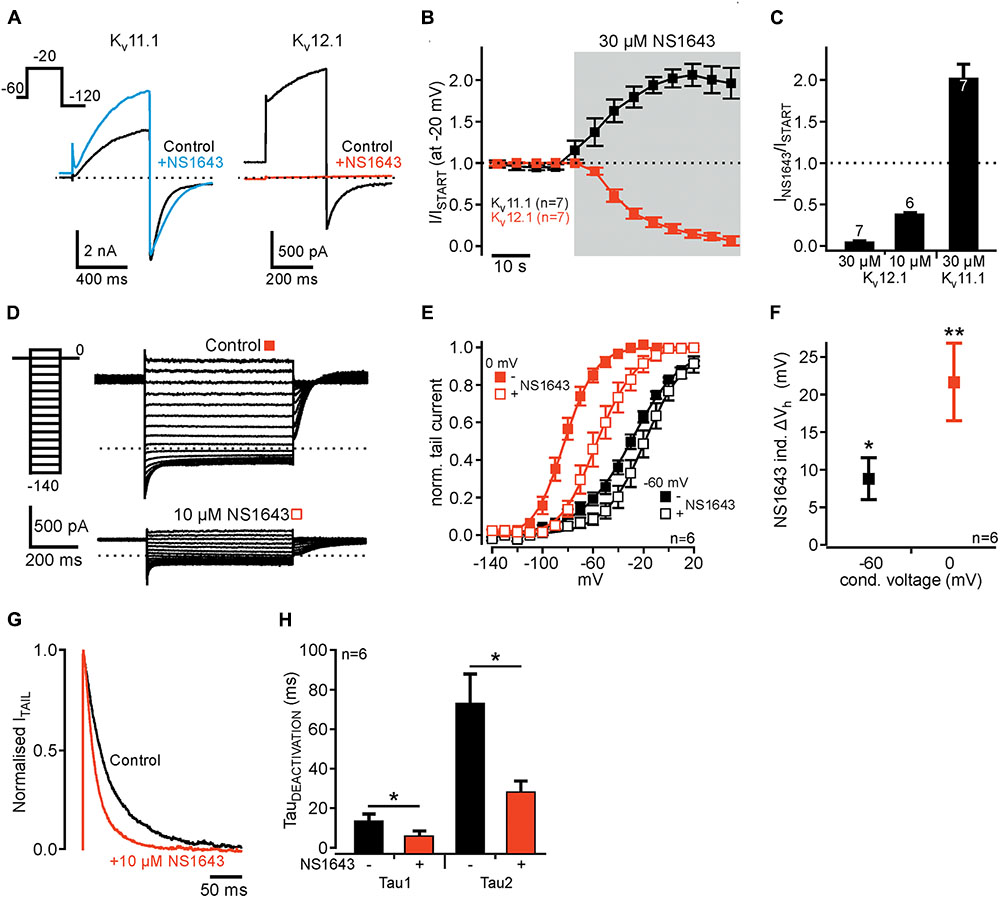
FIGURE 5. Human Kv12.1 channels are inhibited by NS1643. (A) Representative recordings of recombinant Kv11.1 (left) and Kv12.1 (right) channels before (black) and after (colored) application of 30 μM NS1643 (voltage protocol as indicated). (B) Averaged time course of the amplitudes of outward currents mediated by Kv11.1 (black) and Kv12.1 (red) channels upon application of 30 μM NS1643. NS1643 activated Kv11.1 channels [as previously reported; (Casis et al., 2006; Hansen et al., 2006)], but completely inhibited Kv12.1 channels (data were derived from recordings as in A). (C) Summarized effects of NS1643 on Kv11.1 and Kv12.1 channels. 10 μM NS1643 did not completely inhibit Kv12.1 channels allowing for further analysis of remaining currents. The time course of Kv12.1 current inhibition by 10 μM NS1643 is shown in Supplementary Figure 4. (D–F) Voltage dependence of Kv12.1 channels changes through application of 10 μM NS1643. (D) Representative recordings of Kv12.1 channels after conditioning potential of 0 mV before (top) and after (bottom) application of 10 μM NS1643 (for clarity only these recordings are shown). (E) 10 μM NS1643 significantly shifted voltage dependence of Kv12.1 channels to depolarized potentials (solid lines represent Boltzmann fits to averaged data). (F) Shows averaged shifts of Vh (ΔVh) induced by 10 μM NS1643 for currents conditioned at –60 and 0 mV. (G,H) NS1643 significantly accelerated Kv12.1 channel deactivation. (G) Representative normalized tail currents elicited at –120 mV after activation at –20 mV before (control, black) and after (red) application of 10 μM NS1643. (H) Shows averaged time constants of deactivation before and after application of NS1643 (10 μM) (∗P ≤ 0.05; ∗∗P ≤ 0.01).
Identification of Kv12.1 Currents in Cells Expressing Different K+ Currents
Our results suggested that voltage-clamp protocols designed to detect mode shift in combination with pharmacology using 4-AP or NS1643 should provide a robust approach for isolation of Kv12.1-mediated currents in native cell types. As proof of principle, we sought to isolate Kv12.1 channel activity in cells expressing different K+ channels (Figure 6). To this end, we co-expressed Kv12.1 channels together with Kv11.1 and typical neuronal K+ channels (Kv7.2, Kv7.3 and Kir2.1). In these experiments, CHO cells were transiently transfected with equal amounts of plasmid DNA encoding the channel subunits (see section “Materials and Methods” for details). We first analyzed whether we could isolate Kir2.1- and Kv7-mediated currents in those cells. In all cells tested, we found large inward currents at hyperpolarized potentials and XE991-sensitive outward currents at depolarized potentials demonstrating expression of functional Kir2.1 and Kv7 channels, respectively (Supplementary Figure 5). As measure for abundance of Kv11.1 and Kv12.1 channels, we then tested whether we could detect mode shift of voltage dependence in the mix of K+ currents. As determined from whole cell currents, Vh was -6.7 ± 1.3 mV and -80.0 ± 2.1 mV after conditioning potentials of -60 and 0 mV, respectively (n = 7; Figures 6A–C). Thus, mode shift of channels expressed in these cells (Kv11.1 and Kv12.1 channels) was readily detectable even among a complement of different voltage-dependent K+ channels. We next attempted to isolate Kv12.1-mediated currents among the mixture of K+ channel by making use of the pharmacological profile established above. 4-AP (3 mM) shifted the voltage dependence of whole cell currents by -8.5 ± 0.9 mV (n = 7; P ≤ 0.001) and by -12.3 ± 2.3 mV (n = 7; P ≤ 0.05) to hyperpolarized potentials after conditioning voltages of -60 and 0 mV, respectively (Figures 6B,C). Thus, although being less pronounced than for Kv12.1 channels alone (Figure 6C), the 4-AP-induced shift of voltage dependence was readily detectable in the mixed K+ current situation. At the same time, 4-AP (3 mM) potentiated whole cell currents elicited at -60 and 0 mV to about 210% and 140% of baseline amplitudes, respectively (Figures 6A,D–F). Finally, we applied 30 μM NS1643 (on top of 4-AP) to the same cells and found that the substance inhibited outward currents and inward tail currents (Figures 6A,D,E). NS1643 also slowed deactivation kinetics of remaining currents (c.f. Figure 6A) indicating that NS1643-sensitive and functional Kv11.1 channels were expressed in these cells.
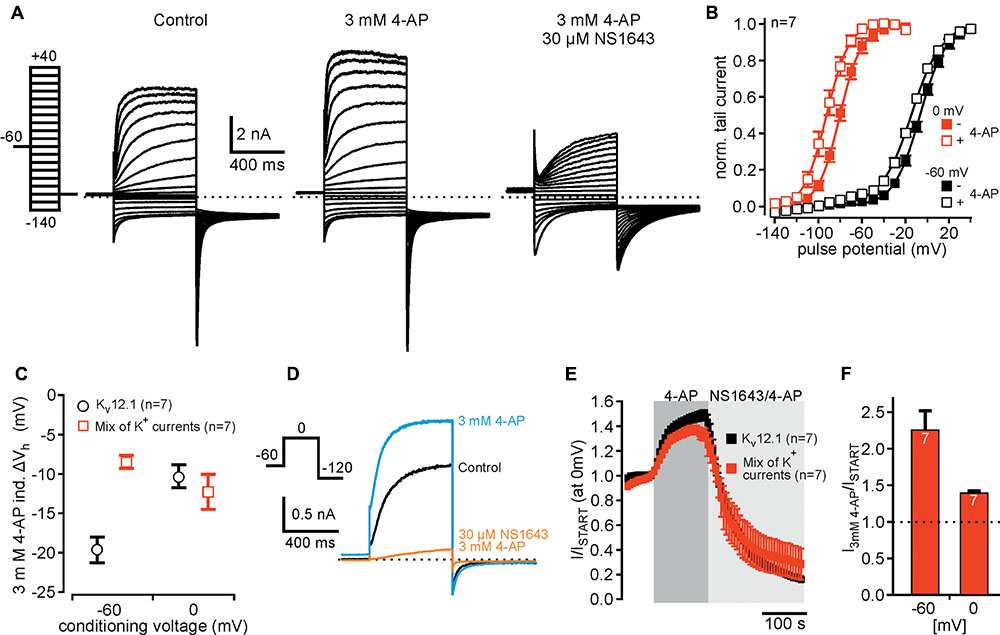
FIGURE 6. Detection of Kv12.1 channel activity in cells expressing various K+ currents. (A) Representative recordings of a CHO cell expressing different K+ channel subunits (Kv7.2, Kv7.3, Kv11.1 and Kir2.1) together with Kv12.1 under control conditions (left), upon application of 3 mM 4-AP (middle) and of 4-AP together with 30 μM NS1643 (right) (voltage protocol as indicated). (B) Voltage dependence of whole cell currents in cells expressing the mix of K+ channel subunits. Vh of whole cell currents was –6.7 ± 1.3 mV (black filled squares) after conditioning potential of –60 mV. When cells were held for 200 ms at a conditioning potential of 0 mV before the activation pulses, Vh of whole cell currents was –80.0 ± 2.1 mV (red filled squares). Application of 3 mM 4-AP shifted voltage dependence to hyperpolarized potentials for both conditioning potentials (open squares; graph displays Boltzmann fits to averaged data as solid lines). (C) 4-AP (3 mM)-induced shift of Vh to hyperpolarized potentials shown for cells expressing the mix of K+ channels (red squares) in comparison to cells expressing only Kv12.1 (black circles). The 4-AP-induced shift of voltage dependence after conditioning voltage of –60 mV was smaller than in cells expressing only Kv12.1 channels. Data for Kv12.1 alone were derived from experiments also shown in Figure 3E. (D,E) Summary of time course and current amplitudes in cells expressing a mix of K+ channels during during application of 4-AP and NS1643 (together with 4-AP). (D) Representative current traces recorded in cells expressing a mix of K+ channels before (black), during application of 3 mM 4-AP (blue) and during application of 3 mM 4-AP together with 30 μM NS1643 (orange). (E) Averaged time course of current amplitudes upon application of 3 mM 4-AP and 30 μM NS1643 for CHO cells expressing Kv12.1 channels alone (black) and the mix of K+ channels (red) (data obtained from recordings as presented in D). (F) Summarized 4-AP-induced potentiation of steady-state outward currents for cells expressing Kv12.1 channels together with a mix of neuronal K+ channels. Data were derived from recordings as presented in (D), and currents elicited at –60 mV and at 0 mV were analyzed.
Taken together, using abovementioned experimental protocols we could unequivocally identify properties of Kv12.1 channels (mode shift, 4-AP and NS1643 sensitivity) among a mixture of K+ currents. These results therefore suggest that the same protocols may be used to assign K+ current components to Kv12.1 channels in complex (native) cellular/neuronal settings.
Discussion
The three members of the ether-à-go-go-like channel family of voltage-gated K+ channels (Kv12.1-Kv12.3) are predominantly expressed in neurons, as shown by mRNA transcripts in several brain areas including cerebral cortex and hippocampus (Miyake et al., 1999; Trudeau et al., 1999; Saganich et al., 2001; Zou et al., 2003), as well as in sympathetic ganglia (Shi et al., 1998). However, neuronal current components and their physiological relevance have been resolved for Kv12.2 subunits exclusively: as demonstrated by application of a selective inhibitor, Kv12.2 activity controls resting membrane potential and spontaneous firing of pyramidal neurons in hippocampus (Zhang et al., 2010). Also, in Kv12.2 knock-out mice it was shown that Kv12.2 channels regulate hippocampal excitability (Zhang et al., 2010) and may be important for processing of spatial working memory (Miyake et al., 2009). However, it is not known whether Kv12 channels mediate relevant currents in any other physiological system.
Here, we focused on recombinant human Kv12.1 to identify distinct biophysical and pharmacological properties allowing for attribution of native currents to these channels.
Human Kv12.1 Channels Exhibit Mode Shift of Activation
Mode shift of activation was recently demonstrated for Kv12.1 channel isoforms of humans and zebrafish heterologously expressed in Xenopus laevis oocytes (Li et al., 2015; Dai and Zagotta, 2017). However, mode shift of these channels has not been characterized in mammalian cells in detail yet. We found that mode shift of human Kv12.1 was readily induced by depolarized holding potentials between -60 and 0 mV and that it manifested by slowed channel deactivation and a striking shift of voltage dependence to hyperpolarized potentials. Deceleration of deactivation is generally considered a biophysical hallmark of mode shift, as channels undergo additional (time-consuming) transitions from this “relaxed” (metastable) open state into deactivation (Bezanilla et al., 1982; Villalba-Galea et al., 2008; Corbin-Leftwich et al., 2016; Villalba-Galea, 2017). Similar to Kv11.1 (Tan et al., 2012) and zebrafish Kv12.1 (Dai and Zagotta, 2017), mode shift of human Kv12.1 channels manifested by a striking -60 mV shift of voltage dependence. Thus, mode shift-dependent hyperpolarizing shifts of voltage dependence were qualitatively and quantitatively comparable for these three ion channels. Of note, mode shift of Kv12.1 channels was readily induced using different voltage protocols, albeit the extent of the hyperpolarizing shift of activation potentials varied considerably with duration of hyperpolarized voltage steps. This demonstrated high sensitivity of Kv12.1 channels to the holding potential, but also highlighted that voltage dependence of native Kv12.1 currents might also vary with the used voltage protocols. In contrast to zebrafish Kv12.1 (Dai and Zagotta, 2017), activation of human Kv12.1 channels did not exhibit prominent double exponential kinetics that might indicate transition into a more stable open conformation. However, human Kv11.1 channels that beyond doubt exhibit mode shift do not display such kinetics neither (Tan et al., 2012; Goodchild et al., 2015). This indicates that either kinetics were masked by channel inactivation, or transition was too fast in the human channel isoforms.
Time course of transition into the relaxed state varies considerably between channels requiring depolarization for minutes in Nav (Bezanilla et al., 1982), seconds in Shaker (Olcese et al., 1997) and some hundreds of milliseconds in Kv11.1 channels (Piper et al., 2003). For human Kv12.1, we found that also some hundreds of milliseconds of conditioning depolarization was sufficient for significant alterations of voltage dependence and kinetics. Thus, time course of development of Kv12.1 mode shift was well in the range of that published for Kv11.1 channels (Piper et al., 2003). As Kv12.1 channels are highly sensitive to changes of the holding potential, voltage dependence of the channels strongly depends on the employed voltage protocols. In fact, such protocol differences could account for considerable variations in voltage dependence of Kv12.1 channels reported in different studies (Vh close to 0 mV in Shi et al., 1998;Vh of about -60 mV in Zou et al., 2003).
Kv12.1 Channels Are Activated by 4-AP, an Established Kv Channel Inhibitor
4-AP is a rather selective blocker of voltage-gated K+ channels: At micromolar concentrations, it reversibly inhibits activity of Kv1 and Kv3 family members, but at higher concentrations (in the millimolar range) 4-AP also blocks other Kv channels (e.g., Kv11) (Gutman et al., 2005; Alexander et al., 2015). Thus, it was somewhat surprising to find that 4-AP activates Kv12.1 channels. As an early study mentioned potentiation of Kv12.3 through 4-AP without, however, showing any recordings (named rELK1; Engeland et al., 1998), such 4-AP sensitivity may constitute a general feature of the Kv12 family.
Earlier, it was shown that currents through Kv2.1 channels that are normally inhibited by 4-AP were potentiated by the substance, but only when Kv6.4 subunits were co-expressed (Stas et al., 2015). As Kv2.1 and Kv6.4 co-assemble into functional channels (reviewed in Bocksteins, 2016), this suggested that Kv6.4 largely determined altered 4-AP sensitivity of the resulting heteromeric channels. 4-AP suppressed closed state inactivation of Kv2.1/Kv6.4 resulting in exclusive current potentiation of currents through those heteromers (Stas et al., 2015). In contrast, Kv12.1 channels did not inactivate in our experiments, and thus 4-AP probably does not potentiate Kv12.1-mediated currents through a similar mechanism. As 4-AP, in contrast to Kv2.1/Kv6.4 heteromers, also modulated voltage dependence of Kv12.1, actions of 4-AP are probably even more complex for these channels. Interestingly, a recent study demonstrated 4-AP-dependent activation of Kv7.4 channels (Khammy et al., 2017) indicating that 4-AP-dependent activation of voltage-gated K+ channels may constitute a more frequent phenomenon than expected. However, further work is needed to elucidate whether other members of the Kv channel superfamily also exhibit this special 4-AP sensitivity.
Yet, we do not know whether 4-AP directly activates Kv12.1 channels or whether an auxiliary subunit endogenously expressed in CHO cells confers 4-AP activation to Kv12.1. Unfortunately, at present nothing is known about physiologically relevant interaction partners of Kv12.1 channels. As Kv channels (with Kv2.1 as exception) normally do not form functional channels with members of other Kv families, heteromerization of Kv12.1 channels with another pore forming α subunit apart from Kv12.2 or Kv12.3 is quite unlikely (Zou et al., 2003). Accordingly, a mechanism as shown for Kv2.1 is rather implausible, and it is hard to imagine how a non-pore-forming auxiliary subunit could reverse 4-AP sensitivity from inhibition to activation. Furthermore, any endogenously expressed auxiliary subunit would need to be expressed at high abundance to saturate overexpressed Kv12.1 channels. Hence, a straightforward model proposes direct activation of Kv12.1 channels through 4-AP. However, we want to point-out that we cannot exclude indirect actions on the channels at the moment.
NS1643, a “Specific” Activator of Kv11 Channels, Inhibits Kv12.1
NS1643 is a well-characterized partial agonist of Kv11 channels that slows deactivation, increases tail-current amplitude, and shifts voltage dependence of activation to hyperpolarized potentials and voltage dependence of C-type inactivation to depolarized potentials (Casis et al., 2006; Hansen et al., 2006). At the same time, NS1643 exhibits weak antagonistic effects on Kv11 channels as evident by an attenuation of drug-induced current increase at higher concentrations (Casis et al., 2006; Schuster et al., 2011). Kv11.3 channels are even inhibited by higher concentrations of NS1643. For Kv12.1 channels, NS1643 accelerated deactivation, inhibited outward and tail currents and shifted voltage dependence of activation to depolarized potentials (c.f. Figure 5). Thus, NS1643 affected Kv11 and Kv12.1 channels exactly in opposite directions. Similar to Kv11.3 channels, NS1643 increased slope factor of voltage dependence of Kv12.1 channels, even though already at lower concentrations. This suggests that NS1643 similarly affects Kv11 and Kv12.1 channels, but antagonistic effects might dominate over activation for Kv12.1. In line, the concentration range of NS1643 effects was similar for these channel isoforms (Casis et al., 2006). However, so far we cannot tell whether NS1643 also exhibits weak agonistic effects on Kv12.1 channels or whether NS1643 binds to homologous regions in Kv11 and Kv12.1 channels.
Conclusion and Outlook
As pharmacological tools and appropriate mouse models are currently missing, identification of native Kv12.1-mediated currents critically depends on identification of unique biophysical and pharmacological properties. Indeed, native currents were successfully attributed to related Kv10 and Kv11 channels by exploiting their unique activation kinetics (c.f. Cole-Moore shift; e.g., Meyer and Heinemann, 1998) or exclusive Na+ sensitivity and pharmacology (Hirdes et al., 2005, 2009; Hardman and Forsythe, 2009), respectively. Here, we present distinctive pharmacological properties, and straightforward experimental protocols that may be employed to isolate Kv12.1 channel activity in native tissue. As mode shift readily manifested in cells expressing various neuronal K+ currents, this experimental protocol may be used to demonstrate expression of channels with mode shift in native cell types. In neurons expressing various K+ current components, such changes of voltage dependence may be easier to detect than associated changes of deactivation kinetics. Such experiments may not provide definite proof for Kv12 channel expression. However, expression of mode shift may be employed together with expression analyses, Na+ sensitivity, activation through 4-AP and inhibition by NS1643 to narrow down (or exclude) contribution of Kv12.1 channels to whole cell currents. Identification of the combination of these properties would provide strong evidence for expression and thus potential physiological relevance of Kv12.1 channels.
Materials and Methods
Cell Culture and Transfection
Chinese hamster ovary (CHO) dhFR- cells were maintained as previously reported (Leitner et al., 2016). Cells were kept in MEM Alpha Medium supplemented with 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (pen/strep) (all Invitrogen GmbH, Darmstadt, Germany) in a humidified atmosphere at 5% CO2 and 37°C. Transient transfection of cells was done with jetPEI transfection reagent (Polyplus Transfection, Illkirch, France). All experiments were performed 24–48 h after transfection at room temperature (22–25°C). The expression vectors used were: Kv7.2-pBK-CMV (gene: human KCNQ2; UniProt accession number: O43526), Kv7.3-pBK-CMV (human KCNQ3; O43525), Kv7.4-pBK-CMV (human KCNQ4; P56696) Kv11.1 (Erg1)-pcDNA3.1 (rat KCNH2; O08962), Kv12.1(Elk1)-pcDNA3.1-IRESeGFP (human KCNH8; Q96L42), Kir2.1-pBK-CMV (human KCNJ2; P63252), and pEGFP-C1 (Addgene, Teddington, United Kingdom). For recordings presented in Figure 6 and Supplementary Figure 5, CHO cells were transiently transfected with identical amounts of plasmids encoding Kv7.2, Kv7.3, Kv11.1, Kv12.1, and Kir2.1 (0.6 μg of plasmid DNA for every subunit).
Electrophysiological Recordings
Electrophysiological recordings were performed in the whole cell configuration with an Axopatch 200B amplifier (Molecular Devices, Union City, CA, United States) or an HEKA EPC10 USB patch clamp amplifier HEKA (Elektronik, Lambrecht, Germany) in voltage-clamp mode (Leitner et al., 2011). All recordings were low-pass filtered at 2 kHz and sampled at 5 kHz. Currents were elicited by voltage protocols indicated in the figures. Dashed lines in representative recordings highlight zero current. Borosilicate glass patch pipettes (Sutter Instrument Company, Novato, CA, United States) were used with an open pipette resistance of 2–3 MΩ after back-filling with intracellular solution. Liquid junction potentials were not compensated (approximately -4 mV). Series resistance (Rs) typically was below 6 MΩ and compensated throughout the recordings (80–90%). Whole cell currents are presented normalized to the cell capacitance (current density; pA/pF) or as normalized to baseline current amplitude (I/IStart). Extracellular solutions for most experiments contained (mM) 144 NaCl, 5.8 KCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 10 HEPES and 5.6 D-glucose, pH 7.4 (with NaOH), 305–310 mOsm/kg. In some experiments, NaCl in the extracellular solution was substituted by N-methyl-D-glucamine (NMDG; Sigma–Aldrich) (c.f. Figure 2). The standard intracellular solution contained (mM) 135 KCl, 2.41 CaCl2 (100 nM free Ca2+), 3.5 MgCl2, 5 HEPES, 5 EGTA, 2.5 Na2ATP, pH 7.3 (with KOH), 290–295 mOsm/kg (Wilke et al., 2014).
Substances
Tetraethylammonium (TEA, Sigma), NMDG (Sigma), 4-aminopyridine (≥99%; Sigma and Tocris Bioscience, Bristol, United Kingdom), 2-aminopyridine (2-AP; Sigma), 3-aminopyridine (3-AP; Sigma), 3,4-diaminopyridine (3,4-DAP; Sigma), 1,3-Bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS 1643, Tocris), 10,10-bis(4-Pyridinylmethyl)-9(10H)-anthracenone dihydrochloride (XE991, Tocris), and N-[4-[[1- [2-(6-Methyl-2-pyridinyl)ethyl]-4-piperidinyl]carbonyl]phenyl] methanesulfonamide dihydrochloride (E-4031, Tocris) were diluted in extracellular solution to concentrations indicated in “Results.” All substances were applied locally via a glass capillary through a custom-made application system.
Note on 4-AP Solutions
4-AP did not significantly change the pH of the extracellular solution at concentrations below 3 mM. After dilution of the substance, pH of the extracellular solution containing 5 mM or 10 mM 4-AP typically was about 8.1 or 9.0, respectively. As indicated in “Results,” in some experiments the pH of solutions containing 5 and 10 mM 4-AP was adjusted to 7.4 after dilution of 4-AP. At the concentration applied in the present study (3 mM), isomeric aminopyridines did not change pH of the extracellular solution (c.f. Figure 4).
Data Analysis
Patch clamp recordings were analyzed with PatchMaster (HEKA) and IgorPro (Wavemetrics, Lake Oswego, OR, United States). Voltage dependence of activation was derived from tail current amplitudes using voltage protocols indicated: Tail currents were fitted with a two-state Boltzmann function with I = Imin + (Imax - Imin)/(1 + exp((V -Vh)/s)), where I is current, V is the membrane voltage, Vh is the voltage at half maximal activation, and s describes the steepness of the curve. Results are shown as conductance-voltage curves, obtained by normalizing to (Imax -Imin), obtained from fits to data of individual experiments. Time constants of deactivation were derived from double-exponential fits to deactivating current components at indicated potentials. For dose-response relations, current potentiation at 0 mV (normalized to baseline) was fitted to a Hill equation with I/Ib = Ib + (Imax-Ib)/(1 + (EC50/[S])nH), where I is the (normalized) current, Ib and Imax denote minimal and maximal currents at low and high drug concentrations, EC50 is the concentration at the half maximal effect, [S] is the drug concentration and nH is the Hill coefficient (Leitner et al., 2012).
Statistical Analysis
Isolated cells under investigation were randomly assigned to different treatment groups. Data recordings and analysis for experiments presented were not performed in a blinded manner. For some experiments, single recordings were normalized to base line values individually to account for baseline variations between cells. Statistical analysis was performed using two-tailed Student’s t-test/Wilcoxon–Mann–Whitney test, and when appropriate comparisons between multiple groups were performed with ANOVA followed by Dunnett test. Significance was assigned at P ≤ 0.05 (∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001). Data subjected to statistical analysis have n over 5 per group and data are presented as mean ± SEM. In electrophysiological experiments, n represents the number of individual cells and accordingly the number of independent experiments (no pseudo-replication).
Author Contributions
MD, SE, BW, and ML planned and performed the experiments and analyzed the data. ML conceived study, designed the experiments, and wrote the paper together with MD. All authors revised and approved the final version of the manuscript. All experiments were conducted at the Institute of Physiology and Pathophysiology at the Philipps-University Marburg (Germany).
Funding
This work was funded by Research Grants of the University Medical Center Giessen und Marburg (UKGM 17/2013; UKGM 13/2016 to ML) and by the German Research Foundation (DFG Priority Program 1608: “Ultrafast and temporally precise information processing: normal and dysfunctional hearing,” [LE 3600/1-1 to ML]).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the kind gift of plasmids for Kv7 (KCNQ) channels from Dr. T. Jentsch, Kv12.1 (Elk) from Dr. T. Jegla, and Kv11.1 (Erg) from Dr. C. Bauer. They thank Olga Ebers and Neslihan Özen for superb technical assistance and Dr. Dominik Oliver (Marburg) for helpful comments and discussion.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00011/full#supplementary-material
References
Alexander, S. P., Catterall, W. A., Kelly, E., Marrion, N., Peters, J. A., Benson, H. E., et al. (2015). The concise guide to PHARMACOLOGY 2015/16: voltage-gated ion channels. Br. J. Pharmacol. 172, 5904–5941. doi: 10.1111/bph.13349
Bauer, C. K., and Schwarz, J. R. (2001). Physiology of EAG K+ channels. J. Membr. Biol. 182, 1–15. doi: 10.1007/s00232-001-0031-3
Bezanilla, F., Taylor, R. E., and Fernandez, J. M. (1982). Distribution and kinetics of membrane dielectric polarization. 1. Long-term inactivation of gating currents. J. Gen. Physiol. 79, 21–40. doi: 10.1085/jgp.79.1.21
Bocksteins, E. (2016). Kv5, Kv6, Kv8, and Kv9 subunits: no simple silent bystanders. J. Gen. Physiol. 147, 105–125. doi: 10.1085/jgp.201511507
Bruening-Wright, A., and Larsson, H. P. (2007). Slow conformational changes of the voltage sensor during the mode shift in hyperpolarization-activated cyclic-nucleotide-gated channels. J. Neurosci. 27, 270–278. doi: 10.1523/JNEUROSCI.3801-06.2007
Casis, O., Olesen, S. P., and Sanguinetti, M. C. (2006). Mechanism of action of a novel human ether-a-go-go-related gene channel activator. Mol. Pharmacol. 69, 658–665. doi: 10.1124/mol.105.019943
Corbin-Leftwich, A., Mossadeq, S. M., Ha, J., Ruchala, I., Le, A. H., and Villalba-Galea, C. A. (2016). Retigabine holds KV7 channels open and stabilizes the resting potential. J. Gen. Physiol. 147, 229–241. doi: 10.1085/jgp.201511517
Curran, M. E., Splawski, I., Timothy, K. W., Vincent, G. M., Green, E. D., and Keating, M. T. (1995). A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803. doi: 10.1016/0092-8674(95)90358-5
Dai, G., and Zagotta, W. N. (2017). Molecular mechanism of voltage-dependent potentiation of KCNH potassium channels. Elife 6:e26355. doi: 10.7554/eLife.26355
Engeland, B., Neu, A., Ludwig, J., Roeper, J., and Pongs, O. (1998). Cloning and functional expression of rat ether-a-go-go-like K+ channel genes. J. Physiol. 513(Pt 3), 647–654.
Goodchild, S. J., Macdonald, L. C., and Fedida, D. (2015). Sequence of gating charge movement and pore gating in HERG activation and deactivation pathways. Biophys. J. 108, 1435–1447. doi: 10.1016/j.bpj.2015.02.014
Gutman, G. A., Chandy, K. G., Grissmer, S., Lazdunski, M., Mckinnon, D., Pardo, L. A., et al. (2005). International union of pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol. Rev. 57, 473–508. doi: 10.1124/pr.57.4.10
Hadley, J. K., Noda, M., Selyanko, A. A., Wood, I. C., Abogadie, F. C., and Brown, D. A. (2000). Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br. J. Pharmacol. 129, 413–415. doi: 10.1038/sj.bjp.0703086
Hansen, R. S., Diness, T. G., Christ, T., Demnitz, J., Ravens, U., Olesen, S. P., et al. (2006). Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643). Mol. Pharmacol. 69, 266–277.
Hardman, R. M., and Forsythe, I. D. (2009). Ether-a-go-go-related gene K+ channels contribute to threshold excitability of mouse auditory brainstem neurons. J. Physiol. 587, 2487–2497. doi: 10.1113/jphysiol.2009.170548
Hirdes, W., Napp, N., Wulfsen, I., Schweizer, M., Schwarz, J. R., and Bauer, C. K. (2009). Erg K+ currents modulate excitability in mouse mitral/tufted neurons. Pflugers. Arch. 459, 55–70. doi: 10.1007/s00424-009-0709-4
Hirdes, W., Schweizer, M., Schuricht, K. S., Guddat, S. S., Wulfsen, I., Bauer, C. K., et al. (2005). Fast erg K+ currents in rat embryonic serotonergic neurons. J. Physiol. 564, 33–49. doi: 10.1113/jphysiol.2004.082123
Ji, H., Tucker, K. R., Putzier, I., Huertas, M. A., Horn, J. P., Canavier, C. C., et al. (2012). Functional characterization of ether-a-go-go-related gene potassium channels in midbrain dopamine neurons - implications for a role in depolarization block. Eur. J. Neurosci. 36, 2906–2916. doi: 10.1111/j.1460-9568.2012.08190.x
Kazmierczak, M., Zhang, X., Chen, B., Mulkey, D. K., Shi, Y., Wagner, P. G., et al. (2013). External pH modulates EAG superfamily K+ channels through EAG-specific acidic residues in the voltage sensor. J. Gen. Physiol. 141, 721–735. doi: 10.1085/jgp.201210938
Khammy, M. M., Kim, S., Bentzen, B. H., Lee, S., Choi, I., Aalkjaer, C., et al. (2017). 4-aminopyridine: a pan Kv channel inhibitor that enhances Kv7.4 currents and inhibits noradrenaline-mediated contraction of rat mesenteric small arteries. Br. J. Pharmacol. doi: 10.1111/bph.14097 [Epub ahead of print].
Kortum, F., Caputo, V., Bauer, C. K., Stella, L., Ciolfi, A., Alawi, M., et al. (2015). Mutations in KCNH1 and ATP6V1B2 cause zimmermann-laband syndrome. Nat. Genet. 47, 661–667. doi: 10.1038/ng.3282
Leitner, M. G., Feuer, A., Ebers, O., Schreiber, D. N., Halaszovich, C. R., and Oliver, D. (2012). Restoration of ion channel function in deafness-causing KCNQ4 mutants by synthetic channel openers. Br. J. Pharmacol. 165, 2244–2259. doi: 10.1111/j.1476-5381.2011.01697.x
Leitner, M. G., Halaszovich, C. R., and Oliver, D. (2011). Aminoglycosides inhibit KCNQ4 channels in cochlear outer hair cells via depletion of phosphatidylinositol(4,5)bisphosphate. Mol. Pharmacol. 79, 51–60. doi: 10.1124/mol.110.068130
Leitner, M. G., Michel, N., Behrendt, M., Dierich, M., Dembla, S., Wilke, B. U., et al. (2016). Direct modulation of TRPM4 and TRPM3 channels by the phospholipase C inhibitor U73122. Br. J. Pharmacol. 173, 2555–2569. doi: 10.1111/bph.13538
Li, X., Anishkin, A., Liu, H., Van Rossum, D. B., Chintapalli, S. V., Sassic, J. K., et al. (2015). Bimodal regulation of an Elk subfamily K+ channel by phosphatidylinositol 4,5-bisphosphate. J. Gen. Physiol. 146, 357–374. doi: 10.1085/jgp.201511491
Mannikko, R., Pandey, S., Larsson, H. P., and Elinder, F. (2005). Hysteresis in the voltage dependence of HCN channels: conversion between two modes affects pacemaker properties. J. Gen. Physiol. 125, 305–326. doi: 10.1085/jgp.200409130
Marcotti, W., Johnson, S. L., Holley, M. C., and Kros, C. J. (2003). Developmental changes in the expression of potassium currents of embryonic, neonatal and mature mouse inner hair cells. J. Physiol. 548, 383–400. doi: 10.1113/jphysiol.2002.034801
Meyer, R., and Heinemann, S. H. (1998). Characterization of an eag-like potassium channel in human neuroblastoma cells. J. Physiol. 508(Pt 1), 49–56. doi: 10.1111/j.1469-7793.1998.049br.x
Miyake, A., Mochizuki, S., Yokoi, H., Kohda, M., and Furuichi, K. (1999). New ether-a-go-go K(+) channel family members localized in human telencephalon. J. Biol. Chem. 274, 25018–25025. doi: 10.1074/jbc.274.35.25018
Miyake, A., Takahashi, S., Nakamura, Y., Inamura, K., Matsumoto, S., Mochizuki, S., et al. (2009). Disruption of the ether-a-go-go K+ channel gene BEC1/KCNH3 enhances cognitive function. J. Neurosci. 29, 14637–14645. doi: 10.1523/JNEUROSCI.0901-09.2009
Numaguchi, H., Johnson, J. P. Jr., Petersen, C. I., and Balser, J. R. (2000). A sensitive mechanism for cation modulation of potassium current. Nat. Neurosci. 3, 429–430. doi: 10.1038/74793
Olcese, R., Latorre, R., Toro, L., Bezanilla, F., and Stefani, E. (1997). Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 110, 579–589. doi: 10.1085/jgp.110.5.579
Pardo, L. A., and Stuhmer, W. (2014). The roles of K(+) channels in cancer. Nat. Rev. Cancer 14, 39–48. doi: 10.1038/nrc3635
Piper, D. R., Varghese, A., Sanguinetti, M. C., and Tristani-Firouzi, M. (2003). Gating currents associated with intramembrane charge displacement in HERG potassium channels. Proc. Natl. Acad. Sci. U.S.A. 100, 10534–10539. doi: 10.1073/pnas.1832721100
Ridley, J. M., Milnes, J. T., Zhang, Y. H., Witchel, H. J., and Hancox, J. C. (2003). Inhibition of HERG K+ current and prolongation of the guinea-pig ventricular action potential by 4-aminopyridine. J. Physiol. 549, 667–672. doi: 10.1113/jphysiol.2003.043976
Robertson, B. E., and Nelson, M. T. (1994). Aminopyridine inhibition and voltage dependence of K+ currents in smooth muscle cells from cerebral arteries. Am. J. Physiol. 267, C1589–C1597. doi: 10.1152/ajpcell.1994.267.6.C1589
Saganich, M. J., Machado, E., and Rudy, B. (2001). Differential expression of genes encoding subthreshold-operating voltage-gated K+ channels in brain. J. Neurosci. 21, 4609–4624.
Sanchez, A., Urrego, D., and Pardo, L. A. (2016). Cyclic expression of the voltage-gated potassium channel KV10.1 promotes disassembly of the primary cilium. EMBO Rep. 17, 708–723. doi: 10.15252/embr.201541082
Sanguinetti, M. C., Jiang, C., Curran, M. E., and Keating, M. T. (1995). A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81, 299–307. doi: 10.1016/0092-8674(95)90340-2
Schuster, A. M., Glassmeier, G., and Bauer, C. K. (2011). Strong activation of ether-a-go-go-related gene 1 K+ channel isoforms by NS1643 in human embryonic kidney 293 and Chinese hamster ovary cells. Mol. Pharmacol. 80, 930–942. doi: 10.1124/mol.111.071621
Sedehizadeh, S., Keogh, M., and Maddison, P. (2012). The use of aminopyridines in neurological disorders. Clin. Neuropharmacol. 35, 191–200. doi: 10.1097/WNF.0b013e31825a68c5
Shi, W., Wang, H. S., Pan, Z., Wymore, R. S., Cohen, I. S., Mckinnon, D., et al. (1998). Cloning of a mammalian elk potassium channel gene and EAG mRNA distribution in rat sympathetic ganglia. J. Physiol. 511(Pt 3), 675–682.
Simons, C., Rash, L. D., Crawford, J., Ma, L., Cristofori-Armstrong, B., Miller, D., et al. (2015). Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat. Genet. 47, 73–77. doi: 10.1038/ng.3153
Stas, J. I., Bocksteins, E., Labro, A. J., and Snyders, D. J. (2015). Modulation of closed-state inactivation in Kv2.1/Kv6.4 heterotetramers as mechanism for 4-AP induced potentiation. PLOS ONE 10:e0141349. doi: 10.1371/journal.pone.0141349
Strupp, M., Teufel, J., Zwergal, A., Schniepp, R., Khodakhah, K., and Feil, K. (2017). Aminopyridines for the treatment of neurologic disorders. Neurol. Clin. Pract. 7, 65–76. doi: 10.1212/CPJ.0000000000000321
Sturm, P., Wimmers, S., Schwarz, J. R., and Bauer, C. K. (2005). Extracellular potassium effects are conserved within the rat erg K+ channel family. J. Physiol. 564, 329–345. doi: 10.1113/jphysiol.2004.078840
Tan, P. S., Perry, M. D., Ng, C. A., Vandenberg, J. I., and Hill, A. P. (2012). Voltage-sensing domain mode shift is coupled to the activation gate by the N-terminal tail of hERG channels. J. Gen. Physiol. 140, 293–306. doi: 10.1085/jgp.201110761
Tilegenova, C., Cortes, D. M., and Cuello, L. G. (2017). Hysteresis of KcsA potassium channel’s activation- deactivation gating is caused by structural changes at the channel’s selectivity filter. Proc. Natl. Acad. Sci. U.S.A. 114, 3234–3239. doi: 10.1073/pnas.1618101114
Trudeau, M. C., Titus, S. A., Branchaw, J. L., Ganetzky, B., and Robertson, G. A. (1999). Functional analysis of a mouse brain Elk-type K+ channel. J. Neurosci. 19, 2906–2918.
Trudeau, M. C., Warmke, J. W., Ganetzky, B., and Robertson, G. A. (1995). HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 269, 92–95. doi: 10.1126/science.7604285
Urrego, D., Movsisyan, N., Ufartes, R., and Pardo, L. A. (2016). Periodic expression of Kv10.1 driven by pRb/E2F1 contributes to G2/M progression of cancer and non-transformed cells. Cell Cycle 15, 799–811. doi: 10.1080/15384101.2016.1138187
Villalba-Galea, C. A. (2017). Hysteresis in voltage-gated channels. Channels 11, 140–155. doi: 10.1080/19336950.2016.1243190
Villalba-Galea, C. A., Sandtner, W., Starace, D. M., and Bezanilla, F. (2008). S4-based voltage sensors have three major conformations. Proc. Natl. Acad. Sci. U.S.A. 105, 17600–17607. doi: 10.1073/pnas.0807387105
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Wilke, B. U., Lindner, M., Greifenberg, L., Albus, A., Kronimus, Y., Bunemann, M., et al. (2014). Diacylglycerol mediates regulation of TASK potassium channels by Gq-coupled receptors. Nat. Commun. 5:5540. doi: 10.1038/ncomms6540
Zhang, X., Bertaso, F., Yoo, J. W., Baumgartel, K., Clancy, S. M., Lee, V., et al. (2010). Deletion of the potassium channel Kv12.2 causes hippocampal hyperexcitability and epilepsy. Nat. Neurosci. 13, 1056–1058. doi: 10.1038/nn.2610
Keywords: Kv10, Kv11, Kv12, HERG, mode shift, voltage-dependent potentiation, 4-aminopyridine, NS1643
Citation: Dierich M, Evers S, Wilke BU and Leitner MG (2018) Inverse Modulation of Neuronal Kv12.1 and Kv11.1 Channels by 4-Aminopyridine and NS1643. Front. Mol. Neurosci. 11:11. doi: 10.3389/fnmol.2018.00011
Received: 10 November 2017; Accepted: 09 January 2018;
Published: 30 January 2018.
Edited by:
Alexandre Mourot, Université Pierre et Marie Curie, FranceReviewed by:
Marco Martina, Northwestern University, United StatesNorelle Christine Wildburger, Washington University in St. Louis, United States
Copyright © 2018 Dierich, Evers, Wilke and Leitner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael G. Leitner, TWljaGFlbC5MZWl0bmVyQGktbWVkLmFjLmF0
†These authors have contributed equally to this work.