 Jereme G. Spiers
Jereme G. Spiers Carlo Breda2
Carlo Breda2 Flaviano Giorgini
Flaviano Giorgini Joern R. Steinert
Joern R. Steinert- 1MRC Toxicology Unit, University of Leicester, Leicester, United Kingdom
- 2Department of Genetics and Genome Biology, University of Leicester, Leicester, United Kingdom
Many neurodegenerative conditions and age-related neuropathologies are associated with increased levels of reactive oxygen species (ROS). The cap “n” collar (CncC) family of transcription factors is one of the major cellular system that fights oxidative insults, becoming activated in response to oxidative stress. This transcription factor signaling is conserved from metazoans to human and has a major developmental and disease-associated relevance. An important mammalian member of the CncC family is nuclear factor erythroid 2-related factor 2 (Nrf2) which has been studied in numerous cellular systems and represents an important target for drug discovery in different diseases. CncC is negatively regulated by Kelch-like ECH associated protein 1 (Keap1) and this interaction provides the basis for a homeostatic control of cellular antioxidant defense. We have utilized the Drosophila model system to investigate the roles of CncC signaling on longevity, neuronal function and circadian rhythm. Furthermore, we assessed the effects of CncC function on larvae and adult flies following exposure to stress. Our data reveal that constitutive overexpression of CncC modifies synaptic mechanisms that positively impact on neuronal function, and suppression of CncC inhibitor, Keap1, shows beneficial phenotypes on synaptic function and longevity. Moreover, supplementation of antioxidants mimics the effects of augmenting CncC signaling. Under stress conditions, lack of CncC signaling worsens survival rates and neuronal function whilst silencing Keap1 protects against stress-induced neuronal decline. Interestingly, overexpression and RNAi-mediated downregulation of CncC have differential effects on sleep patterns possibly via interactions with redox-sensitive circadian cycles. Thus, our data illustrate the important regulatory potential of CncC signaling in neuronal function and synaptic release affecting multiple aspects within the nervous system.
Introduction
One of the major cellular defense mechanisms against oxidative stress is mediated by the mammalian nuclear factor erythroid 2-related factor 2 (Nrf2) and Kelch-like ECH associated protein 1 (Keap1) signaling cascade. The Nrf2/Keap1 pathway regulates gene expression of many cytoprotective and detoxifying enzymes, thus playing a pivotal role in maintaining cellular redox homeostasis. Nrf2 belongs to the cap “n” collar (CncC) subfamily of basic region leucine zipper transcription factors and its regulation and importance in cellular defense mechanisms has been studied in numerous physiological and pathological conditions.
Nrf2 plays a key role in neuronal resistance to oxidative stress mediated by reactive oxygen species (ROS) and glutamate-induced excitotoxicity (He et al., 2014). Balancing oxidative stress by up-regulation of Nrf2 antioxidant defense has been demonstrated to be effective in neurodegenerative disease treatment. Aging is one of the main risk factors for neurodegenerative conditions, but it is also closely associated with a loss of Nrf2 activity. Nrf2 and expression of downstream target genes are decreased in the substantia nigra of aged rats, with Nrf2 overexpression exerting a protective response to neurodegeneration (Habas et al., 2013), including in models of amyotrophic lateral sclerosis (ALS), stroke, Alzheimer’s disease (AD) and Parkinson’s disease (PD). Indeed, Nrf2 activation has been shown to alleviate neurodegenerative symptoms in a Drosophila model of PD (Barone et al., 2011). Furthermore, Nrf2-mediated neuroprotection is primarily conferred by astroglia both in vitro and in vivo (Liddell, 2017) and in AD patients, Nrf2 expression is decreased in both hippocampal neurons and astrocytes (Ramsey et al., 2007) indicating a strong involvement of Nrf2 signaling in neurodegeneration and neuronal function. Previous work has shown that activation of the Nrf2/Keap1 transcriptional pathways can protect hippocampal neurons from Aβ-induced neurodegeneration in an AD mouse model (Lipton et al., 2016) and rescue neuronal deficiencies in various models for PD (Johnson and Johnson, 2015), confirming a protective role in neuronal function with potential for therapeutic treatments. However, the exact targets and mechanisms of the antioxidant activities of Nrf2/Keap1 activation in the modulation of neuronal function are not fully understood.
One important characteristic of neurodegenerative diseases and aging is dysregulation of sleep patterns which has been reported across different species ranging from fly to human (De Lazzari et al., 2018; Vanderheyden et al., 2018). Cumulative evidence demonstrates a close connection between cellular circadian rhythm and redox systems. The circadian clock is involved in the regulation of ROS levels both in vivo and in vitro (Desvergne et al., 2014; Early et al., 2018). In mammals, the circadian clock orchestrates the activities of the antioxidant defense and oxidative stress response systems by mediating Nrf2 signaling. Two proteins involved in circadian rhythm, circadian locomotor output cycles kaput (Clock) and Brain and muscle arnt-like protein-1 (Bmal1) can positively regulate Nrf2 transcription, which in turn drives rhythmic oscillations of antioxidant genes (Xu et al., 2012; Pekovic-Vaughan et al., 2014). Conversely, the cellular redox state is critically important for the regulation of Bmal1 and Clock gene transcriptional activities (Ranieri et al., 2015).
The mechanism underlying the Nrf2 protective response remains obscure and given the limited understanding of Nrf2/Keap1 signaling on neuronal function we utilized the Drosophila model in this study to investigate the effects on aging, synapse function, and circadian activity. Drosophila Keap1 acts as a negative regulator of CncC (Itoh et al., 1999; Sykiotis and Bohmann, 2008; Pitoniak and Bohmann, 2015) and its silencing by RNAi leads to endogenous activation of CncC signaling with flies showing upregulation of the classical antioxidant response element cascade (Sykiotis and Bohmann, 2008), which increases their stress resistance. In particular, CncC activation results in enhanced transcription of the antioxidant and detoxifying enzyme glutathione S-transferase encoded by the Drosophila gstD1 gene (Sawicki et al., 2003; Sykiotis and Bohmann, 2008) which acts in a neuroprotective manner.
We manipulated neuronal antioxidant response ability by either overexpressing CncC or reducing the expression of CncC and Keap1 protein by RNAi. We then investigated longevity, activity, and circadian behavior in adult flies in addition to synaptic function at the larval neuromuscular junction (NMJ). The data show that constitutive overexpression of CncC has important impacts on synaptic release and survival with silencing of the CncC inhibitor, Keap1, inducing beneficial effects on survival and synaptic function. Importantly, application of the antioxidant compounds dithiothreitol (DTT) and glutathione (GSH) produced similar effects to those mediated by CncC overexpression or Keap1 silencing, suggesting that an antioxidant environment boosts synaptic function in a redox-specific manner.
Materials and Methods
Fly Husbandry
Flies were raised on standard maize media at 25°C at a 12-h LD cycle. The elav-GAL4[C155] driver was obtained from the Bloomington Stock Center (Indiana, US). The UAS-RNAi lines [Keap1 (CG3962) and CncC (CG43286)] were purchased from the Vienna Drosophila Resource Centre (VDRC). The UAS-CncC line was kindly provided by Dirk Bohmann, University of Rochester, USA (Sykiotis and Bohmann, 2008; Pitoniak and Bohmann, 2015). The UAS/GAL4 bipartite expression system was utilized to drive pan-neuronal expression. The elav-GAL4 driver (female flies) and the UAS responder lines (male flies) were crossed to obtain offspring expressing the genes of interest. As a control for the RNAi strains, a line carrying an empty RNAi vector inserted in the AttP40 site was used and crossed to the elav-GAL4 driver (referred to as RNAi Ctrl). For CncC overexpressing (OE) lines, experimental lines were compared to control obtained by crossing the GAL4 (elav Ctrl) and UAS lines to w1118. The homozygote w1118 line was used as a control in the pharmacology experiments.
Electrophysiology
TEVC recordings were performed as described previously (Robinson et al., 2018). Sharp-electrode recordings were made from ventral longitudinal m6 in abdominal segments 2 and 3 of third instar larvae using pClamp 10.5, an Axoclamp 900A amplifier and Digidata 1440A (Molecular Devices, US) in hemolymph-like solution 3 (HL-3; Stewart et al., 1994). Recording electrodes (20–50 MΩ) were filled with 3 M KCl. Miniature excitatory junctional currents (mEJCs) were recorded in the presence of 0.5 μM tetrodotoxin (Tocris, UK). All synaptic responses were recorded from muscles with input resistances ≥4 MΩ, holding currents <4 nA at −60 mV and resting potentials more negative than −60 mV at 25°C, as differences in recording temperature cause changes in glutamate receptor kinetics and amplitudes (Postlethwaite et al., 2007). Holding potentials were −60 mV. The extracellular HL-3 contained (in mM): 70 NaCl, 5 KCl, 20 MgCl2, 10 NaHCO3, 115 sucrose, 5 trehalose, 5 HEPES, and 1.5 CaCl2. Average single evoked EJC (eEJC) amplitudes (stimulus: 0.1 ms, 1–5 V) were based on the mean peak eEJC amplitude in response to 10 presynaptic stimuli (recorded at 0.2 Hz). Nerve stimulation was performed with an isolated stimulator (DS2A, Digitimer). All data were digitized at 10 kHz and for miniature recordings, 200-s recordings were analyzed to obtain mean mEJC amplitudes. The quantal content (QC) was estimated for each recording by calculating the ratio of eEJC amplitude/average mEJC amplitude, followed by averaging recordings across all NMJs for a given genotype. mEJC and eEJC recordings were off-line low-pass filtered at 500 Hz and 1 kHz, respectively. Materials were purchased from Sigma-Aldrich (UK).
Cumulative Postsynaptic Current Analysis
The apparent size of the RRP was probed by the method of cumulative eEJC amplitudes (Schneggenburger et al., 1999). Muscles were clamped to −60 mV and eEJC amplitudes during a stimulus train [50 Hz, 500 ms (of a 1-s train)] were calculated as the difference between peak and baseline before stimulus onset of a given eEJC. Receptor desensitization was not blocked as it did not affect eEJC amplitudes, because a comparison of the decay of the first and the last eEJC within a train did not reveal any significant difference in decay kinetics. The number of release-ready vesicles (N) was obtained by back extrapolating a line fit to the linear phase of the 500-ms cumulative eEJC plot (the last 200 ms of the train) to time zero by dividing the cumulative eEJC amplitude at time zero by the mean mEJC amplitude recorded in the same cell. To calculate the QC in the train, we used mean mEJC amplitudes measured before the train.
Heat Shock Protocol
For heat shock survival experiments, methods were adapted from Ishida et al. (2012). Briefly, male adult flies (aged 3–5 days) were transferred to vials containing a moistened filter pad to prevent dehydration. Vials were placed in a 37°C water bath and live flies were counted every 30 min. In larval experiments, heat shock was induced using previously described methods (Robinson et al., 2017). Briefly, age-matched third instar larvae were incubated at 37°C for 1 h and used for electrophysiology 24 h later. These experiments were repeated a minimum of three times for each genotype.
Circadian
Circadian activity and sleep analysis were performed as described previously (Ishida et al., 2012). Briefly, adult male flies (aged 3–5 days) were individually transferred into glass tubes containing food. Single tubes were then loaded into Drosophila Activity Monitor system (Trikinetics). Following a 2 days period of entrainment in incubators kept on a 12:12 light/dark regime at 25°C, locomotor activity was recorded for the consecutive 5 days. Sleep behavior was analyzed by using pySolo software (Gilestro and Cirelli, 2009).
Survival
Groups of 10 newly emerged adult male flies were transferred to new vials containing food and deaths were scored daily. Flies were transferred to new food three times per week and otherwise left undisturbed. Cumulative survival curves are presented and compared using the Log-rank (Mantel-Cox) test.
Geotaxis
Rapid iterative negative geotaxis behavior was performed using methods outlined previously (Rhodenizer et al., 2008; Nichols et al., 2012). Briefly, age-matched adult male flies were collected and groups of 10 were transferred to a clear empty vial without anesthesia at weekly intervals. Tubes were transferred to a quiet room and flies acclimated for 15 min. Tubes were tapped three times on a bench and images were taken after 3 s using a digital camera. A minimum of five trials were conducted per session with an inter-trial interval of 1 min. Average height climbed per vial was calculated from images using Image J software. These experiments were repeated a minimum of three times per genotype.
Crawling Activity
Age-matched third instar male larvae (~100–120 h) were selected, washed and placed onto a moist, food-free surface at a constant temperature of 20°C. Crawling activities were imaged over 10 min using AnyMaze software v4.98 (Stoelting Co., Wood Dale, IL, USA) and data were analyzed off-line as reported previously (Robinson et al., 2014).
Statistics
Statistical analysis was performed with Prism 7 (Graphpad Software Inc., San Diego, CA, USA). Statistical tests were carried out using a one-way ANOVA test when applicable with a posteriori test (with Tukey’s multiple comparisons) or unpaired Student’s t-test [for comparisons between elav × CncC and elav × w1118 (elav Ctrl)]. Data in figures are expressed as mean ± SEM where n is the number NMJs, flies or larvae as indicated, and significance is shown as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
We used this well-characterized expression system and first assessed the effects of down-regulating either CncC or Keap1 protein expression on Drosophila life span as it has long been postulated that oxidative stress contributes to age-related neuronal dysfunction known as the free radical theory of aging. Under unchallenged conditions, the suppression of Keap1 expression (Keap1-RNAi) causes an increase in longevity, whereas CncC silencing (CncC-RNAi) does not show any effects [median life span (in days): RNAi Ctrl (elav GAL4, UAS RNAi Control): 53, CncC-RNAi: 52.5, Keap1-RNAi: 63, p < 0.0001, Log-rank (Mantel-Cox) test, Figure 1A] implicating that the lack of CncC signaling might be compensated by other defense mechanisms, such as the thioredoxin system (Radyuk et al., 2003). Moreover, pan-neuronal overexpression (OE) of CncC did not cause any significant improvements in life span of unstressed flies relative to their controls [median life span (in days): elav × CncC: 36, elav × w1118: 38, w1118 × CncC: 45, p > 0.05, Log-rank (Mantel-Cox) test, Figure 1B] which has previously been reported (Pitoniak and Bohmann, 2015) and could be the result of endogenous suppression of CncC nuclear translocation and transcriptional activity similar to that reported for Nrf1 signaling (Wang and Chan, 2006). In addition to longevity studies, fly aging can also be evaluated by geotaxis-driven activity assays, leading us to assess the activity of adult Drosophila individuals with CncC and Keap1 silencing at 7, 14 and 21 days of age. As CncC OE did not alter longevity we did not further use this genotype in this assay. CncC silencing reduced negative geotaxis-mediated activity (7 days: RNAi Ctrl: 5.9 ± 0.3 cm; CncC-RNAi: 3.8 ± 0.2 cm, Keap1-RNAi: 6.9 ± 0.2 cm, Figure 1C), whereas Keap1-RNAi individuals displayed enhanced performance relative to CncC silencing (14 days: RNAi Ctrl: 5.5 ± 0.2 cm; CncC-RNAi: 4.1 ± 0.2 cm, Keap1-RNAi: 6.6 ± 0.3 cm, 21 days: RNAi Ctrl: 3.7 ± 0.1 cm; CncC-RNAi: 3.0 ± 0.2 cm, Keap1-RNAi: 4.5 ± 0.4 cm, ANOVA).
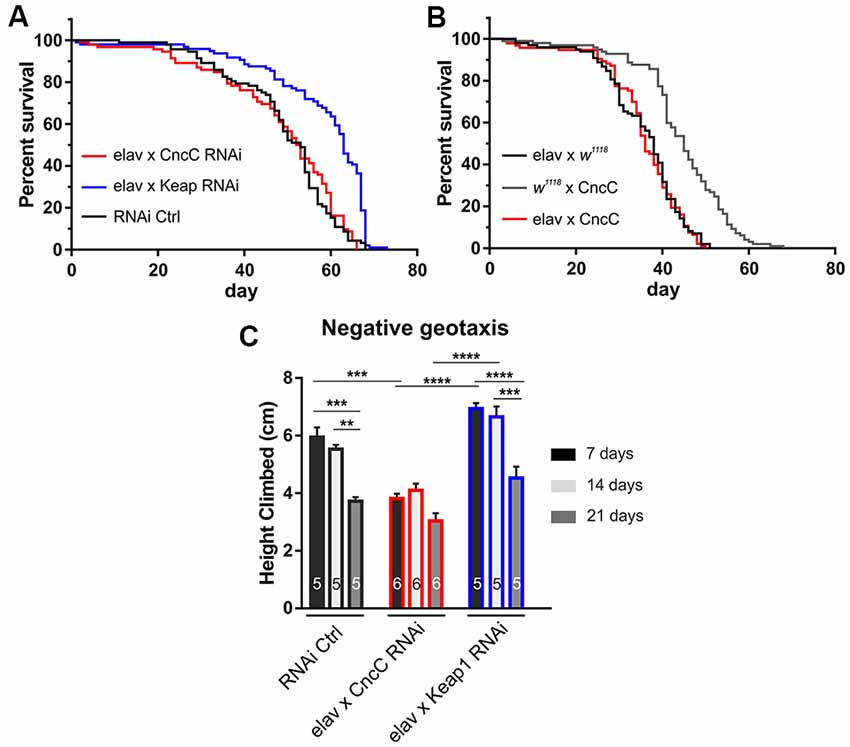
Figure 1. Suppression of Kelch-like ECH associated protein 1 (Keap1) signaling extends life span and promotes negative geotaxis activity. (A) RNAi silencing of Keap1 prolongs life span, whereas CncC downregulation does not affect longevity (n = 60–98 flies, p < 0.0001). (B) Overexpression of CncC does not affect life span (n = 99–72 flies, p > 0.05). Data were compared using the Log-rank (Mantel-Cox) test. (C) Negative geotaxis performance declines with age in control flies (RNAi Ctrl at 21 days, dark gray), however, CncC RNAi expression induces a strong reduction in climbing activity at 7 days (black) with no further effects at older ages. RNAi silencing of Keap1 augments activity decline relative to CncC silencing at seven (black) and 14 (light gray) days (n—number of flies indicated within bars). Data denote mean ± SEM for all data comparisons in (C). One-way ANOVA with post hoc Tukey-Kramer was used for comparisons with **p < 0.01, ***p < 0.001, ****p < 0.0001.
To further characterize the effects on survival and geotaxis-driven activity and their connection to neuronal health, we assessed neuronal function in more detail in electrophysiological experiments of the larval NMJ. This well-studied synapse allows direct assessment of neuronal health and synaptic function by recording single action potential-eEJC, spontaneous release events (mEJC), and total vesicular pool size release. Oxidative stress and aging have been related to compromised neuronal function and diminished synaptic release. However, to our knowledge, the direct effects of CncC/Keap1 signaling on synaptic release have not yet been documented (Besson et al., 2000; Fremeau et al., 2004; Escartin et al., 2011; Wu and Cooper, 2012; Cirillo et al., 2015; Ivannikov and Van Remmen, 2015).
Interestingly, our studies showed that enhancing antioxidant potential via upregulation of CncC signaling by either Keap1 silencing or CncC overexpression (OE), potently increased mEJC amplitudes [RNAi Ctrl: 0.46 ± 0.02 nA, CncC-RNAi: 0.50 ± 0.02 nA, Keap1-RNAi: 0.72 ± 0.09 nA (ANOVA); CncC OE: 0.78 ± 0.04 nA, elav Ctrl: 0.61 ± 0.03 nA (Student’s t-test), Figures 2A,B]. Confirmatory studies further showed treatment with the antioxidants DTT (1 mM DTT; ROS scavenger) or reduced GSH (150 μM GSH) for 45 min significantly enhanced mEJC amplitudes [w1118: 0.62 ± 0.02 nA, DTT: 0.86 ± 0.08 nA, GSH: 1.51 ± 0.11 nA, ANOVA, Figure 2C], further corroborating the effects seen following genetic manipulation of antioxidant signaling pathways and indicating that enhanced antioxidant levels at the synapse promote larger quantal size.
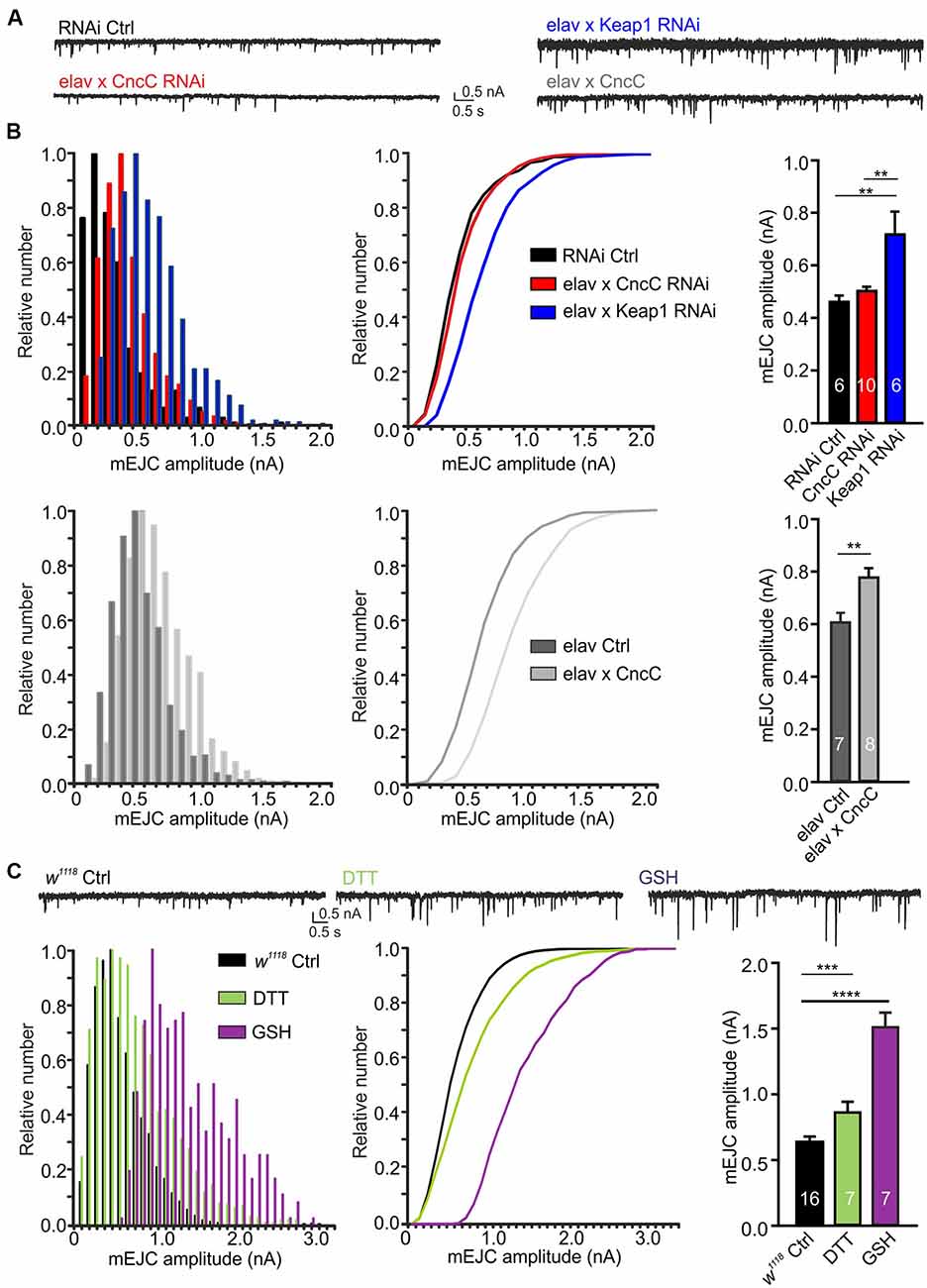
Figure 2. Upregulation of antioxidant signaling enhances spontaneous quantal release at the neuromuscular junction (NMJ) synapse. (A) Example recordings of spontaneous activity at larval NMJs. (B) Overexpression of CncC and silencing of Keap1 enhances the quantal size, illustrated in amplitude frequency plots for miniature excitatory junctional current (mEJC) amplitudes (left), cumulative amplitude frequency plots (middle) and mean bar graphs (right). (C) Exposure to antioxidants dithiothreitol (DTT) and glutathione (GSH) induced a strong increase in quantal size at NMJs of w1118 larvae as illustrated in frequency plots for mEJC amplitudes (left), cumulative amplitude frequency plots (middle) and mean bar graphs (right). One-way ANOVA with post hoc Tukey-Kramer was used for comparisons with **p < 0.01, ***p < 0.001, ****p < 0.0001 [n—number of NMJs (from at least three larvae) indicated within bars].
However, when analyzing evoked EJC (eEJC) amplitudes, we did not detect any increase in amplitudes following genetic induction of antioxidant signaling but unexpectedly a decrease in eEJC amplitudes following CncC OE [RNAi Ctrl: 165 ± 17 nA, CncC-RNAi: 140 ± 9 nA, Keap1-RNAi: 151 ± 12 nA (ANOVA); CncC OE: 99 ± 8 nA, elav Ctrl: 127 ± 12 nA (Student’s t-test), Figures 3A,B]. Although this was somewhat surprising, however, given the fact that this synapse is under high homeostatic regulation (Müller et al., 2012; Frank, 2014; Li et al., 2018), it is likely that elevated quantal size (mEJC amplitude) leads to a reduced QC of the evoked response thereby leaving the evoked amplitudes unchanged or even decreased. This homeostatic feedback pathway regulates release within relatively short time frames (tens of minutes) and provides a rapid physiological regulatory mechanism. In fact, when assessing the QC we found that both CncC OE and knock-down (KD) of Keap1 induced a decrease in QC [RNAi Ctrl: 356 ± 33, CncC-RNAi: 278 ± 16, Keap1-RNAi: 225 ± 32 (ANOVA); CncC OE: 122 ± 8, elav Ctrl: 184 ± 13 (Student’s t-test), Figures 3C,E]. This reduction in release induced a strong effect on the total vesicular release capacity (vesicle pool size) of the NMJ, with the calculated number of releasable quanta per NMJ (as calculated by the cumulative postsynaptic current analysis of a 50 Hz train yielding the cumulative QC) being drastically reduced following CncC OE or Keap1-RNAi expression [RNAi Ctrl: 837 ± 97, CncC-RNAi: 820 ± 72, Keap1-RNAi: 531 ± 110 (ANOVA); CncC OE: 298 ± 34, elav Ctrl: 459 ± 64 (Student’s t-test), Figures 3D,F]. To exclude the possibility that genetic manipulation caused developmental changes which could interfere with our data interpretation, we again assessed the effects of pharmacological manipulation of redox signaling on release parameters of the synapse. Quantification of NMJ evoked responses and QC revealed similar changes using antioxidant supplementation as following genetic manipulations. DTT application for 45 min induced mild effects and GSH application for the same length led to strong effects on the three parameters [eEJC amplitude: w1118: 118 ± 5 nA, DTT: 140 ± 7 nA, GSH: 113 ± 4 nA, QC: w1118: 213 ± 25, DTT: 167 ± 12, GSH: 65 ± 3, cumulative QC: w1118: 441 ± 40, DTT: 247 ± 49, GSH: 63 ± 35, ANOVA, Figures 3G–K] compared to controls (w1118), similar to the changes observed following genetically-induced increase in antioxidant potential suggesting an acute mechanism mediated by a reduction in basal oxidative stress levels. Importantly, CncC KD was without effects indicating that under these unchallenged conditions the deficiency in potential antioxidant capacity did not alter basal synaptic transmission. One important regulatory mechanism with the ability to modulate synaptic release is the control of synaptic release probabilities. The initial release probability can be adjusted in response to various mechanisms. We assessed potential effects on the initial vesicular release probability (pvr) by measuring paired-pulse ratios (PPR) at a 20 ms inter-spike interval. Previously we found that the nitrergic regulation of synaptic release at the NMJ is mediated via a reduction of pvr (Robinson et al., 2014) as manifested in an increased PPR. Paired-pulse experiments at larval NMJs revealed a significant increase in PPR following genetic alterations of Keap1 expression suggesting that pvr is reduced at lower oxidative stress levels [RNAi Ctrl: 0.81 ± 0.05, CncC-RNAi: 0.92 ± 0.02, Keap1-RNAi: 0.99 ± 0.02 (ANOVA); CncC OE: 1.00 ± 0.05, elav Ctrl: 0.88 ± 0.11 (Student’s t-test), Figure 3L]. However, following pharmacological modulation of redox signaling we detected an increase in PPR only after GSH application [w1118: 0.88 ± 0.04, DTT: 0.92 ± 0.04, GSH: 1.12 ± 0.05, ANOVA, Figure 3L]. Importantly, changes in frequency (f) of spontaneous release indicates direct effects on vesicle fusion mediated by the soluble N-ethyl-maleimide-sensitive fusion protein Attachment Protein Receptor (SNARE) and SNARE binding proteins. To investigate the modulation of these mechanisms, we measured mEJC frequencies. The results did not show any differences in f between larvae [RNAi Ctrl: 2.6 ± 0.4 s−1, CncC-RNAi: 3.2 ± 0.3 s−1, Keap1-RNAi: 3.4 ± 0.5 s−1 (ANOVA); CncC OE: 1.4 ± 0.2 s−1, elav Ctrl: 3.14 ± 0.80 (Student’s t-test); w1118: 2.0 ± 0.2 s−1, GSH: 1.2 ± 0.2 s−1, DTT: 3.4 ± 0.3 s−1 (ANOVA) p > 0.05] suggesting that vesicle fusion mechanisms per se are not modulated by changes in the redox level following genetic or pharmacological manipulations.

Figure 3. Increased antioxidant environment allows for reduction of stimulated vesicular release while maintaining synaptic transmission. (A) Recordings of evoked synaptic currents at the larval NMJ. Graphs show means for evoked EJC (eEJC) amplitudes (B), quantal content (QC; C) and cumulative QC (D), QC and cumulative QC graphs of 50 Hz trains for genotypes indicated (E,F). eEJC amplitudes (G), QC (H) and cumulative QC (I) for treatments indicated. (J,K) Graphs of 50 Hz trains from w1118 larvae and w1118 larvae treated with GSH and DTT. (L) Paired-pulse ratios (PPR) were analyzed at 20 ms inter-spike intervals at NMJs from indicated genotypes and/or treatments [ANOVA used for comparison of RNAi expressing strains, Student’s t-test was used to compare elav × CncC with elav Ctrl, n—number of NMJs (from at least three larvae) indicated within bars]. (M) Mean crawling distances of different genotypes over a 10 min imaging period (n—number of larvae indicated within bars). One-way ANOVA with post hoc Tukey-Kramer was used for comparisons with *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Together, our data show that regulation of the redox environment can alter synaptic function with spontaneous release events being positively modulated in a low oxidative stress environment. We next wondered if the observed regulation of synaptic release could translate into changes of larval activity. To test larval activity, we assessed crawling distances of the different genotypes over a period of 10 min (Robinson et al., 2014). Since motoneuronal transmission during crawling activity is predominately related to single motoneuronal action potential–induced synaptic release, which corresponds to a single eEJC event, we would not expect major effects on larval activity. Indeed, neither activation nor suppression of CncC signaling affected larval crawling distances relative to controls [RNAi Ctrl: 0.5 ± 0.03 cm, CncC-RNAi: 0.4 ± 0.02 cm, Keap1-RNAi: 0.5 ± 0.02 cm, CncC OE: 0.4 ± 0.02 cm, elav × w1118: 0.5 ± 0.02 cm, w1118 × CncC: 0.3 ± 0.02 cm, ANOVA, Figure 3M] but the data showed subtle differences between CncC-RNAi and Keap1-RNAi expressing larvae.
Increased oxidative stress levels are caused by altered activities of thiol redox circuits that can result in impaired cell signaling and dysfunctional redox-control (Finkel, 2011). It is linked to several pathological processes including dysfunction of proteostasis and the accumulation of misfolded proteins in the lumen of the endoplasmic reticulum (ER), resulting in ER stress (Braakman and Hebert, 2013). As heat shock is involved in triggering ER stress and ROS signaling, we next wanted to test whether altered expression of CncC and Keap1 would affect Drosophila longevity and synapse function following heat shock challenge. We found that under continual heat stress-challenged conditions, KD of CncC drastically reduced longevity [median life span (in hours): RNAi Ctrl: 4.5, CncC-RNAi: 3, p < 0.0001, Log-rank (Mantel-Cox) test] which can be explained by lack of neuroprotective Nrf2 signaling (Ahmed et al., 2017), while expectedly, Keap1 silencing protected and led to increased survival compared to CncC KD [median life span (in hours): Keap1-RNAi: 4, p < 0.0001, Log-rank (Mantel-Cox) test, Figure 4A]. The neuroprotection mediated by CncC (Nrf2) activation was further assessed in heat shock challenged larvae in which we characterized synaptic responses at the NMJ. The most striking changes at the level of synapse physiology occurred in CncC KD larvae 24 h after a single 1 h of heat shock challenge in which eEJC amplitudes declined drastically. This physiological response was not due to changes in quantal size but rather due to a reduced quantal content [eEJC amplitude: RNAi Ctrl: 112 ± 9 nA, CncC-RNAi: 74 ± 4 nA, Keap1-RNAi: 103 ± 9 nA, QC: RNAi Ctrl: 200 ± 15, CncC-RNAi: 186 ± 33, Keap1-RNAi: 142 ± 15, mEJC amplitude: RNAi Ctrl: 0.56 ± 0.04 nA, CncC-RNAi: 0.54 ± 0.09 nA, Keap1-RNAi: 0.66 ± 0.03 nA, Figures 4B,C,E]. Importantly, control larvae also showed a reduction in QC following challenge with heat shock, with Keap1 KD, however, preventing further neuronal deterioration upon this challenge. These changes were consolidated by measuring the vesicle pool size as cumulative release following synaptic stimulation in trains at 50 Hz [cumulative QC: RNAi Ctrl: 398 ± 64, CncC-RNAi: 423 ± 75, Keap1-RNAi: 272 ± 30, Figure 4D]. The data suggest that following heat shock stimulation, the lack of CncC produces strong phenotypes with regards to longevity and synapse function which were partially observed in controls but abolished following Keap1 KD.
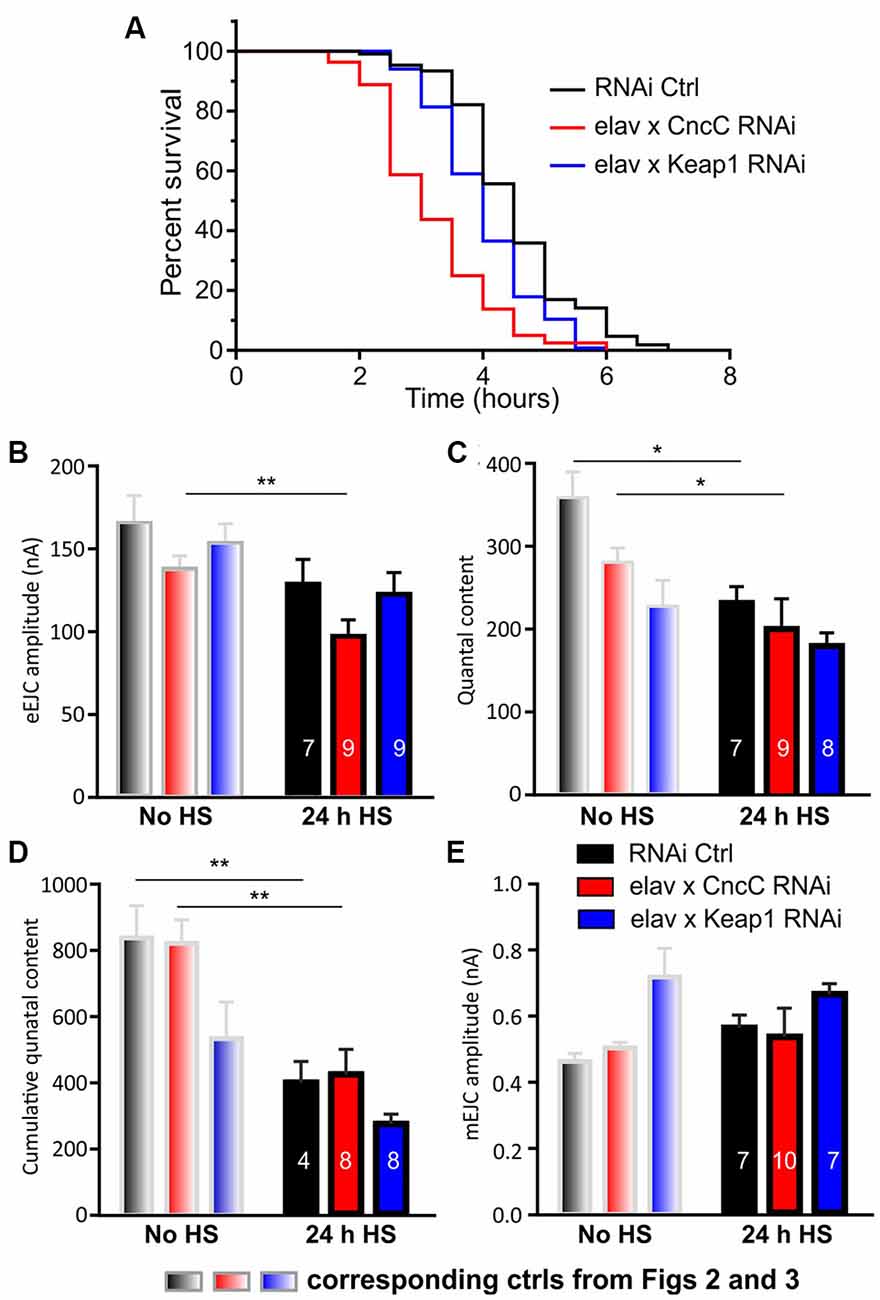
Figure 4. Silencing of Keap1 protects against stress-induced synaptic decline. Life spans were analyzed for indicated lines and survivorship was plotted over time. (A) Survival curves represent an average of three life-span trials (n = 80–106 flies). Data were compared using the Log-rank (Mantel-Cox) test, p < 0.0001). Synaptic function was analyzed showing eEJC amplitudes (B) QC (C), cumulative QC (D) and mEJC amplitudes (E) under control [no heat shock (no HS), gray] and heat shock challenged (24 h HS) conditions. Note that the bars in gray are repeats from Figures 2; 3 and comparisons were made for each genotype before and after HS using the unpaired Student’s t-test with *p < 0.05, **p < 0.01 [n—number of NMJs (from at least three larvae) indicated within bars].
Many neurological conditions including AD and PD exhibit perturbations of the circadian system (sometimes prior to any motor symptoms or clinical manifestation of symptoms), and underlying pathways have been studied in various animal models (Videnovic et al., 2014). Light and temperature are the two most reliable environmental timing cues, referred to as Zeitgeber (ZT), for the resetting of circadian clocks (Pittendrigh, 1960; Buhr et al., 2010; Musiek et al., 2013; Tamaru et al., 2013). Notably, mRNA expression levels for Keap1a/b and Nrf2 vary significantly within 12 h (i.e., between ZT0 and ZT12) implicating their involvement in circadian redox regulation (Zheng et al., 2017). This prompted us to evaluate how changes in ROS levels would impact the circadian behavior of flies with reduced or augmented cellular antioxidant capacity in a 24-h light-dark (LD) cycle. To determine how the circadian system is affected by modulating ROS levels via CncC/Keap1 signaling, we measured activity and sleep pattern as an index of circadian behavior in flies with reduced or augmented cellular antioxidant capacity.
Quantification of sleep episodes in the day and night should give the best overall picture of sleep behavior. We quantified the relative length and number of sleep episodes and found that in the light phase CncC OE reduces the length of sleep but enhances the number of sleep episodes, an effect that was reversed with silencing of CncC (Figures 5A,C). Unexpectedly, following Keap1 KD, sleep behavior was similar to CncC KD flies in this phase. In the scotophase (dark), the overexpression of CncC did not cause any change in sleep parameters which were similar to the controls (Figures 5B,D). Interestingly, the behavior of CncC KD flies in the dark phase was different from the one observed in the photophase (light), showing a decreased length of sleep episodes and an increase in the number of these episodes suggesting that driving redox levels in one direction has opposite effects on sleep depending on the time. Conversely, sleep behavior of Keap1 KD flies was more similar to that observed in the photophase (light), showing only a significant increase in the sleep length episodes (Figures 5C,D). Finally, we measured the total activity of adult flies and found that total activity was only reduced in CncC KD flies in comparison to controls and Keap1 KD (Figures 5E,F). Figure 5G separates the total activity profiles for the studied genotypes into day and night phases showing that CncC but also Keap1 KD reduced activity in the dark phase only. These data imply complex interactions between ROS signaling which causes differential effects over the circadian cycle.
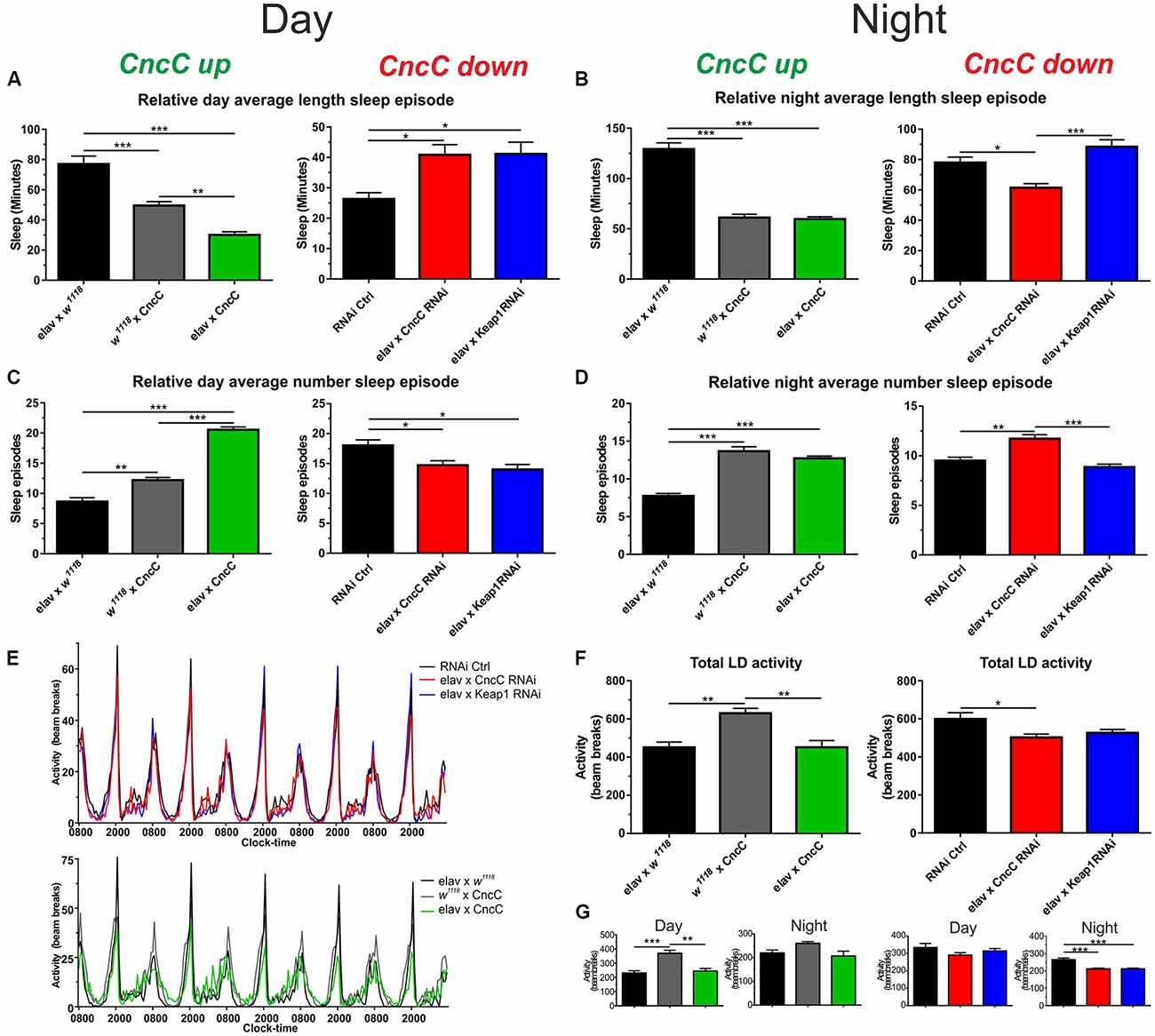
Figure 5. The CncC/Keap1 signaling impacts on sleep and activity patterns. (A,B) Sleep duration (in min) was reduced following CncC OE and enhanced in CncC knock-down (KD) flies in the day time, during the night, CncC KD reduced sleep length. The overexpression of CncC caused fragmented sleep patterns in the day resulting in more but shorter sleep episodes in the night time (B), whereas CncC KD reduces sleep length with more episodes in the night; (C,D) Keap1 silencing reverses the CncC KD effect in the night but not in the day. (D) During the night, both CncC OE and KD induce similar changes resulting in more but shorter sleep episodes. (E–G) Total and night/day activities of genotypes. One-way ANOVA with post hoc Tukey-Kramer was used for comparisons with *p < 0.05, **p < 0.01, ***p < 0.001 [n—numbers: elav × W, 3M (RNAi Ctrl: 38), elav × CncC RNAi: 21, elav × Keap1 RNAi: 44, elav × W1118: 32, W1118 × CncC: 27, elav × CncC: 25].
In summary, our data provide new evidence of how regulation of the redox homeostasis via modulation of CncC/Keap1 signaling can modulate aging, synapse function and sleep behavior. Specifically, the suppression of Keap1 expression induced beneficial effects on survival and synapse function. Equally, overexpression of CncC and pharmacologically enhancing antioxidant signaling resulted in similar phenotypes, with increases in quantal release being a major result of lowered oxidative stress signaling. The changes in synaptic function can further impact redox-sensitive aspects of sleep behavior which is implicated in disease-associated defects of circadian rhythm in neurodegeneration.
Discussion
Drosophila has been instrumental in studying synapse function but also modeling various neurodegenerative diseases, including polyglutamine expansion diseases, α-synuclein-linked PD, and other prionopathies and tauopathies (see review McGurk et al., 2015). We and others have previously shown that expression of huntingtin with polyglutamine expansions, mutant α-synuclein, Aβ40/42 toxicity and prion-mediated pathology suppresses glutamatergic function at the NMJ and causes neurodegenerative phenotypes (Outeiro et al., 2007; Romero et al., 2008; Chakraborty et al., 2011; Steinert et al., 2012; Breda et al., 2015; Vicente Miranda et al., 2016; Fernandez-Funez et al., 2017; Martin-Peña et al., 2017). Furthermore, studies in fly have found that dysfunctional superoxide dismutase 1 (SOD1) activity associated with enhanced oxidative stress can impact upon synapse function. In particular, SOD1 mutant flies exhibit signs of neurodegeneration, locomotor deficits, and shortened life span (Sahin et al., 2017). The Drosophila NMJ specifically offers a unique model synapse to study regulatory mechanisms of vesicular release. However, the direct effects of the redox signaling mediated by the Nrf2/Keap1 cascade have not yet been fully assessed at the level of synapse function and whole animal behavior. Our data present new evidence of how CncC and Keap1 signaling modulates Drosophila longevity, synapse function, and larval and adult fly activities, including effects on circadian sleep patterns. Furthermore, we determined the protective effects of CncC/Keap1 activity under stress-challenged conditions.
Many neurodegenerative diseases are characterized by a slowly progressive loss of neurons. The etiology of these diseases has still not yet been fully elucidated, although elevated levels of oxidative stress have been suggested as one of the potential common factors. One disease, in particular, is associated with defects in the antioxidant system, with mutations in SOD1 being a strong contributor to ALS. Drosophila has been utilized to characterize the effects of ALS-relevant mutant proteins (Milton et al., 2011; Coyne et al., 2017; Kim et al., 2018) and induction of homeostatic neuronal plasticity can reverse ALS-induced degeneration at the NMJ (Kim et al., 2018).
Data indicate that the abnormal excitotoxic glutamate release in the spinal cord of pre-symptomatic ALS mice is mainly based on the increased size of the readily releasable pool of vesicles and release facilitation, supported by plastic changes of specific presynaptic mechanisms (Bonifacino et al., 2016). As ALS is characterized by enhanced cytotoxic oxidative stress, one could speculate that these conditions favor non-physiological release of neurotransmitter. In light of our data which show a strong reduction in the number of evoked quantal release events with an increased quantal size following augmented antioxidant signaling (GSH, Keap1-RNAi, CncC OE), we suggest that low oxidative stress balances release with a reduction in the number of energy-demanding vesicular release events and simultaneously increased quantal size to sustain physiological action potential-evoked synaptic responses. If one considers that a single vesicular release event requires around 60,000 ATP molecules at a glutamatergic synapse in the mammalian central nervous system (CNS), including neurotransmitter refilling, SNARE protein assemble/dissemble, ion pump activities (Attwell and Laughlin, 2001; Harris et al., 2012) and vesicular release represents the highest energy burden to the presynapse (Rangaraju et al., 2014), it is conceivable to suggest that low oxidative stress levels lead to advantageous low QC/high quantal size release parameters.
A great variety of factors controls the level of neurotransmitter within the vesicle and changes in vesicle filling thus have great potential to influence synaptic transmission with vesicular glutamate filling determining quantal size (Karunanithi et al., 2002; Wu et al., 2007; Huang and Trussell, 2014; Choudhury et al., 2016). Increases in Drosophila larval quantal size at the NMJ have been reported following >30 min of enhanced activity (Steinert et al., 2006). Conversely, high-frequency stimulation of the NMJ results in a strong decrease in quantal size (Doherty et al., 1984; Naves and Van der Kloot, 2001) illustrating the ability of this particular synapse which is under strong homeostatic control (Newman et al., 2017), but also others (see review Edwards, 2007), to modulate quantal release. It has been shown that for instance, overexpression of vesicular glutamate transporter (vGLUT) leads to larger quantal sizes and resulting reduced QC (Daniels et al., 2004). Further mechanisms include uptake of transmitter via the Na+-dependent excitatory amino acid transporters (EAATs; Wang and Floor, 1998; Takayasu et al., 2005; Rose et al., 2018), glutamate recycling which includes the glutamine-glutamate cycle, and transmitter transport into the vesicle which involves the exchange of lumenal H+ for cytoplasmic transmitter and hence depends on a H+ electrochemical gradient, that is produced by the vesicular (H+)-vATPase (Cotter et al., 2015). In fact, it has been shown that DTT or reduced GSH reverse H2O2-induced inhibition of the vATPase, suggesting that the mechanism of its inhibition by H2O2 involves oxidation of a reactive cysteine sulfhydryl group in the ATP binding site. Inhibition of vATPase activity would decrease the amount of transmitter stored in synaptic vesicles and thus reduce the quantal size during episodes of oxidative stress (Wang and Floor, 1998).
EAAT transporters contain cysteine-associated sulfhydryl groups sensitive to free radical species. The actions of free radicals result in the formation of cysteine bridges, thereby inhibiting glutamate transport into the cells (Trotti et al., 1998), as demonstrated for superoxide anion, hydrogen peroxide, NO and peroxynitrite (Pogun et al., 1994; Volterra et al., 1994) or into vesicles by reducing vGLUT activities (Wang et al., 2017). Conversely, overexpression of SOD1 protected glutamate transporters from inhibition (Chen et al., 2000). Studies in SOD1 knock-out mice NMJs found reduced quantal size (equal to mEJC) following enhanced oxidative stress leading to weakening of the muscle (Ivannikov and Van Remmen, 2015). Conversely, by reducing oxidative stress in a mouse model of peripheral nerve injury, the authors found increases in expression of vGLUT (Cirillo et al., 2015), the predominate vGLUT responsible for vesicular glutamate filling at the Drosophila NMJ (Wu and Cooper, 2012) and mammalian CNS synapses (Fremeau et al., 2004). Previous studies confirmed that activation of the Nrf2 pathway led to upregulation of the neuronal EAAT3 in mice (Escartin et al., 2011). This transporter has a homologue in Drosophila (Besson et al., 2000) and its upregulation enhances antioxidant activity via increases in glutathione production in addition to allowing sustained presynaptic glutamate levels available for release.
Conceivably, modulation of redox levels might impact on any of the above mechanisms and regulate transmitter release in a negative or positive direction and our data describe how reducing redox stress, either genetically or pharmacologically, leads to enhanced quantal size. However, this increase in quantal size resulted in a reduction of QC, likely due to homeostatic feedback regulation of this highly plastic synapse (Frank, 2014; Li et al., 2018) and future studies will have to evaluate the specific mechanisms by which redox signaling can alter vesicular transmitter release.
The observed changes of neuronal function will have wide implications on animal behavior and together with reported redox-mediated regulation of circadian function, determined by Nrf2/CncC-Keap1 signaling, our data show further evidence how this cascade can influence circadian rhythm. Neurodegeneration causes abnormalities in sleep patterns, partially due to neuronal loss but likely also due to specific dysregulation of circadian circuits. We demonstrated that increases in CncC expression or its silencing results in an opposite alteration in sleep episode length in the day. However, both conditions resulted in similar effects on sleep length during the night time. Total 24-h activity following CncC overexpression was not altered, whereas CncC KD reduced overall activity. These observations are further complicated with the expression of Nrf2 falling under the transcriptional regulation of the Clock/Bmal1-complex (Xu et al., 2012; Pekovic-Vaughan et al., 2014). Clock/Bmal1-dependent Nrf2 regulation gives rise to diurnal patterns in Nrf2 signaling, which underlies the rhythmic expression of antioxidant and metabolic enzymes reported in different cellular systems (Xu et al., 2012; Wang et al., 2018; Ishii et al., 2019). In this context it has been reported that Nrf2 gain- and loss-of-function affect circadian gene expression and rhythmicity in mammalian cellular systems, indicating the coupling of Nrf2 and Clock and the role of Nrf2 to integrate cellular redox status into timekeeping (Wible et al., 2018).
Collectively, this study and previous work illustrate a key mechanistic link between circadian oscillations in redox balance and Clock gene expression rhythms. However, key questions on the complex bidirectional regulation of ROS by circadian activity and vice versa remain to be answered in future studies.
Author Contributions
JRS and JGS designed the study and wrote the manuscript. JRS, JGS, SR and CB conducted and analyzed the data. All authors contributed to, read and approved the final manuscript.
Funding
This research was supported by the Medical Research Council (MRC) UK (JRS, JGS, SR) and MRC project grant MR/N00373X/1 (FG, CB).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahmed, S. M., Luo, L., Namani, A., Wang, X. J., and Tang, X. (2017). Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 585–597. doi: 10.1016/j.bbadis.2016.11.005
Attwell, D., and Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145. doi: 10.1097/00004647-200110000-00001
Barone, M. C., Sykiotis, G. P., and Bohmann, D. (2011). Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Model Mech. 4, 701–707. doi: 10.1242/dmm.007575
Besson, M. T., Soustelle, L., and Birman, S. (2000). Selective high-affinity transport of aspartate by a Drosophila homologue of the excitatory amino-acid transporters. Curr. Biol. 10, 207–210. doi: 10.1016/s0960-9822(00)00339-0
Bonifacino, T., Musazzi, L., Milanese, M., Seguini, M., Marte, A., Gallia, E., et al. (2016). Altered mechanisms underlying the abnormal glutamate release in amyotrophic lateral sclerosis at a pre-symptomatic stage of the disease. Neurobiol. Dis. 95, 122–133. doi: 10.1016/j.nbd.2016.07.011
Braakman, I., and Hebert, D. N. (2013). Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 5:a013201. doi: 10.1101/cshperspect.a013201
Breda, C., Nugent, M. L., Estranero, J. G., Kyriacou, C. P., Outeiro, T. F., Steinert, J. R., et al. (2015). Rab11 modulates alpha-synuclein-mediated defects in synaptic transmission and behaviour. Hum. Mol. Genet. 24, 1077–1091. doi: 10.1093/hmg/ddu521
Buhr, E. D., Yoo, S. H., and Takahashi, J. S. (2010). Temperature as a universal resetting cue for mammalian circadian oscillators. Science 330, 379–385. doi: 10.1126/science.1195262
Chakraborty, R., Vepuri, V., Mhatre, S. D., Paddock, B. E., Miller, S., Michelson, S. J., et al. (2011). Characterization of a Drosophila Alzheimer’s disease model: pharmacological rescue of cognitive defects. PLoS One 6:e20799. doi: 10.1371/journal.pone.0020799
Chen, Y., Ying, W., Simma, V., Copin, J. C., Chan, P. H., and Swanson, R. A. (2000). Overexpression of Cu,Zn superoxide dismutase attenuates oxidative inhibition of astrocyte glutamate uptake. J. Neurochem. 75, 939–945. doi: 10.1046/j.1471-4159.2000.0750939.x
Choudhury, S. D., Mushtaq, Z., Reddy-Alla, S., Balakrishnan, S. S., Thakur, R. S., Krishnan, K. S., et al. (2016). σ2-adaptin facilitates basal synaptic transmission and is required for regenerating endo-exo cycling pool under high-frequency nerve stimulation in Drosophila. Genetics 203, 369–385. doi: 10.1534/genetics.115.183863
Cirillo, G., Colangelo, A. M., Berbenni, M., Ippolito, V. M., De Luca, C., Verdesca, F., et al. (2015). Purinergic modulation of spinal neuroglial maladaptive plasticity following peripheral nerve injury. Mol. Neurobiol. 52, 1440–1457. doi: 10.1007/s12035-014-8943-y
Cotter, K., Stransky, L., McGuire, C., and Forgac, M. (2015). Recent insights into the structure, regulation and function of the V-ATPases. Trends Biochem. Sci. 40, 611–622. doi: 10.1016/j.tibs.2015.08.005
Coyne, A. N., Lorenzini, I., Chou, C. C., Torvund, M., Rogers, R. S., Starr, A., et al. (2017). Post-transcriptional inhibition of Hsc70–4/HSPA8 expression leads to synaptic vesicle cycling defects in multiple models of ALS. Cell Rep. 21, 110–125. doi: 10.1016/j.celrep.2017.09.028
Daniels, R. W., Collins, C. A., Gelfand, M. V., Dant, J., Brooks, E. S., Krantz, D. E., et al. (2004). Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J. Neurosci. 24, 10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004
De Lazzari, F., Bisaglia, M., Zordan, M. A., and Sandrelli, F. (2018). Circadian rhythm abnormalities in parkinson’s disease from humans to flies and back. Int. J. Mol. Sci. 19:E3911. doi: 10.3390/ijms19123911
Desvergne, A., Ugarte, N., Petropoulos, I., and Friguet, B. (2014). Circadian modulation of proteasome activities and removal of carbonylated proteins. Free Radic. Biol. Med. 75:S18. doi: 10.1016/j.freeradbiomed.2014.10.631
Doherty, P., Hawgood, B. J., and Smith, I. C. (1984). Changes in miniature end-plate potentials after brief nervous stimulation at the frog neuromuscular junction. J. Physiol. 356, 349–358. doi: 10.1113/jphysiol.1984.sp015469
Early, J. O., Menon, D., Wyse, C. A., Cervantes-Silva, M. P., Zaslona, Z., Carroll, R. G., et al. (2018). Circadian clock protein BMAL1 regulates IL-1beta in macrophages via NRF2. Proc. Natl. Acad. Sci. U S A 115, E8460–E8468. doi: 10.1073/pnas.1800431115
Edwards, R. H. (2007). The neurotransmitter cycle and quantal size. Neuron 55, 835–858. doi: 10.1016/j.neuron.2007.09.001
Escartin, C., Won, S. J., Malgorn, C., Auregan, G., Berman, A. E., Chen, P. C., et al. (2011). Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J. Neurosci. 31, 7392–7401. doi: 10.1523/JNEUROSCI.6577-10.2011
Fernandez-Funez, P., Sanchez-Garcia, J., and Rincon-Limas, D. E. (2017). Drosophila models of prionopathies: insight into prion protein function, transmission and neurotoxicity. Curr. Opin. Genet. Dev. 44, 141–148. doi: 10.1016/j.gde.2017.03.013
Finkel, T. (2011). Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15. doi: 10.1083/jcb.201102095
Frank, C. A. (2014). Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology 78, 63–74. doi: 10.1016/j.neuropharm.2013.06.015
Fremeau, R. T. Jr., Voglmaier, S., Seal, R. P., and Edwards, R. H. (2004). VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 27, 98–103. doi: 10.1016/j.tins.2003.11.005
Gilestro, G. F., and Cirelli, C. (2009). pySolo: a complete suite for sleep analysis in Drosophila. Bioinformatics 25, 1466–1467. doi: 10.1093/bioinformatics/btp237
Habas, A., Hahn, J., Wang, X., and Margeta, M. (2013). Neuronal activity regulates astrocytic Nrf2 signaling. Proc. Natl. Acad. Sci. U S A 110, 18291–18296. doi: 10.1073/pnas.1208764110
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
He, M., Pan, H., Chang, R. C., So, K. F., Brecha, N. C., and Pu, M. (2014). Activation of the Nrf2/HO-1 antioxidant pathway contributes to the protective effects of Lycium barbarum polysaccharides in the rodent retina after ischemia-reperfusion-induced damage. PLoS One 9:e84800. doi: 10.1371/journal.pone.0084800
Huang, H., and Trussell, L. O. (2014). Presynaptic HCN channels regulate vesicular glutamate transport. Neuron 84, 340–346. doi: 10.1016/j.neuron.2014.08.046
Ishida, Y., Sekine, Y., Oguchi, H., Chihara, T., Miura, M., Ichijo, H., et al. (2012). Prevention of apoptosis by mitochondrial phosphatase PGAM5 in the mushroom body is crucial for heat shock resistance in Drosophila melanogaster. PLoS One 7:e30265. doi: 10.1371/journal.pone.0030265
Ishii, T., Warabi, E., and Mann, G. E. (2019). Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic. Biol. Med. 133, 169–178. doi: 10.1016/j.freeradbiomed.2018.09.002
Itoh, K., Wakabayashi, N., Katoh, Y., Ishii, T., Igarashi, K., Engel, J. D., et al. (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86. doi: 10.1101/gad.13.1.76
Ivannikov, M. V., and Van Remmen, H. (2015). Sod1 gene ablation in adult mice leads to physiological changes at the neuromuscular junction similar to changes that occur in old wild-type mice. Free Radic. Biol. Med. 84, 254–262. doi: 10.1016/j.freeradbiomed.2015.03.021
Johnson, D. A., and Johnson, J. A. (2015). Nrf2–a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 88, 253–267. doi: 10.1016/j.freeradbiomed.2015.07.147
Karunanithi, S., Marin, L., Wong, K., and Atwood, H. L. (2002). Quantal size and variation determined by vesicle size in normal and mutant Drosophila glutamatergic synapses. J. Neurosci. 22, 10267–10276. doi: 10.1523/JNEUROSCI.22-23-10267.2002
Kim, S. H., Stiles, S. G., Feichtmeier, J. M., Ramesh, N., Zhan, L., Scalf, M. A., et al. (2018). Mutation-dependent aggregation and toxicity in a Drosophila model for UBQLN2-associated ALS. Hum. Mol. Genet. 27, 322–337. doi: 10.1093/hmg/ddx403
Li, X., Goel, P., Wondolowski, J., Paluch, J., and Dickman, D. (2018). A glutamate homeostat controls the presynaptic inhibition of neurotransmitter release. Cell Rep. 23, 1716–1727. doi: 10.1016/j.celrep.2018.03.130
Liddell, J. R. (2017). Are astrocytes the predominant cell type for activation of Nrf2 in aging and neurodegeneration? Antioxidants 6:E65. doi: 10.3390/antiox6030065
Lipton, S. A., Rezaie, T., Nutter, A., Lopez, K. M., Parker, J., Kosaka, K., et al. (2016). Therapeutic advantage of pro-electrophilic drugs to activate the Nrf2/ARE pathway in Alzheimer’s disease models. Cell Death Dis. 7:e2499. doi: 10.1038/cddis.2016.389
Martin-Peña, A., Rincon-Limas, D. E., and Fernandez-Funez, P. (2017). Anti-Aβ single-chain variable fragment antibodies restore memory acquisition in a Drosophila model of Alzheimer’s disease. Sci. Rep. 7:11268. doi: 10.1038/s41598-017-11594-2
McGurk, L., Berson, A., and Bonini, N. M. (2015). Drosophila as an in vivo model for human neurodegenerative disease. Genetics 201, 377–402. doi: 10.1534/genetics.115.179457
Milton, V. J., Jarrett, H. E., Gowers, K., Chalak, S., Briggs, L., Robinson, I. M., et al. (2011). Oxidative stress induces overgrowth of the Drosophila neuromuscular junction. Proc. Natl. Acad. Sci. U S A 108, 17521–17526. doi: 10.1073/pnas.1014511108
Müller, M., Liu, K. S., Sigrist, S. J., and Davis, G. W. (2012). RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 32, 16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012
Musiek, E. S., Lim, M. M., Yang, G., Bauer, A. Q., Qi, L., Lee, Y., et al. (2013). Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 123, 5389–5400. doi: 10.1172/JCI70317
Naves, L. A., and Van der Kloot, W. (2001). Repetitive nerve stimulation decreases the acetylcholine content of quanta at the frog neuromuscular junction. J. Physiol. 532, 637–647. doi: 10.1111/j.1469-7793.2001.0637e.x
Newman, Z. L., Hoagland, A., Aghi, K., Worden, K., Levy, S. L., Son, J. H., et al. (2017). Input-specific plasticity and homeostasis at the Drosophila larval neuromuscular junction. Neuron 93, 1388–1404.e10. doi: 10.1016/j.neuron.2017.02.028
Nichols, C. D., Becnel, J., and Pandey, U. B. (2012). Methods to assay Drosophila behavior. J. Vis. Exp. 61:3795. doi: 10.3791/3795
Outeiro, T. F., Kontopoulos, E., Altmann, S. M., Kufareva, I., Strathearn, K. E., Amore, A. M., et al. (2007). Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519. doi: 10.1126/science.1143780
Pekovic-Vaughan, V., Gibbs, J., Yoshitane, H., Yang, N., Pathiranage, D., Guo, B., et al. (2014). The circadian clock regulates rhythmic activation of the NRF2/glutathione-mediated antioxidant defense pathway to modulate pulmonary fibrosis. Genes Dev. 28, 548–560. doi: 10.1101/gad.237081.113
Pitoniak, A., and Bohmann, D. (2015). Mechanisms and functions of Nrf2 signaling in Drosophila. Free Radic. Biol. Med. 88, 302–313. doi: 10.1016/j.freeradbiomed.2015.06.020
Pittendrigh, C. S. (1960). Circadian rhythms and the circadian organization of living systems. Cold Spring Harb. Symp. Quant. Biol. 25, 159–184. doi: 10.1101/sqb.1960.025.01.015
Pogun, S., Dawson, V., and Kuhar, M. J. (1994). Nitric oxide inhibits 3H-glutamate transport in synaptosomes. Synapse 18, 21–26. doi: 10.1002/syn.890180104
Postlethwaite, M., Hennig, M. H., Steinert, J. R., Graham, B. P., and Forsythe, I. D. (2007). Acceleration of AMPA receptor kinetics underlies temperature-dependent changes in synaptic strength at the rat calyx of held. J. Physiol. 579, 69–84. doi: 10.1113/jphysiol.2006.123612
Radyuk, S. N., Sohal, R. S., and Orr, W. C. (2003). Thioredoxin peroxidases can foster cytoprotection or cell death in response to different stressors: over- and under-expression of thioredoxin peroxidase in Drosophila cells. Biochem. J. 371, 743–752. doi: 10.1042/bj20021522
Ramsey, C. P., Glass, C. A., Montgomery, M. B., Lindl, K. A., Ritson, G. P., Chia, L. A., et al. (2007). Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 66, 75–85. doi: 10.1097/nen.0b013e31802d6da9
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Ranieri, D., Avitabile, D., Shiota, M., Yokomizo, A., Naito, S., Bizzarri, M., et al. (2015). Nuclear redox imbalance affects circadian oscillation in HaCaT keratinocytes. Int. J. Biochem. Cell Biol. 65, 113–124. doi: 10.1016/j.biocel.2015.05.018
Rhodenizer, D., Martin, I., Bhandari, P., Pletcher, S. D., and Grotewiel, M. (2008). Genetic and environmental factors impact age-related impairment of negative geotaxis in Drosophila by altering age-dependent climbing speed. Exp. Gerontol. 43, 739–748. doi: 10.1016/j.exger.2008.04.011
Robinson, S. W., Bourgognon, J. M., Spiers, J. G., Breda, C., Campesan, S., Butcher, A., et al. (2018). Nitric oxide-mediated posttranslational modifications control neurotransmitter release by modulating complexin farnesylation and enhancing its clamping ability. PLoS Biol. 16:e2003611. doi: 10.1371/journal.pbio.2003611
Robinson, S. W., Gutierrez Olmo, M., Martin, M. G., Smith, T. M., Moreno, N., and Steinert, J. R. (2017). Endogenous nitric oxide synthase activity regulates synaptic transmitter release. Opera Med. Physiol. 2, 31–38. doi: 10.20388/omp2017.002.0046
Robinson, S. W., Nugent, M. L., Dinsdale, D., and Steinert, J. R. (2014). Prion protein facilitates synaptic vesicle release by enhancing release probability. Hum. Mol. Genet. 23, 4581–4596. doi: 10.1093/hmg/ddu171
Romero, E., Cha, G. H., Verstreken, P., Ly, C. V., Hughes, R. E., Bellen, H. J., et al. (2008). Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 57, 27–40. doi: 10.1016/j.neuron.2007.11.025
Rose, C. R., Ziemens, D., Untiet, V., and Fahlke, C. (2018). Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res. Bull. 136, 3–16. doi: 10.1016/j.brainresbull.2016.12.013
Sahin, A., Held, A., Bredvik, K., Major, P., Achilli, T. M., Kerson, A. G., et al. (2017). Human SOD1 ALS mutations in a Drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics 205, 707–723. doi: 10.1534/genetics.116.190850
Sawicki, R., Singh, S. P., Mondal, A. K., Benes, H., and Zimniak, P. (2003). Cloning, expression and biochemical characterization of one Epsilon-class (GST-3) and ten delta-class (GST-1) glutathione S-transferases from Drosophila melanogaster and identification of additional nine members of the Epsilon class. Biochem. J. 370, 661–669. doi: 10.1042/bj20021287
Schneggenburger, R., Meyer, A. C., and Neher, E. (1999). Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23, 399–409. doi: 10.1016/s0896-6273(00)80789-8
Steinert, J. R., Campesan, S., Richards, P., Kyriacou, C. P., Forsythe, I. D., and Giorgini, F. (2012). Rab11 rescues synaptic dysfunction and behavioural deficits in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 21, 2912–2922. doi: 10.1093/hmg/dds117
Steinert, J. R., Kuromi, H., Hellwig, A., Knirr, M., Wyatt, A. W., Kidokoro, Y., et al. (2006). Experience-dependent formation and recruitment of large vesicles from reserve pool. Neuron 50, 723–733. doi: 10.1016/j.neuron.2006.04.025
Stewart, B. A., Atwood, H. L., Renger, J. J., Wang, J., and Wu, C. F. (1994). Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. A 175, 179–191. doi: 10.1007/bf00215114
Sykiotis, G. P., and Bohmann, D. (2008). Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev. Cell 14, 76–85. doi: 10.1016/j.devcel.2007.12.002
Takayasu, Y., Iino, M., Kakegawa, W., Maeno, H., Watase, K., Wada, K., et al. (2005). Differential roles of glial and neuronal glutamate transporters in Purkinje cell synapses. J. Neurosci. 25, 8788–8793. doi: 10.1523/JNEUROSCI.1020-05.2005
Tamaru, T., Hattori, M., Ninomiya, Y., Kawamura, G., Varès, G., Honda, K., et al. (2013). ROS stress resets circadian clocks to coordinate pro-survival signals. PLoS One 8:e82006. doi: 10.1371/journal.pone.0082006
Trotti, D., Danbolt, N. C., and Volterra, A. (1998). Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 19, 328–334. doi: 10.1016/s0165-6147(98)01230-9
Vanderheyden, W. M., Lim, M. M., Musiek, E. S., and Gerstner, J. R. (2018). Alzheimer’s disease and sleep-wake disturbances: amyloid, astrocytes and animal models. J. Neurosci. 38, 2901–2910. doi: 10.1523/JNEUROSCI.1135-17.2017
Vicente Miranda, H., Gomes, M. A., Branco-Santos, J., Breda, C., Lázaro, D. F., Lopes, L. V., et al. (2016). Glycation potentiates neurodegeneration in models of Huntington’s disease. Sci. Rep. 6:36798. doi: 10.1038/srep36798
Videnovic, A., Lazar, A. S., Barker, R. A., and Overeem, S. (2014). ‘The clocks that time us’–circadian rhythms in neurodegenerative disorders. Nat. Rev. Neurol. 10, 683–693. doi: 10.1038/nrneurol.2014.206
Volterra, A., Trotti, D., Tromba, C., Floridi, S., and Racagni, G. (1994). Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci. 14, 2924–2932. doi: 10.1523/jneurosci.14-05-02924.1994
Wang, W. P., and Chan, J. Y. (2006). Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain-inhibition of nuclear translocation and transacting function. J. Biol. Chem. 281, 19676–19687. doi: 10.1074/jbc.m602802200
Wang, Y., and Floor, E. (1998). Hydrogen peroxide inhibits the vacuolar H+-ATPase in brain synaptic vesicles at micromolar concentrations. J. Neurochem. 70, 646–652. doi: 10.1046/j.1471-4159.1998.70020646.x
Wang, Y., Lv, D., Liu, W., Li, S., Chen, J., Shen, Y., et al. (2018). Disruption of the circadian clock alters antioxidative defense via the SIRT1-BMAL1 pathway in 6-OHDA-induced models of Parkinson’s disease. Oxid. Med. Cell. Longev. 2018:4854732. doi: 10.1155/2018/4854732
Wang, Y., Zhou, Z., Tan, H., Zhu, S., Wang, Y., Sun, Y., et al. (2017). Nitrosylation of vesicular transporters in brain of amyloid precursor protein/presenilin 1 double transgenic mice. J. Alzheimers. Dis. 55, 1683–1692. doi: 10.3233/JAD-160700
Wible, R. S., Ramanathan, C., Sutter, C. H., Olesen, K. M., Kensler, T. W., Liu, A. C., et al. (2018). NRF2 regulates core and stabilizing circadian clock loops, coupling redox and timekeeping in Mus musculus. Elife 7:e31656. doi: 10.7554/eLife.31656
Wu, W. H., and Cooper, R. L. (2012). The regulation and packaging of synaptic vesicles as related to recruitment within glutamatergic synapses. Neuroscience 225, 185–198. doi: 10.1016/j.neuroscience.2012.08.037
Wu, X. S., Xue, L., Mohan, R., Paradiso, K., Gillis, K. D., and Wu, L. G. (2007). The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J. Neurosci. 27, 3046–3056. doi: 10.1523/JNEUROSCI.4415-06.2007
Xu, Y. Q., Zhang, D., Jin, T., Cai, D. J., Wu, Q., Lu, Y., et al. (2012). Diurnal variation of hepatic antioxidant gene expression in mice. PLoS One 7:e44237. doi: 10.1371/journal.pone.0044237
Keywords: redox signaling, Nrf2, Keap1, synaptic release, longevity, sleep, Drosophila neuromuscular junction
Citation: Spiers JG, Breda C, Robinson S, Giorgini F and Steinert JR (2019) Drosophila Nrf2/Keap1 Mediated Redox Signaling Supports Synaptic Function and Longevity and Impacts on Circadian Activity. Front. Mol. Neurosci. 12:86. doi: 10.3389/fnmol.2019.00086
Received: 30 January 2019; Accepted: 20 March 2019;
Published: 16 April 2019.
Edited by:
Giuseppe Filomeni, Danish Cancer Society, DenmarkReviewed by:
Sara Baldelli, Università telematica San Raffaele, ItalyKatia Aquilano, University of Rome Tor Vergata, Italy
Copyright © 2019 Spiers, Breda, Robinson, Giorgini and Steinert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joern R. Steinert, anMzMzNAbGUuYWMudWs=
†Present address: Jereme G. Spiers, Department of Biochemistry and Genetics, La Trobe Institute for Molecular Science, La Trobe University, Bundoora, VIC, Australia Joern R. Steinert, Department of Neuroscience, Psychology and Behaviour, University of Leicester, Leicester, United Kingdom