 Ritu Sampige1*
Ritu Sampige1* Lyra E.A. Seaborn1
Lyra E.A. Seaborn1 Molly Pluenneke1
Molly Pluenneke1 Annika Jyothi1
Annika Jyothi1 Sophie Saland2
Sophie Saland2 Chisom M. Chinedu-Obi3
Chisom M. Chinedu-Obi3 Caroline Keehn1
Caroline Keehn1 Andrew G. Lee1,4,5,6,7,8,9,10
Andrew G. Lee1,4,5,6,7,8,9,10- 1School of Medicine, Baylor College of Medicine, Houston, TX, United States
- 2McGovern Medical School at the University of Texas at Houston, Houston, TX, United States
- 3Tulane University School of Medicine, New Orleans, LA, United States
- 4Department of Ophthalmology, University of Texas Medical Branch, Galveston, TX, United States
- 5Texas A&M School of Engineering Medicine, Bryan, TX, United States
- 6Department of Ophthalmology, Blanton Eye Institute, Houston Methodist Hospital, Houston, TX, United States
- 7The Houston Methodist Research Institute, Houston Methodist Hospital, Houston, TX, United States
- 8Departments of Ophthalmology, Neurology, and Neurosurgery, Weill Cornell Medicine, New York, NY, United States
- 9University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 10Department of Ophthalmology, The University of Iowa Hospitals and Clinics, Iowa City, IA, United States
Autosomal dominant optic atrophy (ADOA) is among the most prevalent inherited optic neuropathies with hallmark symptoms of bilateral, painless, progressive, and typically permanent vision loss over time. ADOA can affect patients’ quality of life with debilitating visual symptoms, and there is a pressing need for effective therapeutics. In this paper, we review the current and future investigational therapies for ADOA, including the use of intravitreal injections of antisense oligonucleotides through Targeted Augmentation of Nuclear Gene Output (TANGO), CRISPR-based therapy, genetic editing, gene replacement approaches, and idebenone, a small-molecule mitochondrial modulator. Additionally, we review clinical trials for ADOA treatment and opportunities for future research on ADOA therapeutics, including the utilization of mitochondria-targeted peptides and antioxidants, NAD+ boosters/metabolic support, mitophagy and fission-fusion modulators, and cell-based regenerative therapy. The use of emerging technology to compensate for OPA1 protein haploinsufficiency provides new and vast avenues for the management of this otherwise vision-altering disease. Increased awareness of therapeutics for ADOA will allow for patient counseling regarding treatment access via clinical trials and for underscoring the importance of genetically testing family members, who may be incidentally identified with ADOA in a timely manner for newly available therapies. While patients with ADOA typically have poor visual prognoses, there are increasing promising therapies with the potential for preserving and improving visual function.
1 Introduction
Autosomal dominant optic atrophy (ADOA), also known as Kjer’s syndrome, affects approximately 1 in 30,000 to 1 in 50,000 people globally, with differing prevalence among specific geographic populations. For example, 1 in 10,000 individuals in Denmark have ADOA due in part to a founder effect (1–3). Patients with ADOA typically develop bilateral loss of vision beginning in childhood, often in the first decade of life, that is subsequently progressive and irreversible (1, 4, 5). However, the onset, course, and severity of ADOA is variable (i.e., variable expressivity) (1). Some patients experience loss of vision from birth, while clinically significant symptoms do not occur in others until early childhood or adulthood (1), and some patients with ADOA are asymptomatic. The symptoms and signs of ADOA include decreased visual acuity in both eyes (OU), reduced color vision, visual field deficits (typically central or cecocentral scotomas OU), and eventual optic disc atrophy OU (2, 6–8). While the conventional form of ADOA manifests with optic atrophy and vision loss OU, recent literature has shed light on additional clinical findings (ADOA+). The broader ADOA+ phenotype encompasses other systemic, non-ocular findings including sensorineural hearing loss, progressive external ophthalmoplegia, myopathy, ataxia, and peripheral neuropathy, potentially occurring in the absence of optic atrophy decades after initial vision loss (3). Additionally, ADOA+ has been associated with more severe visual symptoms (3).
ADOA is genetically heterogeneous, meaning that there is variation in the causative genetic insult (4, 9, 10). The most commonly implicated gene, OPA1, is located on chromosome 3 (3q28) (9). Among individuals with ADOA, the prevalence of OPA1 mutations ranges from 60-90% (2, 11, 12). The OPA1 protein, synthesized by the OPA1 gene, is a mitochondrial GTPase required for mitochondrial homeostasis, including mitochondrial import, fusion and fission, cristae formation and maintenance, respiratory chain complex functioning, and apoptosis (5, 8, 13–16). Mutations in one of the two OPA1 gene copies lead to inadequate amounts of OPA1 protein generation, known as protein haploinsufficiency (17). Thus, mutation of OPA1 is associated with inadequate mitochondrial function, leading to apoptosis of retinal ganglion cells (RGCs) and atrophy of the optic disc (8, 18). Additionally, ADOA from OPA1 mutations may also occur via a dominant-negative effect. For instance, missense mutations in the OPA1 GTPase domain may lead to mutated protein product generation that negatively affects the function of the remaining normal product from the wild-type allele (5, 14, 19). In the dominant-negative variant, OPA1 expression levels are not diminished; instead, there is competition between the protein products of the mutated versus wild-type allele (14). In fact, the dominant-negative variant of OPA1-associated ADOA, from the missense mutation, is associated with more severe multisystem, extraocular symptoms, aligning with the ADOA+ phenotype (5, 14, 19, 20). The pathogenesis of ADOA from OPA1 gene mutations represents a complex interplay of loss-of-function and dominant-negative effects. Furthermore, some OPA1 mutations occur outside the GTPase domain, such as in the BSE α-helix or the GTPase effector domain and may subsequently impair mitochondrial fusion (15, 16).
2 Novel therapeutics for ADOA
To counter the progressive, debilitating effects of the OPA1 gene mutation in ADOA, several therapeutic mechanisms have been proposed (Table 1), including the use of antisense oligonucleotides, gene therapy, and idebenone, with additional investigational therapeutics underway (Table 2) via in vivo studies and clinical trials.
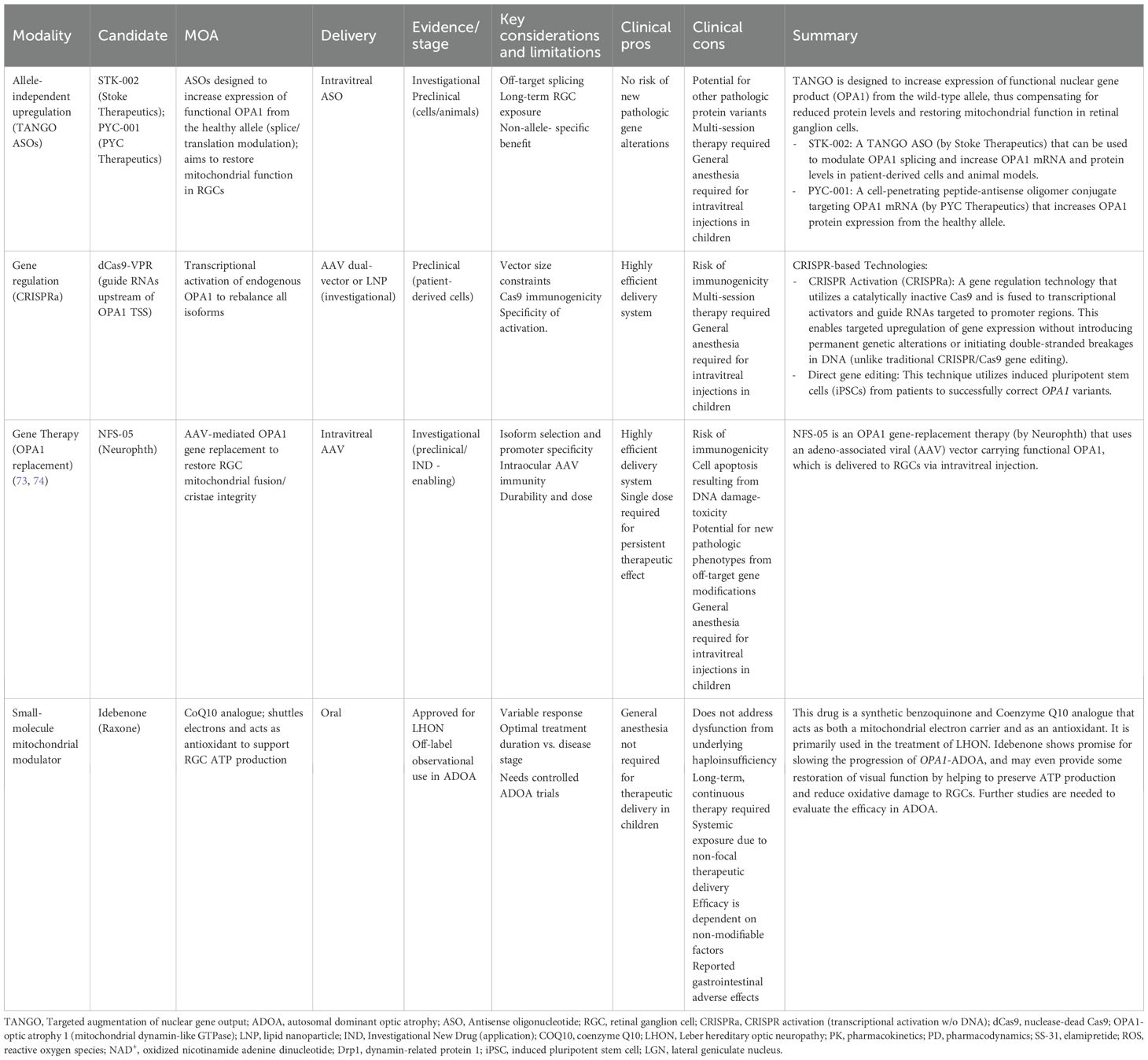
Table 1. Summary of current investigational therapies for autosomal dominant optic atrophy (ADOA).
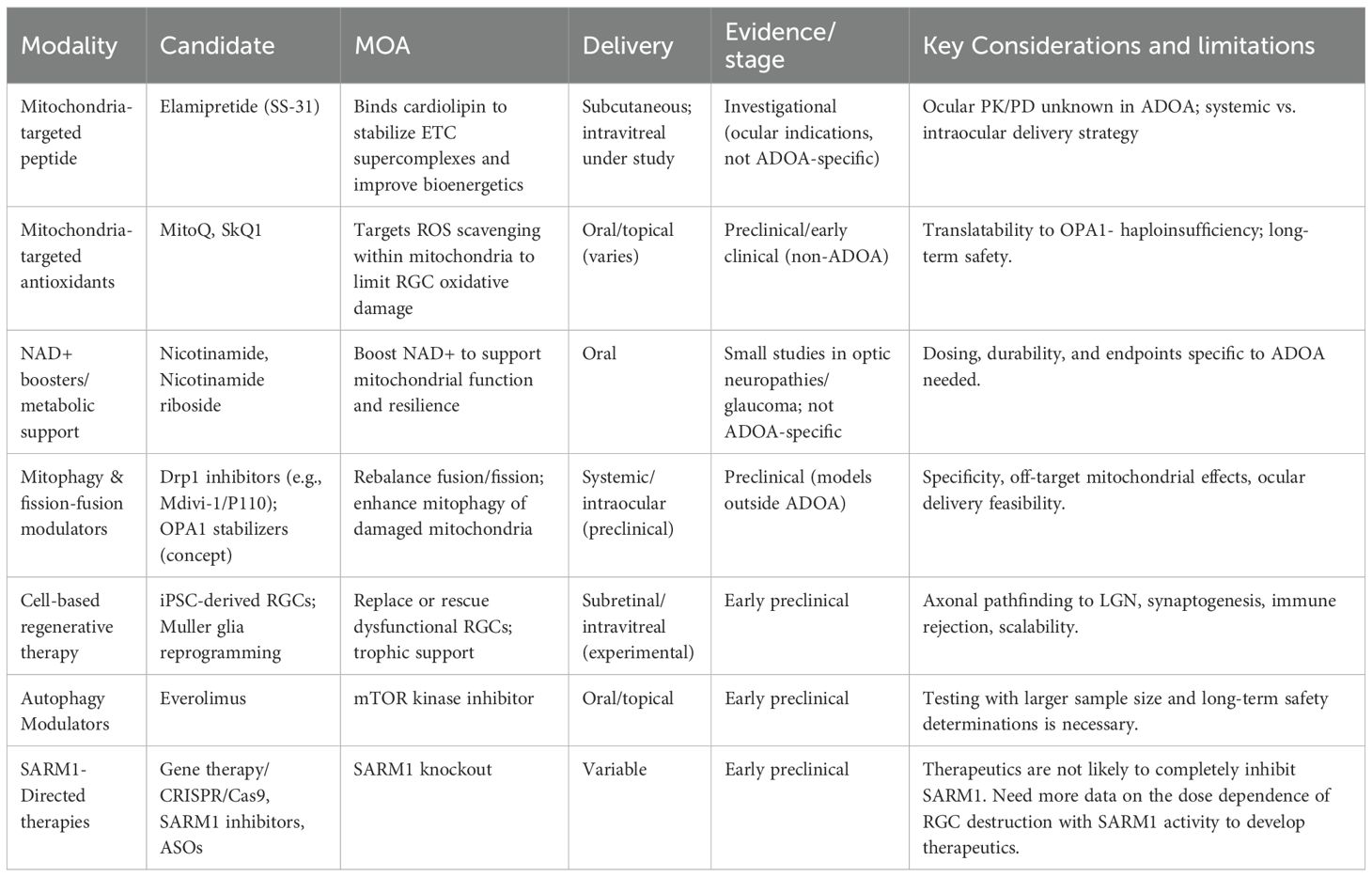
Table 2. Summary of potential investigational therapies for autosomal dominant optic atrophy (ADOA).
2.1 TANGO
Targeted Augmentation of Nuclear Gene Output (TANGO) is a novel and evolving therapeutic modality for individuals with ADOA, specifically those with OPA1 gene mutations (12, 21). TANGO therapy, delivered intravitreally, works by preserving the wild-type (WT) gene’s transcribed products. In ADOA-directed TANGO therapy, the STK-002 compound is the antisense oligonucleotide (ASO) delivered to bind nonsense-mediated decay exons on pre-mRNA transcribed from the WT APO1 gene. Specifically, this ASO attaches to nonsense-mediated decay exons in the WT pre-mRNA, preventing WT transcribed mRNA from degrading prematurely (12, 21, 22). As a result, WT OPA1’s mRNA pre- and post-splice products increase in stability, abundance, and concentration (22). This produces a greater quantity of WT OPA1 protein products compared to baseline. Over time, the higher concentration of stabilized WT mRNA available for translation is greater than the amount of non-stabilized, mutant mRNA able to undergo translation. Consequently, the mutant-OPA1 phenotype seen in OPA1 haploinsufficiency is overcome (12, 21).
While there are currently no Food and Drug Administration (FDA) approved therapeutics for ADOA in the United States, TANGO technology shows promising results in preclinical studies (12, 21, 23, 24). Recent data from Stoke Therapeutics demonstrated, with preclinical primate models, that repeated intravitreal injections of antisense oligonucleotides bilaterally led to a dose-dependent elevation in OPA1 protein levels four weeks after initial injections in Cynomolgus monkeys and persisted eight weeks after injection therapy (23). Additionally, TANGO technology was recently approved for clinical trials, such as the Phase I/II OSPREY study for evaluation of efficacy among patients with confirmed APO1 mutations (22, 25).
2.2 Gene therapies
Beyond traditional gene replacement approaches like NFS-05, which is a type of OPA1 gene-replacement therapy that uses an adeno-associated viral (AAV) vector carrying functional OPA1 for RGCs, clustered regularly interspersed short palindromic repeat (CRISPR)-based technologies offer innovative solutions for addressing OPA1 haploinsufficiency in ADOA. These approaches can be broadly categorized into two strategies: CRISPR activation (CRISPRa), which upregulates OPA1 expression from the wild-type allele, and direct gene editing, which corrects pathogenic mutations at the genomic level.
2.2.1 CRISPR activation
Similar to TANGO, CRISPRa addresses the fundamental problem of OPA1 haploinsufficiency without requiring correction of the underlying mutation; however, CRISPRa accomplishes this by altering gene expressivity. By this mechanism, the Cas9 endonuclease is catalytically inactivated (dCas9) and fused to transcriptional activators (e.g., VP64, p65, Rta), enabling targeted upregulation of gene expression (26). CRISPRa directs guide RNAs upstream of the OPA1 transcription start site near the OPA1 promoter, thus enhancing expression across all OPA1 isoforms and potentially restoring physiological protein concentration and mitochondrial function (26).
This approach offers several theoretical advantages over conventional gene replacement therapy. AAV-based gene therapies are limited by viral packaging capacity and typically deliver only a single isoform of OPA1. In contrast, CRISPRa leverages the endogenous wild-type OPA1 gene, thereby restoring physiologic levels of all eight isoforms. Recent preclinical studies showed promising results in patient-derived fibroblasts carrying the common c.2708_2711del OPA1 variant. Using a dCas9-VPR system with guide RNAs positioned ~150–300 bp upstream of the transcription start site, researchers achieved significant upregulation of OPA1 expression (26). Interestingly, isoform 5 expression was selectively increased while maintaining appropriate splicing regulation, suggesting that cells can preserve homeostatic isoform ratios under CRISPRa stimulation (26).
Functionally, CRISPRa treatment improved the classic phenotypes of OPA1 deficiency, including mitochondrial network fragmentation. Treated cells displayed increased summed branch length and network branching, indicating partial restoration of mitochondrial connectivity. Importantly, CRISPRa constructs can be packaged into AAV vectors for potential intravitreal delivery, raising the possibility of clinical translation-like conventional gene replacement, but with the added advantage of restoring physiologic isoform diversity (26).
2.2.2 Direct gene editing
Another potential CRISPR-based therapy involves directly correcting pathogenic OPA1 mutations. Unlike CRISPRa, which enhances expression from the WT allele, this strategy aims to restore normal gene function by repairing the causative mutation, potentially providing a permanent cure. Studies using patient-derived induced pluripotent stem cells (iPSCs) have shown successful correction of OPA1 variants. For example, correction of the c.1334A>G (p.R445H) mutation with CRISPR-Cas9 restored mitochondrial morphology, normalized oxidative phosphorylation capacity, stabilized mitochondrial DNA, and improved apoptosis resistance, thus reversing many forms of disease-associated cellular dysfunction (27). Similarly, correction of additional OPA1 mutations in iPSC-derived retinal ganglion cells improved mitochondrial bioenergetics and reduced disease-associated phenotypes (28). Advances in base-editing and prime-editing technologies also raise the possibility of correcting OPA1 mutations with higher precision and fewer off-target effects, further expanding therapeutic feasibility (29).
2.3 Idebenone
In addition to gene-based therapies, novel pharmacologic agents are being explored to slow the progression of ADOA. One such drug is idebenone, a synthetic benzoquinone that has already shown promise for improving visual acuity in patients with Leber hereditary optic neuropathy (LHON).
Similar to ADOA, LHON is a mitochondrial disease that causes RGC dysfunction and apoptosis, generally leading to bilateral central vision loss (30). Furthermore, both LHON and ADOA show similar patterns of optic nerve degeneration, although ADOA typically presents earlier in life while LHON onset is more delayed and progression more rapid (31, 32). However, the mutations responsible for LHON affect genes encoding the NADH:ubiquinone oxidoreductase (complex I) subunit, a component of the mitochondrial oxidative phosphorylation system that generates ATP (33). This subunit is implicated in multiple cell signaling pathways and is thought to be one of the main sites of reactive oxygen species (ROS) production (34). Consequently, pathogenic complex I variants result in reduced ATP formation, increased mitochondrial ROS creation, and, ultimately, cell death.
First synthesized in the 1980s, idebenone was initially investigated for use as a potential treatment of Alzheimer’s disease prior to its utilization for mitochondrial disorders (35, 36). Subsequent investigations delved into its efficacy for managing other conditions. Like other benzoquinones, idebenone serves to transfer electrons within the mitochondrial respiratory chain and has been shown to act as a potent antioxidant (37–40). Though idebenone shares a quinone moiety with Coenzyme Q10 (CoQ10), a naturally occurring benzoquinone marketed as a dietary supplement to mitigate a range of conditions from migraines to heart failure and more, the two compounds differ in several key ways (41). Notably, the bioactivation of mitochondrial CoQ10 is dependent on proper mitochondrial function, whereas idebenone is predominantly activated in the cytoplasm (37). Furthermore, in contrast to CoQ10, idebenone can channel electrons beyond the complex I directly to complex III, thus maintaining the production of ATP even in the setting of dysfunctional complex I (38, 42–44). Researchers soon recognized that idebenone’s unique properties could lend potential applications in LHON and other mitochondrial diseases.
In 1992, a case report of LHON successfully treated with idebenone was published in The Lancet (45). However, it was not until June 2015 that the European Medicine Agency (EMA) approved idebenone (Raxone, Santhera Pharmaceuticals, Liestal, Switzerland) for use in LHON patients. Currently, it remains the only approved drug for treating visual impairment in adolescents and adults with LHON (46). Even so, evidence for its efficacy in visual recovery remains limited and controversial (35). Recommendations for idebenone use in LHON vary depending on the chronicity of disease (30, 47). Additionally, treatment effects have been shown to differ with patient age, sex, and mtDNA mutation.
Despite the existence of conflicting data, idebenone’s successes inspired trials of the drug in patients with ADOA. A pilot study of seven participants suggested that treatment with idebenone for at least one year may improve visual acuity in OPA1-mutant ADOA (32). Similarly, a subsequent cohort examination of 87 OPA1-ADOA patients found significantly greater visual stabilization and recovery in patients treated with idebenone compared to untreated subjects, even after controlling for confounders (32). Dosages ranged from 135–675 mg/day, with the majority (48%) of idebenone-treated subjects taking 540 mg/day. Another study of OPA1-ADOA patients treated with 900 mg/day for 12 months found significant improvement in visual acuity, visual field testing, and self-reported vision-related quality of life compared to baseline measures. However, there were no observed changes in color vision or contrast sensitivity (48).
Idebenone is generally considered to be safe and well-tolerated, with most clinical studies demonstrating high compliance with medication intake and low or no idebenone-related adverse events (43, 47–49). A longitudinal analysis of idebenone’s efficacy in LHON patients found that, of the adverse events reported, the majority were mild and gastrointestinal in nature (e.g., diarrhea), with the few observed severe/fatal outcomes considered unrelated to idebenone treatment (46).
Overall, idebenone shows promise for slowing the progression of OPA1-ADOA and may even restore some visual function by helping to preserve ATP production and reduce RGC oxidative damage. Larger-scale trials and prospective investigations are needed to better elucidate the drug’s efficacy. Additionally, it remains unclear whether idebenone may benefit ADOA patients with non-OPA1 mutations. Future studies addressing these gaps could determine appropriate treatment candidates and dosing schedules among ADOA patient populations, potentially leading to new breakthroughs in pharmaceutical management.
2.4 Therapeutics for dominant-negative OPA1 mutations
TANGO and CRISPRa techniques may have limited application for patients with dominant-negative OPA1 mutations compared to the haploinsufficiency form of OPA1 mutations. More specifically, in dominant-negative OPA1 mutations, the protein product of the mutated OPA1 gene negatively affects the function of the protein product of the normal OPA1 allele, thus generating a more severe ADOA phenotype, including mitochondrial dysfunction and extraocular disease presentation, such as ADOA + (5, 14, 19, 20). TANGO technology aims to increase OPA1 protein generation (12, 21); however, by doing so, both the normal and the mutated OPA1 protein is inadvertently increased. Similarly, while CRISPRa may be successful in genetically editing the mutated copy of the OPA1 gene (26), any remaining mutated OPA1 protein has the capacity to disturb the function of the normal OPA1 protein, thus perpetuating the ADOA phenotype. Instead, specific unique strategies are necessary to address dominant-negative OPA1 mutations, such as selective silencing of the mutant allele instead of an overexpression of the wild-type allele. For instance, viral vectors may be considered to block the mutant OPA1 allele (5). Additionally, small interfering RNAs (siRNAs) or ASOs may be harnessed to target certain mRNA sequences for cleavage, degradation, and subsequent selective gene silencing (5).
2.5 Other clinical trials for ADOA therapeutics
Currently, there are no other FDA approved treatments for individuals with ADOA. However, various novel therapies have entered preclinical and clinical testing for ADOA in recent years. They are broadly divided into pharmacological neuroprotection, gene therapy, and cell-based regenerative therapies (5).
PYC-001 is an OPA1-targeting ASO developed by PYC Therapeutics. Specifically, PYC-001 is a cell-penetrating peptide-antisense oligomer conjugate targeting OPA1 mRNA (50). In patient-derived retinal cell models, PYC-001 increased OPA1 protein production to nearly normal levels, restoring the mitochondrial network (50).
A Phase 1b open-label, randomized sequential dose trial has been set to assess safety, tolerability, and optimal dosage of PYC-001 intravitreal injection therapy for patients with confirmed OPA1 mutations. Additional metrics will assess ocular structural and function changes as well as the pharmacokinetics of PYC-001 (51). As of August 2025, the trial has not begun recruiting participants yet.
Gene therapies provide another potential therapeutic option for patients with ADOA. NFS-05, an OPA1 gene-replacement therapy created by the biotech company Neurophth, uses an adeno-associated viral (AAV) vector carrying functional OPA1 that is delivered to RGCs via intravitreal injection. This therapy received approval to begin clinical trials in Australia for the treatment of ADOA (52).
Finally, because RGC loss is central to the pathophysiology of ADOA, several trials have explored cell-based neuroprotection. In the Stem Cell Ophthalmology Treatment Study (SCOTS), autologous bone marrow-derived stem cells (BMSCs) were infused into the eyes of ADOA patients. In a report of 6 patients, 5 of 6 (83%) showed improved visual acuity, with an average improvement for all eyes of 29.5% (53). These findings demonstrate another potential treatment. The NIH-registered SCOTS2 trial is currently recruiting participants to evaluate the safety and efficacy of ocular BMSC in retinal and optic nerve damage and disease (54).
Overall, the recent clinical trials investigating potential therapies for ADAO, including TANGO ASO (STK-002 and PYC-001), gene therapies (NFS-05), idebenone, and cell-based regenerative therapies, collectively show promise toward disease-modifying treatment of ADOA.
3 Discussion
3.1 Challenges associated with novel therapeutics for ADOA
3.1.1 Genetic heterogeneity
The challenges associated with novel therapeutics for ADOA are multifaceted and involve both the biology of the disease and transitional barriers. The first challenge arises from the heterogeneity of OPA1 mutations. More than 400 pathogenic variants have been described, each producing variable effects on protein function and clinical severity (7). These mutations range from missense and nonsense variants to splice-site changes and large deletions, each producing variable effects on OPA1 protein stability, mitochondrial dynamics, and ultimately clinical severity (5). For example, missense mutation in the GTPase domain, such as the dominant-negative variant of OPA1-linked ADOA often results in early-onset, severe optic atrophy, whereas truncating mutations may produce milder or later-onset phenotypes (5, 14). Another example of the genetic heterogeneity of ADOA includes the interplay of mutations within and beyond the GTPase domain (16). In fact, a recent study of patients with ADOA revealed two forms of OPA1 mutations, including the V465F mutation within the GTPase domain (GTPase β-fold) and the V560F mutation outside of the GTPase domain (BSE α-helix) (16). Another study of ADOA pathogenesis showed an association between the deletion of the GTPase effector domain of OPA1 and ADOA development, apart from GTPase domain missense mutations (55). In fact, mutations in the OPA1 GTPase effector domain have been linked with partial defects in mitochondrial fusion and GTPase functioning (15).
This diversity complicates the design of treatments that can be broadly effective across the patient population, since a therapeutic that rescues haploinsufficiency may not adequately correct dominant-negative variants. In addition, variable expressivity within families carrying the same mutation further emphasizes the need for mutation-specific or genotype-stratified therapeutic approaches, yet such precision medicine strategies remain underdeveloped in ADOA research (7). This genetic heterogeneity; therefore, establishes a fundamental barrier for the design of universal therapeutic strategies.
3.1.2 Retinal ganglion cell delivery
Furthermore, a pivotal aspect requiring attention is efficient delivery to retinal ganglion cells (RGCs), which are the primary site of degeneration in ADOA. RGCs are post-mitotic and situated deep in the inner retina, a compartment that is relatively difficult to access with therapeutic agents, making efficient delivery a challenge. Preclinical studies have shown that intravitreal injection of ASOs and adeno-associated viral (AAV) vectors can achieve a selective degree of uptake into RGCs (23). For instance, AAV2 vectors demonstrate preferential tropism for RGCs following intravitreal delivery, and animal models of optic neuropathies have shown partial preservation of visual function with this approach (24). However, translation to durable widespread expression in human RGCs remains an unmet need. Clinical trials of intravitreal AAV-based therapies in other optic neuropathies, such as Leber hereditary optic neuropathy (LHON), emphasize both the promise and limitation of this route, efficacy has been variable and expression levels often subtherapeutic (24). These findings suggest that while vector-based and oligonucleotide-based therapies are biologically feasible, optimizing delivery methods to achieve long-term RGC transduction without compromising safety is still a major challenge.
3.1.3 Modality-specific limitations
Each therapeutic modality introduces its own set of limitations. ASOs, such as those used in the STK-002 TANGO program, require repeated intravitreal dosing to sustain activity, raising concerns about patient compliance and the cumulative, although low, risks associated with repeated ocular injections, such as endophthalmitis, retinal detachment, and repeat general anesthesia required for intravitreal administration in children (12, 21, 23). In parallel, gene therapy approaches that rely on AAV-mediated OPA1 delivery face the inherent limitation of viral packaging capacity, since the OPA1 gene (approximately 100 kb isoforms) exceeds the optimal size for standard AAV vectors (56). Moreover, systemic or intraocular immune responses to the vector can limit transduction efficiency or necessitate immunosuppression (57). Small-molecule therapies such as idebenone, designed to bypass mitochondrial dysfunction and mitigate oxidative stress, have demonstrated modest clinical benefit in subsets of patients (32). For example, idebenone has been shown to improve visual function in certain patients with optic neuropathies, yet responses in ADOA remain inconsistent, possibly reflecting underlying genetic heterogeneity. Taken together, these examples emphasize how each therapeutic modality presents novel challenges that must be balanced against its potential benefits.
Additionally, despite preclinical results regarding CRISPR-based gene therapies, several translational challenges remain. First, homology-directed repair and other editing mechanisms can be less efficient in non-dividing cells, like retinal ganglion cells, which may limit in vivo correction rates. Second, safe and effective delivery of CRISPR constructs into RGCs presents hurdles, including viral packaging constraints, immune response risk, and the need for durable long-term expression. Finally, rigorous in vivo studies are needed to address the durability of gene correction and to ensure minimal off-target genomic modifications.
3.2 Clinical trial design
Finally, the design and execution of clinical trials in ADOA present significant logistical difficulties. Given the rarity of ADOA, patient recruitment in clinical trials is inherently limited and challenged; hence, most studies to date have been small, non-randomized, open-label, or uncontrolled cohorts, weakening the strength of conclusions that can be drawn (22). For instance, several idebenone studies in ADOA included fewer than 20 patients, with heterogeneous outcome measures, making it difficult to establish reproducible efficacy (48). The lack of standardized endpoints further complicates interpretation, visual acuity and optical coherence tomography (OCT) of the retinal nerve fiber layer (RNFL) are commonly used however may not capture subtle disease progression or early treatment effect (58). Moreover, the slow, variable natural history of ADOA progression necessitates lengthy follow-up periods, increasing trial costs and complexity. These challenges suggest not only the difficulty of generating high-quality evidence but also the urgent need for more coordinated, multicenter trial infrastructure to advance ADOA therapeutics from concept to clinical reality.
3.3 Opportunities for further exploration
Future work in ADOA therapeutics is focused on overcoming the challenges outlined above, with the most immediate advances expected from ongoing clinical trials. The OSPREY study, which evaluates the safety and efficacy of STK-002, represents the first controlled human trial of an antisense oligonucleotide (ASO) therapy specifically designed for ADOA (22). Early reports suggest acceptable tolerability and evidence of target engagement; however, longer-term data are needed to confirm efficacy (59, 60). If successful, OSPREY could establish proof-of–concept that modulating OPA1 expression with ASOs is both feasible and clinically meaningful, setting a benchmark for subsequent trials. This would not only validate ASOs as a therapeutic modality in ADOA but may also provide critical insights into trial design, including the feasibility of patient recruitment and utility of different outcome measures in a rare disease context. Positive readouts could therefore catalyze broader investment and accelerate development pipelines across multiple therapeutic platforms.
At the same time, advances in gene editing hold considerable promise for more durable, mutation-specific interventions. Traditional CRISPR-Cas9 approaches face limitations due to double-strand DNA breaks and the risk of off-target mutations. Next-generation platforms, such as base editing and prime editing, offer more precise tools to correct pathogenic OPA1 variants at the nucleotide level without introducing double-strand breaks (26). Preclinical studies in other monogenic retinal diseases have demonstrated the feasibility of these approaches, showing restoration of normal protein function with fewer off-target effects. Although OPA1’s large size and isoform complexity add unique challenges, the rapid evolution of editing tools increases the likelihood that such approaches could be adapted for ADOA. Importantly, combining gene editing with improved delivery platforms may help overcome barriers to efficient RGC targeting, a key limitation of earlier gene therapy efforts.
Parallel to gene editing, refined delivery methods will improve the efficiency of RGC targeting. Conventional AAV vectors are constrained by their limited packaging capacity and immunogenicity, but engineered capsids with enhanced tropism for RGCs are currently in development (56). In addition, non-viral nanoparticle systems, including lipid nanoparticles (LNPs) have shown increasing promise in ocular drug delivery, offering the potential for reduced immune activation and repeat dosing (57). These approaches could broaden the therapeutic window while providing a modality to deliver larger constructs, including those required for full-length OPA1 isoforms or gene editing machinery. Advances in delivery, therefore, represent a pivotal step toward achieving the durable and widespread expression necessary for long-term disease modification in ADOA.
Combination strategies are also being explored to maximize therapeutic benefit. For example, small molecules such as idebenone could be used (48) alongside gene-based therapies to stabilize mitochondrial function during the early treatment period, potentially mitigating cellular stress while genetic correction takes effect. Such multimodal approaches have precedent in other mitochondrial diseases, where pharmacologic and genetic strategies together enhance durability of response. For instance, the combination use of small-molecule inhibitors, ASOs, and gene-therapy approaches may help to further increase OPA1 protein to correct haploinsufficiency and to improve mitochondrial functioning. However, established, robust data is first necessary regarding each of these therapies individually for ADOA treatment to better understand the effects of potential combination. Further testing of combination strategies is necessary to evaluate safety and efficacy as combination therapy does not necessarily guarantee a synergistic effect and may run the risk of toxicity. Beyond therapeutic efficacy, progress in clinical trial infrastructure will play a crucial role in advancing the field. International registries, harmonized outcome measures, and multicenter natural history studies are beginning to emerge, providing the platform necessary for larger and more rigorous randomized controlled trials (58). As these efforts mature, they will enable a shift from small, underpowered exploratory studies to robust investigations capable of generating regulatory-grade evidence. Collectively, these developments point toward a future in which targeted, durable, and accessible therapies may become available for patients with ADOA.
Other emerging approaches potentially include mitochondrial transplantation, which is an experimental strategy aimed at restoring mitochondrial function. Although mitochondrial transplantation has not yet been used to treat clinical ADOA, the replacement of mutated mitochondrial DNA (mtDNA) with wild-type mtDNA via cybrid technology has displayed correction of another mitochondrial disease: Leber’s hereditary optic neuropathy (61). Additionally, mitochondrial transfer has been shown to improve morbidity and mortality in a murine model of Leigh syndrome, another mitochondrial disease (62). Furthermore, autophagy modulation is another promising treatment method for ADOA. In fact, recent studies in patient-derived skin fibroblasts have shown that OPA1 mutations displaying haploinsufficiency may impair autophagy and trigger cellular senescence (19, 63). Thus, treatment of fibroblasts with everolimus, an mTOR kinase inhibitor, was successfully able to restore these phenotypes, suggesting a potential therapeutic strategy for ADOA (63). More specifically, such evidence showed the ability for everolimus to protect OPA-1 mutated fibroblasts from senescence and to restore autophagy (63). However, such treatment may be more specific to OPA1 haploinsufficiency since dominant-negative variants of OPA1 mutations are contrastingly linked with increased autophagy and mitophagy (19). Additionally, another study revealed a role for autophagy in the control of mitochondrial content in axons and in visual loss in a mouse model of ADOA caused by OPA1 deletion (64). More specifically, the deletion of autophagy and mitophagy genes in RGCs with mutated OPA1 is linked with a restoration of mitochondrial content and protection from vision loss (64).
OPA1 mutations are linked with an activation of sterile alpha and TIR motif containing 1 (SARM1), a prodestructive factor that codes for a protein, known as an injury-driven NADase, that breaks down NAD+ into cyclic ADP-ribose (65). SARM1 has a sequence that allows for specific mitochondrial targeting and is activated in degenerative, including neurodegenerative, states, leading to axon degeneration and neuron death (65). A recent study demonstrated that SARM1 knockout mitigates degeneration in an ADOA mouse model (65). In fact, emerging SARM1-directed therapies, such as small-molecule SARM1 inhibitors, ASOs, and gene-therapy approaches that target SARM1), may be applied to the treatment of ADOA due to the role of SARM1 activation in ADOA pathogenesis (65). Current emerging research shows how gene therapy may be harnessed to block SARM1 and the downstream axon degeneration (66), how SARM1 inhibitors can help restore axons that are in moderate injury prior to full degradation (67), and how Sarm1 ASOs can reduce SARM1 levels and impede axon degeneration (68), thus showing potential for potential use in ADOA therapeutics. Furthermore, spliceosome-mediated RNA trans-splicing may be further investigated as a potential therapeutic approach for ADOA. This form of gene therapy targets mutations at the level of transcription (69), which may be harnessed for OPA1 replacement or to block SARM1. For example, a recent study aimed to fix OPA1-associated splice mutations to address haploinsufficiency with engineered U1 snRNA and showed that engineering the U1 binding process can address the OPA1 splice defect and subsequently elevate the level of properly spliced OPA1 transcripts (70). Lastly, retinal or retinal ganglion cell organoids derived from patient-induced pluripotent stem cells with OPA1 mutations have great potential as a platform for future therapy testing, given their ability to recapitulate disease-relevant phenotypes in vitro (71, 72).
4 Conclusion
Although ADOA is a rare genetic disease, it is one of the most common hereditary optic neuropathies and can drastically impact patient quality of life. The increasing use of novel therapies – such as TANGO technology, CRISPR gene editing, and gene replacement therapy to address the OPA1 protein haploinsufficiency; idebenone to stall RGC destruction and protect the Retinal Nerve Fiber Layer; cell-based regenerative therapy to replace dysfunctional RGCs; and targeted mitochondrial peptides and antioxidants, NAD+ boosters, and mitophagy modulators for mitochondrial support – is creating exciting opportunities for the management of this vision-threatening condition.
Ophthalmologists and patients should be aware of these options and the availability of clinical trials for ADOA. Increased knowledge of such therapeutics will open doors for patient counseling regarding interventions and the importance of referring family members for genetic testing. Both will enable pathogenic mutation identification in a timely manner and allow early treatment intervention. In short, research into these therapeutic modalities for hereditary optic neuropathies is ongoing and represents a major advance in ophthalmology.
Author contributions
RS: Writing – original draft, Project administration, Visualization, Conceptualization, Investigation, Writing – review & editing. LS: Writing – review & editing, Writing – original draft, Investigation, Visualization. MP: Writing – original draft, Investigation, Writing – review & editing, Visualization. AJ: Writing – review & editing, Investigation, Writing – original draft, Visualization. SS: Visualization, Investigation, Writing – review & editing, Writing – original draft. CC-O: Investigation, Writing – review & editing, Writing – original draft, Visualization. CK: Writing – review & editing, Investigation, Writing – original draft, Visualization. AL: Conceptualization, Visualization, Project administration, Writing – review & editing, Investigation, Writing – original draft, Supervision.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
AL has served as a consultant for the National Aeronautics and Space Administration NASA, the National Football League NFL, and is a consultant for Amgen, AstraZeneca, Bristol-Myers Squibb, Alexion, Stoke, Ethyreal, Catalyst, Dompe, and Viridian. Dr. Lee is also the Editor in Chief for Frontiers in Ophthalmology.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, et al. Dominant optic atrophy. Orphanet J Rare Diseases. (2012) 7:46. doi: 10.1186/1750-1172-7-46
2. Cohn AC, Toomes C, Potter C, Towns KV, Hewitt AW, Inglehearn CF, et al. Autosomal dominant optic atrophy: penetrance and expressivity in patients with OPA1 mutations. Am J Ophthalmology. (2007) 143:656–62.e1. doi: 10.1016/j.ajo.2006.12.038
3. Skidd PM, Lessell S, and Cestari DM. Autosomal dominant hereditary optic neuropathy (ADOA): A review of the genetics and clinical manifestations of ADOA and ADOA +. Semin Ophthalmol. (2013) 28:422–6. doi: 10.3109/08820538.2013.825296
4. Cohn AC, Toomes C, Hewitt AW, Kearns LS, Inglehearn CF, Craig JE, et al. The natural history of OPA1-related autosomal dominant optic atrophy. Br J Ophthalmology. (2008) 92:1333–6. doi: 10.1136/bjo.2007.134726
5. Wong DCS, Harvey JP, Jurkute N, Thomasy SM, Moosajee M, Yu-Wai-Man P, et al. OPA1 dominant optic atrophy: pathogenesis and therapeutic targets. J Neuro-Ophthalmology. (2023) 43:464–74. doi: 10.1097/WNO.0000000000001830
6. Pesch UEA, Fries JE, Bette S, Kalbacher H, Wissinger B, Alexander C, et al. OPA1, the disease gene for autosomal dominant optic atrophy, is specifically expressed in ganglion cells and intrinsic neurons of the retina. Invest Ophthalmol Visual Science. (2004) 45:4217–25. doi: 10.1167/iovs.03-1261
7. Ham M, Han J, Osann K, Smith M, and Kimonis V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion. (2019) 46:262–9. doi: 10.1016/j.mito.2018.07.006
8. Amati-Bonneau P, Milea D, Bonneau D, Chevrollier A, Ferré M, Guillet V, et al. OPA1-associated disorders: Phenotypes and pathophysiology. Int J Biochem Cell Biol. (2009) 41:1855–65. doi: 10.1016/j.biocel.2009.04.012
9. Eiberg H, Hansen L, Kjer B, Hansen T, Pedersen O, Bille M, et al. Autosomal dominant optic atrophy associated with hearing impairment and impaired glucose regulation caused by a missense mutation in the WFS1 gene. J Med Genet. (2006) 43:435–40. doi: 10.1136/jmg.2005.034892
10. Votruba M, Thiselton D, and Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmology. (2003) 87:48–53. doi: 10.1136/bjo.87.1.48
11. Ranieri M, Del Bo R, Bordoni A, Ronchi D, Colombo I, Riboldi G, et al. Optic atrophy plus phenotype due to mutations in the OPA1 gene: two more Italian families. J Neurol Sci. (2012) 315:146–9. doi: 10.1016/j.jns.2011.12.002
12. Venkatesh A, Li Z, Christiansen A, Lim KH, Kach J, Hufnagel R, et al. Antisense oligonucleotide mediated increase of OPA1 expression using TANGO technology for the treatment of autosomal dominant optic atrophy. Invest Ophthalmol Vis Sci. (2020) 61:2755.
13. Lee H and Yoon Y. Mitochondrial membrane dynamics-functional positioning of OPA1. Antioxidants (Basel). (2018) 7:1–21. doi: 10.3390/antiox7120186
14. Han J, Li Y, You Y, Fan K, and Lei B. Autosomal dominant optic atrophy caused by six novel pathogenic OPA1 variants and genotype-phenotype correlation analysis. BMC Ophthalmol. (2022) 22:322. doi: 10.1186/s12886-022-02546-0
15. Cartes-Saavedra B, Lagos D, Macuada J, Arancibia D, Burté F, Sjöberg-Herrera MK, et al. OPA1 disease-causing mutants have domain-specific effects on mitochondrial ultrastructure and fusion. Proc Natl Acad Sci U S A. (2023) 120:e2207471120. doi: 10.1073/pnas.2207471120
16. Zhang K, Zhang W, Zhang L, Hou X, Tian R, Hu Z, et al. OPA1 mutations in dominant optic atrophy: domain-specific defects in mitochondrial fusion and apoptotic regulation. J Transl Med. (2025) 23:471. doi: 10.1186/s12967-025-06471-w
17. Millet AM, Bertholet AM, Daloyau M, Reynier P, Galinier A, Devin A, et al. Loss of functional OPA1 unbalances redox state: implications in dominant optic atrophy pathogenesis. Ann Clin Transl Neurol. (2016) 3:408–21. doi: 10.1002/acn3.305
18. Venkatesh A, McKenty T, Ali S, Sonntag D, Ravipaty S, Cui Y, et al. Antisense oligonucleotide mediated increase in OPA1 improves mitochondrial function in fibroblasts derived from patients with autosomal dominant optic atrophy (ADOA). Assoc Res Vision Ophthalmology: Stoke Ther. (2021) 1–19.
19. Kane MS, Alban J, Desquiret-Dumas V, Gueguen N, Ishak L, Ferre M, et al. Autophagy controls the pathogenicity of OPA1 mutations in dominant optic atrophy. J Cell Mol Med. (2017) 21:2284–97. doi: 10.1111/jcmm.13149
20. Yao S-Q, Liang J-J, Zhou H, Tan S, Cao Y, Chen C-B, et al. Contrasting pathophysiological mechanisms of OPA1 mutations in autosomal dominant optic atrophy. Cell Death Discovery. (2025) 11:259. doi: 10.1038/s41420-025-02442-8
21. Venkatesh A, Ali S, Oh RS, Sonntag D, Li Z, McKenty T, et al. Antisense oligonucleotide mediated increase in OPA1 improves mitochondrial function in fibroblasts derived from patients with autosomal dominant optic atrophy (ADOA). Invest Ophthalmol Vis Sci. (2021) 62:1482.
22. Stoke Therapeutics I. Stoke Therapeutics Receives Authorization to Initiate a Phase 1/2 Study of STK-002 for Autosomal Dominant Optic Atrophy (ADOA) in the United Kingdom. 45 Wiggins Ave, Bedford, MA 01730: Stoke Therapeutics, Inc (2023).
23. Anderson K, Venkatesh A, McKenty T, Slate D, Lin Q, Ravipaty S, et al. STK-002, an antisense oligonucleotide (ASO) for the treatment of autosomal dominant optic atrophy (ADOA), is taken up by retinal ganglion cells (RGC) and upregulates OPA-1 protein expression after intravitreal administration to non-human primates (NHPs). Invest Ophthalmol Visual Science. (2022) 63:1111.
24. Venkatesh A, McKenty T, Ali S, Sonntag D, Ravipaty S, Cui Y, et al. Antisense oligonucleotide STK-002 increases OPA1 in retina and improves mitochondrial function in autosomal dominant optic atrophy cells. Nucleic Acid Ther. (2024) 34:221–33. doi: 10.1089/nat.2024.0022
25. Votruba M, Yu-Wai-Man P, Saluti K, Wang Y, Ticho B, and Gross S. OSPREY: an open-label study to investigate safety, tolerability, and exposure of the antisense oligonucleotide (ASO) STK-002 in patients with autosomal dominant optic atrophy (ADOA). Invest Ophthalmol Visual Science. (2024) 65:88.
26. Becchi G, Whitehead M, Harvey JP, Sladen PE, Dushti M, Chapple JP, et al. CRISPRa-mediated increase of OPA1 expression in dominant optic atrophy. Int J Mol Sci. (2025) 26:6364. doi: 10.3390/ijms26136364
27. Caruso SM, Quinn PM, da Costa BL, and Tsang SH. CRISPR/Cas therapeutic strategies for autosomal dominant disorders. J Clin Invest. (2022) 132:1–11. doi: 10.1172/JCI158287
28. Sundaresan Y, Yacoub S, Kodati B, Amankwa CE, Raola A, and Zode G. Therapeutic applications of CRISPR/Cas9 gene editing technology for the treatment of ocular diseases. FEBS J. (2023) 290:5248–69. doi: 10.1111/febs.16771
29. Yu W and Wu Z. Ocular delivery of CRISPR/Cas genome editing components for treatment of eye diseases. Adv Drug Delivery Rev. (2021) 168:181–95. doi: 10.1016/j.addr.2020.06.011
30. Carelli V, Carbonelli M, de Coo IF, Kawasaki A, Klopstock T, Lagrèze WA, et al. International consensus statement on the clinical and therapeutic management of leber hereditary optic neuropathy. J Neuro-Ophthalmology. (2017) 37:371–81. doi: 10.1097/WNO.0000000000000570
31. Halawani MA and Badeeb NO. The crossroads of Leber hereditary optic neuropathy and autosomal dominant optic Atrophy: Clinical profiles of patients with coexisting pathogenic genetic variants. Am J Ophthalmol Case Rep. (2025) 38:102346. doi: 10.1016/j.ajoc.2025.102346
32. Romagnoli M, La Morgia C, Carbonelli M, Di Vito L, Amore G, Zenesini C, et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann Clin Transl Neurol. (2020) 7:590–4. doi: 10.1002/acn3.51026
33. Achilli A, Iommarini L, Olivieri A, Pala M, Hooshiar Kashani B, Reynier P, et al. Rare primary mitochondrial DNA mutations and probable synergistic variants in leber’s hereditary optic neuropathy. PloS One. (2012) 7:e42242. doi: 10.1371/journal.pone.0042242
34. Sharma LK, Lu J, and Bai Y. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem. (2009) 16:1266–77. doi: 10.2174/092986709787846578
35. Varricchio C, Beirne K, Heard C, Newland B, Rozanowska M, Brancale A, et al. The ying and yang of idebenone: Not too little, not too much – cell death in NQO1 deficient cells and the mouse retina. Free Radical Biol Med. (2020) 152:551–60. doi: 10.1016/j.freeradbiomed.2019.11.030
36. Sugiyama Y and Fujita T. Stimulation of the respiratory and phosphorylating activities in rat brain mitochondria by idebenone (CV-2619), a new agent improving cerebral metabolism. FEBS Letters. (1985) 184:48–51. doi: 10.1016/0014-5793(85)80650-5
37. Gueven N, Woolley K, and Smith J. Border between natural product and drug: Comparison of the related benzoquinones idebenone and coenzyme Q10. Redox Biol. (2015) 4:289–95. doi: 10.1016/j.redox.2015.01.009
38. Yu-Wai-Man P, Griffiths PG, and Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res. (2011) 30:81–114. doi: 10.1016/j.preteyeres.2010.11.002
39. García-López C, García-López V, Matamoros JA, Fernández-Albarral JA, Salobrar-García E, de Hoz R, et al. The role of citicoline and coenzyme Q10 in retinal pathology. Int J Mol Sci. (2023) 24:1–23. doi: 10.3390/ijms24065072
40. Catalani E, Brunetti K, Del Quondam S, and Cervia D. Targeting mitochondrial dysfunction and oxidative stress to prevent the neurodegeneration of retinal ganglion cells. Antioxidants (Basel). (2023) 12:1–12. doi: 10.3390/antiox12112011
41. Garrido-Maraver J, Cordero MD, Oropesa-Avila M, Vega AF, de la Mata M, Pavon AD, et al. Clinical applications of coenzyme Q10. Front Biosci (Landmark Ed). (2014) 19:619–33. doi: 10.2741/4231
42. Giorgio V, Petronilli V, Ghelli A, Carelli V, Rugolo M, Lenaz G, et al. The effects of idebenone on mitochondrial bioenergetics. Biochim Biophys Acta (BBA) - Bioenergetics. (2012) 1817:363–9. doi: 10.1016/j.bbabio.2011.10.012
43. Ferro Desideri L, Traverso CE, and Iester M. Current treatment options for treating OPA1-mutant dominant optic atrophy. Drugs Today (Barc). (2022) 58:547–52. doi: 10.1358/dot.2022.58.11.3448291
44. D’Esposito F, Zeppieri M, Cordeiro MF, Capobianco M, Avitabile A, Gagliano G, et al. Insights on the genetic and phenotypic complexities of optic neuropathies. Genes (Basel). (2024) 15:1–18. doi: 10.3390/genes15121559
45. Mashima Y, Hiida Y, and Oguchi Y. Remission of Leber’s hereditary optic neuropathy with idebenone. Lancet. (1992) 340:368–9. doi: 10.1016/0140-6736(92)91442-B
46. Catarino CB, von Livonius B, Priglinger C, Banik R, Matloob S, Tamhankar MA, et al. Real-world clinical experience with idebenone in the treatment of leber hereditary optic neuropathy. J Neuro-Ophthalmology. (2020) 40:558–65. doi: 10.1097/WNO.0000000000001023
47. Yu-Wai-Man P, Carelli V, Newman NJ, Silva MJ, Linden A, Van Stavern G, et al. Therapeutic benefit of idebenone in patients with Leber hereditary optic neuropathy: The LEROS nonrandomized controlled trial. Cell Rep Med. (2024) 5:101437. doi: 10.1016/j.xcrm.2024.101437
48. Valentin K, Georgi T, Riedl R, Aminfar H, Singer C, Klopstock T, et al. Idebenone treatment in patients with OPA1-dominant optic atrophy: A prospective phase 2 trial. Neuroophthalmology. (2023) 47:237–47. doi: 10.1080/01658107.2023.2251575
49. Klopstock T, Yu-Wai-Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. (2011) 134:2677–86. doi: 10.1093/brain/awr170
50. Grainok J, Utama S, Chai T, Ong F, Champain D, Lui G, et al. PYC-001: A promising RNA therapeutic for autosomal dominant optic atrophy. Invest Ophthalmol Visual Science. (2024) 65:5305.
51. Safety of Single and Repeat Dose of PYC-001 Eye Injections in People With Autosomal Dominant Optic Atrophy: MedPath. Available online at: https://trial.medpath.com/clinical-trial/1b93e866a4bb012c/nct06970106-pyc-001-opa1-optic-atrophy (Accessed August 14, 2025).
52. ADOA (Autosomal Dominant Optic Atrophy). Neurophth. Available online at: https://www.neurophth.com/en/ProjectD-67.html (Accessed August 14, 2025).
53. Weiss JN and Levy S. Stem Cell Ophthalmology Treatment Study (SCOTS): bone marrow derived stem cells in the treatment of Dominant Optic Atrophy. Stem Cell Investig. (2019) 6:41. doi: 10.21037/sci.2019.11.01
54. Stem Cell Ophthalmology Treatment Study II (SCOTS2). National Library of Medicine 8600 Rockville Pike, Bethesda, MD 20894: NIH National Library of Medicine. Available online at: https://www.clinicaltrials.gov/study/NCT03011541 (Accessed March 20, 2025).
55. Olichon A, Landes T, Arnauné-Pelloquin L, Emorine LJ, Mils V, Guichet A, et al. Effects of OPA1 mutations on mitochondrial morphology and apoptosis: relevance to ADOA pathogenesis. J Cell Physiol. (2007) 211:423–30. doi: 10.1002/jcp.20950
56. Soldatov VO, Kubekina MV, Skorkina MY, Belykh AE, Egorova TV, Korokin MV, et al. Current advances in gene therapy of mitochondrial diseases. J Trans Med. (2022) 20:562. doi: 10.1186/s12967-022-03685-0
57. Zanfardino P, Amati A, Perrone M, and Petruzzella V. The balance of MFN2 and OPA1 in mitochondrial dynamics, cellular homeostasis, and disease. Biomolecules. (2025) 15:433. doi: 10.3390/biom15030433
58. Votruba M, Gross S, Lam BL, Liao YJ, Mudumbai RC, Saluti K, et al. Flavoprotein fluorescence in the 24-month FALCON natural history study of autosomal dominant optic atrophy: A potential biomarker for mitochondrial dysfunction. Invest Ophthalmol Visual Science. (2025) 66:5862.
59. Stoke Therapeutics Second Quarter 2025 Business Update. Stoke Therapeutics 2025. Available online at: https://investor.stoketherapeutics.com/static-files/d954d0f6-8168-47b2-b38e-eec2617957e7 (Accessed August 14, 2025).
60. Stoke Therapeutics Reports Second Quarter 2025 Financial Results and Provides Business Updates (2025). Stoke Therapeutics. Available online at: https://investor.stoketherapeutics.com/news-releases/news-release-details/stoke-therapeutics-reports-second-quarter-2025-financial-results (Accessed August 14, 2025).
61. Wong RCB, Lim SY, Hung SSC, Jackson S, Khan S, Van Bergen NJ, et al. Mitochondrial replacement in an iPSC model of Leber’s hereditary optic neuropathy. Aging (Albany NY). (2017) 9:1341–50. doi: 10.18632/aging.101231
62. Nakai R, Varnum S, Field RL, Shi H, Giwa R, Jia W, et al. Mitochondria transfer-based therapies reduce the morbidity and mortality of Leigh syndrome. Nat Metab. (2024) 6:1886–96. doi: 10.1038/s42255-024-01125-5
63. Zanfardino P, Amati A, Doccini S, Cox SN, Tullo A, Longo G, et al. OPA1 mutation affects autophagy and triggers senescence in autosomal dominant optic atrophy plus fibroblasts. Hum Mol Genet. (2024) 33:768–86. doi: 10.1093/hmg/ddae008
64. Zaninello M, Palikaras K, Naon D, Iwata K, Herkenne S, Quintana-Cabrera R, et al. Inhibition of autophagy curtails visual loss in a model of autosomal dominant optic atrophy. Nat Commun. (2020) 11:4029. doi: 10.1038/s41467-020-17821-1
65. Ding C, Ndiaye PS, Campbell SR, Fry MY, Gong J, Wienbar SR, et al. SARM1 loss protects retinal ganglion cells in a mouse model of autosomal dominant optic atrophy. . J Clin Invest. (2025) 135:1–17. doi: 10.1172/JCI191315
66. Geisler S, Huang SX, Strickland A, Doan RA, Summers DW, Mao X, et al. Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J Exp Med. (2019) 216:294–303. doi: 10.1084/jem.20181040
67. Hughes RO, Bosanac T, Mao X, Engber TM, DiAntonio A, Milbrandt J, et al. Small molecule SARM1 inhibitors recapitulate the SARM1-/- phenotype and allow recovery of a metastable pool of axons fated to degenerate. Cell Rep. (2021) 34:1–12. doi: 10.1016/j.celrep.2020.108588
68. Gould SA, Gilley J, Ling K, Jafar-Nejad P, Rigo F, and Coleman M. Sarm1 haploinsufficiency or low expression levels after antisense oligonucleotides delay programmed axon degeneration. Cell Rep. (2021) 37:1–10. doi: 10.1016/j.celrep.2021.110108
69. Yang Y and Walsh CE. Spliceosome-mediated RNA trans-splicing. Mol Ther. (2005) 12:1006–12. doi: 10.1016/j.ymthe.2005.09.006
70. Jüschke C, Klopstock T, Catarino CB, Owczarek-Lipska M, Wissinger B, and Neidhardt J. Autosomal dominant optic atrophy: A novel treatment for OPA1 splice defects using U1 snRNA adaption. Mol Ther Nucleic Acids. (2021) 26:1186–97. doi: 10.1016/j.omtn.2021.10.019
71. Sladen PE, Jovanovic K, Guarascio R, Ottaviani D, Salsbury G, Novoselova T, et al. Modelling autosomal dominant optic atrophy associated with OPA1 variants in iPSC-derived retinal ganglion cells. Hum Mol Genet. (2022) 31:3478–93. doi: 10.1093/hmg/ddac128
72. Lei Q, Xiang K, Cheng L, and Xiang M. Human retinal organoids with an OPA1 mutation are defective in retinal ganglion cell differentiation and function. Stem Cell Rep. (2024) 19:68–83. doi: 10.1016/j.stemcr.2023.11.004
73. Uddin F, Rudin CM, and Sen T. CRISPR gene therapy: applications, limitations, and implications for the future. Front Oncol. (2020) 10:1387. doi: 10.3389/fonc.2020.01387
Keywords: autosomal dominant optic atrophy, Targeted Augmentation of Nuclear Gene Output (TANGO), OPA1, idebenone, review
Citation: Sampige R, Seaborn LEA, Pluenneke M, Jyothi A, Saland S, Chinedu-Obi CM, Keehn C and Lee AG (2025) IT TAKES TWO TO TANGO: potential novel therapies for autosomal dominant optic atrophy. Front. Ophthalmol. 5:1688232. doi: 10.3389/fopht.2025.1688232
Received: 27 August 2025; Accepted: 23 October 2025;
Published: 05 November 2025.
Edited by:
Denis Plotnikov, Kazan State Medical University, RussiaReviewed by:
Chen Ding, Boston Children’s Hospital and Harvard Medical School, United StatesPaola Zanfardino, University of Bari Aldo Moro, Italy
Copyright © 2025 Sampige, Seaborn, Pluenneke, Jyothi, Saland, Chinedu-Obi, Keehn and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ritu Sampige, cml0dS5zYW1waWdlQGJjbS5lZHU=