1
Division of Cell and Molecular Biology, Imperial College London, London, UK
2
Institute of Medical Sciences, University of Aberdeen, Aberdeen, UK
Although GABAA receptor-mediated inhibition of cerebellar Purkinje cells by molecular layer interneurons (MLIs) has been studied intensely at the cellular level, it has remained unclear how this inhibition regulates cerebellum-dependent behaviour. We have implemented two complementary approaches to investigate the function of the MLI-Purkinje cell synapse on the behavioural level. In the first approach we permanently disrupted inhibitory fast synaptic transmission at the synapse by genetically removing the postsynaptic GABAA receptors from Purkinje cells (PC-Δγ2 mice). We found that chronic disruption of the MLI-Purkinje cell synapse strongly impaired cerebellar learning of the vestibular occular reflex (VOR), presumably by disrupting the temporal patterns of Purkinje cell activity. However, in PC-Δγ2 mice the baseline VOR reflex was only mildly affected; indeed PC-Δγ2 mice show no ataxia or gait abnormalities, suggesting that MLI control of Purkinje cell activity is either not involved in ongoing motor tasks or that the system compensates for its loss. To investigate the latter possibility we developed an alternative genetic technique; we made the MLI-Purkinje cell synapse selectively sensitive to rapid manipulation with the GABAA receptor modulator zolpidem (PC-γ2-swap mice). Minutes after intraperitoneal zolpidem injection, these PC-γ2-swap mice developed severe motor abnormalities, revealing a substantial contribution of the MLI-Purkinje cell synapses to real time motor control. The cell-type selective permanent knockout of synaptic GABAergic input and the fast reversible modulation of GABAergic input at the same synapse illustrate how pursuing both strategies gives a fuller view.
“It seems likely that the cerebellum may be the first fragment of the higher levels of the nervous system to be understood in principle, all the way from peripheral input to peripheral output”. So stated John Eccles in a Review Lecture at a Physiological Society meeting in the early 1970s (Eccles, 1973
). At that time it was already known that the cerebellum was necessary for motor control and produces and stores motor memories. The question was how. Eccles expected the “cerebellar problem” to soon be solved; after all, it was considered the simplest part of the higher brain. During the 1960s, Eccles and collaborators had given Cajal’s famous cerebellar circuit life and colour. They investigated the transmitters and polarity of the synapses, providing a view that has not changed fundamentally to this day and was captured in the slogan: “the cerebellum as a neuronal machine” (Figure 1
A, Eccles et al., 1967a
; Ito, 2006
). Anticipating “systems biology”, mathematicians such as Albus and Marr were captivated, and provided models that continue to guide investigations (Marr, 1969
; Albus, 1971
). Although Eccles’ prophecy has not been fulfilled, reaching an understanding of how neuronal circuits work and what they compute remains a fundamental aim of neuroscience, perhaps even the most fundamental. For the cerebellum we remain in an excellent position to achieve this aim (Hansel et al., 2001
; Linden, 2003
; Nakanishi, 2009
). The prosperous circumstances include: 1, the accurate, almost fully known circuit map (Grillner et al., 2005
); 2, quantifiable behavioural tests for the structure’s performance such as locomotion, vestibular-occular reflex (VOR), eye blink conditioning (Boyden et al., 2004
; De Zeeuw and Yeo, 2005
) and fear conditioning (Scelfo et al., 2008
); 3, precise cell-type specific targeting, permitting interventional analysis of the circuit (e.g. Oberdick et al., 1990
; Smeyne et al., 1995
; Bahn et al., 1997
; Barski et al., 2000
; Aller et al., 2003
; Wulff et al., 2007
, 2009
; Gajendran et al., 2009
; Nakanishi, 2009
); 4, techniques that enable specific cerebellar cell types to be reversibly modulated on a fast time scale in vivo (e.g. Tervo and Karpova, 2007
; Wulff et al., 2007
; Luo et al., 2008
; Arenkiel and Ehlers, 2009
). These techniques allow dynamic analysis of individual circuit components and how they relate to, or produce behaviour. Here we discuss our work on how two precise interventions within the circuit, one a permanent ablation of inhibitory synaptic input onto Purkinje cells, the other a selective fast reversible augmentation of the same inhibitory input have given complimentary insights into the function of molecular layer interneuron (MLI)-Purkinje cell synapses. Both strategies involved the same protein, the γ2 subunit of the GABAA receptor (Pritchett et al., 1989
).
Conserved in its essential organization throughout the 550 million years of vertebrate evolution, from fish through to humans, the cerebellar cortex is a folded sheet of microcircuitry reiterated on a vast scale (Eccles et al., 1967a
; Apps and Garwicz, 2005
; Grillner et al., 2005
; Rokni et al., 2008
). The same circuitry in different areas of the cerebellum probably computes similar operations linked to different parts of, for example, the neocortex (Ito, 2008
). The conservation implies that there has been a selection pressure to maintain a core neural computation performed by this configuration of circuitry (Devor, 2000
; Bell, 2002
; Paulin, 2005
). This core function is still not understood, but it probably measures or predicts time intervals and serves motor control, perception and cognition (Ohyama et al., 2003
; Apps and Garwicz, 2005
; De Zeeuw and Yeo, 2005
; Paulin, 2005
; Bastian, 2006
; Ito, 2008
; Rokni et al., 2008
); in mammals, a notably similar and developmentally related cerebellar-like circuit, with analogous cell types, occurs in the dorsal cochlear nucleus of the hearing system (Devor, 2000
). Specialized parts of the cerebellar cortex receive specific peripheral inputs and participate in defined behaviours e.g. the flocculus contributes to vestibular function and compensatory eye movements, and vermal lobules V-VI (an area of convergence of acoustic and nociceptive stimuli) to fear conditioning (Sacchetti et al., 2002
; Gittis and du Lac, 2006
). Purkinje cells form the only output from the cerebellar cortex, projecting to the deep cerebellar (DCN) or vestibular nuclei (VN) (Figure 1
A). Thus all other cerebellar cortical synapses and cell types must exist with the sole aim of modifying, directly or indirectly, Purkinje cell output to the DCN and VN (Figure 1
A; Pugh and Raman, 2009
). Why are all these cortical components needed? Purkinje cells are spontaneously active at around 50 action potentials/second in the absence of synaptic input (Hausser and Clark, 1997
; Raman and Bean, 1997
; Pugh and Raman, 2009
), so their inhibition or excitation by the surrounding cortical microcircuitry modifies either their firing rate or, for a given frequency, the temporal pattern of Purkinje cell action potentials (spikes).
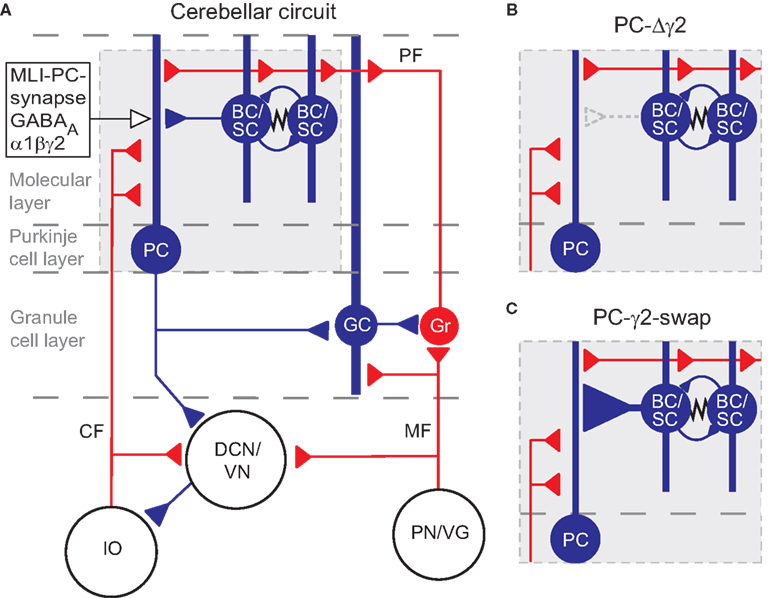
Figure 1. The cerebellar circuitry and the interventions at the MLI Purkinje cell synapses. Excitatory cells are shown in red, inhibitory cells are shown in blue. (A) All cells in the cortex are inhibitory apart from granule cells (Gr) which give rise to parallel fibres (PF), and unipolar brush cells (not shown). The cerebellar cortex receives excitatory input via mossy fibres (MF) and climbing fibres (CF). The only output of the cortex is via Purkinje cells (PC), which project to the deep cerebellar nuclei (DCN) and vestibular nuclei (VN). The activity of Purkinje cells is under inhibitory control from molecular layer basket and stellate cells (BC/SC) through α1βγ2 subunit containing GABAA receptors. Basket/stellate cells mutually inhibit each other and are coupled by gap junctions (zig-zag line). Diagram adapted from Grillner et al. (2005)
. (B) and (C) show the MLI-Purkinje cell synapse after chronic disruption in PC-Δγ2 mice (B) and during rapid enhancement with zolpidem in PC-γ2-swap mice (C). GC, Golgi cell; IO, inferior olive; PN/VG, pontine nuclei and vestibular ganglion.
Inhibition onto Purkinje Cells
A clear feature from the static description of the circuit in Figure 1
A is the dominance of inhibition: the majority of the cortex uses inhibitory synapses and GABAA receptors (Eccles, 1973
; Brickley et al., 1996
; Wisden et al., 1996
; Vicini et al., 2001
; Fritschy and Panzanelli, 2006
); and the Purkinje cells themselves are inhibitory (GABAergic). In the adult circuit, molecular layer interneurons (MLIs) (stellate and basket cells) control Purkinje cells by feed-forward inhibition (Eccles et al., 1967b
; Mittmann et al., 2005
; Smith and Otis, 2005
; Santamaria et al., 2007
; Barmack and Yakhnitsa, 2008
). Basket cells specialize in innervating the Purkinje cell soma and lower one third of the dendritic tree; stellate cells innervate the outer two thirds of the Purkinje cell dendritic tree. In the developing mouse cerebellum subsets of Purkinje cells receive another inhibitory input via Purkinje cell axon collaterals (Watt et al., 2009
). However, these Purkinje-Purkinje contacts are not common in adult mice (Watt et al., 2009
). The cortical cerebellar circuit has a precise geometry, lattice like, with the Purkinje cell dendritic tree flattened out into a plane; and the innervations onto the Purkinje cell dendritic tree arriving orthogonal to this (Eccles et al., 1967a
; Rokni et al., 2008
). Each stellate/basket cell innervates (in the cat) about 20–50 Purkinje cells (Eccles et al.,1967a
); the stellate/basket cells inhibit each other, and are coupled by gap junctions (Mann-Metzer and Yarom 1999
; Mittmann et al., 2005
). Adult Purkinje cells fire complex spikes in response to glutamatergic climbing fibre activity, and simple spikes (conventional action potentials) that reflect the integration of intrinsic pacemaker activity with glutamatergic and GABAergic synaptic inputs from parallel fibres and MLIs respectively (Pugh and Raman, 2009
).
Feed-Forward Inhibition and Plasticity
Feed-forward inhibition emerges when inhibitory interneurons and their target cells receive common excitatory input (Smith and Otis, 2005
): in the cerebellar cortex, parallel fibres excite Purkinje cells and stellate/basket cells (MLIs), and these in turn inhibit Purkinje cells (Figure 1
A); this happens fast, about 1 ms after Purkinje cells are activated (Mittmann et al., 2005
). The main role of feed-forward inhibition could be to sharpen up (reduce) the time window in which excitatory postsynaptic potentials onto the Purkinje cells summate to reach spike threshold, a way of coincidence detection of separate parallel fibre inputs (Mittmann et al., 2005
). Previous studies on cerebellar learning have emphasized the role of glutamatergic excitation for plasticity and learning in the cerebellar cortex, e.g. long term potentiation (LTP) and long term depression (LTD) at the parallel fibre to Purkinje cell synapse. However, MLIs also display plasticity both at the input and output level (Kano et al., 1992
; Jorntell and Ekerot, 2002
; Duguid and Smart, 2004
; Liu and Lachamp, 2006
; Mittmann and Hausser, 2007
; Scelfo et al., 2008
; Lachamp et al., 2009
). Some forms of plasticity at MLI-Purkinje cell synapses are expressed post-synaptically or have a postsynaptic component in their mechanism (Kano et al., 1992
; Duguid and Smart, 2004
; Mittmann and Hausser, 2007
); others are expressed presynaptically (Liu and Lachamp, 2006
; Scelfo et al., 2008
; Lachamp et al., 2009
). Indeed, the strength of MLI-Purkinje cell synapses increases after fear conditioning because of enhanced GABA release (Scelfo et al., 2008
); this happens in parallel with LTP at the parallel fibre-Purkinje cell synapse (Scelfo et al., 2008
). The potentiation of the MLI-Purkinje cell synapses may balance the LTP of the excitatory terminal in a form of scaling to preserve a narrow time window for coincidence detection of parallel fibre inputs. However, for lack of a suitably specific method, it has only recently become possible to study the function of the stellate/basket cell inhibitory network on the whole animal level.
Removal of Synaptic GABAA Receptors from Purkinje Cells Reveals that Fast Feed-Forward Inhibition of Purkinje Cells Contributes to Cerebellar Learning
To understand how a system works, and in addition to simply observing it, one can break, inhibit or activate a component and then see how the system behaves, or in the words of a recent column in Nature: “most sciences are in the habit of poking that which they study to gauge its response” (Nature 458, p1077, 2009 – an editorial). Loss of function gene mutations, whether spontaneous or engineered, have given, and continue to give, essential information about the roles of a protein in the organism. Thus to investigate how MLIs contribute to cerebellum-regulated behaviour, our first approach was to disrupt selectively the GABA-gated chloride channel, the GABAA receptor, responsible for conferring fast inhibition onto Purkinje cells. Most GABAA receptors are complexes of α, β and γ2 subunits (Schofield et al., 1987
; Pritchett et al., 1989
; Seeburg et al., 1990
; Luddens and Wisden, 1991
; Rudolph and Mohler, 2004
; Mohler, 2006
; Goetz et al., 2007
; Olsen and Sieghart, 2009
); Purkinje cells express α1β2β3γ2-type GABAA receptors (Laurie et al., 1992
; Persohn et al., 1992
; Pirker et al., 2000
; Vicini et al., 2001
; Fritschy et al., 2006
). The subunits assemble as a pentamer, with the chloride channel at the centre. Although GABAA receptors will form as lower conductance (12–15 pS) αβ complexes, the γ2 subunit enables a high single channel conductance (25–30 pS) and, importantly, is essential to target the receptor complex to the postsynaptic membrane (Lorez et al., 2000
; Schweizer et al., 2003
; Tretter and Moss, 2008
). Mice with a γ2 gene knockout in every cell die in the first postnatal week, underscoring the importance of this subunit for neuronal function (Gunther et al., 1995
). By crossing the L7Cre line, which has Cre recombinase restrictively expressed in Purkinje cells (Barski et al., 2000
), with a line containing a floxed γ2 (gabrg2) allele (Wulff et al., 2007
), we made mice (PC-Δγ2) where the GABAA receptor γ2 subunit was selectively removed from Purkinje cells (Wulff et al., 2007
, 2009
). This produced a Purkinje cell-selective loss of fast GABAA receptor-mediated inhibitory postsynaptic currents (IPSCs), and thus the functional removal of stellate and basket cells from the cerebellar cortex (Figure 1
B). When tested for motor performance, PC-Δγ2 mice showed no ataxia or gait abnormalities and no impairment in the rotarod test (Wulff et al., 2007
). To test for cerebellar function at a higher resolution, we analysed the VOR. The VOR is a cerebellum-dependent reflex needed to stabilize the retinal image despite head motion by transforming vestibular input (from semicircular canals) into occulomotor output (eye muscle contraction), so that the eyes rotate to counteract the head movement (Boyden et al., 2004
; Broussard and Kassardjian, 2004
; De Zeeuw and Yeo, 2005
).
The criteria which quantitatively assess the VOR are (i) gain, defined as eye velocity/head velocity (to avoid retinal slip, a gain of one is needed) and (ii), phase, defined as time shift in degrees between stimulus and eye velocity (to avoid retinal slip, 0° phase difference is needed) (Boyden et al., 2004
). The VOR requires the flocculus, a specialized region of the cerebellar cortex, and the associated vestibular nuclei (“VN” in Figure 1
; Ito, 2006
). In agreement with the absence of clear motor impairments (see above), PC-Δγ2 mice had only mild abnormalities when tested for baseline VOR performance, suggesting that some compensation took place in the system (Wulff et al., 2007
, 2009
). However, the flocculus is also needed for VOR adaptation, a form of cerebellar learning (Boyden et al., 2004
; Broussard and Kassardjian, 2004
; De Zeeuw and Yeo, 2005
; Gittis and du Lac, 2006
; Ito, 2006
). Unlike baseline VOR, any adaptations in the VOR need vision (via the accessory visual system, which signals errors – retinal slip – via climbing fibres to the Purkinje cells and stellate/basket cells). When a drum (visual stimulus), which surrounds the rotating platform with the head-restrained mouse, rotates in or against the direction of the platform, the baseline VOR will lead to a retinal slip (Boyden et al., 2004
). An error signal is transmitted by the climbing fibres (De Zeeuw and Yeo, 2005
). After repeated trials, the VOR system learns to compensate by moving the eyes at a different speed. If for example, the surrounding drum (visual stimulus) moves repeatedly in the same direction as the head, the VOR system needs to slow down or even cancel the eye movements that would normally occur in the opposite direction (this is termed “gain down” adaptation) (Boyden et al., 2004
). This adaptation can go to extremes: when the drum is rotated with higher velocity but in the same direction as the head, the direction of the eye movements becomes completely reversed (phase shift by 180°). When we assessed cerebellar learning (VOR adaptation) in PC-Δγ2 mice, we found that PC-Δγ2 mice showed a near normal capacity for gain-decrease adaptation when tested right after the training session, but a profound lack of memory consolidation when measurements were resumed the next day (Wulff et al., 2009
). The degree of gain reduction (savings) carried forward from the previous day’s learning was significantly smaller in PC Δγ2 mice. The same prominent differences were observed in the consolidation of phase changes. Indeed phase adaptation was more broadly affected in PC-Δγ2 mice and also included strong deficits during acquisition. Thus, PC-Δγ2 mice were entirely unable to learn VOR phase reversal (i.e. to move their eyes in the same direction as the head instead of the opposite direction) (Wulff et al., 2009
). Accordingly, some “resonance” or ongoing activity of feed forward-inhibition onto Purkinje cells is needed after or during learning for the memory to consolidate (see also Attwell et al., 2002
; Cooke et al., 2004
; Kassardjian et al., 2005
). As the presynaptic form of plasticity at MLI-Purkinje cell synapses, due to enhanced GABA release (Scelfo et al., 2008
; Lachamp et al., 2009
), may be preserved in the PC-Δγ2 mice, we cannot rule out that this plasticity contributes to residual learning by dynamically regulating tonic inhibition. Certainly, PC-Δγ2 Purkinje cells receive inhibitory input from both metabotropic GABAB receptors (coupled to G proteins and second messengers) and extrasynaptic low conductance αβ GABAA receptors (Wulff et al., 2009
). These forms of inhibition are probably too slow to precisely organize the simple spike intervals needed for motor learning.
Since Purkinje cells are the only output of the cerebellar cortex, any behavioural deficits in PC-Δγ2 mice must come from altered Purkinje cell activity. Extracellular recordings of Purkinje cell activity in the flocculus of PC-Δγ2 and control mice showed that the temporal patterns of Purkinje cell simple spike activity were changed in PC-Δγ2 mice. Simple spike activity was much more regular in PC-Δγ2 mice (Wulff et al., 2009
). This is consistent with in vitro data: if Purkinje cells in acute slices are bathed in GABAA receptor blockers, the cells fire action potentials with more regularity (Hausser and Clark, 1997
); without the inhibition, the regular “clockwork” pacemaker dominates. Other parameters of Purkinje cell activity such as simple spike firing frequency, phase modulation or modulation amplitude during optokinetic stimulation were not changed in PC-Δγ2 mice. In summary we found that chronic disruption of the MLI-Purkinje cell synapse leads to changes in the temporal pattern of Purkinje cell simple spike activity and deficits in the consolidation of motor memories. We hypothesize that the MLIs shape temporal activity patterns of Purkinje cells under conditions of increased excitatory plasticity (Wulff et al., 2009
). Ultimately, VOR-based motor memories become stored in the VN (Kassardjian et al., 2005
; Gittis and du Lac, 2006
; Shutoh et al., 2006
). The course of motor memory transfer from the cerebellar cortex to the VN is likely to correspond to the consolidation phase; during this time, the Purkinje cells must contribute to plastic changes in the VN by precise temporal interaction with the excitatory mossy fibre input onto the same cells (Nelson et al., 2003
, 2005
; Pugh and Raman, 2009
). So from our studies on the PC-Δγ2 mice, we hypothesized that the precise spacing of the Purkinje cell action potentials would specify the plastic changes during the transfer from the cortex to the VN; in PC-Δγ2 mice the temporal control over Purkinje cell simple spike activity is impaired so that memory cannot be consolidated (transferred from cortex to the VN). Indeed, many believe that it is the fine scale patterning of Purkinje cell action potentials, combined with pauses in spike trains, that is crucial for information transfer to the deep cerebellar and VN (Walter et al., 2006
; De Schutter and Steuber, 2009
). The PC-Δγ2 mice, with their imposed regularity in Purkinje cell spike firing and failure to consolidate VOR adaptation, support this view.
A potential problem in the subtractive analysis of brain function is compensation (Lomber, 1999
; Wulff and Wisden, 2005
; Marder and Goaillard, 2006
). Quite often genetic knockouts produce clear effects (e.g. no consolidation of VOR adaptation in PC-Δγ2 mice, changes in regularity of Purkinje cell firing; Wulff et al., 2009
). However, if no effect is seen (e.g. no change in rotarod or baseline VOR performance), it is possible that either the component is genuinely not important for that particular task or that its loss has been compensated for (e.g. Brickley et al., 2001
; Figure 2
). For example, a knockout of the α6Δ-GABAA receptor that provides extrasynaptic tonic inhibition in cerebellar granule cells leads to transcriptional upregulation of three K2P potassium channel genes (TASK-1, TASK-3 and THIK-2) encoding leak conductances (Brickley et al., 2001
; Aller and Wisden, 2008
). The raised K+ conductance prevents the cells undergoing any change in excitability (Brickley et al., 2001
). Two alternative solutions to the same problem of maintaining granule cells in a certain range of excitability can be provided by either GABAA receptors or K+ leak channels. Parallel solutions to neural network operations also appear in other circuits (Marder and Goaillard, 2006
). How do we decide if there is really no deficit in a knockout or if there has been compensation? For example, in our PC-Δγ2 mice, is fast inhibition onto Purkinje cells genuinely not used to coordinate in real time e.g. motor performance, and is only used for memory formation? Or can the system develop without this inhibition and generate another solution? Indeed, we found that there is an apparent adaptive change in the PC-Δγ2 Purkinje cells: the ablation of GABAergic synaptic input onto Purkinje cells was accompanied by a reduction in parallel fibre-evoked excitatory responses (Wulff et al., 2009
). This could arise either from an altered postsynaptic AMPA receptor function or from an alteration in presynaptic release.
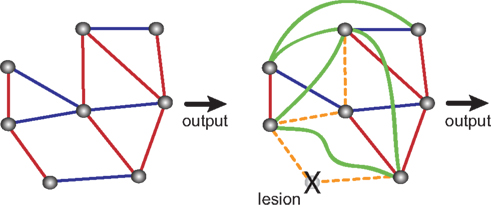
Figure 2. Compensatory changes in an abstract biological network. Blue lines indicate inhibitory interactions, red lines indicate excitatory interactions. Shown are an intact network (left side) and the same network after ablation of one of its nodes. Lesioning one element causes the rest of the system to compensate for the loss. Some interactions are lost (orange dashed lines) and some new interactions emerge (green lines), so that the final output is unchanged. Adapted from Greenspan (2001)
.
The earlier during development we interfere with a system and the longer this interference persists, the larger the probability that the system compensates. Thus we wanted a method for fast (minutes in our case) and reversible modulation of the MLI-Purkinje cell synapse at any chosen time point. Such an approach would also allow us to investigate “when” and for “how long” the synapse is required in a particular process. We have temporal control. These thoughts are not new: reversibly inactivating whole brain parts by cooling (e.g. Eccles applied this for the cerebellum (Eccles et al., 1975
)) or infusion of anesthetics or GABAA receptor agonists and glutamate receptor antagonists into the cerebellum has been used frequently (Lomber, 1999
; Attwell et al., 2002
; Nagao and Kitazawa, 2003
; Cooke et al., 2004
; Shutoh et al., 2006
). Reversible approaches revealed a transfer of memory from cortex to deep cerebellar nuclei: inactivating the cerebellar cortex with muscimol at different times after eyeblink conditioning defined a consolidation time window of 1–2 h post training (Attwell et al., 2002
; Cooke et al., 2004
). Similar experiments with reversible inactivation have been carried out for VOR adaptation (Kassardjian et al., 2005
).
These pharmacological approaches described above reveal what the regions in the cerebellar system do and when. Francis Crick encouraged molecular biologists to go one step beyond this and develop cell type-selective genetic techniques to turn neuronal subtypes on or off with fast (milliseconds) and relatively fast (seconds to minutes) timescales by “remote control” (Crick, 1999
). The first methods for activating neurons by remote control used light, either to activate a retinal like transduction machinery heterologously expressed (Zemelman et al., 2002
) or photo-uncageable capsaicin that activates a ligand-gated ion channel (TRPV1), again heterologoulsy experessed (Zemelman et al., 2003
– this paper, in its title, appears to have the first use of the phrase “remote control” in this context). Just a short while after the introduction of these clever methods came the introduction of the light-activated channel rhodopsins (see Boyden et al., 2005 and Han et al., 2009a
,b
; Sohal et al., 2009
). The extremely easy applicability of the channel rhodopsin system in diverse animals and circuit settings has been a phenomenal breakthrough and captured the imagination of the neuroscience community (O’Connor et al., 2009
; Scanziani and Hausser, 2009
). Light activation has the advantage of high temporal precision (milliseconds) and the potential to deliver specific patterns of stimuli. But we were interested in remote control of the entire population of Purkinje cells. Light at 1 mW mm−2 can illuminate, through an optical fibre, a volume of 0.56 mm3 in the mouse hypothalamus; this is the volume of a typical hypothalamic nucleus (Adamantidis et al., 2007
). Thus, with current optical technology, we would not be able to easily influence many Purkinje cells in vivo with light. Thus for our particular system we decided to build in unique pharmacological sensors into Purkinje cells, such that we could add a ligand into the blood (or orally), that would then cross the blood brain barrier and influence the activity of only Purkinje cells and no other cell type. Such a method of remotely controlling Purkinje cells would have to work reasonably fast (effects within minutes of drug administration) and wear off within an hour or so.
Various ingenious ligand-receptor systems have been applied to selectively inhibit neuronal subtypes in vivo: these include the allatostatin system (Tan et al., 2006
, 2008
), a 5HT1A-receptor system (Tsetsenis et al., 2007
), and the ivermectin-gated Cl− channel system (Lerchner et al., 2007
). Additionally, there is the promising metabotropic clozapine-N-oxide-hM(4)D receptor system, which has so far been published only using in vitro applications (Armbruster et al., 2007
; Nichols and Roth, 2009
). All of these systems rely on the ectopic expression of receptors or ligand-gated channels in the target neurons, and then giving a ligand which can only work on cells expressing the receptor (Wulff and Wisden, 2005
; Arenkiel and Ehlers, 2009
). The allatostatin method consists of a Drosophila peptide-7TM metabotropic receptor pair which promotes opening of GIRK K+ channels. Given that the peptide does not cross the blood brain barrier or easily diffuse within neuronal tissue, it has to be pumped in to the relevant brain area at high concentrations. This method has been used successfully to study inhibitory V1 neurons in mouse spinal cord (Gosgnach et al., 2006
; Tan et al., 2006
) and to selectively silence somatostatin (Sst)-expressing neurons in the preBötzinger complex (Tan et al. 2008
). By contrast, the 5HT1A system has a water soluble agonist (8-OH-DPAT) that can be given systemically and this receptor also promotes opening of GIRK K+ cells, inhibiting neurons reversibly with very good kinetics; on the other hand, a 5HT1A receptor knockout mouse background has to be used, which could cause potential complications. The “5HT method” was applied successfully to inhibit selectively hippocampal dentate granule cells and the central nucleus of the amygdala in vivo and to probe how this affected anxiety-related behaviours (Tsetsenis et al., 2007
). Thus, if expressed selectively in Purkinje cells, triggering either the allatostatin or 5HT1A ligand-receptor systems would probably mimic G-protein-coupled GABAB receptor activation. So these systems would be potentially useful to explore how metabotropic inhibition onto Purkinje cells influences motor memory consolidation. At the other end of the timescale, the Caenorhabdtis elegans ivermectin-gated Cl− channel system, whose α and β subunits are encoded in the same gene superfamily as the GABAA receptor, effectively silences neurons for periods lasting around 8 h, but has slow onset kinetics (Lerchner et al., 2007
). So although this method will aid certain types of experiment, it would not have been appropriate for studying the acute modulation of MLI-Purkinje cell synapses. Nevertheless, because the α and β subunits must co-express to assemble a functional ivermectin receptor, this does give the possibility of applying intersectional genetics to get more selective cell-type targeting (Luan and White, 2007
).
Given our focus on understaning how GABAergic synapses regulate the cerebellar circuit through precisely timed fast inhibition, the obvious and natural strategy for us was to take advantage of drugs which allosterically and selectively modulate GABAA receptors (Schoch et al., 1985
; Luddens and Wisden, 1991
; Korpi et al., 1997
; Rudolph and Mohler, 2004
; Goetz et al., 2007
; Olsen and Sieghart, 2009
). Many drugs bind at sites on the GABAA receptor distinct from the GABA-binding site; these drugs change the shape of the receptor oligomer so that the efficacy of GABA at opening the channel is either increased (positive allosteric agonists, e.g., diazepam or zolpidem) or decreased (negative allosteric agonists, e.g., the β-carboline, DMCM) (Korpi et al., 1997
; Mohler, 2006
; Hanson and Czajkowski, 2008
; Olsen and Sieghart, 2009
). Allosteric modulation of GABAA receptors is a very powerful way to modulate neuronal circuit function, as attested by the universal clinical use of such drugs as benzodiazpeines, steroids and propofol (Yentis et al., 2004
). In the extreme case, by prolonging the duration of GABA-mediated IPSCs, propofol induces full anesthesisa (loss of consciousness); but changing the IPSC kinetics can profoundly alter most aspects of brain function. If we could make these drugs work at specific cell types (or more accurately at specific GABAergic synapses), without affecting other cell types, this would be an effective way to explore how an isolated neuronal pathway contributes to a behaviour. Thus we and our colleagues developed a method for fast reversible modulation of selected GABAergic synapses and applied it to the specific modulation with zolpidem of the stellate/basket cell inhibitory inputs to Purkinje cells (Figures 1
C and 3
; Wulff et al., 2007
). Before we explain this method, we first describe the “benzodiazepine site”.
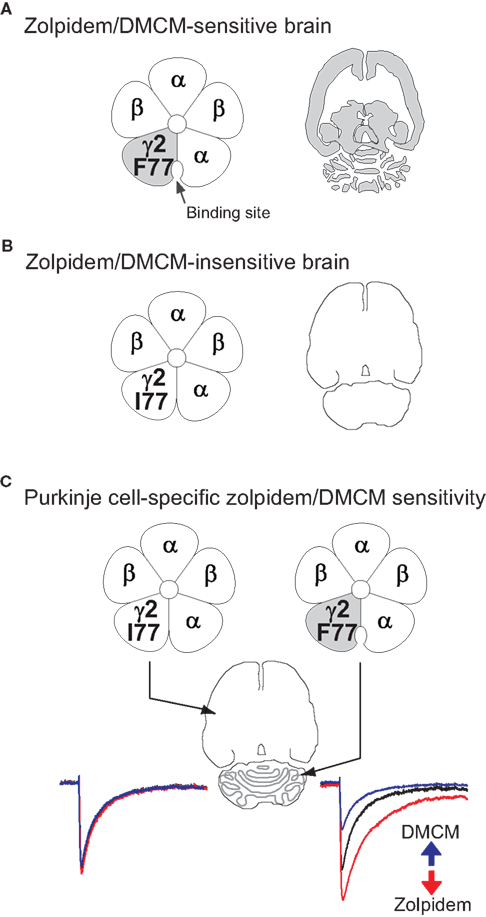
Figure 3. The strategy for bi-directional modulation of selected GABAergic synapses. (A) shows the wild-type situation with a phenylalanine (F) at position 77 in the γ2 subunit of the GABAA receptor. Zolpidem/DMCM binding occurs at the interface of the α and the γ2 subunits and is widely distributed throughout the brain (indicated in grey in the cartoon of a horizontal brain section). In a first step (B) we have changed the phenylalanine at position 77 to isoleucine (I) by homologous recombination in mouse embryonic stem cells. In these mice zolpidem/DMCM binding is abolished. In the last step (C) we have reintroduced the drug-sensitive wild-type γ2 subunit under the control of the Purkinje cell-specific L7 promoter to selectively restore zolpidem/DMCM-sensitivity by a Cre-mediated Purkinje cell-specific swap of γ2 subunits (PC-γ2-swap mice). In these mice expression of zolpidem/DMCM sensitive GABAA receptors is restricted to Purkinje cells of the cerebellum (grey). Note the difference to wild-type mice in (A), which show drug binding throughout the cerebellar cortex. In Purkinje cells zolpidem (red trace) and DMCM (blue trace) now selectively enhance or reduce, respectively, GABAA receptor-mediated inhibitory post synaptic currents, illustrated by the artificial traces on the right (GABA alone: black trace). All other neurons in the brain are insensitive to these drugs (left traces).
Depending on the ligand, the “benzodiazepine site” can mediate different allosteric effects. These drugs require αβγ2-type receptors (Pritchett et al., 1989
; Seeburg et al., 1990
); the drug-binding site resides between the α and γ2 subunits (Sigel, 2002
; Sancar et al., 2007
). It is important to emphasise the modulator aspect of the benzodiazepine site. This is not the same site where GABA binds the receptor; and furthermore, the benzodiazepines and related drugs will not alter Cl− gating unless GABA is also present as well. So the drugs modulate active GABAergic synapses, and also extrasynaptic GABAA receptors (if GABA is also present outside the synapse). Benzodiazepine antagonists like flumazenil (Ro 15-1788), which bind at the same site as the agonists, inhibit the effects of both agonists (positive allosteric modulators) and inverse agonists (negative allosteric modulators); this property, for example, makes flumazenil useful for diagnosing and treating benzodiazepine intoxication e.g. for treating prolonged sedation with benzodiazepines in the intensive care unit (Korpi et al., 1997
; Yentis et al., 2004
). The amino acids contributing to the binding sites of the benzodiazepines and similar drugs, found at the interface between the α and γ2 subunits, have been studied in detail (Wieland et al., 1992
; Sigel, 2002
; Sancar et al., 2007
; Hanson and Czajkowski, 2008
; Olsen and Sieghart, 2009
). The best known is histidine at position 101 in the α1, α2, α3 and α5 subunits; mutating this histidine to arginine produces αβγ2 GABAA receptors insensitive to most benzodiazepine ligands without changing any other property of the receptor (Wieland et al., 1992
). Placing this mutation into the α subunit genes in mice (knock-in mice) allowed the multiple actions of benzodiazepine ligands on the whole animal (sedation, hypnosis, anxyiolysis, muscle relaxing) to be explained in terms of particular GABAA receptor subtypes (Rudolph et al., 1999
; Crestani et al., 2000
; McKernan et al., 2000
; Rudolph and Mohler, 2004
).
As well as amino acid residues on the α subunits, key side chains that form or influence the benzodiazepine binding pocket are found in the γ2 subunit: replacing phenylalanine (F) by isoleucine (I) at position 77 of γ2 abolishes binding of zolpidem, DMCM and flumazenil in αβγ2 complexes (Buhr et al., 1997
; Wingrove et al., 1997
; Cope et al., 2004
; Ogris et al., 2004
). Zolpidem modulates GABA’s action at three main types of GABAA receptor (α1βγ2 – comprising 43% of receptors in the brain, α2βγ2 – 18% of receptors, and α3βγ2 – 17% of receptors, where the β can be one of three variants); together, these α1−α3 receptors account for 78% of the total number of brain GABAA receptors, i.e. the majority of GABAA receptors will be sensitive to zolpidem modulation in the brain (McKernan and Whiting, 1996
). Of these three receptor types, the most abundant and widely expressed in the brain, α1β2/3γ2, has the highest affinity for zolpidem, with an inhibition constant (Ki) of 20 nM (Niddam et al., 1987
; Pritchett et al., 1989
; Wisden et al., 1992
; Duncan et al., 1995
). At higher concentrations, zolpidem modulates receptors, and thus circuits, containing α2βγ2 and α3βγ2 receptors (Ki = 400 nM).
The γ2 subunit gene is widely expressed (Persohn et al., 1992
; Wisden et al., 1992
; Pirker et al., 2000
), and contributes to most GABAA receptors (see above), and therefore most circuit functions in the brain. We reasoned that by engineering all neurons to express a γ2I77 gene (encoding zolpidem-insensitive receptors) and then reintroducing a zolpidem sensitive γ2F77 subunit back into selected cell types, we could explore the function of a variety of brain circuits by selectively manipulating the GABA input onto selected cell types. By homologous recombination at the γ2 subunit gene locus we made knock-in mice (γ2I77lox) such that all neuronal types had GABAA receptors insensitive to zolpidem (Figure 3
), as assessed by behavioural, electrophysiological and pharmacological assays (Cope et al., 2004
, 2005
; Ogris et al., 2004
). Normally, zolpidem is an effective hypnotic and induces sleep, presumably by enhancing GABAA receptor inhibition in sleep regulatory areas (Crestani et al., 2000
). Thus on a rotarod task, normal mice when given zolpidem fall off the rod from a mixture of ataxia and sedation (Cope et al., 2004
); however, γ2I77 mice showed no impairment even after high doses of zolpidem (Cope et al., 2004
). As expected from recombinant studies (Buhr et al., 1997
; Wingrove et al., 1997
), the I77 mutation did not influence GABA’s action at its receptor: in γ2I77 mice, mIPSCs from hippocampal and cerebellar neurons had normal rise times, amplitudes and decay kinetics (Cope et al., 2004
, 2005
; Wulff et al., 2007
). We then arranged it by a cell type-specific genetic swap of the γ2 subunit gene, that only cerebellar Purkinje cells expressed the zolpidem-sensitive γ2F77 subunit, whereas all other neurons in the brain expressed the insensitive γ2I77 subunit. In other words, only GABAA receptors on Purkinje cells were sensitive to zolpidem – these mice were termed PC γ2 swap (Figures 1
C and 3
). We could thus rapidly modulate a class of synapses (MLI-Purkinje cell synapses) in isolation from all other synapses in the brain to analyse their relevance for cerebellar tasks. Several minutes after intraperitoneal injection of zolpidem, PC-γ2-swap mice became ataxic and showed strong impairments in the rotarod and horizontal beam motor tasks. Thus rapid intervention at the MLI-Purkinje cell synapse revealed the involvement of MLIs in the control of real-time motor coordination. Accordingly, the chronic disruption of the MLI-Purkinje cell synapse in PC-Δγ2 mice must have induced compensatory mechanisms (Wulff et al., 2007
).
Bidirectional Modulation of GABAergic Synapses
The opposite of inhibition, activation, is also important for understanding how specific neuronal cell types contribute to an animal’s behaviour and/or perception or the function of a circuit (e.g. Boyden et al., 2005
; Lima and Miesenböck, 2005
; O’Connor et al., 2009
; Zimmermann et al., 2009
). In mice, cell type-specific activation in vivo can be done with light-activated channels (Adamantidis et al., 2007
; O’Connor et al., 2009
), capsaicin-activated TRPV1 channels (Arenkiel et al., 2008
) and a synthetically evolved Gq-coupled metabotropic receptor (hM3Dq) activated by the otherwise inert ligand clozapine-N-oxide (Alexander et al., 2009
; Nichols and Roth, 2009
). Bidirectional modulation allows the exploration of a wider range of function (or potential function) of a particular cell type (or synaptic input). Ideally both approaches should be applied to the same cell. This can be done with light by co-expressing in neurons the genes coding for the blue light-activated cation channelrhodopsin-2 and the yellow light-driven chloride pump halorhodopsin (NpHR), and illuminating the cells with light pulses (Han and Boyden, 2007
; Zhang et al., 2007
; Han et al., 2009a
; Scanziani and Hausser, 2009
; Sohal et al., 2009
). By delivering precisely timed blue and yellow light pulses, this enables the details of neural coding (e.g. spike timing) to be explored, a process termed “informational lesioning” by Han et al. (2009a)
(see also Sohal et al., 2009
).
Manipulating GABAA receptors with allosteric modulators also permits a bidirectional modulation (Figure 3
). The mutation of the γ2 subunit that abolishes zolpidem binding also removes the β-carboline binding site (e.g. DMCM) (Buhr et al., 1997
; Wingrove et al., 1997
): we have shown that GABAA receptors in γ2I77 mice are about 1000-fold less sensitive to DMCM (Ogris et al., 2004
). As DMCM is an inverse agonist at the benzodiazepine binding site, it decreases the effectiveness with which GABA opens the receptor and/or speeds the rate of channel deactivation. This means that DMCM is an excitatory agent; it decreases inhibition. In wild-type (γ2F77) mice DMCM (at low doses) causes severe seizures within minutes of an i.p. injection (Leppa et al., 2005
). However, γ2I77 mice can receive high doses of DMCM without behavioural effects; at extremely high doses, there are still no seizures or neurological defects (e.g. ataxia), but some mild anxiolytic effects, most likely caused by another (undefined) GABAA receptor binding site or subtype (Leppa et al., 2005
). An example of DMCM’s utility or application with the γ2I77/F77 system could be as follows: after fear conditioning, the MLI-Purkinje cell synapses undergo long-term potentiation (Scelfo et al., 2008
); we predict that selectively decreasing the strength at these synapses using DMCM in PC-γ2-swap mice would either undermine learning if given during acquisition, or disrupt consolidation if given directly after acquisition.
Advantages and Disadvantages of the GABAA Receptor System to Control Neural Activity Cell-Type Selectively
(1) As demonstrated by their continuing wide and successful clinical use, drugs that allosterically modulate GABAA receptors have a well-proven effectiveness in modulating all aspects of central nervous system function, from pain perception through to arousal and cognition (Korpi et al., 1997
; Yentis et al., 2004
; Mohler, 2006
).
(2) Since zolpidem works on all α1, α2, α3, β, γ2 subunit containing GABAA receptors (it is selective for α1βγ2 receptors at low nM concentrations, but in the low μM range it modulates in addition α2βγ2 and α3βγ2 receptors), we can target many types of GABAergic synapses in the brain.
(3) Because the GABAA receptor is an ion channel, there are no complications with multiple second messenger pathways having different effects depending on cell type, or even in the same cell type.
(4) GABAergic circuit operations can be modulated bi-directionally with zolpidem (increase in amplitude and prolonged decay of inhibitory postsynaptic currents) and DMCM (decrease in amplitude and faster decay of inhibitory postsynaptic currents). The effect of both drugs can be, in principle, rapidly terminated with flumazenil, as this drug is an antagonist of the benzodiazepine binding site and blocks the actions of agonist benzodiazepine drugs if simultaneously present (Lauven et al., 1985
). Indeed, the behavioural effects produced by zolpidem in PC-γ2–swap mice were blocked by pre-administration of flumazenil (Wulff et al., 2007
).
(5) All drugs are water-soluble molecules and cross the blood brain barrier. Systemic drug applications (i.p. injections) work well for zolpidem, DMCM and flumazenil. For our cerebellar experiments targeting Purkinje cells, no surgery or catheters were needed – but for other situation or brain regions/nuclei the drugs could be given more locally using catheters. After i.p. injection, drug effects are produced within several minutes – the half life of zolpidem after i.p. injection in rodents is about 20 min (Benavides et al., 1988
).
(6) Manipulations with these drugs can modulate the inhibitory synapses on a widely distributed population of neurons (e.g. Purkinje cells, subtypes of hippocampal and neocortical interneurons). With current technology, light delivered through optical cables could only reach few cells. Admittedly, activating even a few cells with light can produce defined outputs: for the hypothalamus a whole nucleus can be light activated to produce a change in arousal (Adamantidis et al., 2007
), or dopaminergic cells in the ventral tegmental area can be selectively light-stimulated to cause changes in behavioural conditioning (Tsai et al., 2009
). Indeed, inducing 14 action potentials (APs) in a single neuron using conventional microstimulation in rat barrel somatosensory cortex is detected by the animal (Houweling and Brecht, 2008
). But some situations may require manipulating many more cells. Han et al. (2009a)
suggest that tiling the brain with arrays of LED- or laser-coupled optical fibres will solve this problem.
(7) Altering fast GABAergic input with GABAA modulators uses the endogenous transmitter pathways, and differs in its effects from sudden activation (e.g. with light) or total silencing. We are thus modulating the natural “rhythm” of GABAergic communication at the synapse, which is a more subtle approach.
(8) The temporal resolution of our pharmacological manipulation is unsuited to probing events on the millisecond range, and it could not be used to deliver precise patterns of stimuli, but drug manipulations of selected cell types with zolpidem and DMCM are suited for reversibly manipulating neuronal activity during periods of a few minutes to hours to study processes like learning, which may require many repetitions over minutes to hours so that biochemical/structural changes can occur at the relevant synapses. Extended continuous modulation of this process by inhibition with zolpidem or excitation with DMCM may be useful.
(9) Because of the requirement for the γ2I77 background, selective manipulation with zolpidem can only be done in transgenic mice; it cannot, unlike the simple ligand-G protein-coupled receptor systems such as the allatostatin or the hM3Dq receptors (Tan et al., 2006
; Alexander et al., 2009
) or light-activated channels and pumps (Boyden et al., 2005
; Han et al., 2009a
,b
), be applied in diverse species. We are, however, currently developing a viral and a “γ2 switch mouse” system that will make the “zolpidem system” more accessible in the mouse.
One of the most enjoyable aspects of researching the brain is that so many methods and approaches can be used to study it (Wulff and Wisden, 2005
; Arenkiel and Ehlers, 2009
; O’Connor et al., 2009
; Scanziani and Hausser, 2009
). There is no “one size fits all” solution for the problem of which particular technique to use for investigating circuits. For the cerebellar system, we have used two complementary genetic approaches to investigate how MLIs regulate Purkinje cells and contribute to cerebellum-dependent behaviour at the whole animal level. Whereas chronic disruption of the stellate/basket cell to Purkinje cell synapse (PC-Δγ2 mice) did not produce any obvious deficits in motor coordination (Wulff et al., 2007
, 2009
), remote control of Purkinje cells by a reversible and selective enhancement of GABAergic transmission at the same synapse (PC-γ2-swap mice) following i.p. administration of a drug, zolpidem, revealed the involvement of interneurons in on-line motor control (Wulff et al., 2007
). These results suggest that the circuit can compensate for the loss of MLIs despite their genuine involvement in motor coordination. Extended analysis of PC-Δγ2 mice, however, showed that MLI are indispensable for a different cerebellar task: motor learning (Wulff et al., 2009
). By combining these two approaches we were thus able to delineate dispensable contributions and indispensable requirements of MLIs depending on the behavioural context. Reversible modulation of selected GABAergic synapses can now be applied to analyse the time window in which MLI activity is required for cerebellar learning. Modulating the inhibitory drive onto distributed populations of selected cell types with zolpidem could also assist the study of other neural circuits.
The authors declare that this review article was written in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by MRC grant G0800399 to Peer Wulff and William Wisden. We thank our colleagues A. Badura, D. Cope, M. Farrant, Z. Gao, T. Goetz, F. E. Hoebeek, E. Korpi, E. Leppa, A. M. Linden, M. Renzi, M. Sassoè-Pognetto, M. Schonewille, W. Sieghart, E. Sigel, P. Somogyi, J. Swinny, S. van Dorp, O. Y. Vekovischeva, L. Viltono and C. I. De Zeeuw for their contributions to the published work summarized in this review; and N. P. Franks for helpful discussion.
Alexander, G. M., Rogan, S. C., Abbas, A. I., Armbruster, B. N., Pei, Y., Allen, J. A., Nonneman, R. J., Hartmann, J., Moy, S. S., Nicolelis, M. A., McNamara, J. O., and Roth, B. L. (2009). Remote control of neuronal activity in transgenic mice expressing evolved G protein-coupled receptors. Neuron 63, 27–39.
Benavides, J., Peny, B., Dubois, A., Perrault, G., Morel, E., Zivkovic, B., and Scatton, B. (1988). In vivo interaction of zolpidem with central benzodiazepine (BZD) binding sites (as labeled by [3H]Ro 15-1788) in the mouse brain. Preferential affinity of zolpidem for the omega 1 (BZD1) subtype. J. Pharmacol. Exp. Ther. 245, 1033–1041.
Cope, D. W., Halbsguth, C., Karayannis, T., Wulff, P., Ferraguti, F., Hoeger, H., Leppa, E., Linden, A. M., Oberto, A., Ogris, W., Korpi, E. R., Sieghart, W., Somogyi, P., Wisden, W. and Capogna, M. (2005). Loss of zolpidem efficacy in the hippocampus of mice with the GABAA receptor γ2 F77I point mutation. Eur. J. Neurosci. 21, 3002–3016.
Cope, D. W., Wulff, P., Oberto, A., Aller, M. I., Capogna, M., Ferraguti, F., Halbsguth, C., Hoeger, H., Jolin, H. E., Jones, A., Mckenzie, A. N. J., Ogris, W., Poeltl, A., Sinkkonen, S. T., Vekovischeva, O. Y., Korpi, E. R., Sieghart, W., Sigel, E., Somogyi, P. and Wisden, W. (2004). Abolition of zolpidem sensitivity in mice with a point mutation in the GABAA receptor γ2 subunit. Neuropharmacology 47, 17–34.
Gunther, U., Benson, J., Benke, D., Fritschy, J. M., Reyes, G., Knoflach, F., Crestani, F., Aguzzi, A., Arigoni, M., Lang, Y., Bluethmann, H., Mohler, H. and Luscher, B. (1995). Benzodiazepine-insensitive mice generated by targeted disruption of the γ2 subunit gene of γ-aminobutyric acid type A receptors. Proc. Natl. Acad. Sci. U.S.A. 92, 7749–7753.
McKernan, R. M., Rosahl, T. W., Reynolds, D. S., Sur, C., Wafford, K. A., Atack, J. R., Farrar, S., Myers, J., Cook, G., Ferris, P., Garrett, L., Bristow, L., Marshall, G., Macaulay, A., Brown, N., Howell, O., Moore, K. W., Carling, R. W., Street, L. J., Castro, J. L., Ragan, C. I., Dawson, G. R., and Whiting, P. J. (2000). Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABAA receptor α1 subtype. Nat. Neurosci. 3, 587–592.
Schofield, P. R., Darlison, M. G., Fujita, N., Burt, D. R., Stephenson, F. A., Rodriguez, H., Rhee, L. M., Ramachandran, J., Reale, V., Glencorse, T. A., Seeburg, P. H., and Barnard, E. A. (1987). Sequence and functional expression of the GABAA receptor shows a ligand-gated receptor super-family. Nature 328, 221–227.
Wulff, P., Goetz, T., Leppa, E., Linden, A. M., Renzi, M., Swinny, J. D., Vekovischeva, O. Y., Sieghart, W., Somogyi, P., Korpi, E. R., Farrant, M., and Wisden, W. (2007). From synapse to behavior: rapid modulation of defined neuronal types with engineered GABAA receptors. Nat. Neurosci. 10, 923–929.
Wulff, P., Schonewille, M., Renzi, M., Viltono, L., Sassoe-Pognetto, M., Badura, A., Gao, Z., Hoebeek, F. E., van Dorp, S., Wisden, W., Farrant, M. and De Zeeuw, C. I. (2009). Synaptic inhibition of Purkinje cells mediates consolidation of vestibulo-cerebellar motor learning. Nat. Neurosci. 12, 1042–1049.