 Jing-Yang Wang1,2,3
Jing-Yang Wang1,2,3 Bin Tang1,2Wen-Xiang Sheng1,2Li-Dong Hua4Yang Zeng1,2Cui-Xia Fan1,2Wei-Yi Deng1,2Mei-Mei Gao1,2Wei-Wen Zhu3
Bin Tang1,2Wen-Xiang Sheng1,2Li-Dong Hua4Yang Zeng1,2Cui-Xia Fan1,2Wei-Yi Deng1,2Mei-Mei Gao1,2Wei-Wen Zhu3 Na He3
Na He3 Tao Su1,2*
Tao Su1,2*
- 1Institute of Neuroscience, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 2Key Laboratory of Neurogenetics and Channelopathies, Ministry of Education of China, Guangzhou, China
- 3Department of Neurology, The Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
- 4Translational Medicine Center, Maternal and Child Health Research Institute, Guangdong Women and Children’s Hospital, Guangzhou, China
Objective: Naturally occurring in-frame deletion is a unique type of genetic variations, causing the loss of one or more amino acids of proteins. A number of in-frame deletion variants in an epilepsy-associated gene SCN1A, encoding voltage gated sodium channel alpha unit 1.1 (Nav1.1), have been reported in public database. In contrast to the missense and truncation variants, the in-frame deletions in SCN1A remains largely uncharacterized.
Methods: We summarized the basic information of forty-four SCN1A in-frame deletion variants and performed further analysis on six variants identified in our cases with epilepsy. Mutants of the six in-frame deletions and one truncating variant used as comparison were generated and co-transfected with beta-1 and -2 subunits in tsA201 cells, followed by patch clamp recordings.
Results: Reviewing all the in-frame deletions showed that they spread over the entire Nav1.1 protein, without obvious “hot spots.” The dominant type (54%) was single residue loss. There was no obvious relationship between the length or locations of deletions and their clinical phenotypes. The six in-frame deletions were two single residue deletions (p.M400del and p.I1772del), one microdeletion (p.S128_F130del) and three macrodeletions (p.T303_R322del, p.T160_Y202del, and p.V1335_V1428del). They scatter and affect different functional domains, including transmembrane helices, pore region, and P-loop. Electrophysiological recordings revealed no measurable sodium current in all of the six mutants. In contrast, the truncating mutant p.M1619Ifs*7 that loses a long stretch of peptides retains partial function.
Significance: The complete loss-of-function in these shortened, abnormal mutants indicates that Nav1.1 protein is a highly accurate structure, and many of the residues have no redundancy to ion conductance. In-frame deletions caused particularly deleterious effect on protein function possibly due to the disruption of ordered residues.
Introduction
Voltage-gated sodium channels (Nav) are responsible for the generation and propagation of action potentials in excitable membrane. These channels are complexes of one α subunit in association with two auxiliary β subunits. In humans, there are nine functional α subunits (Nav1.1-Nav1.9) encoded by the genes SCN1A-SCN11A, with different patterns of tissue expression and biophysical properties. The α subunit of ∼2000 amino acid (AA) residues is organized in four homologous domains but non-identical domains (DI-DIV), each of which contains six transmembrane segments (S1–S6) and an additional membrane re-entrant pore loop (P-loop). Four transmembrane domains (DI–DIV) are connected by intracellular loop structures (Catterall, 2000; Goldin et al., 2000; Eijkelkamp et al., 2012).
Variants in the gene SCN1A encoding Nav1.1 α subunit have been associated with a spectrum of epilepsy disorders ranging from the relatively benign generalized epilepsy with febrile seizures plus (GEFS+) to the devastating disorder, severe myoclonic epilepsy of infancy (SMEI) (Escayg et al., 2000, 2001; Marini et al., 2009). To date, more than 1,800 epilepsy associated variants annotated for SCN1A have been reported in different databases, such as SCN1A database1, the Human Gene Mutation Database (HGMD), and the ClinVar database of NCBI. Most of these are missense variants that lead to a single amino acid substitution, while there are also a significant number of in-frame deletions and premature truncations that lacks one or more amino acids. Previous studies focused on characterizing the biophysical properties of missense and truncating variants (Lossin et al., 2003; Yamakawa, 2006; Meng et al., 2015). The in-frame deletions occurred in SCN1A have not been well characterized, except that an in-frame deletion (p.F1289del), located at DIII S3, was reported with no measurable sodium current (Ohmori et al., 2006). The naturally occurring in-frame deletions would be unique and useful models to explore the underlying biology of Nav1.1, and the genotype-phenotype relationship as well.
Here we first collected a total of 44 in-frame deletion variants in SCN1A from the HGMD and SCN1A database and characterized their features of clinical phenotypes and locations. To gain insights into sub-molecular gating network of Nav1.1, six in-frame deletions identified in our laboratory were further subject to site-directed mutagenesis experiments to determine the functional features of the shortened Nav1.1.
Materials and Methods
Public Data Collection
All available in-frame deletion variants in SCN1A (a total of 44 variants) were retrieved from the SNP database of the NCBI2, the SCN1A database (see text footnote 1) and the HGMD. The NCBI database was queried with amino acid sequences of human Nav1.1 to obtain the corresponding information of DNA locus, and related functional regions.
Genetic Testing
Diagnose and treatments of the patients were conducted in our Epilepsy Center (Guangzhou, China). Clinical data including medical records, standardized questionnaires, and EEG recordings were available. The probands were assessed using a standardized protocol after providing written informed consent. This study was approved by the Research Ethics Board of the Hospital. Genomic DNAs were prepared from ethylenediaminetetraacetic acid (EDTA)-treated whole blood samples. SCN1A were screened for genetic abnormalities. Primers were designed to amplify all exons and the flanking intronic splice sites of the gene. The purified PCR products of polymerase chain reaction were directly sequenced. The variant was verified by a second targeted PCR and sequencing. A total of six in-frame deletion variants in SCN1A were identified in our genetic testing, among which three were novel and three variants (c.383 + 1A > G/p.S128_F130del, c.602 + 1G > A/p.T160_Y202del, and c.1200_1202delGAT/p.M400del) were previously reported (Depienne et al., 2009; Selmer et al., 2009).
Mutagenesis and Heterologous Expression
To reconstitute the native brain sodium channel complex, SCN1A was co-expressed heterologously with human accessory β1 and β2 subunits in human tsA201 cells. The expression vectors of wild-type (WT) human sodium channel Nav1.1, pCMV-SCN1A-WT, pCD8-IRES-hβ1, and pGFP-IRES-hβ2 that express α, β1, and β2 subunits, were kindly donated by Professor Alfred L. George Jr. To improve the monitoring of transfection, pCD8-IRES-hβ1 had been modified into pDsred-IRES-hβ1 with red fluorescence; whereas pGFP-IRES-hβ2 expression is recognized by green fluorescence. The mutant vectors were generated from corresponding WT vectors using Quick-change site-directed mutagenesis (Stratagene, Cedar Creek, TX, United States) according to the manufacturer’s protocol. All constructs were verified by resequencing before being transfected to human tsA201 cells. The cells were grown in 1:1 Ham’s F-12 and Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, and 100 μg/ml streptomycin. The cells were maintained in a humidified incubator at 37°C with 5% CO2. Cells were then co-transfected with pCMV-SCN1A, pCD8-IRES-hβ1, and pGFP-IRES-hβ2, using Lipofectamine 3000 reagent Kit from Thermo Fisher Scientific. After incubation for 12–15 h, cells were replated in 35-mm culture dishes.
Patch Clamp Analysis
Electrophysiological studies were performed 20–48 h after transfection, according to our previous report (Chen et al., 2015). Cells displaying green and red fluorescence were chosen for recording. According to our previous experience, almost all the cells (>90%) that had green and red fluorescence were expressing a complex of co-transfected α, β1 and β2 subunits. Whole-cell patch clamp was performed according to previous reports. Sodium currents were recorded from tsA201 cells at room temperature (22–24°C). Series resistance (2.0–3.0 MΩ) was compensated 85–95% to assure that the command potential was reached within microseconds and with a voltage error of <4 mV. All data were acquired at 10–50 kHz and low-pass filtered at 5 kHz. The pipette solution contained (in mM): NaF 10, CsF 110, CsCl 20, EGTA 2, and HEPES 10, with a pH of 7.35 and osmolarity of 310 mOsM/kg. The extracellular solution contained (in mM): NaCl 145, KCl 4, CaCl2 1.8, MgCl2 1, and HEPES 10, with a pH of 7.35 and osmolarity of 310 mOsM/kg. Sodium currents were recorded with EPC10 amplifiers (HEKA Elektronik, Lambrecht, Germany). Sodium currents were recorded at various test potentials from a holding potential of −120 mV. The inward currents were validated by Nav blocker tetrodotoxin. Sodium conductance (G) was calculated according to the equation G = Ipeak/(Vtest-Vrev), where Ipeak is the peak inward current, Vtest is the test potential, and Vrev is the reversal potential for Na+. To compare voltage dependence of activation, data were fitted to a Boltzmann function, according to the equation G/Gmax = 1−1/{1 + exp[(Vm–V1/2)/k]}, where Gmax is the maximum conductance, Vm is the potential of individual step pulses, V1/2 is the average half activation potential (at which G is one-half maximal), and k is the slope factor. The voltage dependence of channel availability was assessed following a prepulse to various potentials followed by 20-ms pulse to −10 mV. The normalized current was plotted against the voltage and the inactivation curves were fit with Boltzmann functions (I/Imax = 1/(1 + exp[(Vm−V1/2)/k]) to determine the voltage for half-maximal channel inactivation (V1/2) and slope factor (k). Recovery from inactivation was determined using a two-pulse protocol. The peak current from the test pulse was normalized to the peak current from a prepulse and plotted against the recovery period. Data were fit with the two exponential function, I/Imax = Af [1−exp(−t/τf)] + As [1−exp(−t/τs)], where τf and τs denote time constants (fast and slow components, respectively).
Structural Modeling
The structures of the WT Nav1.1 and fragmental peptides (about 600 AA, deletion locus was covered) of the deletion variants were modeled to predict the effect of deletion mutations on protein structure by using I-Tasser3. PyMOL 2.3 software was used for three-dimensional protein structure visualization and analysis.
Statistical Analysis
All data were analyzed using a combination of Fit master v2.53 (HEKA Electronics, Lambrecht, Germany), Excel 2003 (Microsoft, Seattle, WA, United States), and OriginPro 8.0 (OriginLab, Northampton, MA, United States) software. For statistical evaluation, results are shown as means ± SEM, and differences between WT and mutant channels were assessed by Student’s t-test. One-way analysis of variance (ANOVA) was used to compare means among different groups. Significance was assigned at P < 0.05.
Results
Features of In-Frame Deletion Variants in SCN1A
Six in-frame deletions in SCN1A were identified in our cases with SMEI, generalized epilepsy with febrile seizures plus (GEFS+), partial epilepsy with febrile seizures plus (PEFS+) and Lennox–Gastaut syndrome (LGS) (Table 1). One variant (c.383 + 1A > G/p.S128_F130) associated with LGS and two variants (c.602 + 1G > A/p.T160_Y202del and c.1200_1202delGAT/p.M400del) associated with SMEI were reported in the ClinVar database or previous studies (Depienne et al., 2009; Selmer et al., 2009), while the other variants were novel. Residues affected by these variants are located in several distinct protein domains including the DI/S1 (c.383 + 1A > G/p.S128_F130del), DI/S2-S3 (c.602 + 1G > A/p.T160_Y202del), pore loop in DI (c.909A > G/p.T303_R322del, c.1200_1202delGAT/p.M400del), cytoplasmic DIII/S4-S5 linker to pore loops in DIII (c.4284 + 2T > C/p.V1335_V1428del), and DIV/S6 (c.5313_5315delCAT/p.I1772del) (illustrated in Figure 1).
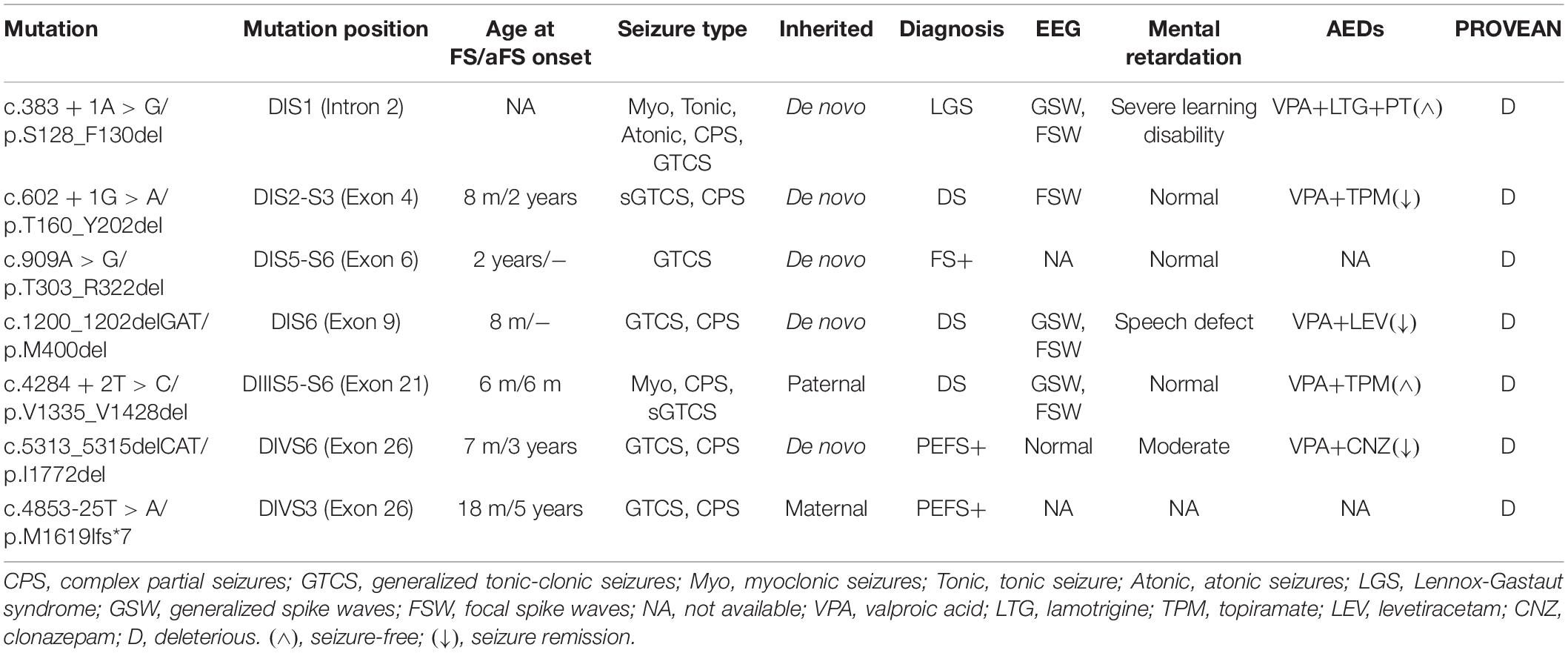
Table 1. Clinical features associated with six identified in-frame deletion and one truncation variants in SCN1A.
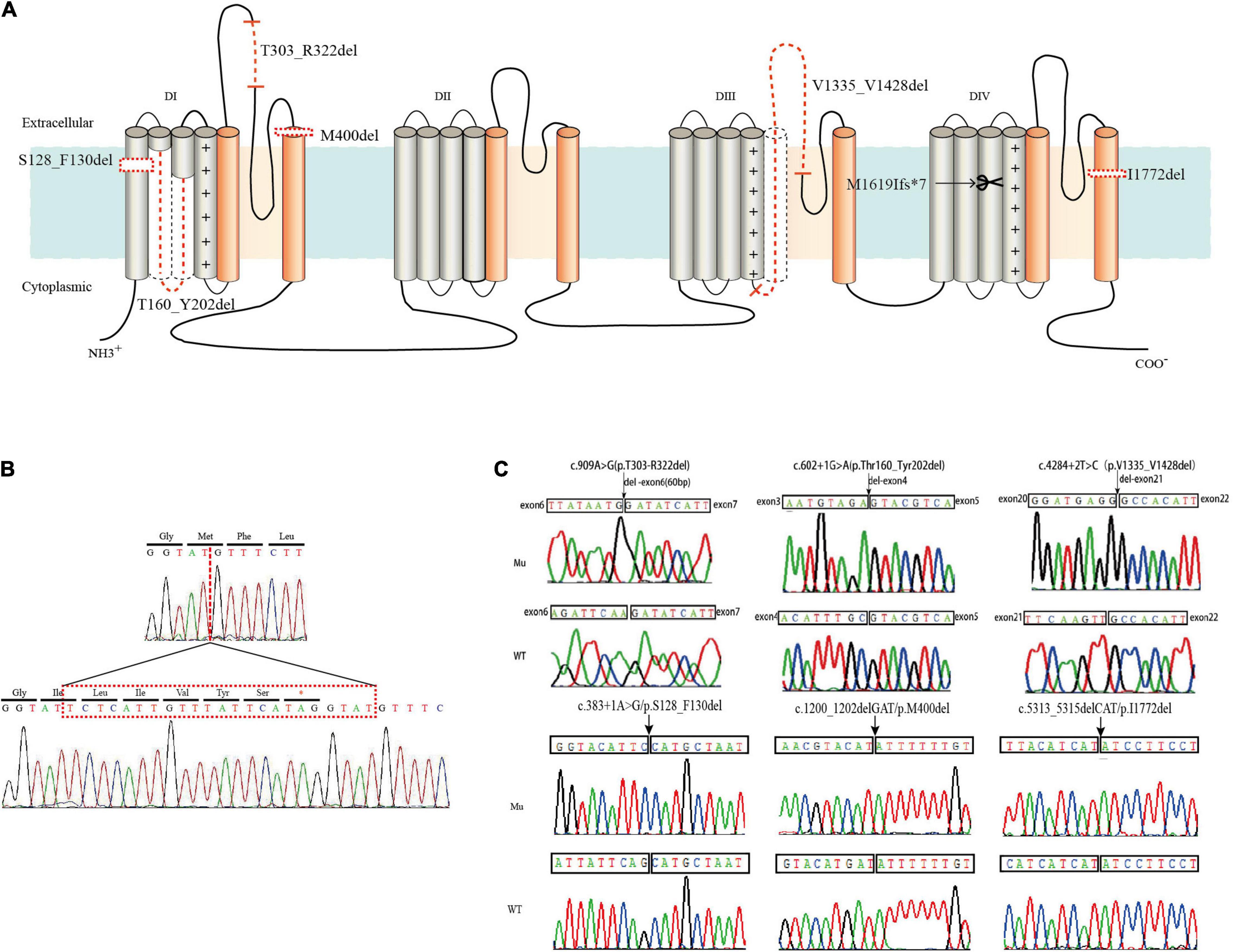
Figure 1. Locations of six in-frame deletion and one truncating variants in SCN1A and sequence identification. (A) Schematic location of the variants identified. The in-frame deletion variants were marked with dotted lines and the truncating variant was marked with a scissor. (B) DNA sequencing of the truncating variant (c.4853-25T > A/p.M1619Ifs*7). (C) DNA sequencing of the in-frame deletion variants.
We summarized all the in-frame deletion variants (a total of 44, together with the six variants identified by us) from the databases, and found that most of them (38/44) were reported as SMEI, developmental and epileptic encephalopathy (DEE) and LGS (Table 2). These variants spread over six functional domains of Nav1.1 (Figures 2A,B). Approximately 54% of the in-frame deletions are single deletions of 1 AA, followed by microdeletions (2–6 AA, 30%) and macrodeletions (>6 AA, 16%) (Figure 2C). Almost all the single deletions are associated with SMEI, DEE, except of one in DIV/S6 presented with PEFS+ Figure 2D). One microdeletion located in DI S5-S6 linker is associated with febrile seizure plus (FS+). There is no clear relationship between clinical phenotypes and the length of deletions.
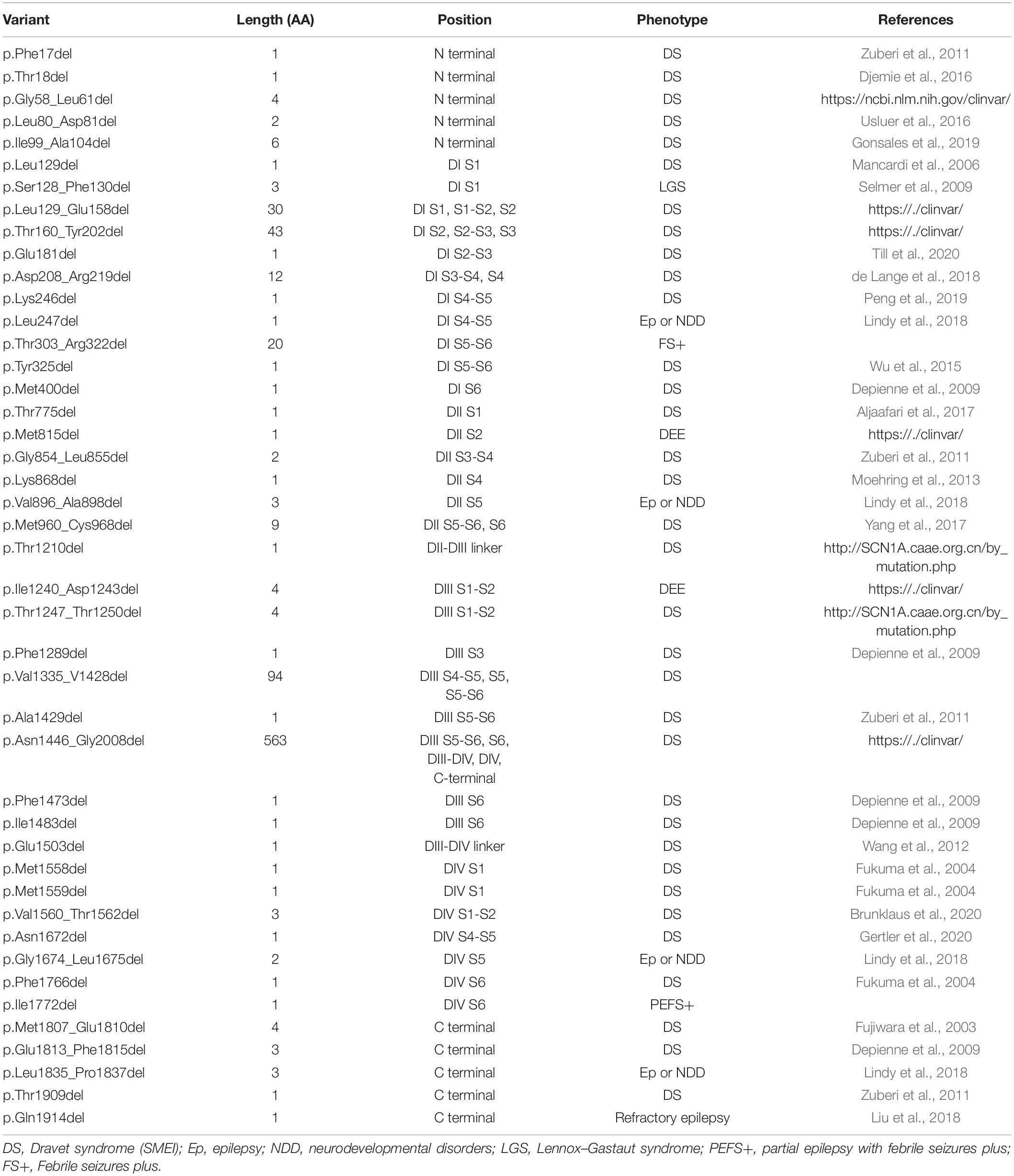
Table 2. In-frame deletion variants of SCN1A.
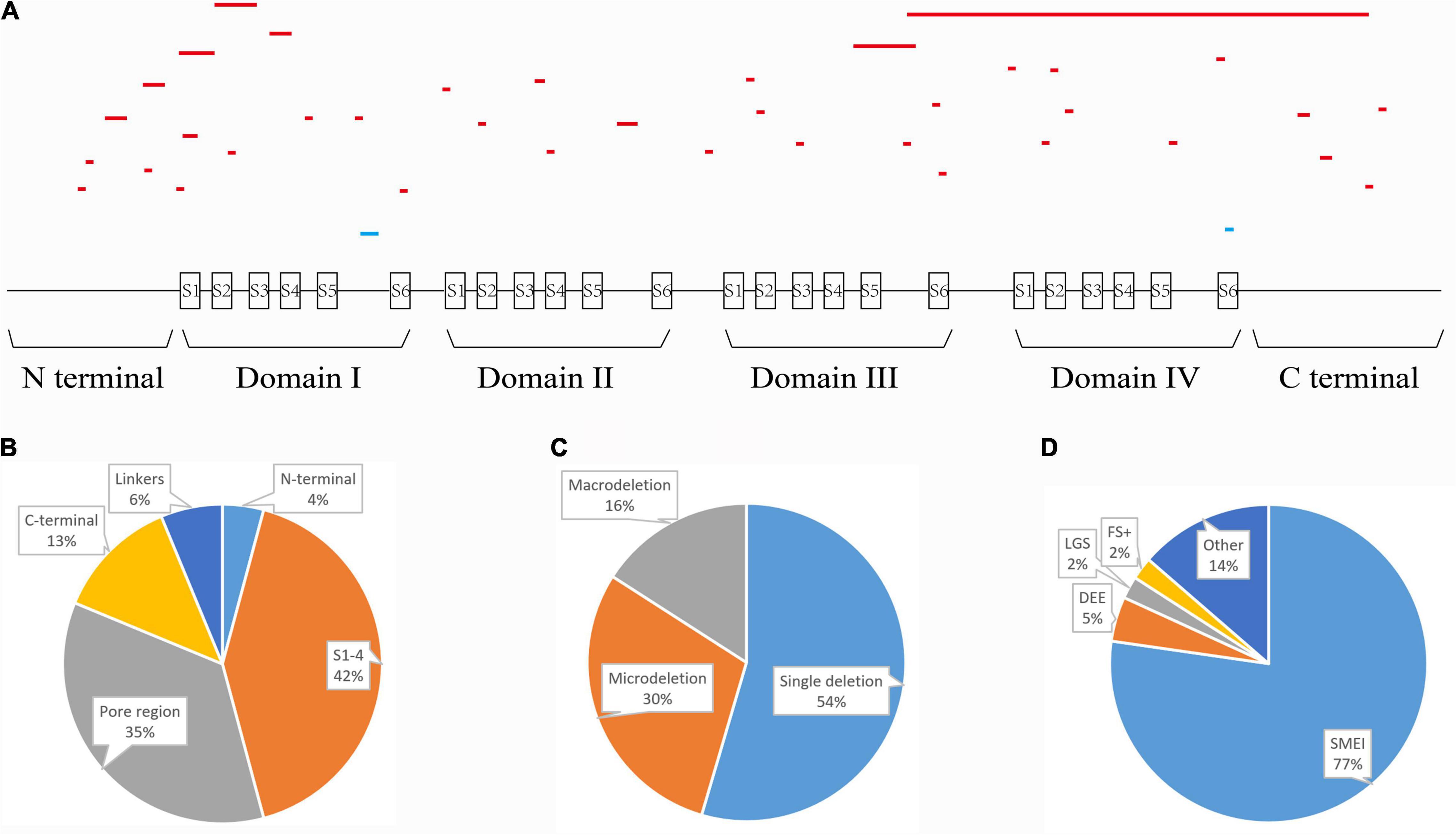
Figure 2. Statistics of all in-frame deletions in SCN1A. (A) Schematic picture showing the length, location, and phenotypes of the 44 variants. Lines indicate different variants. Severe phenotypes (SMEI, DEE, and LGS) were highlighted as red, while mild phenotypes (PEFS+ and FS+) were blue. (B) Distribution of the variants on different functional domains. (C) The proportions of single deletion, microdeletions, and macrodeletions. (D) Phenotypic spectrum.
In-Frame Deletion Mutants Exhibit Complete Loss-of-Function
The WT and six in-frame deletion mutant alpha-subunits were expressed transiently in human tsA201 cells. We performed electrophysiological recordings for the transfected neurons and the non-transfected neurons as negative control. In contrast to the WT Nav1.1 with apparent current recorded (>90 pA/pF), the six in-frame deletion mutants all exhibited tiny raw currents (usually <20 pA/pF) (Figures 3A,B). The tiny currents were demonstrated as endogenous current, which was also recorded in non-transfected cells. Therefore, we confirmed that the in-frame deletion mutants were not able to generate Na+ current and fail to exhibit sufficient sodium current for biophysical analysis.
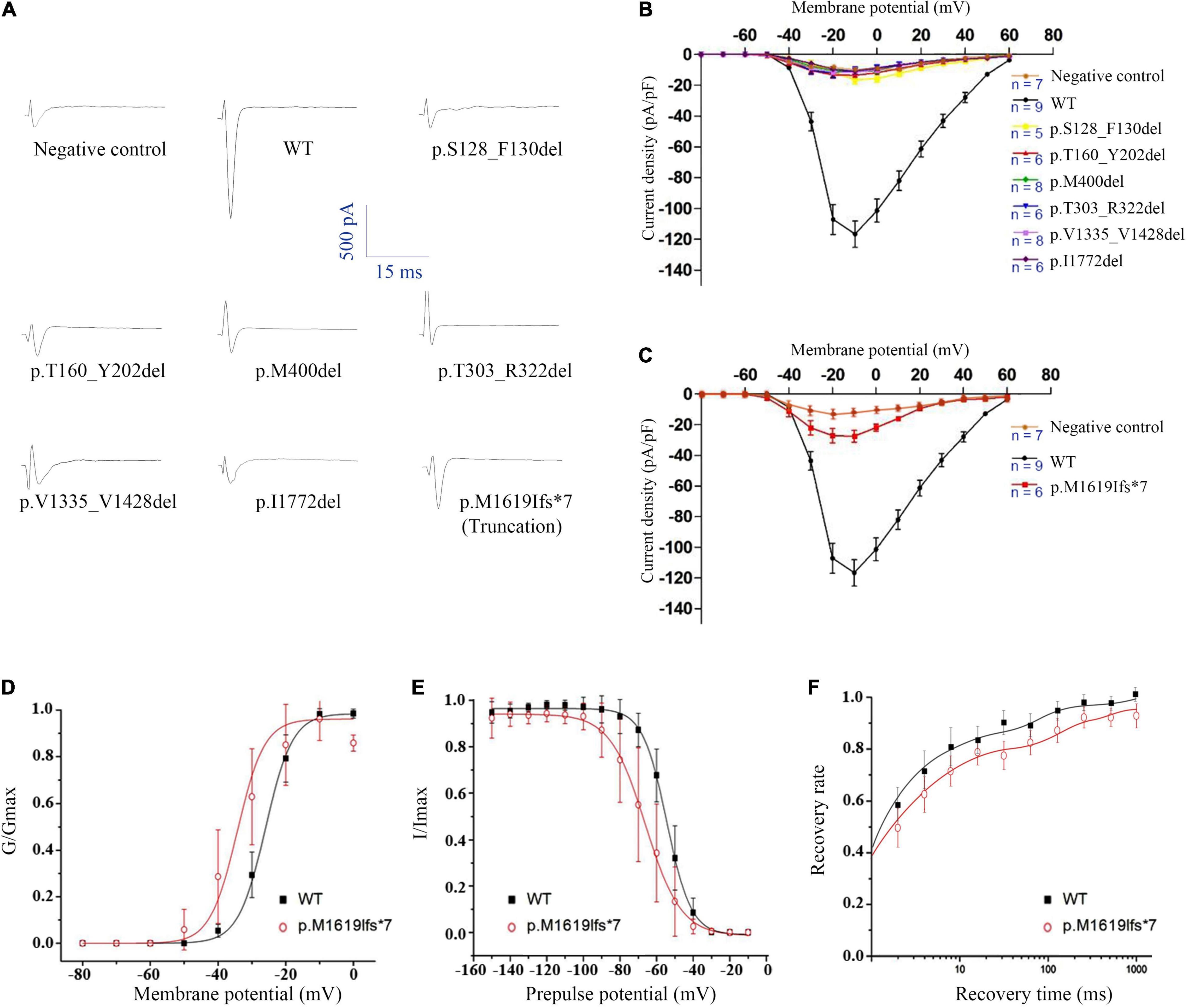
Figure 3. Electrophysiological analysis of the SCN1A in-frame deletion and truncated mutants. (A) Representative traces of sodium current evoked at 0 mV. (B) Current-voltage relationship of six in-frame deletion mutants. (C) Current-voltage relationship of the truncated mutant. (D) G-V activation curve, (E) steady-state inactivation curve, and (F) recovery from inactivation, all for the truncated mutant p.Met1619Ilefs*7. Non-transfected cells were used as negative control.
A Truncated Mutant Retains Partial Channel Function
We noticed that the in-frame deletion mutant causing a single loss of isoleucine at the S6 of DIV (I1772) resulted in complete loss-of-function. This raised an interesting question about whether a truncation mutant that was much shortened still functioned. A truncating variant c.4853-25T > A/p.M1619Ifs*7 was previously identified in our patient with PEFS+. The premature truncation is expected to have seven AA substitutions from p.M1619-E1626, with a loss of peptides from S3 of DIV to c-terminal. Interestingly, the truncated mutant exhibited partial loss-of-function, with reduced peak current density (−27.47 ± 8.46 pA/pF vs. −116.60 ± 25.60 pA/pF in the WT control, 23.6% of the WT) (Figure 3C and Table 3). The current-voltage relation analysis showed that the p.M1619Ifs*7 mutant exhibited a less steep conductance-voltage (G-V) curve with the smaller slopes of steady-state availability, but remained unchanged in the half-maximal activation and inactivation (V1/2), suggesting a slight disruption of the voltage sensor (Figures 3D,E and Table 3). There was no significant difference in the time constants in the kinetics of recovery from inactivation (Figure 3F and Table 3).
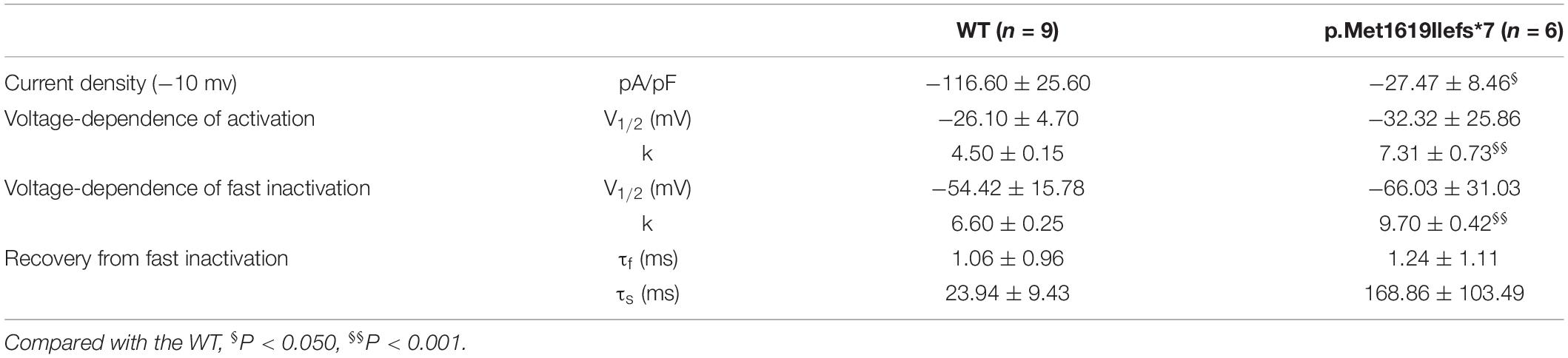
Table 3. Biophysical parameters of truncating mutant p.Met1619Ilefs*7.
Structure Modeling
To explore structural changes as a result of the in-frame deletions, we performed tertiary structure prediction. The comparative analysis of the predicted structures showed that the local tertiary folding differed strongly between the WT and mutants, especially for the macrodeletions (p.T303_R322del, p.T160_Y202del, and p.V1335_V1428del). The local structures of the macrodeletion mutants were deformed and the surrounding segments were relocated (Figure 4). The p.T160_Y202del deletion with two transmembrane segments involved has the most remarkable difference in protein folding, in which the expected segments (S2 and S3) disappear and moreover the adjacent S1 and S4 are relocated to the different sites. Both the single deletion (p.M400del) and microdeletion (p.S128_F130del) that have lost their hydrophobic residues including isoleucine, methionine and phenylalanine, were predicted to turn the coiled coil alpha helices into flexible loops. The other deletion (p.I1772del) at S6 exhibited a smaller difference in the alpha helix, but had a larger difference in the angle of the neighboring units compared to the WT.
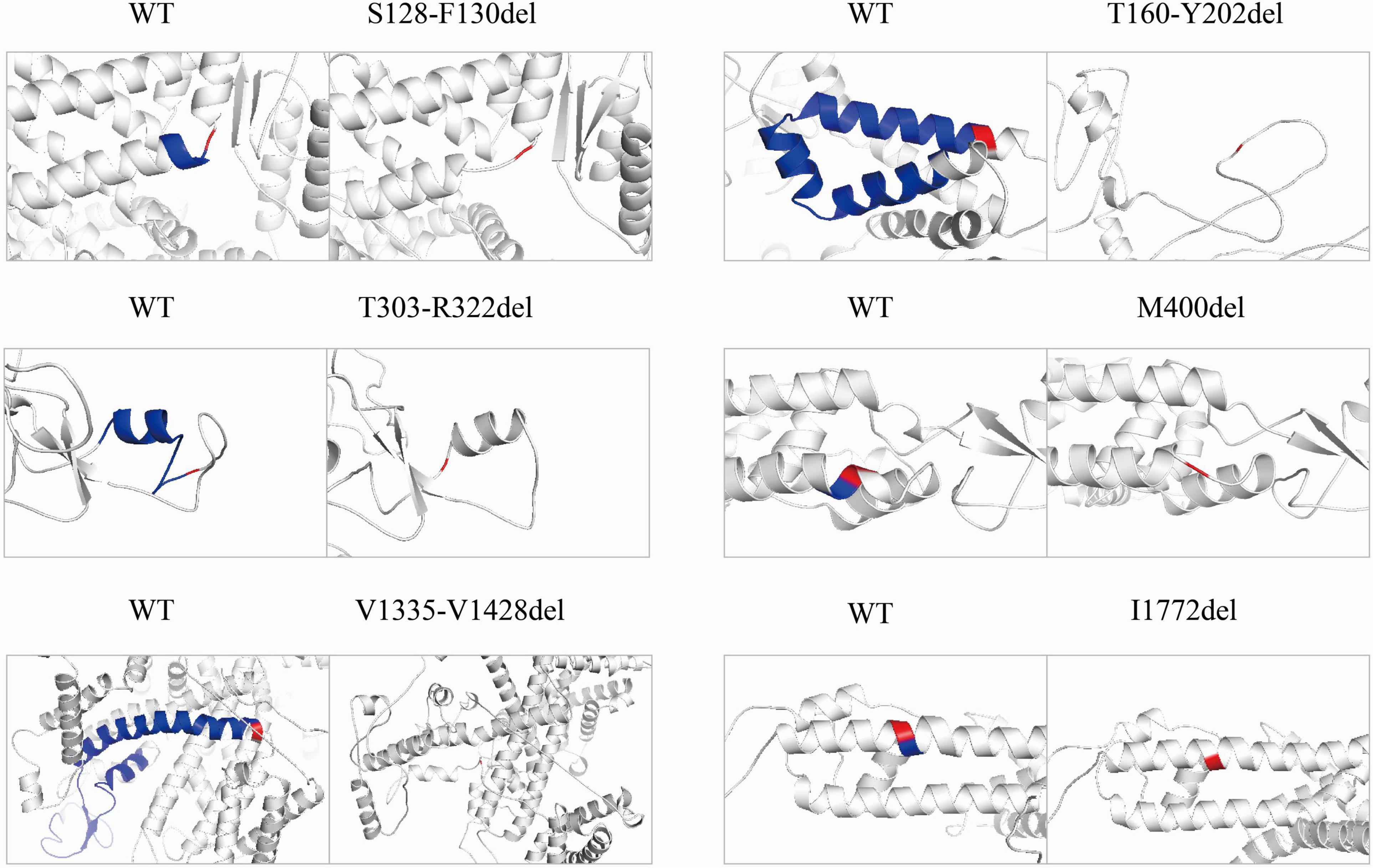
Figure 4. Pairwise comparison of the predicted local structure features between wild-type and in-frame deletions. Location of deleted residues is indicated in blue, and their next residues are in red.
Discussion
Genetic defects in the Nav α subunit can lead to various excitability diseases in brain, muscle, and heart, such as muscle paralysis, cardiac arrhythmias, and epileptic disorders (Meisler and Kearney, 2005; Lee et al., 2009; Remme and Bezzina, 2010; Kasperaviciute et al., 2013). The particular importance of Nav1.1 channel has continually motivated researchers to identify structural or functional residues responsible for protein stability and activity. So far, more than 1800 SCN1A variants have been identified in epilepsy, but few studies have investigated the function of these genetic defects. Functional analysis in coding regions of Nav1.1 channel might help to gain insights into the intramolecular gating network.
In this study, we report the phenotypic relevance and biophysical characterization of seven SCN1A variants, including six SCN1A in-frame deletions and one truncating variant. The six in-frame deletions scatter and affect different functional domain, including transmembrane helices, pore region, and P-loop. There are two single residue deletions (p.M400del, p.I1772del), one microdeletion (p.S128_F130del) and three macrodeletions (p.T303_R322del, p.T160_Y202del, and p.V1335_V1428del). In accordance to the features of all in-frame deletion variants summarized from public databases, the six deletion variants were associated with severe phenotypes such as SMEI, DEE, and LGS, except that two cases presented with milder phenotypes, FS+ and PEFS+. In spite of different locations, residue length of deletions, and associated phenotypes, the six in-frame deletion mutants were found to consistently lose their ion conductance, showing barely detectable inward sodium currents in the heterologous expression experiments.
It is conceivable that the macrodeletions with the loss of a large stretch of peptides would have impact on protein structure and serious functional effect. By contrast, the fact that the microdeletion and single deletion mutants also resulted in the complete loss-of-function is more striking, especially for the two single deletions (p.M400del and p.I1772del) locating at transmembrane domain DIV/S6. Analogous to p.I1772del, an in-frame deletion in the identical DIV/S6 in Nav1.6 (p.I1750del), which is a spontaneous mouse variant, was reported to be associated with a chronic movement disorder with early onset tremor and adult onset dystonia (Jones et al., 2016). The removal of isoleucine in mouse Nav1.6 also exhibited no measurable current in the functional studies (Jones et al., 2016). In addition, no measurable current was previously reported in a DS-associated in-frame deletion variant p.F1289del (Thompson et al., 2012), in which phenylalanine in the transmembrane segment DIII/S3 in Nav1.1 was removed. In our case, the microdeletion p.S128_F130del with a combined loss of phenylalanine, leucine, and serine in S1 led to complete loss-of-function. These transmembrane residues, including isoleucine, methionine, phenylalanine and leucine are all hydrophobic and critical for the interaction between transmembrane helices and lipid membrane. However, an exceptional case was found in an in-frame deletion of leucine (p.L955del) within DII/S6 of Nav1.7, which showed a gain-of-function with a robust hyperpolarizing shift of activation and slow inactivation (Yang et al., 2013). More efforts are needed to understand the functional architecture of voltage-gated sodium channels.
The variant p.T303_R322del is expected to shorten the membrane-reentrant P-loop rather than affect transmembrane helices. The P-loop is the linker between S5 and S6, forming a selectivity filter—a narrow pathway that determines which ion will pass the pore (Catterall et al., 2007; Yang et al., 2009). Functional loss in the p.T303_R322del mutant indicates the non-redundant role of the P-loop for ion conductance. In support of this postulation, it was shown that a five amino acids in-frame deletion of P-loop in a p.R1370-L1374del of Nav1.7, which is associated with channelopathy-associated insensitivity to pain disorder, also resulted in a normally expressed but non-functional channel (Cox et al., 2010). Likewise in Nav1.5, a heterozygous in-frame deletion p.N1380del that was associated with cardiac conduction disturbance and ventricular tachycardia exhibited no detectable current (Yang et al., 2017), even though only a single amino acid was removed. Together with previous experimental mutagenesis and clinical studies (Terlau et al., 1991; Favre et al., 1996; Ishii et al., 2017), it has well established that P-loops are critical determinants of catalytic permeation properties of Na+ channels, but their precise structure-function and deletion-phenotype relationships remain largely unknown.
The most puzzling result in the study is that the truncating mutant (p.M1619Ifs*7), that is expected to delete the S4–S6 pore-forming segments in DIV and the whole C-terminal tail, retained partial channel function. According to several expression and functional studies in the truncated Nav, most of the truncation variants would fail to produce functional channel (e.g., hNav1.1-p.R1234*), except those occurring at the C-terminal tail (e.g., hNav1.1-p.R1892*, hNav1.5-p.R1860Gfs*12) (Sugawara et al., 2003; Bechi et al., 2012; Brunklaus et al., 2020). However, the Na+ current in the p.M1619Ifs*7 transfected cell was observed in this study, although the current remarkably decreased to 24% of the WT. The evidence proved that the truncated channel without last pore-forming segments still retained the basic biophysics of Nav1.1. A possible explanation could be that new assembly of the remaining segments is capable of forming a functional channel.
Several mechanisms might underlie the non-functional Nav1.1 channel. First, as Nav channel opening is determined by a series of gating checkpoints in the transmembrane and cytosolic regions, disruptions caused by missense or deletion variants at these gating checkpoints would definitely impact on the functioning of Nav1.1 channel. For example, the residues in the pore regions are highly conservative and determine ion selectivity and permeation, and the voltage-sensing domains determine the right response to membrane potential. Secondly, integrity of the functional motifs that determine mRNA expression, splicing, and protein trafficking has been destroyed. Thirdly, a local misfolding of residues disturbs the topology of the adjoining functional motifs and misleads gating motions. In our study, p.T303_R322del, p.M400del, p.V1335_V1428del, and p.I1772del affect the integrity of the pore-forming regions. This is consistent with the roughly loss-of-function changes found in missense variants in these gate checkpoints (Ohmori et al., 2006). However, both p.S128_F130del and p.T160_Y202del transformed the α helices into flexible loops according to the structural modeling (Figure 4), which would have impacts on overall arrangements of different segments. As functional motifs of a protein are usually a fixed order of the residues, deletions could be at higher risk of disrupting the order, thereby causing more deleterious effects on function or structure, when compared to missense or truncation mutations.
It has been suggested that changes in amino acid sequence have variable functional effects on sodium channels, with a mixture of loss-of-function and gain-of-function effects (Yamakawa, 2006; Gataullina and Dulac, 2017). The SMEI-associated SCN1A variants seem to be more closely correlated with haploinsufficiency for Nav1.1, which caused by deleterious nonsense and frameshift variants in SCN1A (Gambardella and Marini, 2009; de Jonghe, 2011), and missense variants exhibiting remarkably attenuated or barely detectable sodium currents (Sugawara et al., 2003). Studies on heterozygous SCN1A± mice demonstrated that substantially reduced Na+ current density with a loss of sustained high-frequency firing of action potentials were found in hippocampal and cortical interneurons, possibly responsible for the spontaneous seizure phenotypes in the SCN1A± mice (Yu et al., 2006). In agreement with the previous result, the six in-frame deletion mutants in this study were non-functional and mostly associated with SMEI, except that two variants, p.T160_Y202del and p.I1772del, which affect pore regions, were associated with FS+ and PEFS+, respectively. The variable phenotypes might be explained by the role of genetic modifiers and molecular interactions. It remains a challenging conundrum for researchers about how the common loss-of-function in Nav1.1 results in different phenotypes.
This study provides some new insights into the effect of in-frame deletion on the biological functions of Nav1.1, suggesting a complete loss-of-function of channel and their associations with clinical phenotypes including SMEI, FS+, and PEFS+. However, we only characterized the functional properties in the six out of 44 in-frame deletions identified to date. With more deletion variants identified and functionally characterized, the relationship among deletion pattern, functional changes, and clinical phenotypes would become more clear.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of The Second Affiliated Hospital of Guangzhou Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
TS and NH contributed to the conceptualization and funding acquisition. TS and J-YW designed the study, while the other authors performed the experiments and/or analyzed the data, and contributed to the manuscript draft. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the National Natural Science Foundation of China (Grant Nos. 81271197 to TS, 81971216 to NH, and 82171439 to Yi-Wu Shi), Science and Technology Project of Guangdong Province (Grant No. 2017B030314159 to Wei-Ping Liao), Guangdong Basic and Applied Basic Research Foundation (Grant Nos. 2020A1515011048 to NH and 2021A1515010986 to Yi-Wu Shi), and Science and Technology Project of Guangzhou (Grant Nos. 201904010292 to NH and 201904020028 to Wei-Ping Liao). The funders had no role in study design, data collection and analysis, and decision to publish or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are deeply grateful to the patients and clinicians who participated in this study.
Footnotes
- ^ http://scn1a.caae.org.cn/
- ^ http://www.ncbi.nlm.nih.gov/
- ^ https://zhanglab.ccmb.med.umich.edu/I-TASSER/
References
Aljaafari, D., Fasano, A., Nascimento, F. A., Lang, A. E., and Andrade, D. M. (2017). Adult motor phenotype differentiates Dravet syndrome from Lennox-Gastaut syndrome and links SCN1A to early onset parkinsonian features. Epilepsia 58, e44–e48. doi: 10.1111/epi.13692
Bechi, G., Scalmani, P., Schiavon, E., Rusconi, R., Franceschetti, S., and Mantegazza, M. (2012). Pure haploinsufficiency for Dravet syndrome Na(V)1.1 (SCN1A) sodium channel truncating mutations. Epilepsia 53, 87–100. doi: 10.1111/j.1528-1167.2011.03346.x
Brunklaus, A., Du, J., Steckler, F., Ghanty, I. I., Johannesen, K. M., Fenger, C. D., et al. (2020). Biological concepts in human sodium channel epilepsies and their relevance in clinical practice. Epilepsia 61, 387–399. doi: 10.1111/epi.16438
Catterall, W. A. (2000). From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26, 13–25. doi: 10.1016/s0896-6273(00)81133-2
Catterall, W. A., Cestele, S., Yarov-Yarovoy, V., Yu, F. H., Konoki, K., and Scheuer, T. (2007). Voltage-gated ion channels and gating modifier toxins. Toxicon 49, 124–141. doi: 10.1016/j.toxicon.2006.09.022
Chen, Y. J., Shi, Y. W., Xu, H. Q., Chen, M. L., Gao, M. M., Sun, W. W., et al. (2015). Electrophysiological differences between the same pore region mutation in SCN1A and SCN3A. Mol. Neurobiol. 51, 1263–1270. doi: 10.1007/s12035-014-8802-x
Cox, J. J., Sheynin, J., Shorer, Z., Reimann, F., Nicholas, A. K., Zubovic, L., et al. (2010). Congenital insensitivity to pain: novel SCN9A missense and in-frame deletion mutations. Hum. Mutat. 31, E1670–E1686. doi: 10.1002/humu.21325
de Jonghe, P. (2011). Molecular genetics of Dravet syndrome. Dev. Med. Child. Neurol. 53, 7–10. doi: 10.1111/j.1469-8749.2011.03965.x
de Lange, I. M., Koudijs, M. J., VanTSlot, R., Gunning, B., Sonsma, A. C. M., Van Gemert, L., et al. (2018). Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia 59, 690–703. doi: 10.1111/epi.14021
Depienne, C., Trouillard, O., Saint-Martin, C., Gourfinkel-An, I., Bouteiller, D., Carpentier, W., et al. (2009). Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J. Med. Genet. 46, 183–191. doi: 10.1136/jmg.2008.062323
Djemie, T., Weckhuysen, S., von Spiczak, S., Carvill, G. L., Jaehn, J., Anttonen, A. K., et al. (2016). Pitfalls in genetic testing: the story of missed SCN1A mutations. Mol. Genet. Genomic Med. 4, 457–464. doi: 10.1002/mgg3.217
Eijkelkamp, N., Linley, J. E., Baker, M. D., Minett, M. S., Cregg, R., Werdehausen, R., et al. (2012). Neurological perspectives on voltage-gated sodium channels. Brain 135, 2585–2612. doi: 10.1093/brain/aws225
Escayg, A., De Waard, M., Lee, D. D., Bichet, D., Wolf, P., Mayer, T., et al. (2000). Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am. J. Hum. Genet. 66, 1531–1539. doi: 10.1086/302909
Escayg, A., Heils, A., MacDonald, B. T., Haug, K., Sander, T., and Meisler, M. H. (2001). A novel SCN1A mutation associated with generalized epilepsy with febrile seizures plus–and prevalence of variants in patients with epilepsy. Am. J. Hum. Genet. 68, 866–873. doi: 10.1086/319524
Favre, I., Moczydlowski, E., and Schild, L. (1996). On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys. J. 71, 3110–3125. doi: 10.1016/S0006-3495(96)79505-X
Fujiwara, T., Sugawara, T., Mazaki-Miyazaki, E., Takahashi, Y., Fukushima, K., Watanabe, M., et al. (2003). Mutations of sodium channel alpha subunit type 1 (SCN1A) in intractable childhood epilepsies with frequent generalized tonic-clonic seizures. Brain 126, 531–546. doi: 10.1093/brain/awg053
Fukuma, G., Oguni, H., Shirasaka, Y., Watanabe, K., Miyajima, T., Yasumoto, S., et al. (2004). Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB). Epilepsia 45, 140–148. doi: 10.1111/j.0013-9580.2004.15103.x
Gataullina, S., and Dulac, O. (2017). From genotype to phenotype in Dravet disease. Seizure 44, 58–64. doi: 10.1016/j.seizure.2016.10.014
Gertler, T. S., Calhoun, J., and Laux, L. (2020). A single-center, retrospective analysis of genotype-phenotype correlations in children with Dravet syndrome. Seizure 75, 1–6. doi: 10.1016/j.seizure.2019.12.009
Goldin, A. L., Barchi, R. L., Caldwell, J. H., Hofmann, F., Howe, J. R., Hunter, J. C., et al. (2000). Nomenclature of voltage-gated sodium channels. Neuron 28, 365–368.
Gonsales, M. C., Montenegro, M. A., Preto, P., Guerreiro, M. M., Coan, A. C., Quast, M. P., et al. (2019). Multimodal Analysis of SCN1A missense variants improves interpretation of clinically relevant variants in dravet syndrome. Front. Neurol. 10:289. doi: 10.3389/fneur.2019.00289
Ishii, A., Watkins, J. C., Chen, D., Hirose, S., and Hammer, M. F. (2017). Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome. Epilepsia 58, 282–290. doi: 10.1111/epi.13639
Jones, J. M., Dionne, L., Dell’Orco, J., Parent, R., Krueger, J. N., Cheng, X., et al. (2016). Single amino acid deletion in transmembrane segment D4S6 of sodium channel Scn8a (Nav1.6) in a mouse mutant with a chronic movement disorder. Neurobiol. Dis. 89, 36–45. doi: 10.1016/j.nbd.2016.01.018
Kasperaviciute, D., Catarino, C. B., Matarin, M., Leu, C., Novy, J., Tostevin, A., et al. (2013). Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain 136, 3140–3150. doi: 10.1093/brain/awt233
Lee, S. C., Kim, H. S., Park, Y. E., Choi, Y. C., Park, K. H., and Kim, D. S. (2009). Clinical Diversity of SCN4A-Mutation-associated skeletal muscle sodium channelopathy. J. Clin. Neurol. 5, 186–191. doi: 10.3988/jcn.2009.5.4.186
Lindy, A. S., Stosser, M. B., Butler, E., Downtain-Pickersgill, C., Shanmugham, A., Retterer, K., et al. (2018). Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 59, 1062–1071. doi: 10.1111/epi.14074
Liu, J., Tong, L., Song, S., Niu, Y., Li, J., Wu, X., et al. (2018). Novel and de novo mutations in pediatric refractory epilepsy. Mol. Brain 11:48.
Lossin, C., Rhodes, T. H., Desai, R. R., Vanoye, C. G., Wang, D., Carniciu, S., et al. (2003). Epilepsy-associated dysfunction in the voltage-gated neuronal sodium channel SCN1A. J. Neurosci. 23, 11289–11295. doi: 10.1523/JNEUROSCI.23-36-11289.2003
Mancardi, M. M., Striano, P., Gennaro, E., Madia, F., Paravidino, R., Scapolan, S., et al. (2006). Familial occurrence of febrile seizures and epilepsy in severe myoclonic epilepsy of infancy (SMEI) patients with SCN1A mutations. Epilepsia 47, 1629–1635. doi: 10.1111/j.1528-1167.2006.00641.x
Marini, C., Scheffer, I. E., Nabbout, R., Mei, D., Cox, K., Dibbens, L. M., et al. (2009). SCN1A duplications and deletions detected in Dravet syndrome: implications for molecular diagnosis. Epilepsia 50, 1670–1678. doi: 10.1111/j.1528-1167.2009.02013.x
Meisler, M. H., and Kearney, J. A. (2005). Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Invest. 115, 2010–2017. doi: 10.1172/JCI25466
Meng, H., Xu, H. Q., Yu, L., Lin, G. W., He, N., Su, T., et al. (2015). The SCN1A mutation database: updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum. Mutat. 36, 573–580. doi: 10.1002/humu.22782
Moehring, J., von Spiczak, S., Moeller, F., Helbig, I., Wolff, S., Jansen, O., et al. (2013). Variability of EEG-fMRI findings in patients with SCN1A-positive Dravet syndrome. Epilepsia 54, 918–926. doi: 10.1111/epi.12119
Ohmori, I., Kahlig, K. M., Rhodes, T. H., Wang, D. W., and George, A. L. Jr. (2006). Nonfunctional SCN1A is common in severe myoclonic epilepsy of infancy. Epilepsia 47, 1636–1642. doi: 10.1111/j.1528-1167.2006.00643.x
Peng, J., Pang, N., Wang, Y., Wang, X. L., Chen, J., Xiong, J., et al. (2019). Next-generation sequencing improves treatment efficacy and reduces hospitalization in children with drug-resistant epilepsy. CNS Neurosci. Ther. 25, 14–20. doi: 10.1111/cns.12869
Remme, C. A., and Bezzina, C. R. (2010). Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc. Ther. 28, 287–294. doi: 10.1111/j.1755-5922.2010.00210.x
Selmer, K. K., Lund, C., Brandal, K., Undlien, D. E., and Brodtkorb, E. (2009). SCN1A mutation screening in adult patients with Lennox-Gastaut syndrome features. Epilepsy. Behav. 16, 555–557. doi: 10.1016/j.yebeh.2009.08.021
Sugawara, T., Tsurubuchi, Y., Fujiwara, T., Mazaki-Miyazaki, E., Nagata, K., Montal, M., et al. (2003). Nav1.1 channels with mutations of severe myoclonic epilepsy in infancy display attenuated currents. Epilepsy. Res. 54, 201–207. doi: 10.1016/s0920-1211(03)00084-6
Terlau, H., Heinemann, S. H., Stuhmer, W., Pusch, M., Conti, F., Imoto, K., et al. (1991). Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 293, 93–96. doi: 10.1016/0014-5793(91)81159-6
Thompson, C. H., Porter, J. C., Kahlig, K. M., Daniels, M. A., and George, A. L. Jr. (2012). Nontruncating SCN1A mutations associated with severe myoclonic epilepsy of infancy impair cell surface expression. J. Biol. Chem. 287, 42001–42008. doi: 10.1074/jbc.M112.421883
Till, A., Zima, J., Fekete, A., Bene, J., Czako, M., Szabo, A., et al. (2020). Mutation spectrum of the SCN1A gene in a Hungarian population with epilepsy. Seizure 74, 8–13. doi: 10.1016/j.seizure.2019.10.019
Usluer, S., Salar, S., Arslan, M., Yis, U., Kara, B., Tekturk, P., et al. (2016). SCN1A gene sequencing in 46 Turkish epilepsy patients disclosed 12 novel mutations. Seizure 39, 34–43. doi: 10.1016/j.seizure.2016.05.008
Wang, J. W., Shi, X. Y., Kurahashi, H., Hwang, S. K., Ishii, A., Higurashi, N., et al. (2012). Prevalence of SCN1A mutations in children with suspected Dravet syndrome and intractable childhood epilepsy. Epilepsy. Res. 102, 195–200. doi: 10.1016/j.eplepsyres.2012.06.006
Wu, Y. W., Sullivan, J., McDaniel, S. S., Meisler, M. H., Walsh, E. M., Li, S. X., et al. (2015). Incidence of Dravet Syndrome in a US Population. Pediatrics 136, e1310–e1315.
Yamakawa, K. (2006). Na channel gene mutations in epilepsy–the functional consequences. Epilepsy. Res. 70, S218–S222. doi: 10.1016/j.eplepsyres.2005.11.025
Yang, Y., Estacion, M., Dib-Hajj, S. D., and Waxman, S. G. (2013). Molecular architecture of a sodium channel S6 helix: radial tuning of the voltage-gated sodium channel 1.7 activation gate. J. Biol. Chem. 288, 13741–13747. doi: 10.1074/jbc.M113.462366
Yang, Y. C., Hsieh, J. Y., and Kuo, C. C. (2009). The external pore loop interacts with S6 and S3-S4 linker in domain 4 to assume an essential role in gating control and anticonvulsant action in the Na(+) channel. J. Gen. Physiol. 134, 95–113. doi: 10.1085/jgp.200810158
Yang, Z., Lu, D., Zhang, L., Hu, J., Nie, Z., Xie, C., et al. (2017). p.N1380del mutation in the pore-forming region of SCN5A gene is associated with cardiac conduction disturbance and ventricular tachycardia. Acta Biochim. Biophys. Sin. 49, 270–276. doi: 10.1093/abbs/gmx003
Yu, F. H., Mantegazza, M., Westenbroek, R. E., Robbins, C. A., Kalume, F., Burton, K. A., et al. (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149. doi: 10.1038/nn1754
Keywords: sodium channel, SCN1A, epilepsy, in-frame deletion, variant
Citation: Wang J-Y, Tang B, Sheng W-X, Hua L-D, Zeng Y, Fan C-X, Deng W-Y, Gao M-M, Zhu W-W, He N and Su T (2022) Clinical and Functional Features of Epilepsy-Associated In-Frame Deletion Variants in SCN1A. Front. Mol. Neurosci. 15:828846. doi: 10.3389/fnmol.2022.828846
Received: 04 December 2021; Accepted: 21 February 2022;
Published: 14 March 2022.
Edited by:
Yuwu Jiang, Peking University First Hospital, ChinaReviewed by:
Jing Peng, Central South University, ChinaNazzareno D’Avanzo, Université de Montréal, Canada
Copyright © 2022 Wang, Tang, Sheng, Hua, Zeng, Fan, Deng, Gao, Zhu, He and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Su, c3VAZ3puZXVyb3NjaS5jb20=