 Aaishwariya A. Gulani1
Aaishwariya A. Gulani1 Eric D. Gaier
Eric D. Gaier- 1Hamilton Eye Institute University of Tennessee, Memphis, TN, United States
- 2Massachusetts Eye and Ear, Boston, MA, United States
- 3Specialized Pediatric Eye Care, Beverly, MA, United States
- 4Department of Ophthalmology, Boston Children’s Hospital, Boston, MA, United States
- 5Department of Ophthalmology, Harvard Medical School, Boston, MA, United States
- 6Picower Institute for Learning and Memory, Massachusetts Institute of Technology, Cambridge, MA, United States
WFS1 spectrum disorder is a rare condition, characterized by diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (DIDMOAD). A 2-year-old female patient with a history of sensorineural hearing loss presented with rapid, sequential cataract development. Diabetes mellitus was not manifested at the time but developed 4 years later. While cataracts have been described in this syndrome, rapid acquisition of cataracts in the setting of mild hyperglycemia was unique considering they could not be definitively attributed to diabetes mellitus alone. This case provides real-world evidence that rapid WFS1-related cataract development may result from the underlying condition in conjunction with or independent from WFS1-associated diabetes mellitus.
WFS1 spectrum disorder (WFS1-SD) is a rare genetic condition, characterized by diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (DIDMOAD) (1). Patients typically present with insulin-dependent diabetes mellitus at a median age of 6 years and optic atrophy at 11 years (1). Other manifestations include neurological deficits, primarily ataxia, anterior pituitary dysfunction, and urological complications such as hydroureteronephrosis; most of these present later in life and, thus, are screened for once the genetic diagnosis is made (2). Aside from optic atrophy, ophthalmic manifestations include cataracts, pigmentary retinopathy, and diabetic retinopathy (2). This case provides evidence that rapid WFS1-related cataract development may result from the underlying condition perhaps in conjunction with WFS1-associated diabetes mellitus.
A 2-year-old female patient was transferred from an outside emergency department because of right eye leukocoria and exotropia. Two weeks prior to presentation, the patient’s mother noticed the patient closing the right eye, which was drifting out more with a “cloudy haze” over the pupil. Examination on presentation revealed visual acuities of light perception OD and 20/160 (Teller) OS. She manifested a constant exotropia and left eye fixation preference (Figure 1A). A complete cataract precluded any view to the right fundus. B-scan ultrasound was without retinal detachment. There was optic disc pallor on the left with no significant cataract precluding an adequate view to the fundus.
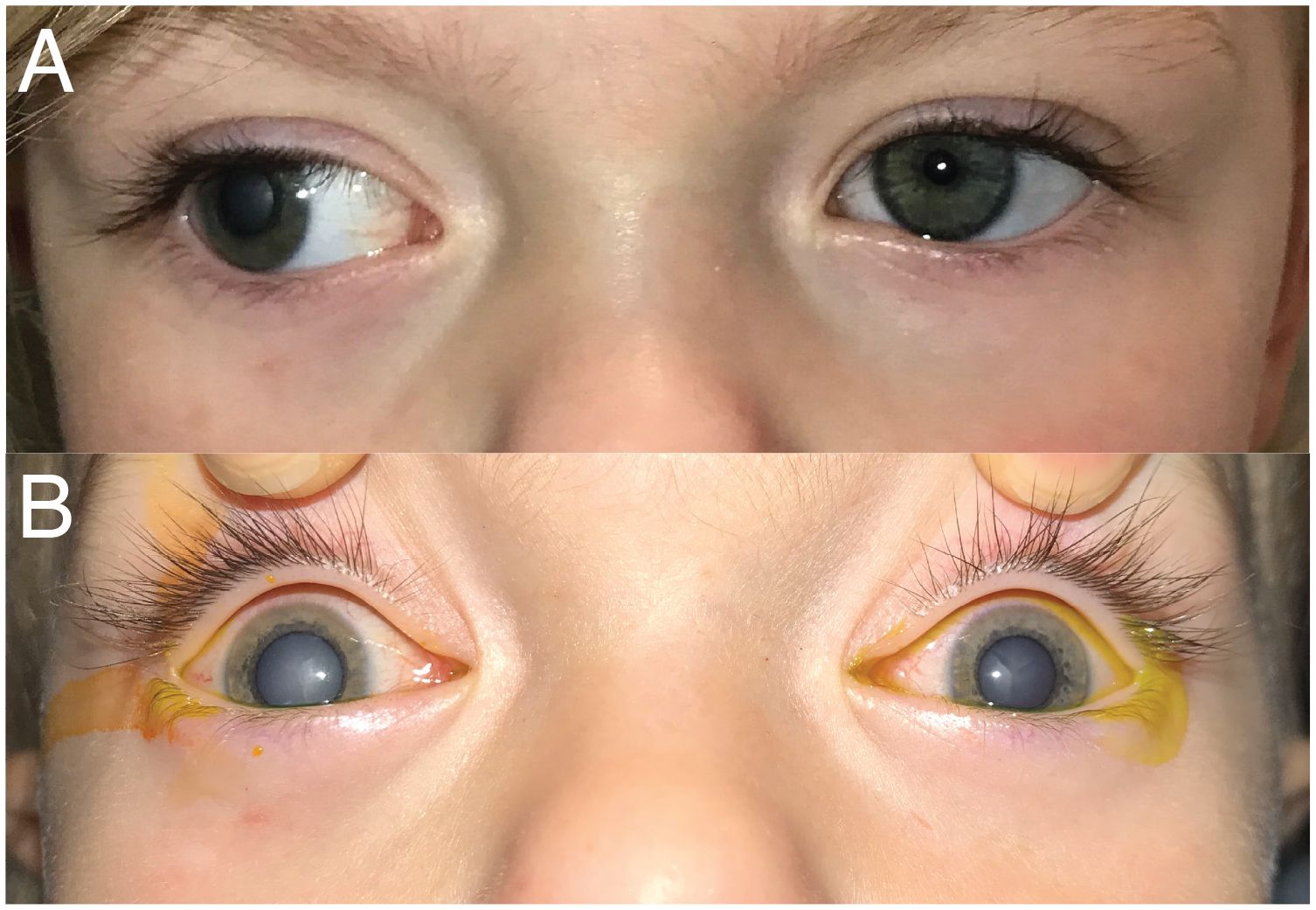
Figure 1. External photographs of exotropia and cataracts. (A) Day 0—first exam/presentation. (B) Day 5—under anesthesia just prior to cataract extraction on the right.
The patient had been born at term via c-section with APGAR scores of 8 and 9. She had a failed auditory screening at birth that prompted further workup. Infectious screening, including cytomegalovirus and rubella, was negative. Magnetic resonance imaging (MRI) pursued as a part of this workup showed bilateral hypoplastic auditory nerves and no comment on the optic nerves. In addition to congenital hearing loss, she had a malpositioned anus, poor weight gain, and short stature and was referred to genetics for further workup. Workup showed high concentrations of amino acids and hyperammonemia, three auricular tags, and normal eyes with reactive pupils. At age 1, an ophthalmologist diagnosed intermittent exotropia; no other ocular abnormalities were reported. She did not have a relative afferent pupillary defect. Family history was significant for insulin-dependent diabetes mellitus in her mother, uncle, and maternal grandfather. Genetic testing of the patient revealed a heterozygous missense variant of likely risk [NM_006005.3(WFS1):c.2430C>G (p.Phe810Leu); rs1553879021] in WFS1 (Otogenome, Laboratory for Molecular Medicine). This variant is listed in ClinVar (3) and absent from gnomAD (4), but a variant with the same translational consequence was recently reported as likely pathogenic (5). Multiple in silico analyses of the variant were suggestive of pathogenicity. The patient’s mother did not carry the variant, and the father, whose family history is unknown, was not available for testing.
The decision was made to remove the cataract. The right eye cataract extraction with intraocular lens (IOL) placement occurred 5 days after initial presentation to the emergency department. Dilated fundus examination under anesthesia after cataract removal revealed a pale right optic disc. During this exam, it was apparent that the left lens had become involved with a smaller, central cataract (Figure 1B). She underwent left cataract extraction with IOL placement 5 weeks later. There was no posterior bulge to either lens to suggest persistent fetal vasculature or posterior lentiglobus.
One-month post-operative assessment confirmed bilateral pallor with a cup-to-disc ratio of 0.8 OD and 0.4 OS. Visual acuities had improved to 20/130 OD and OS, and she was prescribed bifocal glasses to optimize her visual development. Hemoglobin A1c was 6.1%, 5.9%, and 6.5% at 1, 3, and 4 months post-operatively, respectively, with home fasting finger-stick glucose values of 78–90 mg/dL without treatment. Serum osmolality at that time was normal (286 mOsm/kg). Six months post-operatively, the patient began experiencing polyuria and polydipsia and her hemoglobin A1c was found to be 7.2%. She was referred to a local hospital for admission and initiation of insulin therapy. Three years later, visual acuity with correction was 20/41 with both eyes viewing (Teller; patient would not tolerate monocular occlusion).
Early life cataracts have primarily been associated with dominant WFS1-SD (1). Berry et al. isolated a missense mutation in WFS1 (distinct from our patient’s; c.1385A-to-G in exon 8, E462G) that causes congenital nuclear cataracts (6). De Franco et al. described spontaneous heterozygous WFS1 variants associated with neonatal diabetes, deafness, and congenital cataracts within the first year of life, a more severe phenotype than typical of WFS1-SD (7). Our patient already carried a diagnosis of WFS1-SD after genetic testing triggered by identification of sensorineural hearing loss and reportedly had a complete screening eye examination without cataracts. She subsequently developed rapid-onset, sequential cataracts in the setting of prediabetic glycemic screening. Similarly, a recent report describes a Pakistani child who acquired cataracts at age 3, subsequently developed systemic manifestations of diabetes mellitus 2 years later, and harbored a different, novel, heterozygous WFS1 variant (8). While we cannot exclude the fact that our patient had subtle congenital cataracts that progressed, her normal dilated ophthalmic examination prior to presentation suggests they were acquired.
Outside of WFS1-SD, rapid cataract development can be seen in isolated diabetes mellitus, albeit in <1% of cases. However, among pediatric patients who develop diabetes-related cataracts, hyperglycemic severity ranges well beyond those seen in this patient with HbA1c percentages of 9.0% (8.55%–9.38%, p < 0.001) in patients with early cataracts (9). The wolframin protein is expressed in the developing lens (6), and it plays various roles in the regulation of endoplasmic reticulum stress and calcium homeostasis (10). These same cellular functions are implicated in the cataract development in diabetes mellitus (11, 12). Taken together, we posit that increased baseline endoplasmic reticulum stress secondary to compromised wolframin function may impart increased susceptibility to hyperglycemic stress in lens cells to yield early and rapid cataract development in the setting of relatively mild glycemic dysregulation. Runaway, vicious-cycle mechanisms may explain the sequential nature of rapid development seen in this case.
The chronology of cataracts presenting prior to the diagnosis of insulin-dependent diabetes along with the WFS1 variant noted in our patient is unique but aligns with prior studies that identify WFS1 gene dysfunction as a pathway for cataract formation converging with those related to diabetes mellitus. This case suggests that rapidly acquired cataract formation can result from dominant WFS1-SD and should be worked up with genetic testing in certain clinical contexts.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
AG: Writing – original draft, Writing – review & editing. CD: Writing – review & editing, Data curation. DL: Supervision, Writing – review & editing, Data curation. EG: Funding acquisition, Project administration, Supervision, Data curation, Investigation, Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by NIH K08 EY030164 (EDG).
Conflict of interest
EG: Luminopia, Inc. advisor, equity, patent; Stoke Therapeutics, Inc. consultant; Neurofieldz, Inc. consultant.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Barrett TG and Bundey SE. Wolfram (DIDMOAD) syndrome. J Med Genet. (1997) 34:838–41. doi: 10.1136/jmg.34.10.838
2. Rigoli L, Bramanti P, Di Bella C, and De Luca F. Genetic and clinical aspects of Wolfram syndrome 1, a severe neurodegenerative disease. Pediatr Res. (2018) 83:921–9. doi: 10.1038/pr.2018.17
3. ClinVar. Available online at: https://www.ncbi.nlm.nih.gov/clinvar/RCV003128249/.
4. gnomAD browser. Available online at: https://gnomad.broadinstitute.org/gene/ENSG00000109501?dataset=gnomad_r4 (Accessed August 8, 2025).
5. de Muijnck C, Haer-Wigman L, van Everdingen JAM, Lushchyk T, Heutinck PaT, Van Dooren MF, et al. Characteristics of autosomal dominant WFS1-associated optic neuropathy and its comparability to OPA1-associated autosomal dominant optic atrophy. Sci Rep. (2024) 14:22956. doi: 10.1038/s41598-024-74364-x
6. Berry V, Gregory-Evans C, Emmett W, Waseem N, Raby J, Prescott D, et al. Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet. (2013) 21:1356–60. doi: 10.1038/ejhg.2013.52
7. De Franco E, Flanagan SE, Yagi T, Abreu D, Mahadevan J, Johnson MB, et al. Dominant ER stress-inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts. Diabetes. (2017) 66:2044–53. doi: 10.2337/db16-1296
8. Hanif MI, Ahmed H, Ibrahim MN, Raza SJ, and Ahmed SA. A novel de novo likely pathogenic variant of wfs-1 gene in a Pakistani child with non-classic wfs-1 spectrum disorder. J Ayub Med Coll Abbottabad. (2024) 36:433–5. doi: 10.55519/JAMC-02-12379
9. Reiter UM, Eckert AJ, Dunstheimer D, Bechtold-Dalla Pozza S, Lullwitz C, Golembowski S, et al. Cataract in children and adolescents with type 1 diabetes. Insights from the German/Austrian DPV registry. Pediatr Diabetes. (2022) 23:362–9. doi: 10.1111/pedi.13316
10. Rigoli L, Lombardo F, and Di Bella C. Wolfram syndrome and WFS1 gene. Clin Genet. (2011) 79:103–17. doi: 10.1111/j.1399-0004.2010.01522.x
11. Li H and Yang J. Endoplasmic reticulum stress as a regulator of high glucose-induced epithelial-mesenchymal transition in human lens epithelial cells. J Ocul Pharmacol Ther. (2025) 41(6):347–54. doi: 10.1089/jop.2024.0210
Keywords: WFS1 spectrum disorder, Wolfram syndrome, cataract, diabetes mellitus, optic atrophy
Citation: Gulani AA, Duggan C, Ledoux DM and Gaier ED (2025) Case Report: Rapid cataract development preceding diabetes mellitus in WFS1 spectrum disorder. Front. Ophthalmol. 5:1612964. doi: 10.3389/fopht.2025.1612964
Received: 15 May 2025; Accepted: 11 August 2025;
Published: 09 September 2025.
Edited by:
Chiara Bianca Maria Platania, University of Catania, ItalyReviewed by:
Palaiologos Alexopoulos, New York University, United StatesMisbah Hanif, Dow University of Health Sciences, Pakistan
Copyright © 2025 Gulani, Duggan, Ledoux and Gaier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric D. Gaier, ZXJpYy5nYWllckBjaGlsZHJlbnMuaGFydmFyZC5lZHU=
†These authors have contributed equally to this work