 Zhen Tian
Zhen Tian Bin Feng2†
Bin Feng2† Xing-Qin Wang
Xing-Qin Wang- 1College of Pharmaceutical Sciences, Southwest University, Chongqing, China
- 2State Key Laboratory of Military Stomatology, National Clinical Research Center for Oral Diseases, Shaanxi International Joint Research Center for Oral Diseases, Department of Pharmacy, School of Stomatology, Fourth Military Medical University, Xi’an, China
- 3Department of Neurosurgery, Nanfang Hospital, Southern Medical University, Guangzhou, China
- 4Department of Infection, Children’s Hospital of Chongqing Medical University, National Clinical Research Center for Child Health and Disorders, Ministry of Education Key Laboratory of Child Development and Disorders, Chongqing Key Laboratory of Child Infection and Immunity, The First Batch of Key Disciplines On Public Health in Chongqing, Chongqing, China
Cyclin-dependent kinases 5 (Cdk5) is a special member of proline-directed serine threonine kinase family. Unlike other Cdks, Cdk5 is not directly involved in cell cycle regulation but plays important roles in nervous system functions. Under physiological conditions, the activity of Cdk5 is tightly controlled by p35 or p39, which are specific activators of Cdk5 and highly expressed in post-mitotic neurons. However, they will be cleaved into the corresponding truncated forms namely p25 and p29 under pathological conditions, such as neurodegenerative diseases and neurotoxic insults. The binding to truncated co-activators results in aberrant Cdk5 activity and contributes to the initiation and progression of multiple neurological disorders through affecting the down-stream targets. Although Cdk5 kinase activity is mainly regulated through combining with co-activators, it is not the only way. Post-translational modifications of Cdk5 including phosphorylation, S-nitrosylation, sumoylation, and acetylation can also affect its kinase activity and then participate in physiological and pathological processes of nervous system. In this review, we focus on the regulatory mechanisms of Cdk5 and its roles in a series of common neurological disorders such as neurodegenerative diseases, stroke, anxiety/depression, pathological pain and epilepsy.
Introduction
Cyclin-dependent kinases (Cdks) are a group of small proline-directed serine/threonine kinases mainly involved in the modulation of cell cycle, gene transcription and cell differentiation (Malumbres, 2014; Xie et al., 2022). Cyclin dependent kinase 1 (Cdk1) was the first discovered Cdk, which was initially named as cell division cycle 2 (Cdc2) because of its high homology with a yeast fission kinase of the same name (Lee and Nurse, 1987). Until now, twenty-one genes encoding Cdks and five genes encoding Cdk-like kinases have been found in human genome (Malumbres et al., 2009). Generally, Cdks are activated through associating with specific cyclin regulatory subunits and regulated through phosphorylation of specific T-loops by Cdk-activating kinase (Malumbres et al., 2009). Among the family of Cdks, Cdk5 is a special one though it shares significant sequence homology with other members (Hellmich et al., 1992). It can neither be activated by cyclins nor involved in cell division directly. Cdk5 is ubiquitous in kinds of tissues, but its activity is extremely high in post-mitotic neurons because its activators p35 and p39 are selectively expressed in these cells (Tsai et al., 1994; Ko et al., 2001). P35 and p39 are different from cyclins as they lack the typically conserved amino acid sequences. The expression of Cdk5 and p35/p39 transcripts overlaps spatially and temporally in the developing brain, and upon directly binding, they form active complexes and induces the alterations of downstream signal pathways.
In the developing nervous system, Cdk5 seems to be expressed mainly in terminally differentiated neurons but not proliferating cells. Because the expression and kinase activity of Cdk5 was shown to increase progressively from embryonic day 15 (E15) and reaches the peak at E17 in developing mouse forebrain, which period is a critical time widow for neuronal differentiation (Tsai et al., 1993). During the embryonic brain development of rats, the expression pattern of Cdk5 is similar to that of mouse, with a gradual increase from E12 to E18 and reaches a peak at postnatal day 7 (P7), which is then maintained at this level through adulthood and aged phase (up to 18 months old) (Wu et al., 2000). In the adult central nervous system, Cdk5 is distributed throughout the brain and spinal cord, especially in the hippocampus, cerebellum, cerebral cortex, olfactory bulb, amygdala and brain stem. Generally, the level of Cdk5 is higher in the large-sized and medium-sized neurons such as hippocampal and cortical pyramidal cells, cerebellar Purkinje cells, motoneurons in spinal cord (Ino et al., 1994). The expression level of Cdk5 in various brain regions was also compared and quantified. The results showed that there was no obvious difference about Cdk5 level in the cerebral cortex, hippocampus, cerebellum and striatum of rats aged from postnatal day 14 (P14) to 18 months old (18 M) (Wu et al., 2000).
As for p35, the co-activator of Cdk5, its expression becomes detectable in E16 rat brains and gradually increases to its peak level at P7–P14, then declines in the adult and aged rat brains. The Cdk5 kinase activity is low at E12 and it also progressively reaches a peak between P7 and P14, and then declines from 1 to 3 M, a timetable correlating well with the expression pattern of p35 (Wu et al., 2000). Different from the expression pattern of Cdk5 protein, p35 expression and Cdk5 kinase activity were persistently higher in the hippocampus and cerebral cortex than cerebellum or striatum in rats from P14 to 18 M, indicating that Cdk5/p35 complex may have region-specific functions (Wu et al., 2000). P39 is the isoform of p35 with over 50% homology in genomic sequence, the functions of p39 largely overlap with those of p35. However, their spatial and temporal expression patterns are different, with p35 being highly expressed from embryonic to postnatal stage while p39 is scarcely expressed until postnatal (Tsai et al., 1994; Ko et al., 2001). In addition, the expression of p35 is mostly prominent in cerebral cortex while p39 is primarily expressed in brain stem, cerebellum and spinal cord (Ko et al., 2001). Furthermore, the binding affinity of p39 with Cdk5 is lower whereas the Cdk5-p39 complex is more stable compared with Cdk5-p35 (Yamada et al., 2007; Minegishi et al., 2010).
Cyclin-dependent kinases 5 complex plays a very important role in neuronal functions from embryogenesis to postnatal brain modulation. During embryogenesis, Cdk5 is essential for normal brain development and conditional deletion of Cdk5 is lethal (Ohshima et al., 1996; Shah and Lahiri, 2014). It is because that Cdk5 governs multiple steps of cortical neuronal migration, including the formation of multipolar processes (Kawauchi et al., 2006), gain of neuronal polarity (Ohshima et al., 2007) and the locomotion mode (Nishimura et al., 2010). Cdk5 deficiency will lead to the lack of cerebral cortical laminar structure and cerebellar foliation in mice (Ohshima et al., 1996). Mice lacking p35 show less severe corticogenesis defect and only suffer from sporadic adult lethality, which may be attributed to functional compensation by another Cdk5 activator, p39 (Chae et al., 1997). Although loss of 39 is not lethal, it attenuates overall Cdk5 activity in neurons as well as leads to aberrant axonal growth and impaired dendritic spine formation through affecting Cdk5 targets governing neuronal differentiation and network formation (Li et al., 2016). In adult brain, emerging evidences have indicated that Cdk5 complex regulated multiple neuronal functions including neuronal survival, neurite and axon outgrowth, synaptic plasticity, neurotransmission (Ye et al., 2014; Sasamoto et al., 2017; Takahashi et al., 2022). Because of its extensive and crucial roles in nervous system, the dysfunction of Cdk5 is critically involved in numerous neurological disorders such as neurodegenerative diseases, stroke, psychiatric disorders, pathological pain, epilepsy and so on. The multifaceted roles of Cdk5 make it to be an attractive target for therapeutic intervention. An updated review on the relationship between Cdk5 and nervous system dysfunction can help us better understand its potential clinical value. Therefore, we comprehensively summarized the role of Cdk5 in a series of common neurological disorders in this paper. The synthesis of these scientific evidences about Cdk5 could facilitate future research to further explore the potentiality of drugs targeting Cdk5 for clinical therapy.
Regulation of cyclin-dependent kinases 5 kinase activity
The aberrant Cdk5 activity is critically involved in the pathogenesis of neurological disorders, so the regulation of kinase activity may be a potential strategy for disease treatment. Cdk5 kinase activity is mainly regulated by the available protein amounts of p35/p39 and their truncated fragments. p35 and p39 levels are determined by the balance between synthesis and degradation, which are cleavage by the ubiquitin-proteasome system with a half-life of about 30 and 120 min for p35 and p39, respectively (Minegishi et al., 2010). Calpain, a Ca2+ activated protease, is involved in multiple physiological processes and plays an important role in maintaining normal neuronal function. The activation of calpain depends on Ca2+ concentration, which makes it vulnerable to changes of Ca2+ homeostasis (Metwally et al., 2021). Following neurotoxic insults, sustained Ca2+ elevation leads to the over-activation of calpain, which in turn cleaved p35 and p39 into N-terminal p10 and C-terminal p25 or p29 truncated fragments (Lee et al., 2000). However, the half-life of truncated protein fragment is much longer than that of mother molecule and it can form a more stable complex with Cdk5, resulting in the prolonged activation of Cdk5 (Patrick et al., 1999; Minegishi et al., 2010). In addition, the N-terminal p10 region of p39 and p35 contains a second Gly for myristoylation and Lys clusters, the localization motifs helping to anchor the active complex at membrane. However, the truncated forms derived from cleavage of p35 or p39 lost the myristoylation signal that normally maintains Cdk5 at the membrane, leading to accumulation of the complex in nuclear and perinuclear regions (Asada et al., 2008). This aberrant subcellular localization of Cdk5/co-activator complex disturbs its substrate specificity and makes it target not only physiological substrates but also non-physiological substrates, resulting in neurotoxicity and cell death (Mushtaq et al., 2016). Thus, the hyperactivation and mislocalization of Cdk5 caused by p25 accumulation lead to the dysfunction of Cdk5, which contributes to the pathogenesis of various neurological diseases.
Although binding with the regulatory subunits is sufficient to activate Cdk5, its kinase activity can also be regulated by other pathways, such as phosphorylation, S-nitrosylation, sumoylation, and acetylation. Thr14, Tyr15, and Ser159 are the three phosphorylation sites of Cdk5 found so far. Phosphorylation at the sites of Thr14 and Ser159 produces opposite outcomes on Cdk5 activity, with the former inhibiting Cdk5 activity and the latter increasing its activity (Lee et al., 2014). The effect of phosphorylation at Tyr15 on kinase activity is still controversial. Some studies found that Cdk5 activity was enhanced by phosphorylation at Tyr15 while other study got the negative results. For example, Sasaki et al. (2002) reported that phosphorylation of Tyr15 by a non-receptor Src family tyrosine kinase Fyn increased Cdk5 enzymatic activity. However, Kobayashi et al. (2014) found that phosphorylation of Tyr15 had no effect on the activation of Cdk5. Thus, future studies are still needed to further clarify the function of Tyr15 phosphorylation as it is helpful for uncovering new regulatory pathway of Cdk5 activity.
Besides phosphorylation, the S-nitrosylation of Cdk5 is another mechanism to regulate kinase activity. It has been shown that the S-nitrosylation at cysteine residues (Cys) 83 and 157 contributed to the elevation of Cdk5 kinase activity (Qu et al., 2011, 2012). The S-nitrosylation of Cdk5 will cause transnitrosylation, a process of transferring NO group to dynamin-related protein 1 (Drp1), a protein controlling the process of mitochondrial fission (Westermann, 2010). S-nitrosylated Drp1 then stimulates excessive mitochondrial fission and damages the synapses, resulting in dendritic spine loss and synaptic failure finally (Cho et al., 2009). However, it is worth noting that the effect of S-nitrosylation at Cys83 on Cdk5 activity may be determined by the levels of exogenous NO. Cys83 S-nitrosylation by very high levels (non-physiological level) of NO could conversely inhibit Cdk5 activity though the concentrations of NO donors are never reached in vivo (Zhang et al., 2010). The cysteine residues of p35 can also be S-nitrosylated, it was found that S-nitrosylation of p35 at Cys92 by NO signaling led to its ubiquitination and degradation, which resulted in the reduction of Cdk5 activity (Zhang et al., 2015).
Sumoylation and acetylation are two other ways of post-translational modifications of Cdk5 activity. It has been shown that p35 is a novel sumoylation target and the sumoylation of p35 can enhance the activity of Cdk5/p35 complex (Buchner et al., 2015). However, it is still unclear whether Cdk5 can be sumoylated directly. With regard to acetylation, Cdk5 can be acetylated at the lysine residue site 33 (K33), a residue comprising the ATP binding pocket. The acetylation of Cdk5 at K33 causes an impairment of kinase activity due to the loss of ATP binding ability. The Cdk5 acetylation is negatively regulated by Sirtuin-1 (SIRT1), inhibition of SIRT1 will enhance nuclear Cdk5 acetylation whereas SIRT1 activation results in deacetylation of nuclear Cdk5 (Lee et al., 2018). Whether there are any other acetylation sites remains unclear, and the role of Cdk5 acetylation in neurological diseases is also still needed to be further investigated.
Cyclin-dependent kinases 5: A culprit in neurological disorders
Cyclin-dependent kinases 5 dysfunction triggers Parkinson’s disease
Parkinson’s disease (PD) is a common neurodegenerative movement disorder characterized by widespread degeneration of dopaminergic neurons located in the substantia nigra pars compacta (SNpc) and subsequent loss of dopamine reaching striatal projecting neurons (Obeso et al., 2010). The formation of intra-neuronal inclusions called Lewy bodies (LB) is a pathologic hallmark of PD and often correlates with the degree of cognitive decline (Schneider et al., 2012). Cdk5 and p35 has been found to be localized in LB in the SNpc of postmortem patients brains with PD (Nakamura et al., 1997). There is also sustained calpain-dependent conversion of p35 to p25 and enhanced Cdk5 activity in the brain of PD patients and animal models (Smith et al., 2003; Alvira et al., 2008). Quantitative phosphoproteomic analysis of α-synuclein transgenic mice showed that elevated Cdk5/p25 pathway activity contributed to SNpc dopaminergic neuronal death in model mice (Stillwell and Myers, 1988), which was also observed in mice administrated with 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP), a toxin used to induce PD model through destroying the nigrostriatal dopaminergic pathway (Binukumar et al., 2015). TFP5, a specific inhibitory peptide of Cdk5/p25 complex, suppressed the dopaminergic neuronal loss in SNpc and striatum, improved motor function of PD mice (Binukumar et al., 2015).
The mitochondrial dysfunction and subsequent oxidative stress are important mechanisms underlying the pathogenesis of PD. Abnormal Cdk5 activity contributes to mitochondrial dysfunction in PD by aberrant modulation of several key proteins involved in mitochondrial function. Drp1 is a GTPase that controls the process of mitochondrial fission. Drp1 is translocalized from the cytosol to the mitochondrial outer membrane (MOM) and then assembled into ring-like structures wrapping around MOM. Following GTP hydrolysis, Drp1 incises the membrane and instigates mitochondrial fission (Westermann, 2010). In non-human primate PD model, Cdk5 hyperactivity was shown to increase the phosphorylation of Drp1 at Ser616, which in turn increased GTPase activity and accelerated mitochondrial fission, ultimately induced dopaminergic neuronal loss in the SNpc (Park et al., 2019). Inhibition of Cdk5 activity prevented Drp1-dependent mitochondrial fission and neuron death following MPP+ treatment (Yang et al., 2020). Dysfunction of E3 ubiquitin ligases has a close relationship with mitochondrial defects and disruption of protein degradation. Parkin is an E3 ubiquitin ligase and plays an important role in maintaining mitochondrial function. It is able to prevent cell death from toxicity elicited by diverse insults (Petrucelli et al., 2002). However, the aggregation of insoluble Parkin will inhibit its catalytic activity and lead to dopaminergic neuron death caused by accumulation of toxic Parkin substrates, such as p38 (Avraham et al., 2007), ultimately contributing to the pathogenesis of PD (Rubio de la Torre et al., 2009). Cdk5 was demonstrated to phosphorylate Parkin at Ser131 both in vitro and in vivo, decreased its solubility, induced its accumulation and subsequently reduced E3 ubiquitin-ligase activity (Avraham et al., 2007). Consistent with this, enhanced p25 level and elevated Parkin phosphorylation were also found in several brain areas of PD patients (Rubio de la Torre et al., 2009). Glycoprotein 78 (GP78), another E3 ubiquitin ligase, is involved in regulating mitochondrial function. Cdk5 was found to directly phosphorylate GP78 at Ser516, which promoted its ubiquitination and degradation, ultimately caused neuronal death in both cellular and animal PD models (Wang et al., 2018).
Mitochondrial dysfunction and aggregation of damaged mitochondria can cause remarkable oxidative stress indicating by excessive accumulation of cellular reactive oxygen species (ROS). Peroxidases are a series of antioxidant enzymes with the capacity to catalyze hydrogen peroxide into stable non-toxic molecules. Cdk5 was demonstrated to phosphorylate peroxidases 2 (Prx2) at Thr89 and this phosphorylation decreased its peroxidase activity and induced ROS over-generation and dopaminergic neuron loss in the SNc following MPTP insult (Qu et al., 2007). Increased Prx2 phosphorylation in nigral neurons was also observed in brain tissue of postmortem PD patients. Interestingly, the insults-induced cleavage of p35 could also generate p10, a pro-survival N-terminal domain of p35 with the ability of preventing Cdk5/p25-mediated Prx2 phosphorylation and ROS accumulation (Zhang L. et al., 2012). Neuroinflammation is another outcome of mitochondrial dysfunction. Cdk5-mediated inflammasomes activation was observed in the SNpc of PD mouse model and in cerebrospinal fluid of PD patients. Inhibition or deletion of Cdk5 both could inhibit the inflammasome activation and delay the progression of PD in animal models (Zhang P. et al., 2016).
Excessive autophagy is closely related with neuronal death and there is over-production of autophagy markers in PD brains (Anglade et al., 1997). Knockdown of Cdk5 inhibited autophagy-mediated α-synuclein aggregation and promoted the functional recovery in PD mice (Su et al., 2015). Endophilin B1 (EndoB1) has been reported to be implicated in autophagy induction (Takahashi et al., 2007). In MPTP-induced PD model, elevated Cdk5 activity led to EndoB1 phosphorylation at Thr145 and then promoted EndoB1 dimerization, beclin-1 recruitment and autophagy induction, resulting in neuronal loss. EndoB1 or Cdk5 knockdown both remarkably attenuated the neuronal death in PD through inhibiting aberrant autophagy (Wong et al., 2011). Besides autophagy, several other cell death pathways mediated by Cdk5 were also shown to be involved in PD. Myocyte enhancer factor 2 (MEF2), one member of transcription factors family, is an endpoint for diverse signaling pathways that control cellular survival and apoptosis (Mao et al., 1999). Abnormal Cdk5/MEF2 signaling pathway contributes to dopaminergic neuronal death. Upon MPTP insult, the hyperactivity of Cdk5/p25 inactivated MEF2 and led to dopaminergic neuronal loss in the SNpc and formation of LB. Inhibition of Cdk5/p25 complex with TFP5 decreased the levels of MEF2 inactive form, thus significantly suppressed dopaminergic neuronal death (Zhang Q. et al., 2016). Nur77 is one important downstream survival effector of MEF2. Nur77 deficient mice are more sensitive to dopaminergic neuronal loss and exhibit more serious nigrostriatal damage in response to MPTP treatment (Mount et al., 2013). Cdk5-mediated MEF2 phosphorylation led to its inactivation and subsequently resulted in markedly Nur77 reduction in the nigrostriatal region following MPTP injection (Mount et al., 2013). These results imply that Cdk5-MEF2-Nur77 pathway is involved in the dopaminergic neuronal loss. Raf kinase inhibitor protein (RKIP) is able to block Ras/Raf/MEK/ERK signaling pathway by inhibiting Raf1-mediated MEK1 phosphorylation (Yeung et al., 1999). Downregulation of RKIP will lead to over-activation of ERK/MAPK pathway, which is resulted from losing control of Raf-1 (Granovsky and Rosner, 2008). Cdk5 was reported to phosphorylate RKIP at Thr42 and promote the degradation of RKIP. In PD animal models and post-mortal PD patients, Cdk5-mediated RKIP phosphorylation and degradation were demonstrated to occur with excessive activation of the ERK/MAPK cascade followed by cell cycle re-entry and neuron death (Wen et al., 2014).
The pathways related with Cdk5 in PD are summarized and presented as Figure 1.
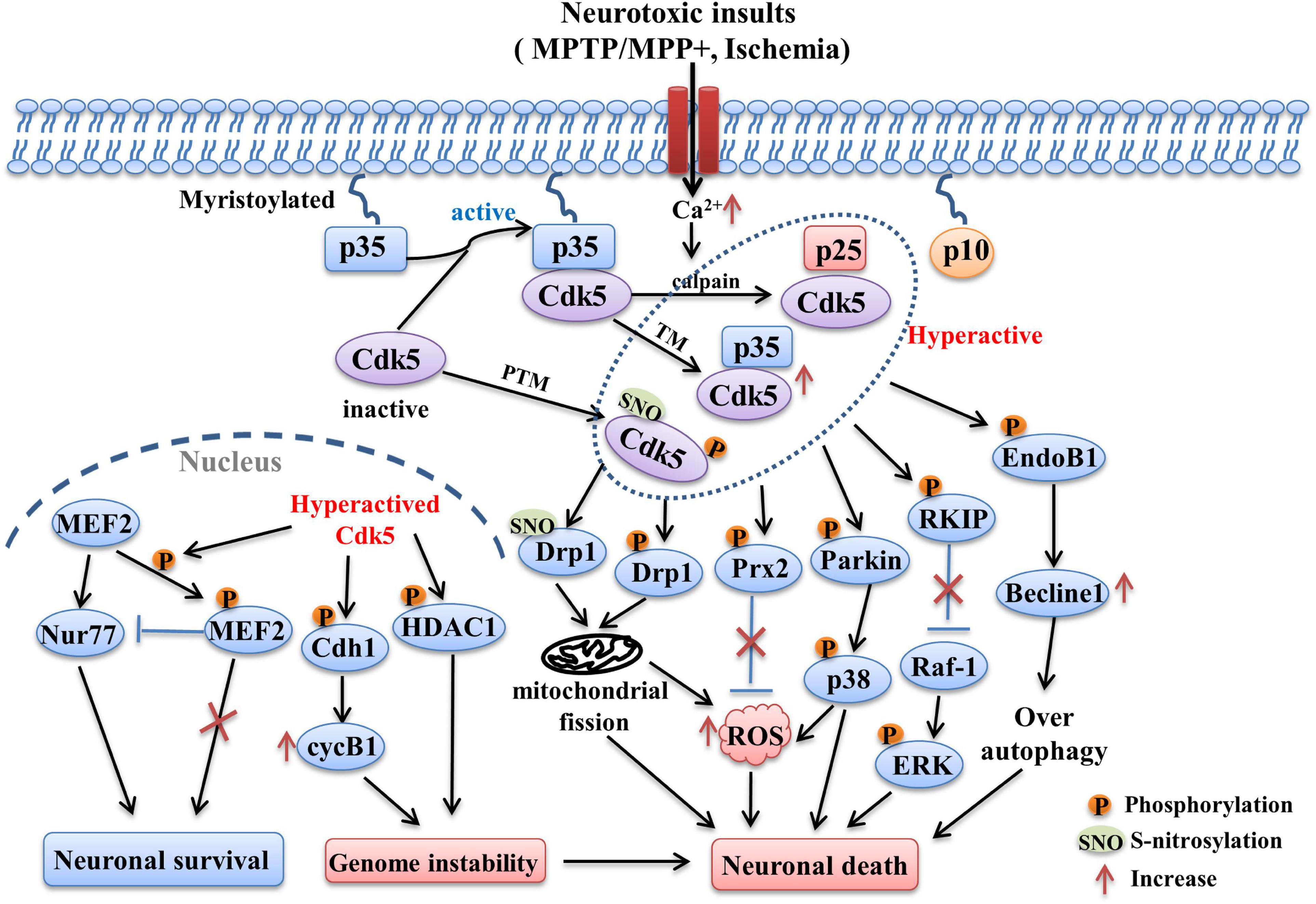
Figure 1. The Cdk5-related pathways involved in PD and stroke. Following neurotoxic insults, sustained Ca2+ elevation induces the over-activation of calpain, which in turn cleaves p35 into p25 and p10 fragments. The half-life of p25 is much longer than p35, it can form more stable complex with Cdk5 and lead to abnormally elevated Cdk5 activity. Its activity can also be regulated by post-translational modulations (PTM). The phosphorylation and S-nitrosylation of certain specific sites are able to hyperactive Cdk5 (e.g., phosphorylation at Ser159, S-nitrosylation at Cys83 and Cys157). In addition, transcriptional modulation (TM) of Cdk5 and p35 can also increase their expression and activity, which is mediated by some related transcription factors (e.g., Fos, CREB). Hyper-activation of Cdk5 then triggers the phosphorylation of multiple substrates including EndoB1, RKIP, Parkin, Prx2, Drp1, MEF2, Cdh1, and HDAC1. Changes in the phosphorylation state of these key substrates induced the occurrence of detrimental events such as mitochondrial dysfunction, oxidative stress, over-autophagy, which in turn contribute to the neuronal loss in PD and stroke.
Cyclin-dependent kinases 5 facilitates the initiation and progression of Alzheimer’s disease
Alzheimer’s disease (AD) is the primary reason for dementia among people over the age of 65. It has two main pathological characteristics, one is senile plaques mainly composed by amyloid-β (Aβ) peptides which is resulted from aberrant amyloid precursor protein (APP) cleavage, and the other is neurofibrillary tangles (NFTs) basically consisting of paired helical filaments (PHFs) which are derived from hyperphosphorylated tau proteins (Lau and Ahlijanian, 2003). These two proteinopathies are the basic changes underlying the progressive neuronal degeneration and cognitive dysfunction in AD patients. In a population-based study, it was found that there was a correlation between the genetic variation of Cdk5 gene and the risk of AD (Arias-Vasquez et al., 2008). Elevated p25/p35 ratio was observed in multiple brain areas including frontal cortex, inferior parietal cortex and hippocampus in human AD brains (Tseng et al., 2002). P25 transgenic mouse model showed AD-like pathological changes such as tau hyperphosphorylation, NFTs, and neuronal loss. Transgenic expression of Cdk5 inhibitory peptide before and after the insult of p25 could both suppress hippocampal tau phosphorylation and neuronal death, improve the cognitive dysfunction of model mice (Xu et al., 2019; Huang et al., 2020). Inhibition of Cdk5/p25 complex also reduced hippocampal Aβ production and tau hyperphosphorylation, improved learning and memory abilities of AD model mice (Zhao et al., 2021).
Amyloid-β peptides are produced from the sequential cleavage of APP by β-amyloid cleavage enzyme 1 (BACE1) and γ-secretase. Aβ40 and Aβ42 are two main forms of Aβ peptide, the longer Aβ42 has a much stronger tendency to oligomerize and aggregate than the shorter Aβ40, and exhibits more neurotoxic effect. Accumulation of oligomerized Aβ peptides forms amyloid plaques, which is closely related with neuronal loss and cognitive decline in AD. Cdk5/p25 activity was upregulated and significantly correlated with BACE1 elevation in brains of AD patients and transgenic mouse models (Sadleir and Vassar, 2012). Wen et al. (2008) found that Cdk5 facilitated Aβ generation through modulation of BACE1 synthesis via signal transducer and activator of transcription 3 (STAT3), who binded to BACE1 promoter and phosphorylated it at Ser727. In cultured primary rat hippocampal neurons, Cdk5 activation triggered peroxisome proliferators-activated receptor γ (PPARγ) phosphorylation at Ser273, then increased BACE1 activity and Aβ production (Quan et al., 2019). Presenilin 1 gene mutation is the main reason for autosomal dominant familial forms of AD (Fraser et al., 2000). As the core subunit of γ-secretase, presenilin-1 (PS1) is essential for the catalytic activity of γ-secretase. Presenilin-1 dysfunction will result in the increase of Aβ42/Aβ40 ratio, which is more toxic than the rising of Aβ42 absolute amounts (Chavez-Gutierrez et al., 2012). Cdk5/p35 is reported to regulate PS1 stability and metabolism through phosphorylating PS1 at Thr354 (Lau et al., 2002), and the aberrant Cdk5 activity will cause γ-secretase dysfunction and the alteration of Aβ42/Aβ40 ratio.
Tau is a microtubule-associated protein primarily distributed in axons. Tauopathy is characterized by abnormal tau aggregation and NFT formation in the brain, which is a hallmark of AD. Cdk5/p25 can promote tau dimerization through phosphorylating it at different sites, which subsequently aggregate into PHF-like filament, resulting in microtubule collapse and neurite retraction (Paudel, 1997). The Cdk5 and hyperphosphorylated tau are found to be co-existed in the neocortical pyramidal neurons and cerebellar neurons of AD (Pei et al., 1998). Moreover, enhanced Cdk5 immunoreactivity was observed in neurons undergoing early stage of NFTs degeneration and plaque-neurites in several regions of human AD brain (Pei et al., 1998). Cdk5 hyperactivation was also found to induce the accumulation of hyperphosphorylated tau, Aβ plaques, and neuronal loss in animal models. TFP5, an inhibitory peptide of Cdk5/p25, significantly reduced tau hyperphosphorylation, neurofilament accumulation and restored synaptic function and behavior abnormalities in transgenic AD model mice as well as the mice overexpressing p25 through attenuating Cdk5 hyperactivity (Shukla et al., 2013, 2017). Long-term and short-term Cdk5 knockdown also both inhibited insoluble tau aggregation in the hippocampus of 3xTg-AD mice and improved their spatial memory (Castro-Alvarez et al., 2014). Collapsin response mediating protein-2 (CRMP2) is capable of promoting microtubule assembly through binding tubulin heterodimers (Fukata et al., 2002). It was reported that Cdk5 phosphorylated CRMP2 at Ser522 and reduced its binding to tubulin (Uchida et al., 2005). And this hyperphosphorylated CRMP2 was demonstrated to be a component of PHF (Yoshida et al., 1998), and were observed in the brain of AD transgenic mouse model (Cole et al., 2007). Cdk5 was also reported to activate microtubule affinity-regulating kinases 4 (MARK4) and increased tau phosphorylation and accumulation through increasing the phosphorylation of MARK4 at Ser262 (Saito et al., 2019).
Besides the above-mentioned Aβ generation and NFT formation, CNS inflammation caused by microglia activation is another main feature of AD, which correlates with the plaque accumulation and may exacerbate the pathology of AD (Kitazawa et al., 2005). Aβ oligomers caused long-lasting activation of microglia and profound neuroinflammation in the hippocampus, which was mediated by over-activation of Cdk5/p25 complex. Cdk5 inhibitor roscovitine suppressed the inflammatory processes evoked by Aβ (Wilkaniec et al., 2018). Increased phospholipase A2 (PLA2) activity is centrally involved in inflammatory responses associated with several neurological disorders including AD (Farooqui et al., 2006). Soluble lipid mediator lysophosphatidylcholine (LPC) is generated through PLA2-mediated phosphatidylcholine hydrolysis (Steinbrecher et al., 1984). It has been demonstrated that p25 overexpression lead to glia activation, neuroinflammation and neurodegeneration partially through cytosolic PLA2-mediated LPC release in neurons (Sundaram et al., 2012). In addition, hyperactivation of Cdk5 mediated by p25 accumulation also accounts for exacerbation of tau pathology caused by lipopolysaccharide, a toxin used to induce nervous system inflammation (Kitazawa et al., 2005).
Oxidative stress and dysfunctional mitochondria appear at the early stages of AD. Cdk5 is an upstream activator of mitochondrial dysfunction in AD. The mitochondrial damage results in more ROS generation and Ca2+ level elevation, which in turn lead to even higher Cdk5 activity, then shapes a vicious circle and contributes to neuron loss in AD (Sun et al., 2008). A variety of molecules and signaling pathways are involved in the oxidation caused by aberrant Cdk5. Sun et al. (2008) reported that over-activation of Cdk5 led to mitochondrial damage and ROS accumulation through inactivating Prx1 and Prx2. Drp1 is a direct target of Cdk5, and Cdk5-mediated phosphorylation of Drp1 at Serine 579 regulates Aβ1-42 induced mitochondrial fission and neuronal toxicity. Inhibition of Cdk5 attenuated Aβ1-42 induced mitochondrial fission by inhibiting Drp1 phosphorylation in primary cultured neurons (Guo et al., 2018). In cultured hippocampus HT-22 cells, Cdk5 inhibition also blocked mitochondrial fragmentation and AD-like hallmarks induced by streptozotocin through suppressing Drp1 phosphorylation (Park et al., 2020). JNK and p38 MAPK signaling pathways are able to be activated by oxidative stress and they are critically involved in Aβ-induced neurotoxicity and NFT formation (Yoshida et al., 2004). Enhanced JNK activity and increased p38 expression were observed in the affected brain areas of AD patients (Pei et al., 2001). Cdk5 interacts with p38/JNK pathways and contributes to the progressive Aβ deposits and AD development. In transgenic AD mouse model, co-immunoprecipitation of p-JNK and p-p38 with Cdk5 was significantly enhanced (Otth et al., 2003). Dysregulated Cdk5 caused elevation of p38 activity by increasing ROS in response to β-amyloid neurotoxic stimuli (Chang et al., 2010). As the major substrate involved in JNK-induced neurotoxicity, c-Jun is also over-activated in the several brain regions of AD patients. Cdk5 is reported to activate c-Jun through phosphorylating it at Ser63 and Ser73 via ROS-mediated activation of JNK (Sun et al., 2009). Myeloid cell leukemia sequence 1 (Mcl-1) is a member of the B-cell lymphoma 2 (Bcl-2) family, which is essential for neuronal survival (Arbour et al., 2008). The disease severity of AD patients was shown to be inversely correlated with Mcl-1 levels. Mcl-1 could be phosphorylated at Thr92 by Cdk5 following neurotoxic insults and this phosphorylation in turn caused Mcl-1 degradation and mitochondrial dysfunction, which promoted the neurodegenerative process of AD (Nikhil and Shah, 2017).
The aberrant Cdk5 activity seen in the progressive neurodegeneration of AD can also be caused by other mechanisms besides the dysfunction of its co-activators. Cancino et al. (2011) reported that Aβ-induced c-Abelson tyrosine kinase (c-Abl) activation facilitated tau phosphorylation via phosphorylating Cdk5 at Tyr15 in vitro. In the brains of AD mice, there was also an elevated Tyr15 phosphorylation of Cdk5 correlating with increased c-Abl levels (Cancino et al., 2011). Cdk5 could be S-nitrosylated by endogenously generated NO (Foster et al., 2009) and S-nitrosylated Cdk5 (SNO-Cdk5) in turn transnitrosylated Drp1 and resulted in mitochondrial fission in dendritic spines, which contributed to dendritic spine loss following Aβ treatment (Qu et al., 2011). Glutathione-S-transferase pi 1 (GSTP1) is able to dislodge p25/p35 and clear ROS accumulation and negatively regulates Cdk5 activity. There was a significant correlation between reduced GSTP1 levels and hyper-activation of Cdk5 in prefrontal cortex (PFC) of human AD brain (Sun et al., 2011). Furthermore, GSTP1 is capable of suppressing the activation of JNK/c-Jun pathway whereas it will lose its control following oxidative or chemical stress (Adler et al., 1999). GSTP1 was also shown to relieve the inhibition of Prx1 mediated by Cdk5 and reactivate it directly (Ralat et al., 2006).
The summation of above molecular pathways related with Cdk5 in AD is presented in Figure 2.
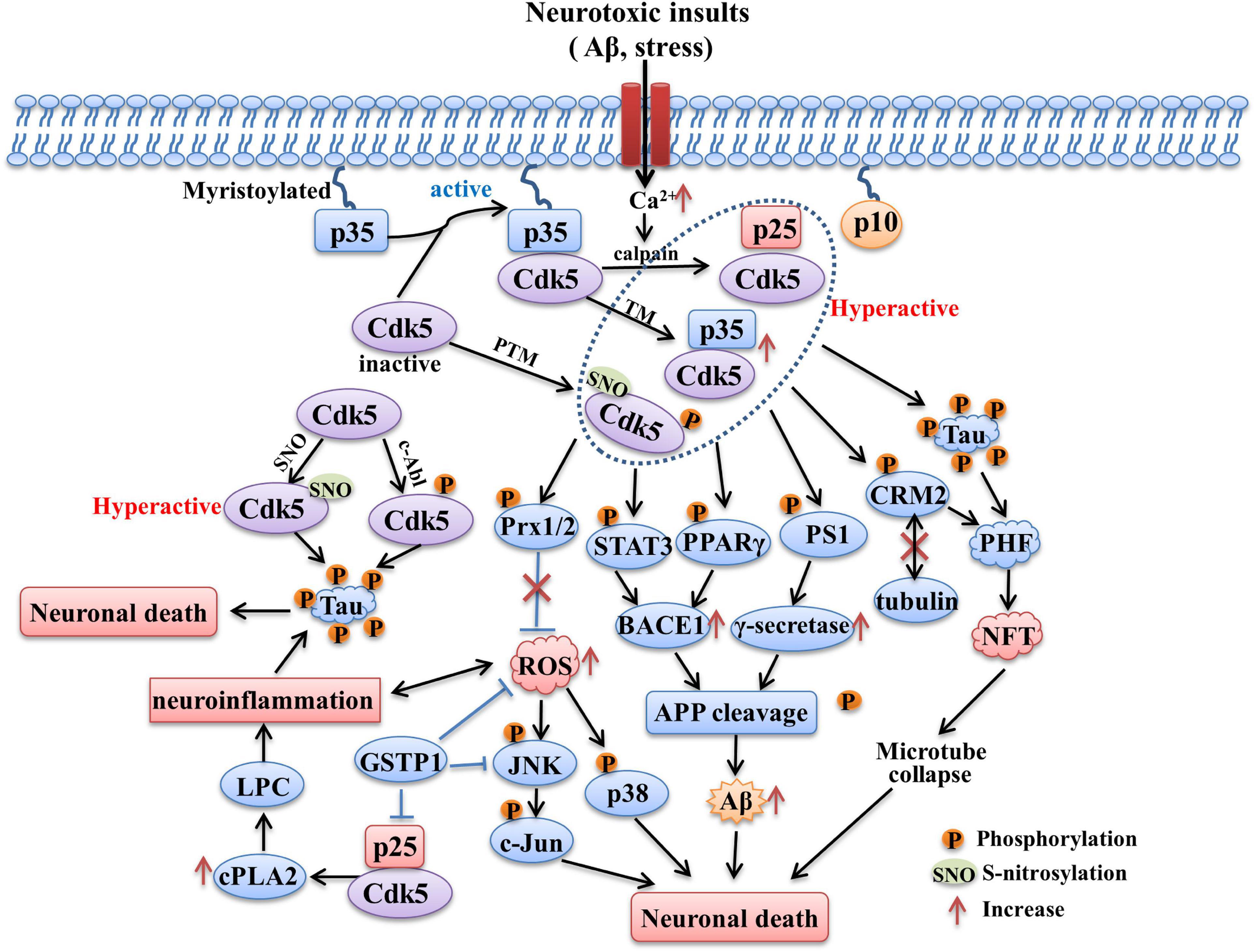
Figure 2. The role of Cdk5 in AD. Sustained activation of Cdk5 hyperphosphorylated tau protein and contributes to formation of PHF and NFT. CdK5 over-activation also induces intracellular Aβ accumulation through increasing APP processing mediated by BACE1 and γ-secretase. The phosphorylation of STAT3, PPARγ and PS1 contributed to the above process. Phosphorylation of Prx2 caused by aberrant Cdk5 activity inhibits its ROS scavenging ability and induces the activation of p38 MAPK and JNK pathways. Hyperactivation of Cdk5 also leads to neuroinflammation mediated by LPC accumulation through increasing cPLA2 activity. In addition, Cdk5 activity could be upregulated through being S-nitrosylated by endogenously generated NO and phosphorylated by c-Abl, both of process contribute to tau hyperphosphorylation. The tau hyperphosphorylation, NFT formation, intracellular Aβ generation, oxidative stress and neuroinflammation caused by aberrant Cdk5 activity all contribute to the neuronal death and neurodegeneration in AD.
The role of cyclin-dependent kinases 5 in Huntington’s disease
Huntington’s disease (HD) is a progressive neurodegenerative disease characterized by chorea, personality changes and dementia (Kaminosono et al., 2008). It is mainly caused by selective loss of striatal medium-sized spiny neurons, resulted from abnormal polyglutamine (polyQ) expansion in the protein huntingtin (htt) (MacDonald et al., 2003). The role of Cdk5 and its co-activators in HD is still controversial. Some studies have reported their neuroprotective effects, while others have shown the opposite results.
It has been demonstrated that htt co-localized with Cdk5 in cellular membrane (Luo et al., 2005). Cdk5 could phosphorylate htt at Ser1181 and Ser1201 both in primary striatal cultures and in the mouse striatum following DNA damage, which prevented neuronal death and exerted neuroprotective effects. In the brain of HD mouse model, decreased Cdk5/p35 association at the late stage resulting from sustained DNA damage led to htt dephosphorylation and polyQ-induced p53 mediated neuronal death (Anne et al., 2007). Low levels of Cdk5 and p35 have been found in the striatum of postmortem HD patients and in HdhQ111 mutant mice (Luo et al., 2005; Paoletti et al., 2008). Consistent with this, there was an accumulation of p25 and increased p25/p35 ration in rat striatum following delivery of quinolinic acid (QA) (Park et al., 2012), a neurotoxin used to make experimental model of HD.
Phosphorylation of huntingtin at Ser421 has been shown to be neuroprotective against intrastriatal QA injection (Metzler et al., 2010). In contrary to the above results, Park et al. (2017) found that single p35 allele deletion resulted in a 17% enhancement of htt phosphorylation at Ser421, indicating that genetic reduction of p35 may be neuroprotective in the context of HD. Moreover, p35 hemizygous knockout mice also showed a much smaller striatal lesion size induced by QA compared with control (Park et al., 2012). Cherubini et al. (2015) reported that the increased activity and translocation of Drp1 to the mitochondria mediated by Cdk5 contributed to striatal neurodegeneration in HD. Inhibition of Cdk5 activity could attenuate mitochondrial fragmentation in striatal cells through modulating Drp1 activity and subcellular location. Cdk5 was also shown to be involved in the hippocampal and corticostriatal-dependent learning and memory impairment relevant to HD. Genetic reduction of Cdk5 alleviated corticostriatal-dependent learning deficits by increasing GluN2B surface levels in the cortex and striatum of HD model mice. Cdk5 knockdown enhanced Rac1 activity and subsequently increased hippocampal dendritic spine density, which may be the mechanism underlying the improvement of hippocampal-dependent memory (Alvarez-Periel et al., 2018).
These findings suggest the multifaceted roles of Cdk5 in HD. Future studies are still needed to further clarify the relationship between Cdk5 dysfunction and HD pathogenesis.
Cyclin-dependent kinases 5 dysfunction contributes to stroke
Stoke is an acute neurologic event caused by dramatic reduction of cerebral blood flow to a localized part of brain (Bacigaluppi and Hermann, 2008). According to the affected brain regions, the cerebral ischemia can be divided into focal and global ischemia stroke (Traystman, 2003). A study analyzed the causes of death in Chinese residents from 1990 to 2017 and surprisingly found that stroke ranked the first, not cancer (Zhou M. et al., 2019). Many patients are disabled or died because of the untimely treatment due to the narrow time window of thrombolytic therapy. In addition, quite a few patients are not suitable for thrombolysis. Therefore, it is still necessary to further identify the pathological mechanisms underlying stroke and search for the potential pharmacological targets.
Disruption of the tight regulation of Cdk5 has been observed in stoke-affected brain tissue of patients (Mitsios et al., 2007), as well as in animal and cellular models of stroke (Meyer et al., 2014). The analysis of Cdk5 expression in human post-mortem brain showed that Cdk5- and p-Cdk5-positive neurons and microvessels increased dramatically in stroke-affected regions, accompanied by irregular arrangement and clumped in the cytoplasm (Mitsios et al., 2007). Ischemia induced either by oxygen glucose deprivation (OGD) in brain slices or middle cerebral artery occlusion (MCAO) in vivo both induced the cleavage of p35 into p25 (Meyer et al., 2014). Pharmacological inhibition or Cdk5 knockdown using the approach of peptide-directed lysosomal degradation prevented neuronal death, reduced infarction size and promoted the recovery of neurological functions following MCAO in mice (Meyer et al., 2014; Zhou Y. F. et al., 2019). In rat hypoxia/ischemia (HI) injury model, Cdk5 activity and p25 level were also significantly increased, and inhibiting Cdk5 with small peptide reduced cerebral infarct volume and promoted functional recovery (Tan et al., 2015).
The mitochondrial dysfunction and oxidative stress are also critically involved in the pathogenesis of stroke. Mitochondrial fusion and fission are two important ways to regulate mitochondrial function, balanced fusion/fission is essential for maintaining normal cellular physiology and fusion/fission imbalance will cause mitochondrial dysfunction and degeneration. Drp1 is a GTPase that controls the process of mitochondrial fission, and the elevated Drp1 phosphorylation will cause excessive mitochondrial fission. Hyper-activation of Cdk5 could increase the phosphorylation of Drp1 at Ser616, which in turn increased GTPase activity and accelerated mitochondrial fission, ultimately contributed to mitochondrial dysfunction and neuronal death following OGDR insults (Chen et al., 2021). Inhibition of Cdk5 hyperactivity could attenuate mitochondrial fragmentation and neuronal loss through reducing Drp1 phosphorylation at Ser616 (Chen et al., 2021). In human body, the oxidative/antioxidative balance is kept by a large group of antioxidant enzymes under physiological condition, which can catalyze hydrogen peroxide into stable non-toxic molecules (Apel and Hirt, 2004). Prx2 is such an antioxidant enzyme. Cdk5 phosphorylates Prx2 at Thr89 and this phosphorylation decreases Prx2 peroxidase activity and lowers its capacity to eliminate ROS which contributes to neuron loss subsequently (Qu et al., 2007). In both focal and global ischemia, elevated phosphorylation of Prx2 at Thr89 and ROS accumulation were found due to upregulated cytoplasmic Cdk5 activity (Rashidian et al., 2009). And p35 knockout mice displayed remarkably smaller infarct size following stroke insult due to a lower level of Prx2 phosphorylation (Rashidian et al., 2009).
The dysfunction of N-methyl-D-aspartate receptors (NMDARs) contributed to a variety of neurological diseases including stroke. Phosphorylation of NMDAR subunits at the cytoplasmic carboxyl termini is an important way to regulate NMDAR function. It has been shown that Cdk5 can phosphorylate NMDAR at multiple sites. The phosphorylation of GluN2A at Ser1232 by Cdk5 enhanced receptor function and promoted CA1 pyramidal neuron death following transient forebrain ischemia (Wang et al., 2003). The phosphorylation of GluN2B at Tyr1472 by Cdk5 promoted receptor internalization (Zhang et al., 2008), similarly phosphorylation of regulatory site Ser1116 also inhibited the membrane trafficking of GluN2B-containing NMDARs (Plattner et al., 2014). Cdk5 was also shown to phosphorylate GluN2B at Ser1284, which may contribute to the ischemic injury in both cultured neurons suffered from OGD-reperfusion and mice subjected to transient global ischemia (Lu et al., 2015). The anaphase-promoting complex/cyclosome (APC/C) is a E3 ubiquitin ligase that controls cell cycle progression (Peters, 2002). Cdh1 is the activator of APC/C and cyclin B1 is one substrate of APC/C–Cdh1, whose nuclear level is increased in affected neurons of stroke patients (Love, 2003). It is well known that abnormal release of glutamate caused by NMDAR overactivation is involved in stroke (Bossy-Wetzel et al., 2004). Cdk5 contributes to NMDAR-mediated neuronal excitotoxicity by phosphorylating Cdh1 at residues Ser40, Thr121, and Ser163, resulting in Cdh1 inactivation and cyclinB1 accumulation in the nucleus (Maestre et al., 2008).
Cyclin-dependent kinases 5 can modulate the process of cell apoptosis and cell cycle through regulating the function of transcription-related molecules, which may be another mechanism involved in stroke. MEF2 is a factor critical for neuronal survival. Activating MEF2 is pro-survival, whereas hyperphosphorylation of MEF2 contributes to neuronal apoptosis (Mao and Wiedmann, 1999). MEF2 can be phosphorylated at Ser408 and Ser444 directly by Cdk5 and this phosphorylation will lead to reduced MEF2 function due to the inhibition of MEF2 transactivation activity (Gong et al., 2003). Nuclear Cdk5 contributes to neuronal death following focal ischemia but not global ischemia by phosphorylating MEF2 (Tang et al., 2005). Histone deacetylase 1 (HDAC1) is critically involved in suppressing transcription of cell cycle genes and regulating the cell cycle (Lagger et al., 2002). Cdk5 was shown to inhibit HDAC1 function and induce double-strand DNA breaks and cell cycle reentry, resulting in neurodegeneration and neurologic defects following ischemia (Kim et al., 2008).
The Cdk5-modulated pathways involved in stroke are shown in Figure 1.
Cyclin-dependent kinases 5 dysfunction triggers anxiety/depression
Anxiety is the emotional response to threatening or potentially threatening stimuli (Sandford et al., 2000) and it is beneficial for the animals to cope with the stress. However, excessive anxious state or pathological anxiety is harmful, characterized by hypervigilance to threatening stimuli or negative events (Rosen and Schulkin, 1998). Depression is mainly manifested by anhedonia, loss of motivation and abnormal neurovegetative functions (Nestler and Carlezon, 2006). Anxiety and depression have become major public health issues and bring multiple challenges to the society. The current clinical strategies for depression and anxiety mainly act through modulating serotonergic system or GABAergic system. However, these medical approaches often have inadequate efficacy and can’t meet clinical needs. Therefore, an urgent need to find new potential therapeutic targets emerges.
More and more evidences have shown that Cdk5 dysfunction plays important roles in psychiatric disorders. Analyzing postmortem brains of patients with major depression showed that Cdk5 activity was significantly increased in Brodmann’s area 25, a subregion of the PFC, which was implicated in major depression and treatment response (Papadopoulou et al., 2015). In rat depressive model induced by chronic mild stress, Cdk5 activity and p35 trafficking was enhanced in the hippocampal dentate gyrus, which was suppressed by the antidepressant venlafaxine (Zhu et al., 2012). The elevation of Cdk5 activity was also observed in several brain regions of anxious mice including basolateral amygdala, septal and lateral septum (Bignante et al., 2008). Pharmacological inhibition or genetic knockout of Cdk5 both significantly attenuated the depressive-like symptoms or aberrant anxiety-like phenotype in mice (Li G. et al., 2014).
There are multiple mechanisms involved in the regulation of anxiety/depression by Cdk5. The dysfunction of glucocorticoid receptor (GR) signaling critically contributed to the pathology of psychiatric disorders. Cdk5 was shown to phosphorylate GR at serine 232 and there was a significant correlation between elevated Cdk5 activity and enhanced GR phosphorylation in hippocampus and PFC of the depressive rats (Mitic et al., 2013). The mechanism study revealed that the aberrant GR phosphorylation may be attributed to the dysfunction of Cav1.2 subunit of the L-type calcium channel, which was closely related with stress-induced neuropsychiatric conditions. Lower levels of p25 production and GR phosphorylation was observed in the Cav1.2 heterozygous (Cav1.2 + /−) mice compared with control mice following chronic unpredictable stress, correlating with reduced depressive-like and anxiety-like behaviors (Bavley et al., 2017). Depression is the most common psychiatric comorbidity of HD. Inhibition of Cdk5 activity in the nucleus accumbens attenuated the depressive-like behaviors of HD mice through modulating dendritic spine plasticity mediated by dopamine- and cAMP-regulated phosphoprotein 32 (DARPP32)/β-adducin signaling pathway (Brito et al., 2019). Anomalous neurotransmission is an important mechanism underlying anxiety disorder. Cdk5 was shown to regulate anxiety-like behaviors through modulating neurotransmission and neuronal excitability. Conditional deletion of Cdk5 in parvalbumin (PV) interneurons results in increased GABAergic neurotransmission which in turn alleviated the anxiety-like behavior of mice (Rudenko et al., 2015). Optogenetic study showed that Cdk5 activation led to the decreased activation of excitatory neurons in the prelimbic cortex, and Cdk5 knockdown reversed the deactivation of these excitatory neurons and alleviated the anxiety-like behaviors induced by chronic inflammation (Wang et al., 2015).
Cyclin-dependent kinases 5 dysfunction elicits pathological pain
Acute nociceptive pain is the physiological sensation of injury that helps animals survive by promoting them to withdrawal from the harmful stimuli and avoid further contacting with such stimuli. However, pathological and chronic pain seems meaningless and will cause much distress. Allodynia and hyperalgesia are two kinds of behavioral phenotypes indicated by decreased threshold or amplification in the responsiveness to noxious stimulation following chronic pain (Rahn et al., 2013). Mounting evidences have shown that Cdk5 is a key molecule involved in pain modulation. In mice model of peripheral inflammatory pain, there were both an elevation of Cdk5 activity in dorsal root ganglia (DRG) and spinal dorsal horn (Wang et al., 2005; Yang et al., 2007). Roscovitine, a Cdk5 inhibitor, alleviated heat hyperalgesia induced by CFA and formalin in a dose-dependent manner (Wang et al., 2005). Conditional deletion of p35 in mice (p35-/-) resulted in decreased sensitivity to painful thermal stimulation due to reduced Cdk5 activity, whereas mice overexpressing p35 exhibited thermal hyperalgesia (Pareek et al., 2006).
Cyclin-dependent kinases 5 participates in the inflammatory pain-induced hypersensitivity through diverse pathways. Extracellular signal-regulated kinase 1 and 2 (ERK1/2) is fast activated in response to innocuous and noxious stimulation (Ji et al., 2002; Cruz et al., 2005). The levels of phosphorylated ERK1/2 (p-ERK) and phosphorylated Cdk5 at Ser159 (p-Cdk5) are increased in spinal cord dorsal horn following adjuvant-mediated inflammatory pain (Zhang X. et al., 2014). And inhibition of ERK1/2 significantly suppresses the nociceptive responses and enhancement of p-ERK and p-Cdk5 (Zhang X. et al., 2014). Tumor necrosis factor-α (TNF-α), a proinflammatory cytokines, participates in the onset and development of inflammatory pain. The release of TNF-α is elevated following peripheral inflammation, which triggers the activation of ERK1/2 and then results in the induction of early growth response 1 (Egr-1), a member of zinc-finger trans-activators. Subsequently, Egr-1 binds to the promoter region of p35 and increases Cdk5 activity (Utreras et al., 2009). These results indicate that ERK-mediated Cdk5 activation plays a vital role in the hypersensitivity of peripheral inflammatory pain.
Transient receptor potential vanilloid 1 (TRPV1) is mainly expressed in the nociceptive sensory neurons and potentiate pain sensitization in different pain models (Caterina et al., 1997; Davis et al., 2000). The phosphorylation of TRPV1 plays an important role in responding to a pain stimulus by regulating intracellular calcium levels (Pareek et al., 2007). Cdk5 regulates TRPV1 membrane trafficking through phosphorylating TRPV1 at the site of Thr407. Conditional Cdk5 knockout in small diameter sensory neurons (C-fibers) abolishes TRPV1 phosphorylation and leads to hypoalgesia (Pareek et al., 2006, 2007). Transforming growth factor-β1 (TGF-β1) signaling pathway is involved in modulating Cdk5-mediated TRPV1 phosphorylation under the condition of inflammation pain. TGF-β1 treatment increased Cdk5-mediated phosphorylation of TRPV1 at Thr407 in vitro. The conditional TGF-β1 knockout reduced Cdk5 activity and Cdk5-dependent TRPV1 phosphorylation with attenuation of thermal hyperalgesia in mice following inflammatory pain (Utreras et al., 2012). TNF-α was shown to increase p35 expression, causing Cdk5-mediated TRPV1 phosphorylation and ROS production in nociceptive neurons and increased pain sensation (Sandoval et al., 2018). These results indicate that TRPV1 and TGF-β signaling participate in the modulation of pain sensation by Cdk5. BDNF-tyrosine kinase, type 2 (TrkB) signaling pathway has been shown to be critically involved in pain modulation. TrkB activation contributes to initiation and maintenance of both heat and mechanical hypersensitivity produced by tissue injury (Wang et al., 2009). Cdk5 is demonstrated to phosphorylate and activate TrkB at Ser478 residue (Lai et al., 2012). Inhibiting Cdk5 attenuates CFA-induced hypersensitivity through suppressing TrkB expression and blocking BDNF/TrkB signaling pathway, implying that the interaction between Cdk5 and BDNF/TrkB also plays a role in pain hypersensitivity induced by inflammation (Zhang H. H. et al., 2014).
Besides its involvement in inflammatory pain, Cdk5 has been also shown to modulate several other kinds of pain such as neuropathic pain (Li K. et al., 2014; Yang et al., 2014), bone cancer pain (Zhang R. et al., 2012), visceral pain (Chang et al., 2011), post-operative pain (Liu et al., 2014), orofacial pain (Prochazkova et al., 2013; Hu et al., 2022). For example, in the mouse neuropathic pain model induced by spinal nerve ligation, the expression of Cdk5 and its activators in DRG was elevated, which contributed to the mechanical allodynia (Gomez et al., 2020). Cdk5 inhibition attenuated mechanical allodynia and thermal hyperalgesia through suppressing mGluRs and (or) NMDAR phosphorylation in both bone cancer pain (Zhang R. et al., 2012) and post-operative pain (Liu et al., 2014) models. Depletion or overexpression of p35 caused mice to be less or more sensitive to orofacial mechanical stimulation, respectively (Prochazkova et al., 2013). Inhibition of Cdk5 activity could also alleviate facial pain by suppressing calcium-mediated trigeminal peripheral sensory neurons activation (Hu et al., 2022). The crucial roles of Cdk5 in pain modulation make it a promising non-opioid target for pain treatment.
Cyclin-dependent kinases 5 dysfunction contributes to epilepsy
Epilepsy affects over 70 million people worldwide, which is mainly manifested by recurrent seizures and brings a variety of physiological and psychosocial consequences (Thijs et al., 2019). The levels of Cdk5, p-Cdk5 and its kinase activity were significantly increased in the anterior temporal lobe samples from the patients with mesial temporal lobe epilepsy accompanied by hippocampal sclerosis (Banerjee et al., 2021). Hippocampal sclerosis (HS), characterized by segmental neuronal loss and gliosis, is the most common cause of refractory epilepsy in adults. The ratio of p25 to p35 and activity of Cdk5/p25 complex increased remarkably in the diseased hippocampi compared with the adjacent normal temporal lobe, indicating a pathological role of Cdk5/p25 in HS (Sen et al., 2006).
Sustained endoplasmic reticulum (ER) stress triggers the regional specific astroglial responses, which contributed to the status epilepticus (SE). Cdk5 phosphorylation was shown to be upregulated in the astrocytes within the hippocampus CA1 region and dentate gyrus following ER stress. Inhibition of Cdk5 with roscovitine markedly suppressed hippocampal astroglial response induced by ER stress (Lee and Kim, 2021), which may be mediated by reducing PKA activity and Drp1 phosphorylations (Hyun et al., 2017). Roscovitine was also shown to suppress SE-induced neuroinflammation mediated by glial responses via p38 MAPK inhibition in rat frontoparietal cortex (Kim et al., 2019). Blood–brain barrier (BBB) dysfunction is critically involved in epilepsy. Specific deletion of Cdk5 in the endothelial cells of BBB resulted in spontaneous seizures in mice, which may be attributed to the decreased astrocytic glutamate reuptake and elevated glutamatergic synaptic function (Liu et al., 2020).
Cyclin-dependent kinases 5 substrates
As one up-stream kinase in the nervous system, Cdk5 triggers the down-stream signaling pathways through interacting with its substrates. Numerous Cdk5 substrates have been detected and their abnormal alterations following aberrant Cdk5 activity are closely related with initiation and progression of neurological disorders. In this review, some important substrates of Cdk5 are listed in Table 1. The readers can be referred to prior reviews with a more comprehensive list of Cdk5 substrates (Dhavan and Tsai, 2001; Su and Tsai, 2011; Pao and Tsai, 2021).
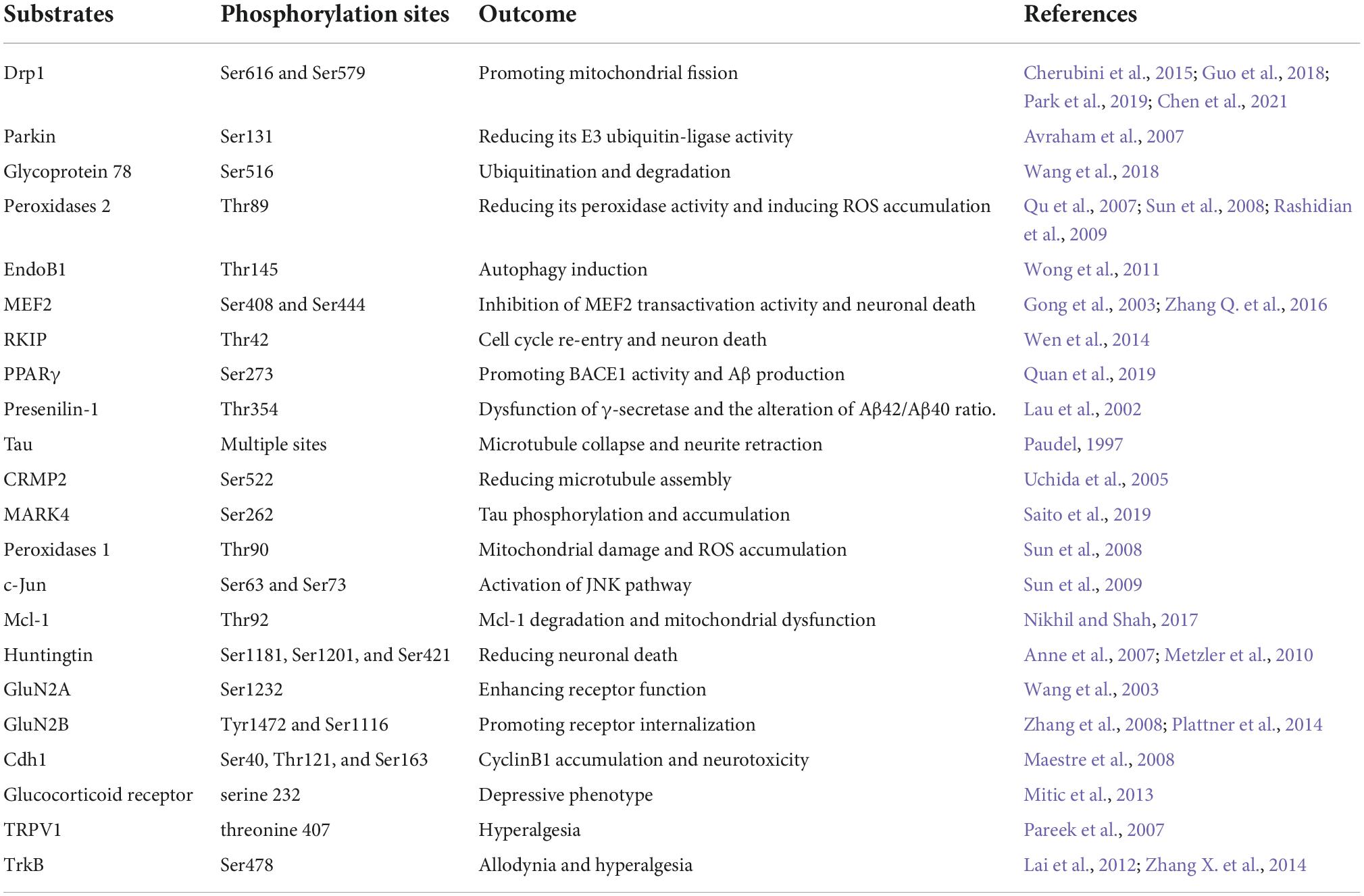
Table 1. List of major Cdk5 substrates reviewed in neurological disorders.
Conclusion
As a kinase distributed extensively in the nervous system, Cdk5 plays crucial roles in various kinds of neuronal function. Aberrant activation of Cdk5 appears to be a driving force for the initiation and progression of multiple neurological disorders. Numerous molecular targets and pharmacological mechanisms are involved in the Cdk5-mediated neurodysfunction (Figure 3). The extensive actions of Cdk5 discussed in this review suggest that it is a promising therapeutic target for neurological diseases. In fact, many small-molecule inhibitors and peptides have been synthetized and their beneficial effects in nervous system have been examined. Although quite a few inhibitors exhibit good neuroprotective properties in various preclinical studies, no selective Cdk5 kinase inhibitors have ever entered clinical trials for therapeutic intervention. A successful Cdk5 inhibitor for neurological diseases should be able to pass the BBB and have a high Cdk5 selectivity. Non-selective Cdk inhibitors are generally unfavorable and ineffective due to the potential off-target toxic and side effects. For example, roscovitine, one of the best known Cdk inhibitors, have been shown promising protective effects in nervous system through inhibiting Cdk5. However, roscovitine can also inhibit Cdk1, Cdk2, Cdk7, and Cdk9, the low selectivity for Cdk5 reduces its potential as a drug candidate targeting neurological disorders.
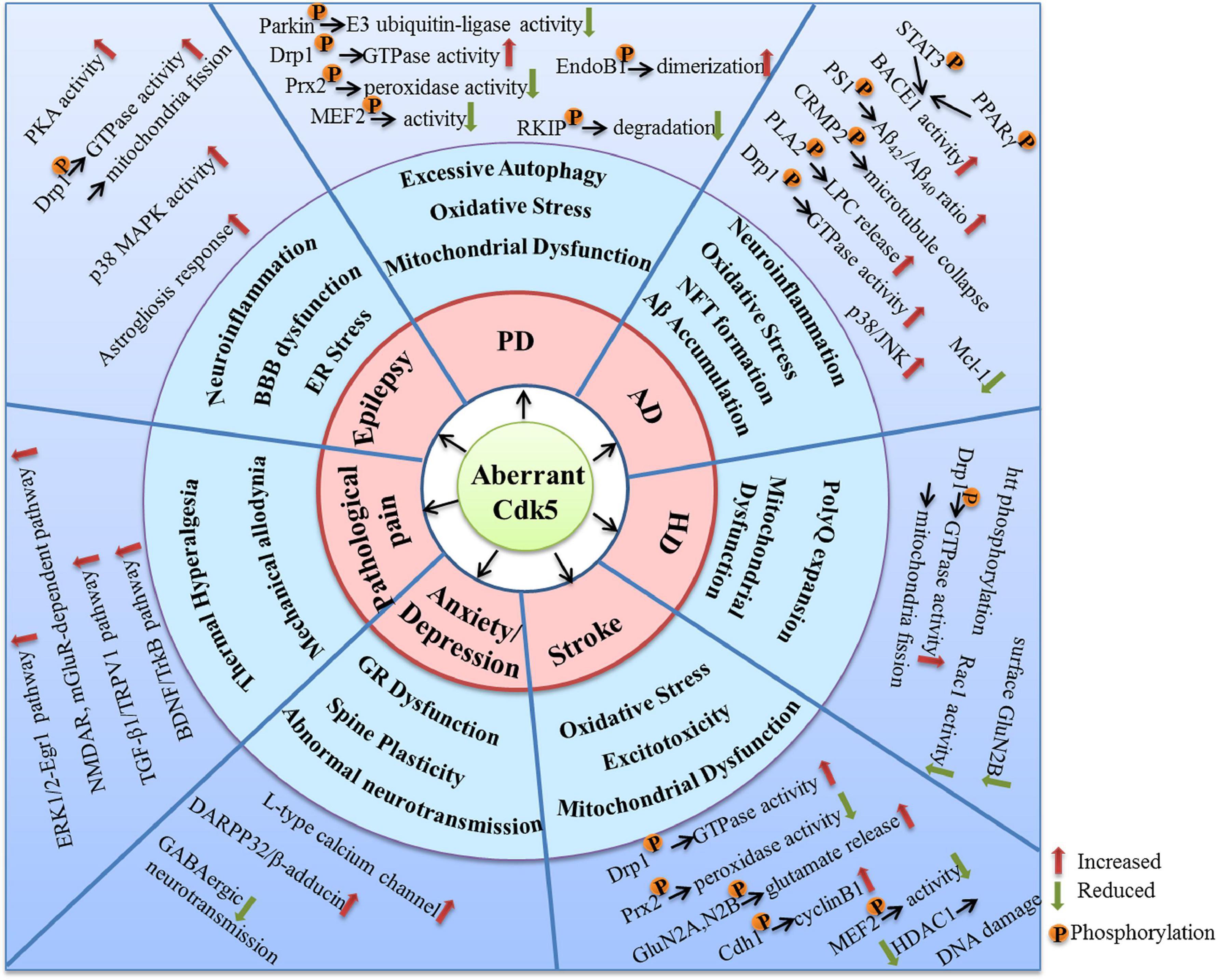
Figure 3. The molecular targets and mechanisms involved in the neurological disorders caused by aberrant Cdk5 activity.
As the aberrant Cdk5 activity is generally caused by p25 accumulation, so small molecule protein-protein interaction (PPI) inhibitors targeted to disturb the interactions between Cdk5 and p25 may represent a promising strategy. It can not only eliminate Cdk5/p25 complex-induced hyper-activation but also retain the physiological function of Cdk5/p35. In addition, there is no concern about the selectivity, as the inhibitors are developed to target the specific PPI site of Cdk5 and p25 without affecting other Cdks kinase activity. However, it can be predicted that the development of any kind of drugs, including the small molecule PPI inhibitors, will not always be smooth. But it may shed light upon the treatment of Cdk5-related neurological diseases. Future extensive studies are still needed to define more precisely the role of Cdk5 hyperactivation in brain function impairments, and it will facilitate the discovery of more effective therapeutic strategies targeting Cdk5 aberrant activity implicated in neurological disorders.
Author contributions
ZT and JT designed the review. JT and X-QW searched the literature. ZT and BF made the figures and table. ZT, X-QW, BF, and JT wrote and edited the original draft. All authors have read and agreed to the published version of the manuscript.
Funding
Work in our laboratories was supported by National Natural Science Foundation of China (No. 81801329), Chongqing Natural Science Foundation (No. cstc2021jcyj-msxmX0610), Science and Technology Research Project of Chongqing Education Commission (No. KJQN202100213), Fundamental Research Funds for the Central Universities (No. SWU-KQ22018) to ZT, and Guangdong Basic and Applied Basic Research Foundation (No. 2020A1515110564) to X-QW.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adler, V., Yin, Z., Fuchs, S. Y., Benezra, M., Rosario, L., Tew, K. D., et al. (1999). Regulation of JNK signaling by GSTp. EMBO J. 18, 1321–1334. doi: 10.1093/emboj/18.5.1321
Alvarez-Periel, E., Puigdellivol, M., Brito, V., Plattner, F., Bibb, J. A., Alberch, J., et al. (2018). Cdk5 contributes to Huntington’s disease learning and memory deficits via modulation of brain region-specific substrates. Mol. Neurobiol. 55, 6250–6268. doi: 10.1007/s12035-017-0828-4
Alvira, D., Ferrer, I., Gutierrez-Cuesta, J., Garcia-Castro, B., Pallas, M., and Camins, A. (2008). Activation of the calpain/cdk5/p25 pathway in the girus cinguli in Parkinson’s disease. Parkinsonism Relat. Disord. 14, 309–313. doi: 10.1016/j.parkreldis.2007.09.005
Anglade, P., Vyas, S., Javoy-Agid, F., Herrero, M. T., Michel, P. P., Marquez, J., et al. (1997). Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 12, 25–31.
Anne, S. L., Saudou, F., and Humbert, S. (2007). Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J. Neurosci. 27, 7318–7328. doi: 10.1523/JNEUROSCI.1831-07.2007
Apel, K., and Hirt, H. (2004). Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 55, 373–399. doi: 10.1146/annurev.arplant.55.031903.141701
Arbour, N., Vanderluit, J. L., Le Grand, J. N., Jahani-Asl, A., Ruzhynsky, V. A., Cheung, E. C., et al. (2008). Mcl-1 is a key regulator of apoptosis during CNS development and after DNA damage. J. Neurosci. 28, 6068–6078. doi: 10.1523/JNEUROSCI.4940-07.2008
Arias-Vasquez, A., Aulchenko, Y. S., Isaacs, A., van Oosterhout, A., Sleegers, K., Hofman, A., et al. (2008). Cyclin-dependent kinase 5 is associated with risk for Alzheimer’s disease in a Dutch population-based study. J. Neurol. 255, 655–662. doi: 10.1007/s00415-008-0770-5
Asada, A., Yamamoto, N., Gohda, M., Saito, T., Hayashi, N., and Hisanaga, S. (2008). Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J. Neurochem. 106, 1325–1336. doi: 10.1111/j.1471-4159.2008.05500.x
Avraham, E., Rott, R., Liani, E., Szargel, R., and Engelender, S. (2007). Phosphorylation of Parkin by the cyclin-dependent kinase 5 at the linker region modulates its ubiquitin-ligase activity and aggregation. J. Biol. Chem. 282, 12842–12850. doi: 10.1074/jbc.M608243200
Bacigaluppi, M., and Hermann, D. M. (2008). New targets of neuroprotection in ischemic stroke. ScientificWorldJournal 8, 698–712. doi: 10.1100/tsw.2008.94
Banerjee, J., Srivastava, A., Sharma, D., Dey, S., Tripathi, M., Sharma, M. C., et al. (2021). Differential regulation of excitatory synaptic transmission in the hippocampus and anterior temporal lobe by cyclin dependent kinase 5 (Cdk5) in mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE-HS). Neurosci. Lett. 761:136096. doi: 10.1016/j.neulet.2021.136096
Bavley, C. C., Fischer, D. K., Rizzo, B. K., and Rajadhyaksha, A. M. (2017). Cav1.2 channels mediate persistent chronic stress-induced behavioral deficits that are associated with prefrontal cortex activation of the p25/Cdk5-glucocorticoid receptor pathway. Neurobiol. Stress 7, 27–37. doi: 10.1016/j.ynstr.2017.02.004
Bignante, E. A., Rodriguez Manzanares, P. A., Mlewski, E. C., Bertotto, M. E., Bussolino, D. F., Paglini, G., et al. (2008). Involvement of septal Cdk5 in the emergence of excessive anxiety induced by stress. Eur. Neuropsychopharmacol. 18, 578–588. doi: 10.1016/j.euroneuro.2008.02.007
Binukumar, B. K., Shukla, V., Amin, N. D., Grant, P., Bhaskar, M., Skuntz, S., et al. (2015). Peptide TFP5/TP5 derived from Cdk5 activator P35 provides neuroprotection in the MPTP model of Parkinson’s disease. Mol. Biol. Cell 26, 4478–4491. doi: 10.1091/mbc.E15-06-0415
Bossy-Wetzel, E., Schwarzenbacher, R., and Lipton, S. A. (2004). Molecular pathways to neurodegeneration. Nat. Med. 10, Suppl S2–S9. doi: 10.1038/nm1067
Brito, V., Giralt, A., Masana, M., Royes, A., Espina, M., Sieiro, E., et al. (2019). Cyclin-dependent kinase 5 dysfunction contributes to depressive-like behaviors in Huntington’s disease by altering the DARPP-32 phosphorylation status in the nucleus accumbens. Biol. Psychiatry 86, 196–207. doi: 10.1016/j.biopsych.2019.03.001
Buchner, A., Krumova, P., Ganesan, S., Bahr, M., Eckermann, K., and Weishaupt, J. H. (2015). Sumoylation of p35 modulates p35/cyclin-dependent kinase (Cdk) 5 complex activity. Neuromolecular Med. 17, 12–23. doi: 10.1007/s12017-014-8336-4
Cancino, G. I., Perez de Arce, K., Castro, P. U., Toledo, E. M., von Bernhardi, R., and Alvarez, A. R. (2011). c-Abl tyrosine kinase modulates tau pathology and Cdk5 phosphorylation in AD transgenic mice. Neurobiol. Aging 32, 1249–1261. doi: 10.1016/j.neurobiolaging.2009.07.007
Castro-Alvarez, J. F., Uribe-Arias, S. A., Kosik, K. S., and Cardona-Gomez, G. P. (2014). Long- and short-term CDK5 knockdown prevents spatial memory dysfunction and tau pathology of triple transgenic Alzheimer’s mice. Front. Aging Neurosci. 6:243. doi: 10.3389/fnagi.2014.00243
Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T. A., Levine, J. D., and Julius, D. (1997). The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 389, 816–824. doi: 10.1038/39807
Chae, T., Kwon, Y. T., Bronson, R., Dikkes, P., Li, E., and Tsai, L. H. (1997). Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron 18, 29–42. doi: 10.1016/s0896-6273(01)80044-1
Chang, C. H., Peng, H. Y., Wu, H. C., Lai, C. Y., Hsieh, M. C., and Lin, T. B. (2011). Cyclophosphamide induces NR2B phosphorylation-dependent facilitation on spinal reflex potentiation. Am. J. Physiol. Renal Physiol. 300, F692–F699. doi: 10.1152/ajprenal.00531.2010
Chang, K. H., de Pablo, Y., Lee, H. P., Lee, H. G., Smith, M. A., and Shah, K. (2010). Cdk5 is a major regulator of p38 cascade: Relevance to neurotoxicity in Alzheimer’s disease. J. Neurochem. 113, 1221–1229. doi: 10.1111/j.1471-4159.2010.06687.x
Chavez-Gutierrez, L., Bammens, L., Benilova, I., Vandersteen, A., Benurwar, M., Borgers, M., et al. (2012). The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274. doi: 10.1038/emboj.2012.79
Chen, C., Peng, X., Tang, J., Hu, Z., Tan, J., and Zeng, L. (2021). CDK5 inhibition protects against OGDR induced mitochondrial fragmentation and apoptosis through regulation of Drp1S616 phosphorylation. Life Sci. 269:119062. doi: 10.1016/j.lfs.2021.119062
Cherubini, M., Puigdellivol, M., Alberch, J., and Gines, S. (2015). Cdk5-mediated mitochondrial fission: A key player in dopaminergic toxicity in Huntington’s disease. Biochim. Biophys. Acta 1852(10 Pt A) 2145–2160. doi: 10.1016/j.bbadis.2015.06.025
Cho, D. H., Nakamura, T., Fang, J., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105. doi: 10.1126/science.1171091
Cole, A. R., Noble, W., van Aalten, L., Plattner, F., Meimaridou, R., Hogan, D., et al. (2007). Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. J. Neurochem. 103, 1132–1144. doi: 10.1111/j.1471-4159.2007.04829.x
Cruz, C. D., Neto, F. L., Castro-Lopes, J., McMahon, S. B., and Cruz, F. (2005). Inhibition of ERK phosphorylation decreases nociceptive behaviour in monoarthritic rats. Pain 116, 411–419. doi: 10.1016/j.pain.2005.05.031
Davis, J. B., Gray, J., Gunthorpe, M. J., Hatcher, J. P., Davey, P. T., Overend, P., et al. (2000). Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405, 183–187. doi: 10.1038/35012076
Dhavan, R., and Tsai, L. H. (2001). A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2, 749–759. doi: 10.1038/35096019
Farooqui, A. A., Ong, W. Y., and Horrocks, L. A. (2006). Inhibitors of brain phospholipase A2 activity: Their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol. Rev. 58, 591–620. doi: 10.1124/pr.58.3.7
Foster, M. W., Forrester, M. T., and Stamler, J. S. (2009). A protein microarray-based analysis of S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 106, 18948–18953. doi: 10.1073/pnas.0900729106
Fraser, P. E., Yang, D. S., Yu, G., Levesque, L., Nishimura, M., Arawaka, S., et al. (2000). Presenilin structure, function and role in Alzheimer disease. Biochim. Biophys. Acta 1502, 1–15. doi: 10.1016/S0925-4439(00)00028-4
Fukata, Y., Itoh, T. J., Kimura, T., Menager, C., Nishimura, T., Shiromizu, T., et al. (2002). CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583–591. doi: 10.1038/ncb825
Gomez, K., Calderon-Rivera, A., Sandoval, A., Gonzalez-Ramirez, R., Vargas-Parada, A., Ojeda-Alonso, J., et al. (2020). Cdk5-dependent phosphorylation of CaV3.2 T-type channels: Possible role in nerve ligation-induced neuropathic allodynia and the compound action potential in primary afferent C fibers. J. Neurosci. 40, 283–296. doi: 10.1523/JNEUROSCI.0181-19.2019
Gong, X., Tang, X., Wiedmann, M., Wang, X., Peng, J., Zheng, D., et al. (2003). Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 38, 33–46. doi: 10.1016/s0896-6273(03)00191-0
Granovsky, A. E., and Rosner, M. R. (2008). Raf kinase inhibitory protein: A signal transduction modulator and metastasis suppressor. Cell Res. 18, 452–457. doi: 10.1038/cr.2008.43
Guo, M. Y., Shang, L., Hu, Y. Y., Jiang, L. P., Wan, Y. Y., Zhou, Q. Q., et al. (2018). The role of Cdk5-mediated Drp1 phosphorylation in Abeta1-42 induced mitochondrial fission and neuronal apoptosis. J. Cell Biochem. 119, 4815–4825. doi: 10.1002/jcb.26680
Hellmich, M. R., Pant, H. C., Wada, E., and Battey, J. F. (1992). Neuronal cdc2-like kinase: A cdc2-related protein kinase with predominantly neuronal expression. Proc. Natl. Acad. Sci. U.S.A. 89, 10867–10871. doi: 10.1073/pnas.89.22.10867
Hu, M., Doyle, A. D., Yamada, K. M., and Kulkarni, A. B. (2022). Visualization of trigeminal ganglion sensory neuronal signaling regulated by Cdk5. Cell Rep. 38:110458. doi: 10.1016/j.celrep.2022.110458
Huang, Y., Huang, W., Huang, Y., Song, P., Zhang, M., Zhang, H. T., et al. (2020). Cdk5 inhibitory peptide prevents loss of neurons and alleviates behavioral changes in p25 transgenic mice. J. Alzheimers Dis. 74, 1231–1242. doi: 10.3233/JAD-191098
Hyun, H. W., Min, S. J., and Kim, J. E. (2017). CDK5 inhibitors prevent astroglial apoptosis and reactive astrogliosis by regulating PKA and DRP1 phosphorylations in the rat Hippocampus. Neurosci. Res. 119, 24–37. doi: 10.1016/j.neures.2017.01.006
Ino, H., Ishizuka, T., Chiba, T., and Tatibana, M. (1994). Expression of CDK5 (PSSALRE kinase), a neural cdc2-related protein kinase, in the mature and developing mouse central and peripheral nervous systems. Brain Res. 661, 196–206. doi: 10.1016/0006-8993(94)91197-5
Ji, R. R., Befort, K., Brenner, G. J., and Woolf, C. J. (2002). ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J. Neurosci. 22, 478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002
Kaminosono, S., Saito, T., Oyama, F., Ohshima, T., Asada, A., Nagai, Y., et al. (2008). Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability. J. Neurosci. 28, 8747–8755. doi: 10.1523/JNEUROSCI.0973-08.2008
Kawauchi, T., Chihama, K., Nabeshima, Y., and Hoshino, M. (2006). Cdk5 phosphorylates and stabilizes p27kip1 contributing to actin organization and cortical neuronal migration. Nat. Cell Biol. 8, 17–26. doi: 10.1038/ncb1338
Kim, D., Frank, C. L., Dobbin, M. M., Tsunemoto, R. K., Tu, W., Peng, P. L., et al. (2008). Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 60, 803–817. doi: 10.1016/j.neuron.2008.10.015
Kim, J. E., Park, H., Choi, S. H., Kong, M. J., and Kang, T. C. (2019). Roscovitine Attenuates microglia activation and monocyte infiltration via p38 MAPK inhibition in the rat frontoparietal cortex following status epilepticus. Cells 8:746. doi: 10.3390/cells8070746
Kitazawa, M., Oddo, S., Yamasaki, T. R., Green, K. N., and LaFerla, F. M. (2005). Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 25, 8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005
Ko, J., Humbert, S., Bronson, R. T., Takahashi, S., Kulkarni, A. B., Li, E., et al. (2001). p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J. Neurosci. 21, 6758–6771. doi: 10.1523/JNEUROSCI.21-17-06758.2001
Kobayashi, H., Saito, T., Sato, K., Furusawa, K., Hosokawa, T., Tsutsumi, K., et al. (2014). Phosphorylation of cyclin-dependent kinase 5 (Cdk5) at Tyr-15 is inhibited by Cdk5 activators and does not contribute to the activation of Cdk5. J. Biol. Chem. 289, 19627–19636. doi: 10.1074/jbc.M113.501148
Lagger, G., O’Carroll, D., Rembold, M., Khier, H., Tischler, J., Weitzer, G., et al. (2002). Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 21, 2672–2681. doi: 10.1093/emboj/21.11.2672
Lai, K. O., Wong, A. S., Cheung, M. C., Xu, P., Liang, Z., Lok, K. C., et al. (2012). TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat. Neurosci. 15, 1506–1515. doi: 10.1038/nn.3237
Lau, K. F., Howlett, D. R., Kesavapany, S., Standen, C. L., Dingwall, C., McLoughlin, D. M., et al. (2002). Cyclin-dependent kinase-5/p35 phosphorylates Presenilin 1 to regulate carboxy-terminal fragment stability. Mol. Cell Neurosci. 20, 13–20. doi: 10.1006/mcne.2002.1108
Lau, L. F., and Ahlijanian, M. K. (2003). Role of cdk5 in the pathogenesis of Alzheimer’s disease. Neurosignals 12, 209–214. doi: 10.1159/000074622
Lee, D. S., and Kim, J. E. (2021). Regional specific activations of ERK1/2 and CDK5 differently regulate astroglial responses to ER stress in the rat hippocampus following status epilepticus. Brain Res. 1753:147262. doi: 10.1016/j.brainres.2020.147262
Lee, J., Ko, Y. U., Chung, Y., Yun, N., Kim, M., Kim, K., et al. (2018). The acetylation of cyclin-dependent kinase 5 at lysine 33 regulates kinase activity and neurite length in hippocampal neurons. Sci. Rep. 8:13676. doi: 10.1038/s41598-018-31785-9
Lee, J., Yun, N., Kim, C., Song, M. Y., Park, K. S., and Oh, Y. J. (2014). Acetylation of cyclin-dependent kinase 5 is mediated by GCN5. Biochem. Biophys. Res. Commun. 447, 121–127. doi: 10.1016/j.bbrc.2014.03.118
Lee, M. G., and Nurse, P. (1987). Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327, 31–35. doi: 10.1038/327031a0
Lee, M. S., Kwon, Y. T., Li, M., Peng, J., Friedlander, R. M., and Tsai, L. H. (2000). Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364. doi: 10.1038/35012636
Li, G., Liu, T., Kong, X., Wang, L., and Jin, X. (2014). Hippocampal glycogen synthase kinase 3beta is critical for the antidepressant effect of cyclin-dependent kinase 5 inhibitor in rats. J. Mol. Neurosci. 54, 92–99. doi: 10.1007/s12031-014-0254-2
Li, K., Zhao, G. Q., Li, L. Y., Wu, G. Z., and Cui, S. S. (2014). Epigenetic upregulation of Cdk5 in the dorsal horn contributes to neuropathic pain in rats. Neuroreport 25, 1116–1121. doi: 10.1097/WNR.0000000000000237
Li, W., Allen, M. E., Rui, Y., Ku, L., Liu, G., Bankston, A. N., et al. (2016). p39 is responsible for increasing Cdk5 activity during postnatal neuron differentiation and governs neuronal network formation and epileptic responses. J. Neurosci. 36, 11283–11294. doi: 10.1523/JNEUROSCI.1155-16.2016
Liu, X. X., Yang, L., Shao, L. X., He, Y., Wu, G., Bao, Y. H., et al. (2020). Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2-mediated reactive astrogliosis. J. Exp. Med. 217:e20180992. doi: 10.1084/jem.20180992
Liu, X., Liu, Y., Zhang, J., Zhang, W., Sun, Y. E., Gu, X., et al. (2014). Intrathecal administration of roscovitine prevents remifentanil-induced postoperative hyperalgesia and decreases the phosphorylation of N-methyl-D-aspartate receptor and metabotropic glutamate receptor 5 in spinal cord. Brain Res. Bull. 106, 9–16. doi: 10.1016/j.brainresbull.2014.04.008
Love, S. (2003). Neuronal expression of cell cycle-related proteins after brain ischaemia in man. Neurosci. Lett. 353, 29–32. doi: 10.1016/j.neulet.2003.09.004
Lu, W., Ai, H., Peng, L., Wang, J. J., Zhang, B., Liu, X., et al. (2015). A novel phosphorylation site of N-methyl-D-aspartate receptor GluN2B at S1284 is regulated by Cdk5 in neuronal ischemia. Exp. Neurol. 271, 251–258. doi: 10.1016/j.expneurol.2015.06.016
Luo, S., Vacher, C., Davies, J. E., and Rubinsztein, D. C. (2005). Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: Implications for mutant huntingtin toxicity. J. Cell Biol. 169, 647–656. doi: 10.1083/jcb.200412071
MacDonald, M. E., Gines, S., Gusella, J. F., and Wheeler, V. C. (2003). Huntington’s disease. Neuromolecular Med. 4, 7–20.
Maestre, C., Delgado-Esteban, M., Gomez-Sanchez, J. C., Bolanos, J. P., and Almeida, A. (2008). Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 27, 2736–2745. doi: 10.1038/emboj.2008.195
Malumbres, M., Harlow, E., Hunt, T., Hunter, T., Lahti, J. M., Manning, G., et al. (2009). Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 11, 1275–1276.
Mao, Z., and Wiedmann, M. (1999). Calcineurin enhances MEF2 DNA binding activity in calcium-dependent survival of cerebellar granule neurons. J. Biol. Chem. 274, 31102–31107. doi: 10.1074/jbc.274.43.31102
Mao, Z., Bonni, A., Xia, F., Nadal-Vicens, M., and Greenberg, M. E. (1999). Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286, 785–790.
Metwally, E., Zhao, G., and Zhang, Y. Q. (2021). The calcium-dependent protease calpain in neuronal remodeling and neurodegeneration. Trends Neurosci. 44, 741–752.
Metzler, M., Gan, L., Mazarei, G., Graham, R. K., Liu, L., Bissada, N., et al. (2010). Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J. Neurosci. 30, 14318–14329. doi: 10.1523/JNEUROSCI.1589-10.2010
Meyer, D. A., Torres-Altoro, M. I., Tan, Z., Tozzi, A., Di Filippo, M., DiNapoli, V., et al. (2014). Ischemic stroke injury is mediated by aberrant Cdk5. J. Neurosci. 34, 8259–8267. doi: 10.1523/JNEUROSCI.4368-13.2014
Minegishi, S., Asada, A., Miyauchi, S., Fuchigami, T., Saito, T., and Hisanaga, S. (2010). Membrane association facilitates degradation and cleavage of the cyclin-dependent kinase 5 activators p35 and p39. Biochemistry 49, 5482–5493. doi: 10.1021/bi100631f
Mitic, M., Simic, I., Djordjevic, J., Radojcic, M. B., and Adzic, M. (2013). Gender-specific effects of fluoxetine on hippocampal glucocorticoid receptor phosphorylation and behavior in chronically stressed rats. Neuropharmacology 70, 100–111. doi: 10.1016/j.neuropharm.2012.12.012
Mitsios, N., Pennucci, R., Krupinski, J., Sanfeliu, C., Gaffney, J., Kumar, P., et al. (2007). Expression of cyclin-dependent kinase 5 mRNA and protein in the human brain following acute ischemic stroke. Brain Pathol. 17, 11–23. doi: 10.1111/j.1750-3639.2006.00031.x
Mount, M. P., Zhang, Y., Amini, M., Callaghan, S., Kulczycki, J., Mao, Z., et al. (2013). Perturbation of transcription factor Nur77 expression mediated by myocyte enhancer factor 2D (MEF2D) regulates dopaminergic neuron loss in response to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). J. Biol. Chem. 288, 14362–14371. doi: 10.1074/jbc.M112.439216
Mushtaq, G., Greig, N. H., Anwar, F., Al-Abbasi, F. A., Zamzami, M. A., Al-Talhi, H. A., et al. (2016). Neuroprotective mechanisms mediated by CDK5 inhibition. Curr. Pharm. Des. 22, 527–534.
Nakamura, S., Kawamoto, Y., Nakano, S., Akiguchi, I., and Kimura, J. (1997). p35nck5a and cyclin-dependent kinase 5 colocalize in Lewy bodies of brains with Parkinson’s disease. Acta Neuropathol. 94, 153–157. doi: 10.1007/s004010050687
Nestler, E. J., and Carlezon, W. A. Jr. (2006). The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 59, 1151–1159.
Nikhil, K., and Shah, K. (2017). The Cdk5-Mcl-1 axis promotes mitochondrial dysfunction and neurodegeneration in a model of Alzheimer’s disease. J. Cell Sci. 130, 3023–3039. doi: 10.1242/jcs.205666
Nishimura, Y. V., Sekine, K., Chihama, K., Nakajima, K., Hoshino, M., Nabeshima, Y., et al. (2010). Dissecting the factors involved in the locomotion mode of neuronal migration in the developing cerebral cortex. J. Biol. Chem. 285, 5878–5887. doi: 10.1074/jbc.M109.033761
Obeso, J. A., Rodriguez-Oroz, M. C., Goetz, C. G., Marin, C., Kordower, J. H., Rodriguez, M., et al. (2010). Missing pieces in the Parkinson’s disease puzzle. Nat. Med. 16, 653–661.
Ohshima, T., Hirasawa, M., Tabata, H., Mutoh, T., Adachi, T., Suzuki, H., et al. (2007). Cdk5 is required for multipolar-to-bipolar transition during radial neuronal migration and proper dendrite development of pyramidal neurons in the cerebral cortex. Development 134, 2273–2282. doi: 10.1242/dev.02854
Ohshima, T., Ward, J. M., Huh, C. G., Longenecker, G., Veeranna, Pant, H. C., et al. (1996). Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. U.S.A. 93, 11173–11178. doi: 10.1073/pnas.93.20.11173
Otth, C., Mendoza-Naranjo, A., Mujica, L., Zambrano, A., Concha, I. I., and Maccioni, R. B. (2003). Modulation of the JNK and p38 pathways by cdk5 protein kinase in a transgenic mouse model of Alzheimer’s disease. Neuroreport 14, 2403–2409. doi: 10.1097/00001756-200312190-00023
Pao, P. C., and Tsai, L. H. (2021). Three decades of Cdk5. J. Biomed. Sci. 28:79. doi: 10.1186/s12929-021-00774-y
Paoletti, P., Vila, I., Rife, M., Lizcano, J. M., Alberch, J., and Gines, S. (2008). Dopaminergic and glutamatergic signaling crosstalk in Huntington’s disease neurodegeneration: The role of p25/cyclin-dependent kinase 5. J. Neurosci. 28, 10090–10101. doi: 10.1523/JNEUROSCI.3237-08.2008
Papadopoulou, A., Siamatras, T., Delgado-Morales, R., Amin, N. D., Shukla, V., Zheng, Y. L., et al. (2015). Acute and chronic stress differentially regulate cyclin-dependent kinase 5 in mouse brain: Implications to glucocorticoid actions and major depression. Transl. Psychiatry 5:e578. doi: 10.1038/tp.2015.72
Pareek, T. K., Keller, J., Kesavapany, S., Agarwal, N., Kuner, R., Pant, H. C., et al. (2007). Cyclin-dependent kinase 5 modulates nociceptive signaling through direct phosphorylation of transient receptor potential vanilloid 1. Proc. Natl. Acad. Sci. U.S.A. 104, 660–665. doi: 10.1073/pnas.0609916104
Pareek, T. K., Keller, J., Kesavapany, S., Pant, H. C., Iadarola, M. J., Brady, R. O., et al. (2006). Cyclin-dependent kinase 5 activity regulates pain signaling. Proc. Natl. Acad. Sci. U.S.A. 103, 791–796.
Park, J., Seo, J., Won, J., Yeo, H. G., Ahn, Y. J., Kim, K., et al. (2019). Abnormal mitochondria in a non-human primate model of MPTP-induced Parkinson’s disease: Drp1 and CDK5/p25 signaling. Exp. Neurobiol. 28, 414–424. doi: 10.5607/en.2019.28.3.414
Park, J., Won, J., Seo, J., Yeo, H. G., Kim, K., Kim, Y. G., et al. (2020). Streptozotocin induces Alzheimer’s disease-like pathology in hippocampal neuronal cells via CDK5/Drp1-mediated mitochondrial fragmentation. Front. Cell Neurosci. 14:235. doi: 10.3389/fncel.2020.00235
Park, K. H. J., Franciosi, S., Parrant, K., Lu, G., and Leavitt, B. R. (2017). p35 hemizygosity activates Akt but does not improve motor function in the YAC128 mouse model of Huntington’s disease. Neuroscience 352, 79–87. doi: 10.1016/j.neuroscience.2017.03.051
Park, K. H., Lu, G., Fan, J., Raymond, L. A., and Leavitt, B. R. (2012). Decreasing levels of the cdk5 activators, p25 and p35, reduces excitotoxicity in striatal neurons. J. Huntingtons Dis. 1, 89–96.
Patrick, G. N., Zukerberg, L., Nikolic, M., de la Monte, S., Dikkes, P., and Tsai, L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622.
Paudel, H. K. (1997). Phosphorylation by neuronal cdc2-like protein kinase promotes dimerization of Tau protein in vitro. J. Biol. Chem. 272, 28328–28334. doi: 10.1074/jbc.272.45.28328
Pei, J. J., Braak, E., Braak, H., Grundke-Iqbal, I., Iqbal, K., Winblad, B., et al. (2001). Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J. Alzheimers Dis. 3, 41–48. doi: 10.3233/jad-2001-3107
Pei, J. J., Grundke-Iqbal, I., Iqbal, K., Bogdanovic, N., Winblad, B., and Cowburn, R. F. (1998). Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res. 797, 267–277.
Peters, J. M. (2002). The anaphase-promoting complex: Proteolysis in mitosis and beyond. Mol. Cell 9, 931–943.
Petrucelli, L., O’Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., et al. (2002). Parkin protects against the toxicity associated with mutant alpha-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36, 1007–1019. doi: 10.1016/s0896-6273(02)01125-x
Plattner, F., Hernandez, A., Kistler, T. M., Pozo, K., Zhong, P., Yuen, E. Y., et al. (2014). Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron 81, 1070–1083. doi: 10.1016/j.neuron.2014.01.022
Prochazkova, M., Terse, A., Amin, N. D., Hall, B., Utreras, E., Pant, H. C., et al. (2013). Activation of cyclin-dependent kinase 5 mediates orofacial mechanical hyperalgesia. Mol. Pain 9:66. doi: 10.1186/1744-8069-9-66
Qu, D., Rashidian, J., Mount, M. P., Aleyasin, H., Parsanejad, M., Lira, A., et al. (2007). Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 55, 37–52. doi: 10.1016/j.neuron.2007.05.033
Qu, J., Nakamura, T., Cao, G., Holland, E. A., McKercher, S. R., and Lipton, S. A. S. – (2011). Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc. Natl. Acad. Sci. U.S.A. 108, 14330–14335. doi: 10.1073/pnas.1105172108
Qu, J., Nakamura, T., Holland, E. A., McKercher, S. R., and Lipton, S. A. (2012). S-nitrosylation of Cdk5: Potential implications in amyloid-beta-related neurotoxicity in Alzheimer disease. Prion 6, 364–370. doi: 10.4161/pri.21250
Quan, Q., Qian, Y., Li, X., and Li, M. (2019). CDK5 Participates in amyloid-beta production by regulating PPARgamma phosphorylation in primary rat hippocampal neurons. J. Alzheimers Dis. 71, 443–460. doi: 10.3233/JAD-190026
Rahn, E. J., Guzman-Karlsson, M. C., and David Sweatt, J. (2013). Cellular, molecular, and epigenetic mechanisms in non-associative conditioning: Implications for pain and memory. Neurobiol. Learn. Mem. 105, 133–150. doi: 10.1016/j.nlm.2013.06.008
Ralat, L. A., Manevich, Y., Fisher, A. B., and Colman, R. F. (2006). Direct evidence for the formation of a complex between 1-cysteine peroxiredoxin and glutathione S-transferase pi with activity changes in both enzymes. Biochemistry 45, 360–372. doi: 10.1021/bi0520737
Rashidian, J., Rousseaux, M. W., Venderova, K., Qu, D., Callaghan, S. M., Phillips, M., et al. (2009). Essential role of cytoplasmic cdk5 and Prx2 in multiple ischemic injury models, in vivo. J. Neurosci. 29, 12497–12505. doi: 10.1523/JNEUROSCI.3892-09.2009
Rosen, J. B., and Schulkin, J. (1998). From normal fear to pathological anxiety. Psychol. Rev. 105, 325–350.
Rubio de la Torre, E., Luzon-Toro, B., Forte-Lago, I., Minguez-Castellanos, A., Ferrer, I., and Hilfiker, S. (2009). Combined kinase inhibition modulates parkin inactivation. Hum. Mol. Genet. 18, 809–823. doi: 10.1093/hmg/ddn407
Rudenko, A., Seo, J., Hu, J., Su, S. C., de Anda, F. C., Durak, O., et al. (2015). Loss of cyclin-dependent kinase 5 from parvalbumin interneurons leads to hyperinhibition, decreased anxiety, and memory impairment. J. Neurosci. 35, 2372–2383. doi: 10.1523/JNEUROSCI.0969-14.2015
Sadleir, K. R., and Vassar, R. (2012). Cdk5 protein inhibition and Abeta42 increase BACE1 protein level in primary neurons by a post-transcriptional mechanism: Implications of CDK5 as a therapeutic target for Alzheimer disease. J. Biol. Chem. 287, 7224–7235. doi: 10.1074/jbc.M111.333914
Saito, T., Oba, T., Shimizu, S., Asada, A., Iijima, K. M., and Ando, K. (2019). Cdk5 increases MARK4 activity and augments pathological tau accumulation and toxicity through tau phosphorylation at Ser262. Hum. Mol. Genet. 28, 3062–3071. doi: 10.1093/hmg/ddz120
Sandford, J. J., Argyropoulos, S. V., and Nutt, D. J. (2000). The psychobiology of anxiolytic drugs. Part 1: Basic neurobiology. Pharmacol. Ther. 88, 197–212. doi: 10.1016/s0163-7258(00)00082-6
Sandoval, R., Lazcano, P., Ferrari, F., Pinto-Pardo, N., Gonzalez-Billault, C., and Utreras, E. (2018). TNF-alpha increases production of reactive oxygen species through Cdk5 activation in nociceptive neurons. Front. Physiol. 9:65. doi: 10.3389/fphys.2018.00065
Sasaki, Y., Cheng, C., Uchida, Y., Nakajima, O., Ohshima, T., Yagi, T., et al. (2002). Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron 35, 907–920. doi: 10.1016/s0896-6273(02)00857-7
Sasamoto, K., Nagai, J., Nakabayashi, T., He, X., and Ohshima, T. (2017). Cdk5 is required for the positioning and survival of GABAergic neurons in developing mouse striatum. Dev. Neurobiol. 77, 483–492. doi: 10.1002/dneu.22424
Schneider, J. A., Arvanitakis, Z., Yu, L., Boyle, P. A., Leurgans, S. E., and Bennett, D. A. (2012). Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 135(Pt 10) 3005–3014. doi: 10.1093/brain/aws234
Sen, A., Thom, M., Martinian, L., Jacobs, T., Nikolic, M., and Sisodiya, S. M. (2006). Deregulation of cdk5 in Hippocampal sclerosis. J. Neuropathol. Exp. Neurol. 65, 55–66.
Shah, K., and Lahiri, D. K. (2014). Cdk5 activity in the brain–multiple paths of regulation. J. Cell Sci. 127(Pt 11) 2391–2400. doi: 10.1242/jcs.147553
Shukla, V., Seo, J., Binukumar, B. K., Amin, N. D., Reddy, P., Grant, P., et al. (2017). TFP5, a peptide inhibitor of aberrant and hyperactive Cdk5/p25, attenuates pathological phenotypes and restores synaptic function in CK-p25Tg mice. J. Alzheimers Dis. 56, 335–349. doi: 10.3233/JAD-160916
Shukla, V., Zheng, Y. L., Mishra, S. K., Amin, N. D., Steiner, J., Grant, P., et al. (2013). A truncated peptide from p35, a Cdk5 activator, prevents Alzheimer’s disease phenotypes in model mice. FASEB J. 27, 174–186. doi: 10.1096/fj.12-217497
Smith, P. D., Crocker, S. J., Jackson-Lewis, V., Jordan-Sciutto, K. L., Hayley, S., Mount, M. P., et al. (2003). Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 100, 13650–13655. doi: 10.1073/pnas.2232515100
Steinbrecher, U. P., Parthasarathy, S., Leake, D. S., Witztum, J. L., and Steinberg, D. (1984). Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc. Natl. Acad. Sci. U.S.A. 81, 3883–3887.
Stillwell, T. J., and Myers, R. P. (1988). Adenomatous polyp in defunctionalized colonic segment used as a urinary bladder. Urology 32, 538–540. doi: 10.1016/s0090-4295(98)90038-3
Su, L. Y., Li, H., Lv, L., Feng, Y. M., Li, G. D., Luo, R., et al. (2015). Melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA/alpha-synuclein aggregation. Autophagy 11, 1745–1759. doi: 10.1080/15548627.2015.1082020
Su, S. C., and Tsai, L. H. (2011). Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 27, 465–491.
Sun, K. H., Chang, K. H., Clawson, S., Ghosh, S., Mirzaei, H., Regnier, F., et al. (2011). Glutathione-S-transferase P1 is a critical regulator of Cdk5 kinase activity. J. Neurochem. 118, 902–914. doi: 10.1111/j.1471-4159.2011.07343.x
Sun, K. H., de Pablo, Y., Vincent, F., and Shah, K. (2008). Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J. Neurochem. 107, 265–278. doi: 10.1111/j.1471-4159.2008.05616.x
Sun, K. H., Lee, H. G., Smith, M. A., and Shah, K. (2009). Direct and indirect roles of cyclin-dependent kinase 5 as an upstream regulator in the c-Jun NH2-terminal kinase cascade: Relevance to neurotoxic insults in Alzheimer’s disease. Mol. Biol. Cell 20, 4611–4619. doi: 10.1091/mbc.e09-05-0433
Sundaram, J. R., Chan, E. S., Poore, C. P., Pareek, T. K., Cheong, W. F., Shui, G., et al. (2012). Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J. Neurosci. 32, 1020–1034. doi: 10.1523/JNEUROSCI.5177-11.2012
Takahashi, M., Nakabayashi, T., Mita, N., Jin, X., Aikawa, Y., Sasamoto, K., et al. (2022). Involvement of Cdk5 activating subunit p35 in synaptic plasticity in excitatory and inhibitory neurons. Mol. Brain 15:37. doi: 10.1186/s13041-022-00922-x
Takahashi, Y., Coppola, D., Matsushita, N., Cualing, H. D., Sun, M., Sato, Y., et al. (2007). Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142–1151.
Tan, X., Chen, Y., Li, J., Li, X., Miao, Z., Xin, N., et al. (2015). The inhibition of Cdk5 activity after hypoxia/ischemia injury reduces infarct size and promotes functional recovery in neonatal rats. Neuroscience 290, 552–560. doi: 10.1016/j.neuroscience.2015.01.054
Tang, X., Wang, X., Gong, X., Tong, M., Park, D., Xia, Z., et al. (2005). Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J. Neurosci. 25, 4823–4834.
Thijs, R. D., Surges, R., O’Brien, T. J., and Sander, J. W. (2019). Epilepsy in adults. Lancet 393, 689–701.
Tsai, L. H., Delalle, I., Caviness, V. S. Jr., Chae, T., and Harlow, E. (1994). p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371, 419–423.
Tsai, L. H., Takahashi, T., Caviness, V. S. Jr., and Harlow, E. (1993). Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development 119, 1029–1040.
Tseng, H. C., Zhou, Y., Shen, Y., and Tsai, L. H. (2002). A survey of Cdk5 activator p35 and p25 levels in Alzheimer’s disease brains. FEBS Lett. 523, 58–62.
Uchida, Y., Ohshima, T., Sasaki, Y., Suzuki, H., Yanai, S., Yamashita, N., et al. (2005). Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: Implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes Cells 10, 165–179. doi: 10.1111/j.1365-2443.2005.00827.x
Utreras, E., Futatsugi, A., Rudrabhatla, P., Keller, J., Iadarola, M. J., Pant, H. C., et al. (2009). Tumor necrosis factor-alpha regulates cyclin-dependent kinase 5 activity during pain signaling through transcriptional activation of p35. J. Biol. Chem. 284, 2275–2284. doi: 10.1074/jbc.M805052200
Utreras, E., Keller, J., Terse, A., Prochazkova, M., Iadarola, M. J., and Kulkarni, A. B. (2012). Transforming growth factor-beta1 regulates Cdk5 activity in primary sensory neurons. J. Biol. Chem. 287, 16917–16929.
Wang, C. H., Chou, W. Y., Hung, K. S., Jawan, B., Lu, C. N., Liu, J. K., et al. (2005). Intrathecal administration of roscovitine inhibits Cdk5 activity and attenuates formalin-induced nociceptive response in rats. Acta Pharmacol. Sin. 26, 46–50. doi: 10.1111/j.1745-7254.2005.00007.x
Wang, G. Q., Cen, C., Li, C., Cao, S., Wang, N., Zhou, Z., et al. (2015). Deactivation of excitatory neurons in the prelimbic cortex via Cdk5 promotes pain sensation and anxiety. Nat. Commun. 6:7660.
Wang, J., Liu, S., Fu, Y., Wang, J. H., and Lu, Y. (2003). Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat. Neurosci. 6, 1039–1047. doi: 10.1038/nn1119
Wang, Q., Jiao, F., Zhang, P., Yan, J., Zhang, Z., He, F., et al. (2018). CDK5-mediated phosphorylation-dependent ubiquitination and degradation of E3 ubiquitin ligases GP78 accelerates neuronal death in Parkinson’s disease. Mol. Neurobiol. 55, 3709–3717. doi: 10.1007/s12035-017-0579-2
Wang, X., Ratnam, J., Zou, B., England, P. M., and Basbaum, A. I. (2009). TrkB signaling is required for both the induction and maintenance of tissue and nerve injury-induced persistent pain. J. Neurosci. 29, 5508–5515. doi: 10.1523/JNEUROSCI.4288-08.2009
Wen, Y., Yu, W. H., Maloney, B., Bailey, J., Ma, J., Marie, I., et al. (2008). Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron 57, 680–690. doi: 10.1016/j.neuron.2008.02.024
Wen, Z., Shu, Y., Gao, C., Wang, X., Qi, G., Zhang, P., et al. (2014). CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson’s disease. Neurobiol. Aging 35, 2870–2880. doi: 10.1016/j.neurobiolaging.2014.05.034
Westermann, B. (2010). Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884.
Wilkaniec, A., Gassowska-Dobrowolska, M., Strawski, M., Adamczyk, A., and Czapski, G. A. (2018). Inhibition of cyclin-dependent kinase 5 affects early neuroinflammatory signalling in murine model of amyloid beta toxicity. J. Neuroinflammation 15:1. doi: 10.1186/s12974-017-1027-y
Wong, A. S., Lee, R. H., Cheung, A. Y., Yeung, P. K., Chung, S. K., Cheung, Z. H., et al. (2011). Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat. Cell Biol. 13, 568–579. doi: 10.1038/ncb2217
Wu, D. C., Yu, Y. P., Lee, N. T., Yu, A. C., Wang, J. H., and Han, Y. F. (2000). The expression of Cdk5, p35, p39, and Cdk5 kinase activity in developing, adult, and aged rat brains. Neurochem. Res. 25, 923–929. doi: 10.1023/a:1007544106645
Xie, Z., Hou, S., Yang, X., Duan, Y., Han, J., Wang, Q., et al. (2022). Lessons learned from past cyclin-dependent kinase drug discovery efforts. J. Med. Chem. 65, 6356–6389. doi: 10.1021/acs.jmedchem.1c02190
Xu, M., Huang, Y., Song, P., Huang, Y., Huang, W., Zhang, H. T., et al. (2019). AAV9-mediated Cdk5 inhibitory peptide reduces hyperphosphorylated tau and inflammation and ameliorates behavioral changes caused by overexpression of p25 in the brain. J. Alzheimers Dis. 70, 573–585. doi: 10.3233/JAD-190099
Yamada, M., Saito, T., Sato, Y., Kawai, Y., Sekigawa, A., Hamazumi, Y., et al. (2007). Cdk5–p39 is a labile complex with the similar substrate specificity to Cdk5–p35. J. Neurochem. 102, 1477–1487. doi: 10.1111/j.1471-4159.2007.04505.x
Yang, L., Gu, X., Zhang, W., Zhang, J., and Ma, Z. (2014). Cdk5 inhibitor roscovitine alleviates neuropathic pain in the dorsal root ganglia by downregulating N-methyl-D-aspartate receptor subunit 2A. Neurol. Sci. 35, 1365–1371. doi: 10.1007/s10072-014-1713-9
Yang, S. H., Huang, C. Y., Hsieh, C. Y., and Chuang, J. I. (2020). CDK4 and CDK5 inhibition have comparable mild hypothermia effects in preventing Drp1-Dependent mitochondrial fission and neuron death induced by MPP. Mol. Neurobiol. 57, 4090–4105. doi: 10.1007/s12035-020-02014-0
Yang, Y. R., He, Y., Zhang, Y., Li, Y., Li, Y., Han, Y., et al. (2007). Activation of cyclin-dependent kinase 5 (Cdk5) in primary sensory and dorsal horn neurons by peripheral inflammation contributes to heat hyperalgesia. Pain 127, 109–120. doi: 10.1016/j.pain.2006.08.008
Ye, T., Ip, J. P., Fu, A. K., and Ip, N. Y. (2014). Cdk5-mediated phosphorylation of RapGEF2 controls neuronal migration in the developing cerebral cortex. Nat. Commun. 5:4826. doi: 10.1038/ncomms5826
Yeung, K., Seitz, T., Li, S., Janosch, P., McFerran, B., Kaiser, C., et al. (1999). Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 401, 173–177.
Yoshida, H., Hastie, C. J., McLauchlan, H., Cohen, P., and Goedert, M. (2004). Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J. Neurochem. 90, 352–358. doi: 10.1111/j.1471-4159.2004.02479.x
Yoshida, H., Watanabe, A., and Ihara, Y. (1998). Collapsin response mediator protein-2 is associated with neurofibrillary tangles in Alzheimer’s disease. J. Biol. Chem. 273, 9761–9768.
Zhang, H. H., Zhang, X. Q., Xue, Q. S., Yan, L., Huang, J. L., Zhang, S., et al. (2014). The BDNF/TrkB signaling pathway is involved in heat hyperalgesia mediated by Cdk5 in rats. PLoS One 9:e85536. doi: 10.1371/journal.pone.0085536
Zhang, L., Liu, W., Szumlinski, K. K., and Lew, J. (2012). p10, the N-terminal domain of p35, protects against CDK5/p25-induced neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 109, 20041–20046. doi: 10.1073/pnas.1212914109
Zhang, P., Fu, W. Y., Fu, A. K., and Ip, N. Y. (2015). S-nitrosylation-dependent proteasomal degradation restrains Cdk5 activity to regulate hippocampal synaptic strength. Nat. Commun. 6:8665. doi: 10.1038/ncomms9665
Zhang, P., Shao, X. Y., Qi, G. J., Chen, Q., Bu, L. L., Chen, L. J., et al. (2016). Cdk5-dependent activation of neuronal inflammasomes in Parkinson’s disease. Mov. Disord. 31, 366–376. doi: 10.1002/mds.26488
Zhang, P., Yu, P. C., Tsang, A. H., Chen, Y., Fu, A. K., Fu, W. Y., et al. (2010). S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development. J. Neurosci. 30, 14366–14370. doi: 10.1523/JNEUROSCI.3899-10.2010
Zhang, Q., Xie, H., Ji, Z., He, R., Xu, M., He, Y., et al. (2016). Cdk5/p25 specific inhibitory peptide TFP5 rescues the loss of dopaminergic neurons in a sub-acute MPTP induced PD mouse model. Neurosci. Lett. 632, 1–7. doi: 10.1016/j.neulet.2016.08.023
Zhang, R., Liu, Y., Zhang, J., Zheng, Y., Gu, X., and Ma, Z. (2012). Intrathecal administration of roscovitine attenuates cancer pain and inhibits the expression of NMDA receptor 2B subunit mRNA. Pharmacol. Biochem. Behav. 102, 139–145. doi: 10.1016/j.pbb.2012.03.025
Zhang, S., Edelmann, L., Liu, J., Crandall, J. E., and Morabito, M. A. (2008). Cdk5 regulates the phosphorylation of tyrosine 1472 NR2B and the surface expression of NMDA receptors. J. Neurosci. 28, 415–424. doi: 10.1523/JNEUROSCI.1900-07.2008
Zhang, X., Zhang, H., Shao, H., Xue, Q., and Yu, B. (2014). ERK MAP kinase activation in spinal cord regulates phosphorylation of Cdk5 at serine 159 and contributes to peripheral inflammation induced pain/hypersensitivity. PLoS One 9:e87788. doi: 10.1371/journal.pone.0087788
Zhao, Y., Wang, C., He, W., and Cai, Z. (2021). Ameliorating Alzheimer’s-like pathology by Minocycline via inhibiting Cdk5/p25 signaling. Curr. Neuropharmacol. 20, 1783–1792. doi: 10.2174/1570159X19666211202124925
Zhou, M., Wang, H., Zeng, X., Yin, P., Zhu, J., Chen, W., et al. (2019). Mortality, morbidity, and risk factors in China and its provinces, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 394, 1145–1158.
Zhou, Y. F., Wang, J., Deng, M. F., Chi, B., Wei, N., Chen, J. G., et al. (2019). The Peptide-directed lysosomal degradation of CDK5 exerts therapeutic effects against stroke. Aging Dis. 10, 1140–1145. doi: 10.14336/AD.2018.1225
Keywords: Cdk5, p35, neurological disorders, Parkinson’s disease, Alzheimer’s disease, stroke
Citation: Tian Z, Feng B, Wang X-Q and Tian J (2022) Focusing on cyclin-dependent kinases 5: A potential target for neurological disorders. Front. Mol. Neurosci. 15:1030639. doi: 10.3389/fnmol.2022.1030639
Received: 29 August 2022; Accepted: 25 October 2022;
Published: 10 November 2022.
Edited by:
Federica Bono, University of Brescia, ItalyCopyright © 2022 Tian, Feng, Wang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen Tian, emhlbnRpYW56aEAxNjMuY29t; Jiao Tian, amlhb3RpYW5qaTA3QDE2My5jb20=
†These authors share first authorship