Abstract
Developmental and epileptic encephalopathies (DEEs) have high genetic heterogeneity, and DEE due to the potassium voltage-gated channel subfamily C member 2 (KCNC2) variant remains poorly understood, given the scarcity of related case studies. We report on two unrelated Chinese patients, an 11-year-old boy and a 5-year-old girl, diagnosed with global developmental delay (GDD), intellectual disability (ID), and focal impaired awareness seizure characterized by generalized spike and wave complexes on electroencephalogram (EEG) in the absence of significant brain lesions. Whole-exome sequencing (WES) and electrophysiological analysis were performed to detect genetic variants and evaluate functional changes of the mutant KCNC2, respectively. Importantly, we identified a novel gain-of-function KCNC2 variant, R405G, in both patients. Previously reported variants, V471L, R351K, T437A, and T437N, and novel R405G were found in multiple unrelated patients with DEE, showing consistent genotype–phenotype associations. These findings emphasize that the KCNC2 gene is causative for DEE and facilitates treatment and prognosis in patients with DEE due to KCNC2 mutations.
Introduction
Developmental and epileptic encephalopathy (DEE) is a group of epilepsy syndromes characterized by severe seizures that are drug-resistant and significant developmental delay or regression (Scheffer et al., 2017). Most patients with DEE exhibit onset during infancy with different types of seizures, e.g., epileptic spasms, tonic, or atonic seizures, and myoclonic seizures. Over 50 causative genes have been found in DEE cases, indicating high heterogeneity in both phenotype and etiopathogenesis of DEE (He et al., 2019; Raga et al., 2021). Impairment of potassium ion subunits, e.g., potassium voltage-gated channel subfamily A, B, and Q (KCNA, KCNB, and KCNQ) members, is well-established to play an important role in DEE (Lal et al., 2020). In comparison with potassium voltage-gated channel subfamily C, member 1 (KCNC1) and member 3 (KCNC3), which were previously reported to be associated with DEE, progressive myoclonus epilepsy (MIM: #616187), and spinocerebellar ataxia (MIM: #605259) (Muona et al., 2015; Zhang and Kaczmarek, 2016), respectively, the linkage of the potassium voltage-gated channel subfamily C member 2 (KCNC2) gene and DEE remains poorly understood due to the scarcity of reported cases (Rademacher et al., 2020; Vetri et al., 2020; Rydzanicz et al., 2021; Schwarz et al., 2022).
The KCNC2 gene, also known as KV3.2, together with KCNC1, KCNC3, and KCNC4 (KV3.4), belongs to the Shaw-related potassium channel family (KV3 subfamily). It is well-established that KV3 proteins share the structure of six-transmembrane (6-TM) voltage-gated K + channels, which maintain the potassium ion permeability of excitable membranes by regulating pore penetration in 6-TM in response to voltage changes, thereby maintaining neuronal functions, such as maintenance of membrane potential, regulation of cell volume or neurotransmitter release, and modulation of electrical excitability (González et al., 2012; Kaczmarek and Zhang, 2017). In the human brain, KV3 channels act as potential regulators of neurons and rapidly activate at high-threshold voltages and deactivate very fast, suggesting a role in the excitability of the central nervous system (CNS) by regulating the membrane resting potential and fast action (Rudy et al., 1999; Rudy and McBain, 2001; McDonald and Mascagni, 2006).
The KCNC2 expression is relatively high in the human brain, particularly in the hippocampus, frontal cortex, anterior cingulate cortex, caudate, hypothalamus, basal amygdala, and pituitary, playing a key role in regulating the excitability and neural communication (Chow et al., 1999). Recent human single-cell transcriptome sequencing (scRNA-seq) studies have demonstrated that KCNC2 is enriched in both inhibitory and excitatory neurons.1 An increasing body of evidence suggests that KCNC2 is essential for subserving a fast-spiking firing pattern and sustaining trains of action potentials at high frequencies to maintain an effective synaptic transmission and modulate the excitation (Erisir et al., 1999; Atzori et al., 2000). It has been shown that knockout of Kcnc2 (KV3.2–/–) in mice resulted in a deficit of high-frequency oscillations modulation, distorted cortical rhythmic activity, suppressed cortical inhibition, impaired fast spiking in cortical interneurons, enhanced susceptibility to epileptic seizures and abnormal sleep patterns, and increased anxiety in the open field. However, it can be challenging to discern whether a neurodevelopmental delay has occurred (Lau et al., 2000; Yu et al., 2006). In addition, KCNC2 homolog-associated seizures are found in other animal models: delayed rectifier downregulated channels KV3.1-2 are observed in the hippocampus of adult gerbils during seizures (Lee et al., 2009), while overexpression of KCNC2 in thalamocortical circuits is thought to be the cause of absence epilepsy (Stern et al., 2020). This in vivo evidence suggests that human KCNC2 loci are linked to epilepsies.
In this study, we identified a novel heterozygous KCNC2 variant, c.1213A > G (p. R405G), in two unrelated patients who were both clinically diagnosed with DEE. We demonstrated that the variant R405G yielded higher activity in vitro than the wild-type (WT) KCNC2 on a range of functioning voltages.
Materials and methods
This study was approved by the Ethics Committee of Wuhan Children’s Hospital, Hubei, China (serial number: 2022497). Written informed consent for genetic testing and publication of photographs was obtained from the legal guardians of each subject. All investigations were conducted in accordance with the principles of the Declaration of Helsinki.
Subjects
Two patients from unrelated families were admitted to the Department of Neurology, Wuhan Children’s Hospital. The medical records, including medical history, symptoms and signs, physical examinations, laboratory tests, radiological results, and developmental assessment, were documented by the attending physicians. Genetic tests and analyses were performed by Chigene (Beijing, China).
Whole-exome sequencing
Whole-exome sequencing (WES) was performed on the proband’s genomic DNA extracted from peripheral blood leukocytes using xGen Exome Research Panel version 1.0 (IDT, IA, United States) and paired-end sequencing (2 × 150 bp) on NovaSeq 6000 (Illumina, San Diego, CA, United States) with not less than 99% regions covered at least 10×. Raw sequence readouts were processed by fastp. Paired-end sequence reads were aligned to the human GRCh37/hg19 genome with Burrows–Wheeler Aligner (BWA). Initial mapping process and variant site identification were conducted using GATK. Annotations were retrieved from the database-based online system independently developed by Chigene (Beijing, China),2 including allele frequency in the Single Nucleotide Polymorphism database (dbSNP), 1000 Genomes Project, Exome Aggregation Consortium (ExAC), Genome Aggregation Database (gnomAD), ESP, and Chigene in-house MAFs database; positions and associated clinical phenotypes based on UCSC, RefGene, GENCODE, and ENSEMBL transcripts; LOVD, SWISS, Clinvitae, HGMD, OMIM, and ClinVar databases; and functional and conservational predictions for the amino-acid changes according to software packages (Provean, SIFT, PolyPhen2, MutationTaster, M-CAP, REVEL, and CADD; MaxEntScan, dbscSNV, and GTAG; GERP, phyloP, and phastCons). Variant filtering was done according to rare variants with minor allele frequency (MAF) of <1% and biological impact, gene features and disease mechanism, recessive/dominant/de novo pattern of inheritance and segregation analysis, phenotypic relevance through human phenotype ontology (HPO) terms, and scientific literature. Subsequently, pathogenic or likely pathogenic variants were evaluated according to the American College of Medical Genetics (ACMG) practice guidelines.
Sanger sequencing
The KCNC2 (NM_139137) variant c.1213A > G treated as plausible disease-causing was verified in the probands and their parents by Sanger sequencing performed using the BigDye Terminator version 1.1 Cycle Sequencing Kit on an ABI 3730XL instrument (Applied Biosystems, Waltham, MA, United States) to define the inheritance model. The primers used are 5′-CTGACCATGTTTGGGGGTACA-3′ and 5′-GAAACGGATCCTGCCTTGAC-3′ (product size of 520 bp and annealing temperature of 60°C). PCR amplification was carried out with a KAPA2G Robust HotStart PCR Kit (KAPA Biosystems, Roche, Basel, Switzerland) on a Hema 9600 PCR Thermo Cycler (Zhuhai Hema Medical Instrument Co., Ltd., Zhuhai, China). The DNA sequences were analyzed using the DNASTAR software package.
Molecular modeling
Predicted (AF-Q96PR1-F1) configurations of human KCNC2 in the AlphaFold Protein Structure Database served as the template for developing a structural model that covered all 638 residues (NP_631875.1). Hydrogen-bond networks of the WT and the variant proteins were visualized in PyMOL.
Cell culture and transfection
HEK293 cells were cultured at 37°C in DMEM supplemented with 10% fetal bovine serum (FBS) (AusGeneX, Oxenford, QLD, Australia) in a humidified 5% (v/v) CO2 atmosphere. HEK-293 cells were seeded in six-well plates and subsequently transfected with 2 μg of WT or mutant cDNAs by transient transfection using Lipofectamine 3000 (Invitrogen, Waltham, MA, United States). The day after transfection, cells were seeded into a 24-well plate at a density of 8 × 103 cells per well. Approximately 48 h after transfection, cells were selected based on the intensity of the fluorescent reporter and placed in the recording chamber.
Electrophysiological analysis
The macroscopic current of transiently transfected HEK-293 cells was recorded with an EPC10 amplifier (HEKA, Lambrecht, Germany) and the Patchmaster software using the whole-cell configuration of the patch-clamp technique. The sampling interval was set at 100 ms (20 kHz) in all experiments. Pipettes (Sutter Instruments, United States) were pulled from borosilicate glass and had resistances between 1.5 and 2.5 MΩ when filled with the pipette solution. The glass pipette was pulled using a micropipette puller and manipulated using a micro-manipulator under the microscope. After touching the cell, a slight suction was applied to achieve high seal resistance (GΩ). Fast capacitance (in pF) compensation was made after achieving a high seal, and the membrane was broken. Cell capacitance (in pF) compensation was made from whole-cell capacitance compensation after the whole-cell mode was achieved. No leak subtraction was made. The extracellular solution contained (in mM) 140 sodium chloride (NaCl), 3.5 potassium chloride (KCl), 1 MgCl2.6H2O, 2 CaCl2.2H2O, 10 D-glucose, 10 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 1.25 NaH2PO4.2H2O, and pH adjusted to 7.4 with sodium hydroxide (NaOH). The pipette solution contained (in mM) 20 KCl, 115 K-aspartic, 1 MgCl2.6H2O, 5 ethylene glycol tetraacetic acid (EGTA), 10 HEPES, 2 Na2-ATP, and pH adjusted to 7.2 using potassium hydroxide (KOH). After the break-in, a 5-min waiting period equilibrated the cytoplasm of the cell and the pipette solution. Currents were activated by 500 ms voltage pulses ranging from −80 to +80 mV (in 10 mV increments) from a holding potential of −80 mV at a sweep frequency of 100 ms, calculated by dividing the current (in pA) by the cell capacitance (in pF). Current–voltage curves were converted through a Boltzmann equation (g(V) = gmax/(1 + exp((V-V0.5)/k))), where g, conductance; gmax, maximal conductance; V, voltage; V0.5, voltage of half-maximal activation; and k, slope factor into normalized conductance–voltage curve.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM). At each measured voltage, e.g., −40, +10, and +60 mV, the membrane currents of mutant and WT cells were repeatedly tested three times, taking the mean value and SEM for t-test comparison. Significant differences were assessed by Student’s t-test, with *p < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Literature search
Keywords “KCNC2” or “KV3.2,” “developmental and epileptic encephalopathy,” “DEE,” or “epilepsy” were used for literature search in the PubMed, Medline, and Google Scholar databases.
Results
Case presentation
Patient 1
Patient 1 was a 9-year-old boy admitted to our hospital for intractable epilepsy. He was born to a non-consanguineous Chinese couple with a history of birth asphyxia and hypoxia. The parents claimed no family history of seizures. At the age of 3 months, the patient experienced paroxysmal attacks with rolling back of eyeballs and loss of consciousness several times per day, which led to an initial diagnosis of focal epilepsy at the local hospital. For the following 8 years, he was successively treated with levetiracetam (LEV), valproate acid (VPA), oxcarbazepine (OXC), lamotrigine (LTG), and nitrazepam (NZP), which were ineffective for seizure control. Despite normal growth, the patient had intellectual disability (ID) with language and motor impairment. At the age of 9 years, the patient could not run and jump and had moderate ID (IQ 56). He experienced several focal seizures with episodic atypical absences during his hospitalization. Electroencephalogram (EEG) showed slowing of the basic background rhythm with hypsarrhythmia and generalized multifocal polyspike and slow wave, especially in the sleep stage. During a seizure, EEG showed generalized high amplitude waves at 3–4 Hz and spike-and-slow-wave bursts for 8–18 s. His blood and urine tests and brain MRI were normal. The patient was seizure-free after treatment with a combination of VPA/LTG/NZP. At the last follow-up, the patient (at the age of 11 years) was seizure-free with multifocal slow-wave, sharp-slow-wave, sharp-wave, and spike complexes on awake EEG. Complexes of multifocal polyspike and slow wave, spike and slow-wave, and sharp-wave were conducted by forward modeling EEG during sleep (Figure 1 and Table 1).
FIGURE 1
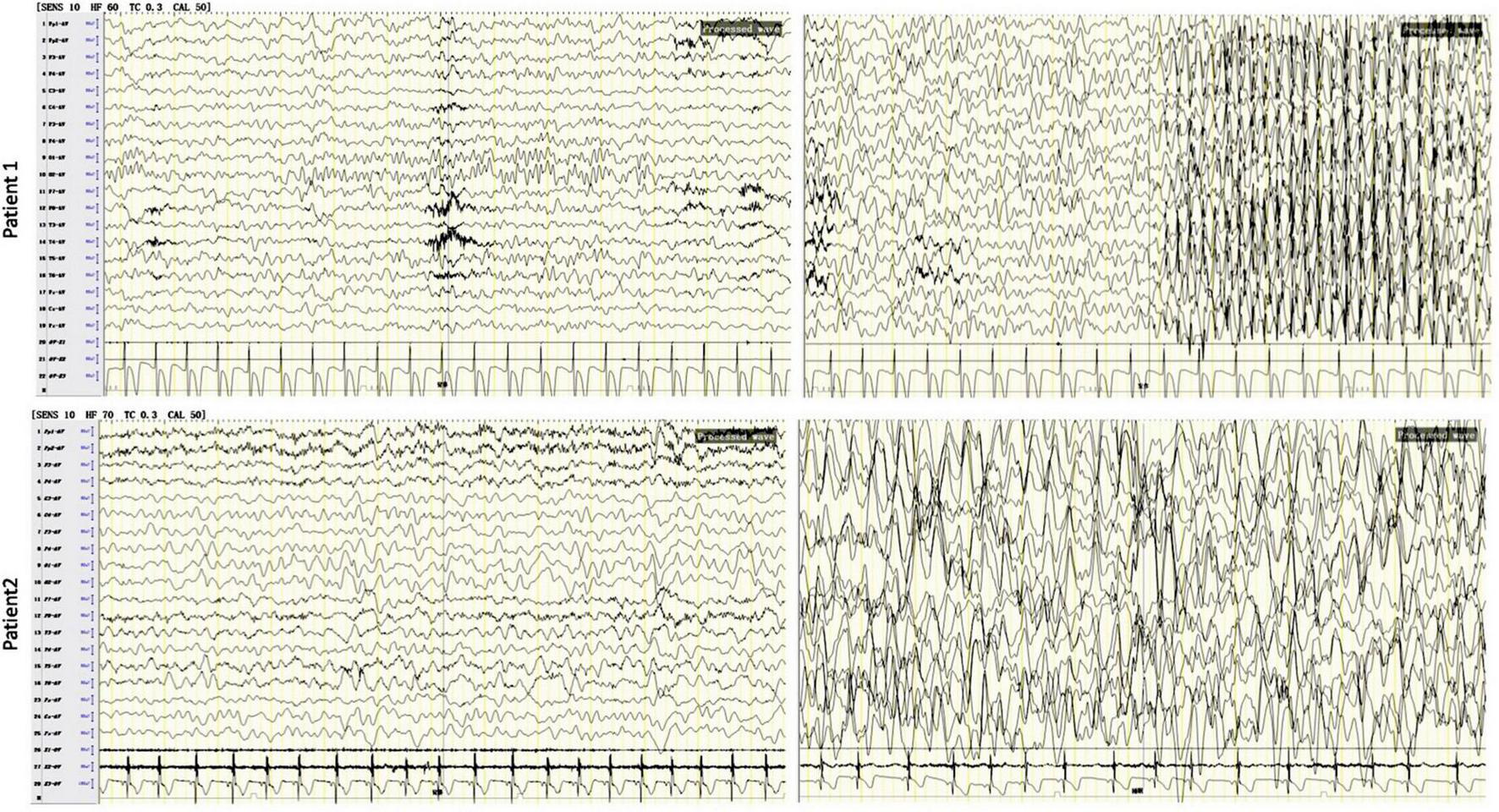
Electroencephalogram (EEG) tests in patients with developmental and epileptic encephalopathie (DEE) due to the KCNC2 R405G variant. The results showed background (left) generalized, diffuse spike, and wave complexes in both patients. In patient 1 (at the age of 9 years), the EEG showed generalized high amplitude waves at 3–4 Hz and spike-and-slow-wave bursts for 8–18 s in an epileptic attack (upper right). In patient 2, a sleep EEG (lower right) showed slowing of the occipital basic rhythm, multifocal generalized complexes of slow wave, spike and slow wave, sharp slow wave, and polyspike and slow wave, especially in occipital, frontal pole, and frontal regions.
TABLE 1
| Pat. | Sex | Age at last examination | Mut., inh. | Clinical diagnosis | Seizure types | EEG | Brain MRI | DD/ID | Anti-epilepsy treatment | References |
| 1 | Male | 11 years | R405G, NA | DEE | Focal unaware, absence | Occipital basic rhythm slowing; multifocal slow-wave, sharp-slow-wave, sharp-wave, and spikes complexes in awake; multifocal polyspike-and-slow-wave, spike-and-slow-wave, and sharp-wave complexes, left forward in sleeping | Normal | Moderate to severe ID, not able to run and jump | SF (VPA/LTG/NZP) | Present study |
| 2 | Female | 4 years | R405G, de novo | DEE | Focal unaware | Occipital basic rhythm slowing; multifocal generalized spikes, spike-and-slow-wave, polyspike- and-slow-wave, and sharp wave complexes | Normal, unsignificant nodular signal | Severe ID, no speech, ataxic gait, not able to run and jump | SF (VPA/LTG) | Present study |
| 3 | Female | 7 years | D167Y, de novo | DEE: WS to LGS | IS, focal unaware, tonic-clonic, atypical Abs, Myo, SE, GTCS | Hypsarrhythmia, multifocal generalized spikes, missing background activity, sleep architecture | Arachnoidal cysts left temporo-basal, delayed myelination, progressive brain atrophy | Moderate to severe ID, ataxic gait, hypotonia, speech disturbance, macrocephaly | DR | Rademacher et al., 2020 |
| 4 | Male | 8 years | V471L, de novo | DEE | IS | Quasi-continuous burst-suppression pattern, generalized and high voltage spike-and-wave complexes | Widespread hypomyelination, hypertrophic frontal lobes | Severe ID, no speech, drooling, spastic tetraplegia, feeding difficulties | DR | Vetri et al., 2020 |
| 5 | Female | 2 years | V471L, de novo | DEE: WS | IS, polymorphic, tonic, tonic-clonic | Multifocal generalized spikes, polyspikes, spike-and-slow-wave, spike, and sharp wave fast complexes | Mild internal and external hydrocephalus | Severe ID, not able to sit, lack head control, hypotonia | Mild improved (lacosamide) | Rydzanicz et al., 2021 |
| 6 | Male | 1.5 ms | C125W, de novo | DEE: EOAE | Abs, Myo, GTCS | G(psw) | Normal | Mild - moderate | SF (VPA/CZP) | Schwarz et al., 2022 |
| 7 | Male | 1 year | E135G, de novo | DEE | Myo, atonic | G | Normal | Mild | DR | Schwarz et al., 2022 |
| 8 | Male | 7 ms | R351K, de novo | DEE: CSWS | GTCS, Myo, focal unaware | CSWS | Normal | Mild | SF (VPA) | Schwarz et al., 2022 |
| 9 | Male | 8 ms | R351K, de novo | DEE: CSWS | GTCS, Myo, Abs, focal unaware | CSWS | Normal | Severe | DR | Schwarz et al., 2022 |
| 10 | Male | 8 ms | T437A, de novo | DEE: EOAE | Abs, tonic | G | Normal | Severe | DR | Schwarz et al., 2022 |
| 11 | Female | 11 ms | T437N, de novo | DEE | FS, Myo | G | Normal | Moderate | DR | Schwarz et al., 2022 |
| 12 | Male | 3 ms | T437N, de novo | DEE | FS, Myo, Abs | G | Normal | Moderate | DR | Schwarz et al., 2022 |
| 13 | Female | 2 years | S333T, mc | DEE | GTCS, Myo, Abs | G | Normal | Mild | SF (VPA) | Schwarz et al., 2022 |
Clinical features and KCNC2 variants in patients with developmental and epileptic encephalopathie (DEE) (n = 13).
The variants are described using the reference sequence NP_631875.1. General information: EEG, electroencephalogram; F, female; inh, inheritance; M, male; mc, maternal non-affected carrier; MRI, magnetic resonance imaging; ms, months; Mut, mutation; Pat, patient; y, year(s). Diagnosis and seizures: Abs, absences; CSWS, continuous spike and wave during sleep; DD, developmental delay; DEE, developmental and epileptic encephalopathy; EOAE, early-onset absence epilepsy; G, generalized; GTCS, generalized tonic-clonic seizure; ID, intellectual disability; IS, infantile spasms; LGS, Lennox–Gastaut syndrome; Myo, myoclonic; NA, not available; psw, poly-spike wave discharges; SE, status epilepticus; WS, West syndrome. Antiepileptic treatment: CZP, clonazepam; DR, drug-resistant; LTG, lamotrigine; NZP, nitrazepam; SF, Seizure free; VPA, valproic acid.
Patient 2
Patient 2 was a girl aged 4 years and 9 months and the first child of a non-consanguineous normal Chinese couple. The patient experienced severely delayed neuromotor development (she was unable to walk independently until 4 years old, with an unsteady gait, and could not understand a single sentence, follow any instructions, or ask), with no ability to speak, and had severe ID (IQ 34). When she was 1 year and 6 months old, the parents found she was subject to spontaneous attacks of eyelid myoclonia (blinking, eyes rolling back to the left, and head moving backward) more than 10 times per day and myoclonic seizures (jerking hands), which usually lasted for 2–3 min and often occurred before sleeping. After the patient was diagnosed with focal onset epilepsy at the local hospital based on an EEG (images were unavailable). on EEG at the local hospital, an antiepileptic treatment was suggested to her. After a poor response to OXC, her seizures were relieved with a combination of LEV/VPA and she was seizure-free last year. At the age of 3 years, the patient came to our hospital, and she was diagnosed with global developmental delay (GDD) with focal epilepsy. EEG showed slowing of the basic occipital rhythm, multifocal generalized complexes of spikes, spike and slow-wave, polyspike and slow-wave, and sharp-wave, significantly at rest and during sleep. Her brain MRI was normal. When the patient was 4 years and 9 months old, upon the last follow-up, the EEG showed the same pattern of slowing of the occipital basic rhythm, multifocal generalized complexes of slow-wave, spike and slow-wave, sharp-slow-wave, and polyspike and slow-wave, especially in occipital, frontal pole, and frontal regions (Figure 1 and Table 1).
Pathogenic or likely pathogenic variant screening and analysis
Whole-exome sequencing was performed on both patients. No pathogenic or likely pathogenic variants were found in well-known DEE-associated genes from the OMIM database (OMIM phenotypic series: PS308350). However, we found a heterozygous KCNC2 variant in both patients, NM_139137.4: c.1213A > G (p.R405G). A review of the literature showed that KCNC2 variants had been documented in several reported DEE cases (Table 1), while R405G had never been documented in public variant or dbSNP. We then performed Sanger sequencing in patient 2 and her parents, which proved the variant was de novo (Figure 2A). Unfortunately, further genetic testing was not available for patient 1. Multiple prediction applications, SIFT, PolyPhen2 (HDIV), PolyPhen2, HVAR, Provean, MutationTaster, M-CAP, and REVEL, revealed KCNC2 R405G as damaging or disease-causing. Thus, KCNC2 R405G was likely pathogenic according to the American College of Medical Genetics and Genomics (ACMG) clinical practice guidelines (PS2 + PM2 + PP3). No pathogenic or likely pathogenic variant was found in other genes associated with developmental disorders in both patients.
FIGURE 2
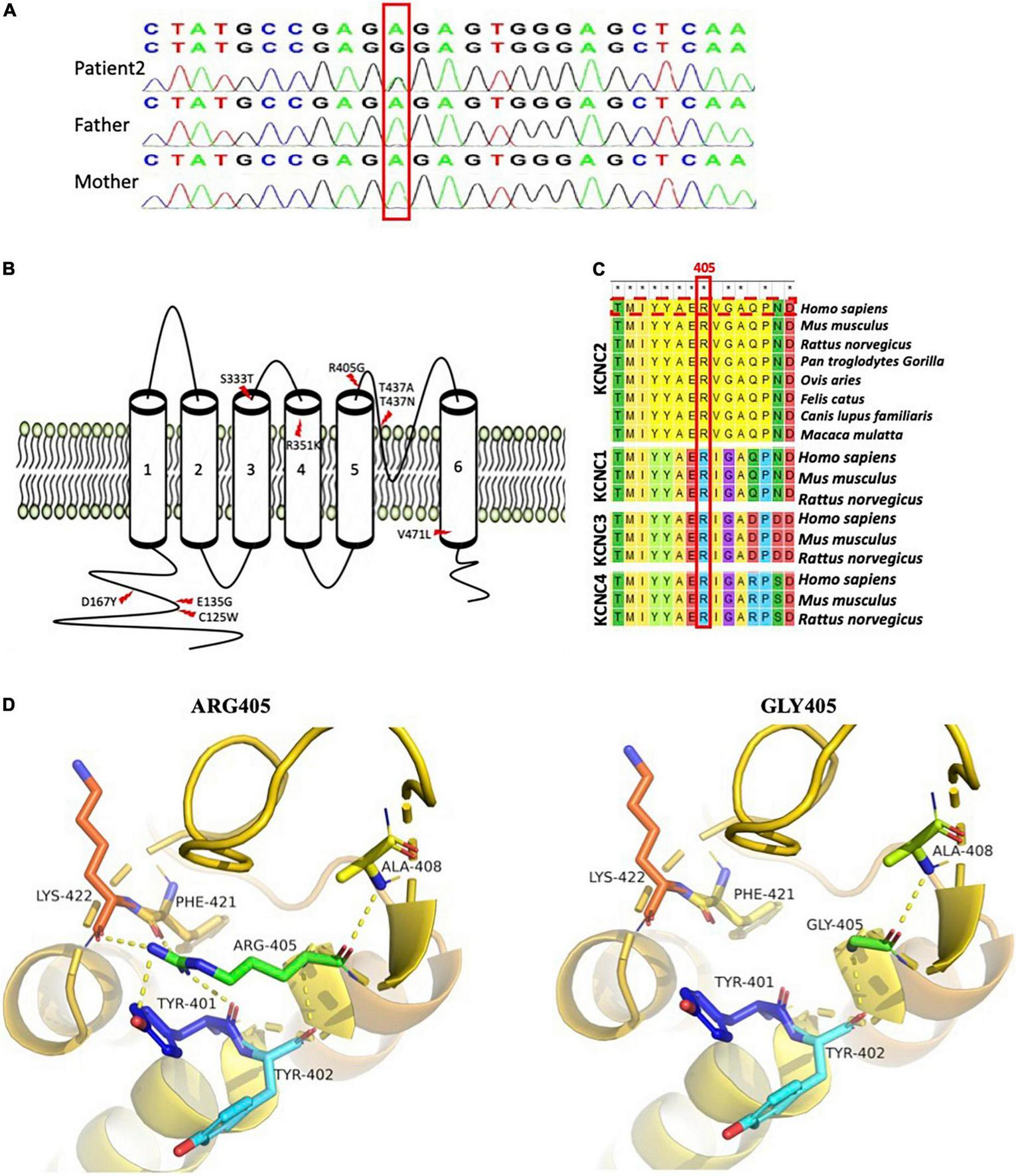
Schematic diagram of known developmental and epileptic encephalopathie (DEE)-linked KCNC2 variants and R405G analysis. (A) Sanger sequencing confirms KCNC2 (NM_139137) c.1213A > G in patient 2 and her parents. (B) Nine well-known DEE-linked variants on eight sites of the KCNC2 amino acid chain. Both previously reported R351K and T437N were found in two unrelated patients with DEE, respectively (Schwarz et al., 2022). (C) Multiple sequence alignment of KCNC1, KCNC2, KCNC3, and KCNC4 subunits across several species is shown by MEGA7 (the R405 residue and KCNC2 human sequence marked in red boxes). Asterisks (*) denote the highly conserved sites. (D) Close view of structural models of the KCNC2 protein (AF-Q96PR1-F1) as ribbons with the p.R405G variant and residues involved in hydrogen-bonding (dotted yellow) network indicated as sticks. (a) Represents wild-type KCNC2 protein with native R405. (b) Represents the mutated KCNC2 with Gly405.
Based on the reviewed structure data (UniProtKB id: Q96PR1), KCNC2 contains cytosolic N- and C-termini and six membrane-spanning segments. KCNC2 R405 is located within the S5-S6 linker, an extracellular loop that forms one of the ion-selective macromolecular protein pores in KCNC2 (Figure 2B). In addition, R405 is strictly conserved in evolution among multiple species and other members of the KV3 subfamily, including KCNC1, KCNC3, and KCNC4 (Figure 2C). Analysis of three-dimensional modeling (AlphaFold id: AF-P35670-F1) has revealed that glycine substitution has disrupted the hydrogen bonds that bind R405 with Tyr401, Phe421, and Lys422 (Figure 2D). As a result, R405G is predicted to change the pore structure.
Electrophysiological characterization
To investigate the impact of R405G in KV3.2 channel functioning, we constructed EGFP-fusion expression vectors expressing KCNC2 WT and R405G cDNA sequences transfected into HEK-293 cells. Compared to WT, R405G caused a significantly higher density current at voltages between −40 and +10 mV (Figures 3A,B). At +60 mV, the normalized current densities of the channel (262.2 ± 61.91) revealed that R405G caused about 40% less normalized density current compared to the WT on average (262.2 ± 61.91 vs. 427.9 ± 137.1), although the difference was not statistically significant (Figure 3C). By measuring the conductance–voltage curves, we found that R405G shifted the curve to the left, indicating a gain-of-function effect compared to the WT (V1/2 = −6.375 mV, k = 49.66 vs. V1/2 = 24.2 mV, k = 21.36) (Figure 3D).
FIGURE 3
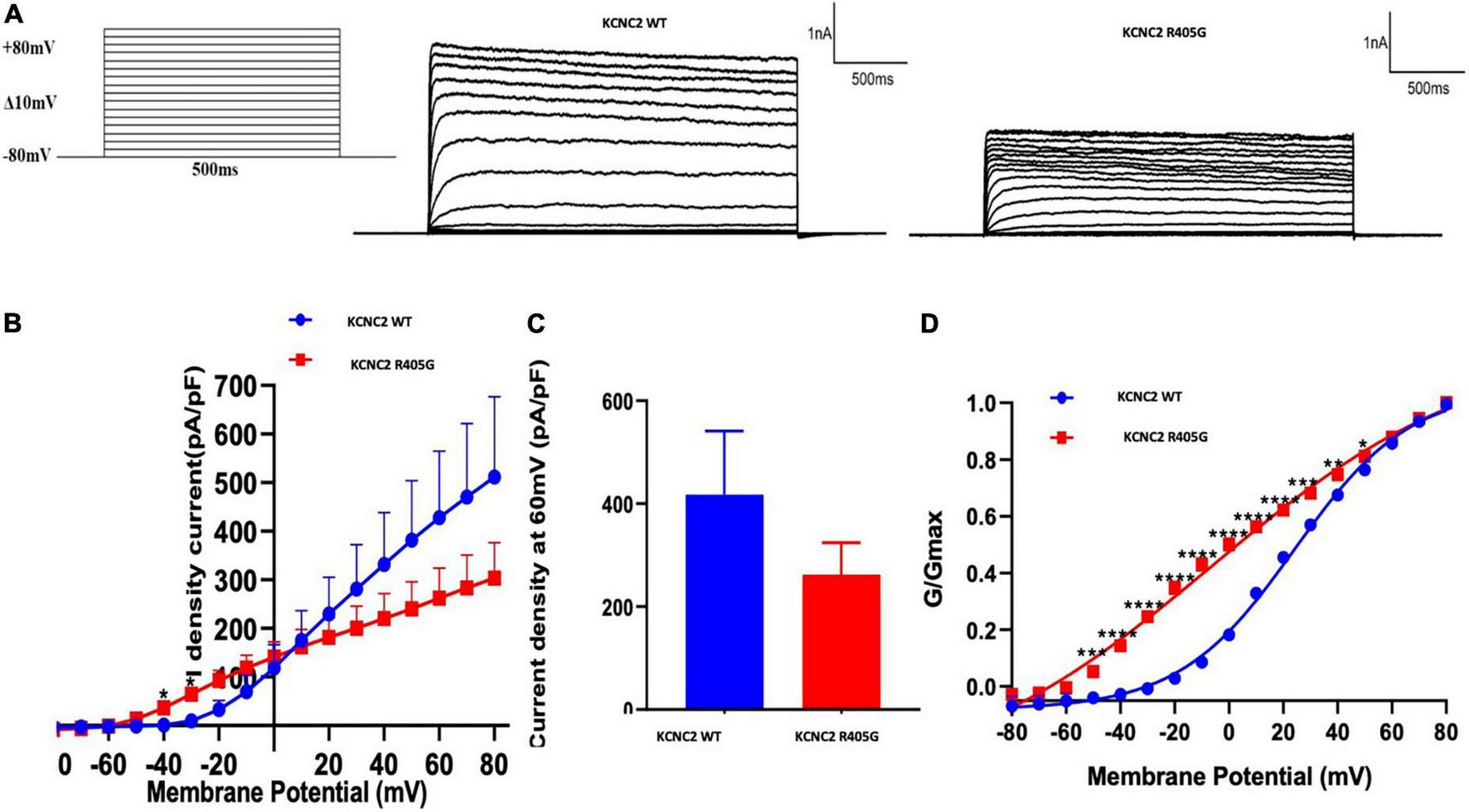
Electrophysiological analysis of wild type and mutant KCNC2 channels. (A) Current outward traces from HEK-293 cells transfected with plasmids encoding wild type (WT) or R405G mutant KCNC2 channels in response to the voltage steps from –80 to +80 mV (with an increment of 10 mV). Current scale: 1,000 pA; time scale: 500 ms. (B) Averaged current density–voltage curves of WT (n = 5) and R405G (n = 7) channels. (C) Normalized current densities at + 60 mV from HEK-293 cells expressing WT (n = 5) or R405 (n = 7) channels. (D) Normalized conductance versus voltage relationships of WT (n = 5) and R405G (n = 7) channels. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Review of the literature
Seven KCNC2 variants were reported in 11 DEE cases in four published research articles, in which V471L, R351K, and T437N were identified in two unrelated patients, respectively, and a T437A was found in a sporadic DEE case (Figure 2B and Table 1). Six out of seven variants were de novo, except S333T in patient 13 (Table 1) was inherited from her unaffected mother, and the authors concluded that S333T was a modifying variant. In addition, none of the seven variants were found in affected individuals without DEE.
Discussion
The clinical findings in both patients were consistent with the diagnostic criteria for DEE. DEE refers to a group of heterogeneous severe progressive disorders consisting of multiform epilepsies, often intractable, accompanied by developmental delay and ID (Hamdan et al., 2017). In this study, both patients had moderate-to-severe GDD, a well-established indicator for CNS developmental disorders or encephalopathy in early childhood. Consistent with the literature (Rademacher et al., 2020; Vetri et al., 2020; Rydzanicz et al., 2021), we found that focal impaired awareness seizure with an EEG of polymorphic generalized spike and wave complexes and infantile spasms with a typical EEG showing hypsarrhythmia were two traits of seizures attributed to KCNC2 defects. In addition, no significant brain lesions were found in both patients in this study, which may account for their better response to antileptic drugs (LEV/VPA were provided in both cases) to a certain extent, than patients with significant findings on brain imaging due to KCNC2-DEE (Rademacher et al., 2020; Vetri et al., 2020; Rydzanicz et al., 2021; Schwarz et al., 2022; Table 1).
Developmental and epileptic encephalopathy is characterized by high genetic heterogeneity with more than 100 genes associated with DEE or syndromes that meet the definition of a DEE (MIM: PS308350). Among them, synaptopathies and neurological channelopathies are two large groups. DEEs caused by synaptopathies gene variants are usually highly heterogeneous and lack clear genotype–phenotype associations, but neurodevelopmental disorders characterized by central vision impairment due to VAMP2 deficiency constitutes the SNARE complex and hand motor loss in Rett-like syndrome, and myoclonic seizures and subsequent ataxia caused by STX1B pathogenic variants are beneficial to clinical differential diagnosis (Spoto et al., 2022); neurological channelopathies have more related disease types. A typical classification includes epilepsy/migraine, neuromuscular disease, and ataxia, which may be related to the expression-specific organization of ion channel genes (Kullmann, 2010). An example related to specific genotype–phenotype associations is the T-type calcium channelopathy study on CACNA1H (Weiss and Zamponi, 2020). Epileptic syndromes caused by CACNA1H defects include a variety of neurodevelopmental disorders and the authors tried to identify the genotypic associations in different phenotypes, including autism spectrum disorder (ASD), immunoglobulin-E (IgE), and neuromuscular disorders. However, the results are not ideal: first, for example, the variant causing ASD may be gain-of-function (GOF) or loss-of-function (LOF). Second, most of the variants listed are only found in a single case. However, a KCNC2-associated DEE is not yet well understood. Recently, eight variants, C125W, E135G, D167Y, R351K, T437N, or T437A, V471L, and S333T, are identified in the KCNC2 gene in DEE cases (Rademacher et al., 2020; Vetri et al., 2020; Rydzanicz et al., 2021; Schwarz et al., 2022), suggesting a subtype of KCNC2-DEE. In addition, Rajakulendran et al. (2013) described three affected members in a family who shared the phenotype of cerebellar ataxia, ID, and motor disability. Interestingly, one had epilepsy with abnormal EEG due to a heterozygous 670 kb deletion of chromosome 12q21 that covers full-length ATXN7L3B and 3–5 exons of KCNC2. Consistently, our findings in two unrelated patients have supported the theory that KCNC2 is a DEE causative gene.
Identifying the same heterozygous variant in unrelated patients can help investigate disease genotype–phenotype associations. In this study, eight of the nine variants, including R405G, were presumed to be linked to DEE, given that none were found in the general population or patients without DEE. Moreover, we compared the core phenotype of DEE in patients harboring KCNC2 variants on four sites, R405, T437, R351, and V471, which found amino acid changes in multiple unrelated individuals. The results revealed a remarkable genotype–phenotype association in these cases. In our study, two unrelated patients shared phenotypes, including focal impaired awareness seizure, motor developmental disorder, severe ID, and good response to antiepileptic drugs; in the case of V471, both patients had infantile spasms, severe ID, and ineffective antiepileptic medication (Vetri et al., 2020; Rydzanicz et al., 2021); DEE due to R351K has been associated with significant heterogeneity in the outcomes of encephalopathy. Schwarz et al. (2022) reported two unrelated male infants (7 years and 8 months old); one had severe DD and intractable seizures, while the other one had mild DD and became seizure-free with VPA treatment. However, both patients had generalized tonic-clonic seizures with myoclonic, complex focal seizures, and an EEG characterized by continuous spike and wave during slow-wave sleep (CSWS) (Schwarz et al., 2022). Moreover, three cases were diagnosed with DEE attributed to a variant on T437 in KCNC2, and two patients with the variant T437N had an identical phenotype: febrile seizure and myoclonic epilepsy with generalized EEG features, which exhibited a poor response to antiepileptic drugs and moderate DD, while T437A caused intractable early-onset absence epilepsy (EOAE) with tonic and severe DD (Schwarz et al., 2022). Interestingly, all patients having variants on R405, T437, or R351 had no significant brain lesions compared to both patients harboring V471L, who had widespread hypomyelination and hypertrophic frontal lobes at the age of 8 years (Vetri et al., 2020) or internal and external hydrocephalus at the age of 2 years (Rydzanicz et al., 2021; Table 1). The pathomechanisms of these recurrent KCNC2 variants remain unclear, although findings of the genotype–phenotype associations would be valuable in clinical practice.
KCNC2 R405 is located in a highly conserved K+ selectivity filter region within the ion transport domain, between transmembrane regions S5 and S6. Although high-resolution structures of KCNC2 channels are still unavailable, modeling based on AlphaFold predicted KCNC2 channels revealed that the R405 residue participates in stabilization by forming hydrogen bonds with the side chains of residues in S5 (Y401 and Y402) and loop (A408, F421, and K422), in which three interactions (Y401, F421, and K422) could not be detected in the G405 mutated protein. Thus, the R405G variant would destabilize the channel by impeding such electrostatic interactions. Recently, the changes described for the R405G channel may differ from D167Y and V471L variants associated with DEE. D167Y is close to the N-terminal region (BTB/POZ domain) and may impair the tetramerization of the channel (Long et al., 2005; Rademacher et al., 2020), while V471L impairs the S6 domain function and alters K+ channel gating as the access to the pore (Liu et al., 1997; Labro and Snyders, 2012). Recently, a novel LOF KCNC2 variant V437A was detected in two siblings diagnosed with autism, epileptic encephalopathy, and mild dysmorphism (Mehinovic et al., 2022). However, there are no other cases of association of V437A with autism or DEE.
We performed an in vitro electrophysiological analysis, and the results revealed that KCNC2 R405G decreased maximal current density but shifted membrane potential to more negative values in the activation curve of KV3.2 channels. Currents carried out by R405G mutant were inhibited, exhibiting reduced selectivity for K+ ions, consistent with the location of the R405 residue in the pore helix before the selectivity filter, which interacted with residues in both S5 and pore helices to modulate the selectivity of KV3.2 channels. During the analysis of the channel kinetics, the R405G mutant displayed a significantly slower deactivation and significantly reduced current compared to WT KCNC2 channels (gain-of-function), which exhibit the same electrophysiological characterization as the D167Y mutant (Rademacher et al., 2020).
In conclusion, we identified a novel KCNC2 variant and provided additional clinical and genetic findings that suggest a subtype of KCNC2-associated DEE syndrome. Comparison of the clinical features in five unrelated DEE cases revealed a consistent genotype–phenotype correlation, while molecular and cellular biology analysis showed that novel variant R405G increased the conductivity at the same voltage, which indicated a gain-of-function alteration due to R405G (Yan et al., 2005).
Statements
Data availability statement
The original contributions presented in this study are publicly available. Information about KCNC2 variant R405G reported in this study can be accessed online using id #0000872152 through the lovd database (https://databases.lovd.nl/shared/variants/0000872152#00010341).
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Wuhan Children’s Hospital, Hubei, China (serial number: 2022497). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
SW: designed and conceptualized the study, contributed to the revision of the manuscript, and editing. YY: designed and conceptualized the study. XW: recruited and phenotyped the patients. XD: contributed to the revision of the manuscript. ZL: drafted the manuscript and prepared the figures. JM: conducted electrophysiological studies. WG: conducted molecular analysis and interpreted the data. DS: conceptualization, visualization, resources, supervision, writing – review & editing, and funding acquisition. All authors reviewed the manuscript and agreed to the published version of the manuscript.
Funding
This work was supported by the General Project of Hubei Provincial Health Commission (No. WJ2021M017), Construction Project of Clinical Medical Research Center for Neurodevelopmental Disabilities in Children in Hubei Province (No. HST2020-19), Project of Wuhan Health and Family Planning Commission (Nos. WX17B12, WX08D17, WX11C26, and WX17C16), and Department of Neurological Diseases, Wuhan Children’s Hospital (No. 2022FEYJS001).
Acknowledgments
We are indebted to the patients and the parents who participated in this study.
Conflict of interest
WG was employed by Chigene (Beijing) Translational Medical Research Center Co. Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1
Atzori M. Lau D. Tansey E. P. Chow A. Ozaita A. Rudy B. et al (2000). H2 histamine receptor-phosphorylation of Kv3.2 modulates interneuron fast spiking.Nat. Neurosci.3791–798. 10.1038/77693
2
Chow A. Erisir A. Farb C. Nadal M. S. Ozaita A. Lau D. et al (1999). K(+) channel expression distinguishes subpopulations of parvalbumin- and somatostatin-containing neocortical interneurons.J. Neurosci.199332–9345. 10.1523/jneurosci.19-21-09332.1999
3
Erisir A. Lau D. Rudy B. Leonard C. S. (1999). Function of specific K(+) channels in sustained high-frequency firing of fast-spiking neocortical interneurons.J. Neurophysiol.822476–2489. 10.1152/jn.1999.82.5.2476
4
González C. Baez-Nieto D. Valencia I. Oyarzún I. Rojas P. Naranjo D. et al (2012). K(+) channels: Function-structural overview.Compr. Physiol.22087–2149. 10.1002/cphy.c110047
5
Hamdan F. F. Myers C. T. Cossette P. Lemay P. Spiegelman D. Laporte A. D. et al (2017). High rate of recurrent de novo mutations in developmental and epileptic encephalopathies.Am. J. Hum. Genet.101664–685. 10.1016/j.ajhg.2017.09.008
6
He N. Lin Z. J. Wang J. Wei F. Meng H. Liu X. R. et al (2019). Evaluating the pathogenic potential of genes with de novo variants in epileptic encephalopathies.Genet. Med.2117–27. 10.1038/s41436-018-0011-y
7
Kaczmarek L. K. Zhang Y. (2017). Kv3 channels: Enablers of rapid firing, neurotransmitter release, and neuronal endurance.Physiol. Rev.971431–1468. 10.1152/physrev.00002.2017
8
Kullmann D. M. (2010). Neurological channelopathies.Annu. Rev. Neurosci.33151–172. 10.1146/annurev-neuro-060909-153122
9
Labro A. J. Snyders D. J. (2012). Being flexible: The voltage-controllable activation gate of kv channels.Front. Pharmacol.3:168. 10.3389/fphar.2012.00168
10
Lal D. May P. Perez-Palma E. Samocha K. E. Kosmicki J. A. Robinson E. B. et al (2020). Gene family information facilitates variant interpretation and identification of disease-associated genes in neurodevelopmental disorders.Genome Med.12:28. 10.1186/s13073-020-00725-6
11
Lau D. Vega-Saenz de Miera E. C. Contreras D. Ozaita A. Harvey M. Chow A. et al (2000). Impaired fast-spiking, suppressed cortical inhibition, and increased susceptibility to seizures in mice lacking Kv3.2 K+ channel proteins.J. Neurosci.209071–9085. 10.1523/jneurosci.20-24-09071.2000
12
Lee S. M. Kim J. E. Sohn J. H. Choi H. C. Lee J. S. Kim S. H. et al (2009). Down-regulation of delayed rectifier K+ channels in the hippocampus of seizure sensitive gerbils.Brain Res. Bull.80433–442. 10.1016/j.brainresbull.2009.07.016
13
Liu Y. Holmgren M. Jurman M. E. Yellen G. (1997). Gated access to the pore of a voltage-dependent K+ channel.Neuron19175–184. 10.1016/s0896-6273(00)80357-8
14
Long S. B. Campbell E. B. Mackinnon R. (2005). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel.Science309897–903. 10.1126/science.1116269
15
McDonald A. J. Mascagni F. (2006). Differential expression of Kv3.1b and Kv3.2 potassium channel subunits in interneurons of the basolateral amygdala.Neuroscience138537–547. 10.1016/j.neuroscience.2005.11.047
16
Mehinovic E. Gray T. Campbell M. Ekholm J. Wenger A. Rowell W. et al (2022). Germline mosaicism of a missense variant in KCNC2 in a multiplex family with autism and epilepsy characterized by long-read sequencing.Am. J. Med. Genet. A1882071–2081. 10.1002/ajmg.a.62743
17
Muona M. Berkovic S. F. Dibbens L. M. Oliver K. L. Maljevic S. Bayly M. A. et al (2015). A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy.Nat. Genet.4739–46. 10.1038/ng.3144
18
Rademacher A. Schwarz N. Seiffert S. Pendziwiat M. Rohr A. van Baalen A. et al (2020). Whole-exome sequencing in NF1-related west syndrome leads to the identification of KCNC2 as a novel candidate gene for epilepsy.Neuropediatrics51368–372. 10.1055/s-0040-1710524
19
Raga S. Specchio N. Rheims S. Wilmshurst J. M. (2021). Developmental and epileptic encephalopathies: Recognition and approaches to care.Epileptic Disord.2340–52. 10.1684/epd.2021.1244
20
Rajakulendran S. Roberts J. Koltzenburg M. Hanna M. G. Stewart H. (2013). Deletion of chromosome 12q21 affecting KCNC2 and ATXN7L3B in a family with neurodevelopmental delay and ataxia.J. Neurol. Neurosurg. Psychiatry841255–1257. 10.1136/jnnp-2012-304555
21
Rudy B. McBain C. J. (2001). Kv3 channels: Voltage-gated K+ channels designed for high-frequency repetitive firing.Trends Neurosci.24517–526. 10.1016/s0166-2236(00)01892-0
22
Rudy B. Chow A. Lau D. Amarillo Y. Ozaita A. Saganich M. et al (1999). Contributions of Kv3 channels to neuronal excitability.Ann. N. Y. Acad. Sci.868304–343. 10.1111/j.1749-6632.1999.tb11295.x
23
Rydzanicz M. Zwolinski P. Gasperowicz P. Pollak A. Kostrzewa G. Walczak A. et al (2021). A recurrent de novo variant supports KCNC2 involvement in the pathogenesis of developmental and epileptic encephalopathy.Am. J. Med. Genet. A1853384–3389. 10.1002/ajmg.a.62455
24
Scheffer I. E. Berkovic S. Capovilla G. Connolly M. B. French J. Guilhoto L. et al (2017). ILAE classification of the epilepsies: Position paper of the ILAE commission for classification and terminology.Epilepsia58512–521. 10.1111/epi.13709
25
Schwarz N. Seiffert S. Pendziwiat M. Rademacher A. V. Brunger T. Hedrich U. B. S. et al (2022). Spectrum of phenotypic, genetic, and functional characteristics in patients with epilepsy with KCNC2 pathogenic variants.Neurology98e2046–e2059. 10.1212/WNL.0000000000200660
26
Spoto G. Valentini G. Saia M. C. Butera A. Amore G. Salpietro V. et al (2022). Synaptopathies in developmental and epileptic encephalopathies: A focus on pre-synaptic dysfunction.Front. Neurol.13:826211. 10.3389/fneur.2022.826211
27
Stern S. Sarkar A. Stern T. Mei A. Mendes A. P. D. Stern Y. et al (2020). Mechanisms underlying the hyperexcitability of CA3 and dentate gyrus hippocampal neurons derived from patients with bipolar disorder.Biol. Psychiatry88139–149. 10.1016/j.biopsych.2019.09.018
28
Vetri L. Cali F. Vinci M. Amato C. Roccella M. Granata T. et al (2020). A de novo heterozygous mutation in KCNC2 gene implicated in severe developmental and epileptic encephalopathy.Eur. J. Med. Genet.63:103848. 10.1016/j.ejmg.2020.103848
29
Weiss N. Zamponi G. W. (2020). Genetic T-type calcium channelopathies.J. Med. Genet.571–10. 10.1136/jmedgenet-2019-106163
30
Yan L. Herrington J. Goldberg E. Dulski P. M. Bugianesi R. M. Slaughter R. S. et al (2005). Stichodactyla helianthus peptide, a pharmacological tool for studying Kv3.2 channels.Mol. Pharmacol.671513–1521. 10.1124/mol.105.011064
31
Yu F. H. Mantegazza M. Westenbroek R. E. Robbins C. A. Kalume F. Burton K. A. et al (2006). Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy.Nat. Neurosci.91142–1149. 10.1038/nn1754
32
Zhang Y. Kaczmarek L. K. (2016). Kv3.3 potassium channels and spinocerebellar ataxia.J. Physiol.5944677–4684. 10.1113/JP271343
Summary
Keywords
developmental and epileptic encephalopathy, whole-exome sequencing, potassium channels, KCNC2, Kv3.2, R405G
Citation
Wang S, Yu Y, Wang X, Deng X, Ma J, Liu Z, Gu W and Sun D (2022) Emerging evidence of genotype–phenotype associations of developmental and epileptic encephalopathy due to KCNC2 mutation: Identification of novel R405G. Front. Mol. Neurosci. 15:950255. doi: 10.3389/fnmol.2022.950255
Received
22 May 2022
Accepted
20 July 2022
Published
25 August 2022
Volume
15 - 2022
Edited by
Sreekanth Puttachary, Oregon State University, United States
Reviewed by
Aleksandra Jezela-Stanek, National Institute of Tuberculosis and Lung Diseases, Poland; Corrado Romano, University of Catania, Italy; Antonio Gennaro Nicotera, University of Messina, Italy; Katrine M. Johannesen, Department of Clinical Genetics, Rigshospitalet, Denmark
Updates
Copyright
© 2022 Wang, Yu, Wang, Deng, Ma, Liu, Gu, Sun.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Sun, Sundan@zgwhfe.com
†These authors have contributed equally to this work
This article was submitted to Brain Disease Mechanisms, a section of the journal Frontiers in Molecular Neuroscience
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.