 Alexey V. Zaitsev
Alexey V. Zaitsev Mark Warren
Mark Warren- Nora Eccles Harrison Cardiovascular Research and Training Institute, University of Utah, Salt Lake City, UT, United States
The traditional view of ventricular excitation and conduction is an all-or-nothing response mediated by a regenerative activation of the inward sodium channel, which gives rise to an essentially constant conduction velocity (CV). However, whereas there is no obvious biological need to tune-up ventricular conduction, the principal molecular components determining CV, such as sodium channels, inward-rectifier potassium channels, and gap junctional channels, are known targets of the “stress” protein kinases PKA and calcium/calmodulin dependent protein kinase II (CaMKII), and are thus regulatable by signal pathways converging on these kinases. In this mini-review we will expose deficiencies and controversies in our current understanding of how ventricular conduction is regulated by stress kinases, with a special focus on the chamber-specific dimension in this regulation. In particular, we will highlight an odd property of cardiac physiology: uniform CV in ventricles requires co-existence of mutually opposing gradients in cardiac excitability and stress kinase function. While the biological advantage of this peculiar feature remains obscure, it is important to recognize the clinical implications of this phenomenon pertinent to inherited or acquired conduction diseases and therapeutic interventions modulating activity of PKA or CaMKII.
Introduction
Traditionally, conduction through the ventricular tissue has been considered an all-or-none event scantly amenable to control. However, as early as 1953 Siebens et al. (1953) showed that sympathetic agonists modestly accelerated ventricle conduction. Using diverse approaches, subsequent studies also showed modest conduction increases in response to adrenergic stimulation (Munger et al., 1994; de Boer et al., 2007; Lang et al., 2015; Ajijola et al., 2017). Of interest, the duration of the QRS complex, which reflects the total time of conduction spread through the ventricles (Nattel and Jing, 1989), may change dynamically in the 24 h cycle (Nakagawa et al., 1998), or in response to exercise (Pilhall et al., 1992). Other studies suggest that sex hormones modulate QRS duration (Macfarlane et al., 1994; Okin et al., 1995). Interestingly, the conductivity of channels involved in the generation and transmission of the ventricular impulse (notably the cardiac Na+ channel, Nav1.5) may be modulated by phosphorylation, and thus are amenable to regulation by protein kinases responding to various neural and hormonal signals, in particular transmitted through activation of G-protein-coupled receptors (GPCRs) (Sato et al., 2015). Prominent in these signaling pathways are the calcium/calmodulin-dependent protein kinase II (CaMKII) (Hund et al., 2010; Ashpole et al., 2012; Glynn et al., 2015; Burel et al., 2017) and the cAMP-activated protein kinase A (PKA) (Murphy et al., 1996; Zhou et al., 2002; Aiba et al., 2014), collectively known as “stress” kinases for their involvement in the “fight or flight” physiological response (Wehrens et al., 2004; Wu et al., 2016). This mini-review will focus on very recent (and still limited) information regarding how the electrical wave propagation through ventricular chambers is regulated by stress kinases. Specifically, we will highlight a largely unknown regional aspect of kinase function in the ventricles, and will discuss its relevance to clinical conditions causing reduced ventricular excitability, such as the Brugada syndrome (BrS). A comprehensive review on the fundamentals of cardiac conduction can be found elsewhere (Veeraraghavan et al., 2014).
Normal Ventricular Conduction Is Sustained by Constitutive Activity of CaMKII and PKA
CaMKII, a serine/threonine-specific protein kinase regulated by the Ca2+/calmodulin complex (Maier and Bers, 2002; Erickson, 2014), modulates the cardiac response to stress by targeting numerous ion channels and transporters (Bers and Grandi, 2009). Importantly, CaMKII functionally regulates the three main components of cardiac excitability: Nav1.5 (Wagner et al., 2006; Yoon et al., 2009; Aiba et al., 2010), inward-rectifier potassium channels underlying the K+ current IK1 (Wagner et al., 2009), and gap junction channels formed by Connexin 43 (Cx43) proteins (Procida et al., 2009; Huang et al., 2011). Recently, CaMKII has garnered attention due to its ability to modulate ion channels in ways that favor afterdepolarizations, and for its prominent role in cardiac disease development (Swaminathan et al., 2012). Increased activity of CaMKII [which may occur due to CaMKII overexpression (Zhang et al., 2002, 2003) or upregulation in the failing heart (Anderson, 2005)] alters Ca2+ homeostasis [including increased Ca2+ entry through ICaL (Anderson et al., 1994); increased Ca2+ release through RyR (Wehrens et al., 2004); and increased Ca2+ reuptake to the SR (Mattiazzi and Kranias, 2014)] and enhances the late sodium current (INaL) (Wagner et al., 2006; Maltsev et al., 2008), both effects promoting abnormal cellular triggered activity and arrhythmia (Anderson, 2005; Vincent et al., 2014). Whereas there is a general consensus on the direction of CaMKII regulation of cellular Ca2+ cycling (Swaminathan et al., 2012), regulation of cellular excitability and conduction by CaMKII remains controversial. On the cellular level, some studies suggested that CaMKII activity favors an overall Na+ current (INa) upregulation (Yoon et al., 2009; Aiba et al., 2010), while others argued that it promotes an overall INa downregulation (Wagner et al., 2006). Yoon et al. (2009) were the first to suggest that baseline CaMKII activity is required for normal ventricular excitation, and that CaMKII inhibition is detrimental. The authors showed that the CaMKII blocker KN93 reduced peak INa, shifted the steady-state inactivation curve to hyperpolarized values, decreased INaL, enhanced intermediate inactivation, and delayed the recovery from fast and slow inactivation. Altered INa kinetics led to a significant suppression of the action potential upstroke velocity (dV/dtmax), a measure of cellular excitability. In terms of the regulation direction, Yoon et al.’s (2009) results are consistent with findings by Aiba et al. (2010), who showed that intracellular delivery of CaMKII (CaMKIIα) to isolated guinea pig myocytes caused upregulation of INa (i.e., changes in kinetics leading to increased availability of INa under physiological conditions). However, the studies by Yoon et al. (2009) and Aiba et al. (2010) contradict the studies performed in isolated adult mouse/rabbit myocytes (Wagner et al., 2006), HEK293 cells (Deschenes et al., 2002; Ashpole et al., 2012), or using simulations (Hund et al., 2008), that showed that overexpression/inhibition of CaMKII suppressed/enhanced INa availability, and inhibitors KN93 (or AIP, a peptide inhibitor) rescued CaMKII overexpression-induced detrimental effects.
CaMKII regulation of ventricular conduction in the whole heart is also controversial. Takanari et al. (2016) reported that a chronic reduction in CaMKII activity in mice following expression of CaMKII-inhibiting autocamtide-3-related peptide (AC3-I) caused an increase in conduction velocity (CV) in both RV and LV. In addition, they showed that inhibition of calmodulin, the upstream regulator of CaMKII, increased ventricular CV, and reduced arrhythmogenicity in isolated rabbit hearts (Takanari et al., 2016). The improved conduction following calmodulin/CaMKII inhibition was attributed to increased localization of Cx43 in the intercalated disk (Takanari et al., 2016). On the physiological level, our own studies yielded strikingly opposite outcomes (Warren and Zaitsev, 2017; Warren et al., 2017; Zaitsev et al., 2019). Specifically, inhibition of either CaMKII or calmodulin slowed down propagation mainly due to inducing severe conduction defects in the right ventricular outflow tract (RVOT, Figure 1A, leftmost and center panels), and this was proarrhythmic (Figure 1B; Warren and Zaitsev, 2017; Zaitsev et al., 2019). Whilst Cx43 channel function was not analyzed, CaMKII blockade reduced dV/dtmax both in myocytes and whole hearts (Warren et al., 2017; Zaitsev et al., 2019), consistent with Yoon et al.’s (2009) data, and suggestive of a reduced INa availability. Moreover, CaMKII inhibition caused highly rate-dependent changes of ventricular conduction and excitability (see more below) (Warren et al., 2017; Zaitsev et al., 2019), readily explained by altered INa inactivation (Yoon et al., 2009), but not by altered localization or conductivity of Cx43. Investigating how CaMKII gain- and loss-of-function alters both myocardial active and passive properties in the same whole-heart animal model will likely resolve the controversy. We invite anyone interested to collaborate on such a study.
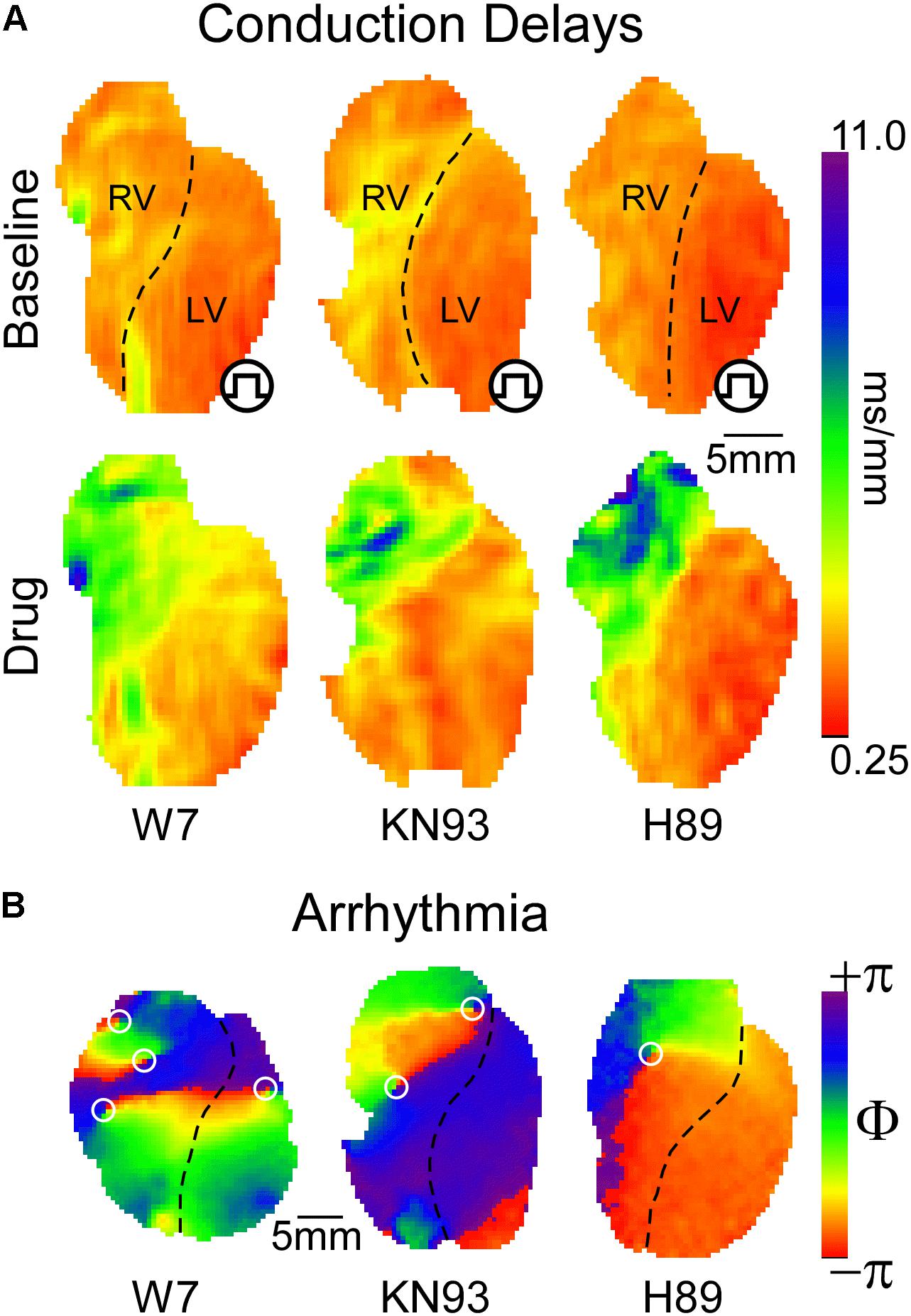
Figure 1. (A) Inhibition of either CaMKII or PKA signaling leads to preferential conduction slowing in the basal RV approximately corresponding to the RVOT. Qualitatively, similar effects are produced by blockade of calmodulin, CaMKII, or PKA with W7, KN93, and H89, respectively. (B) With sufficient concentration and/or time of exposure, all three interventions lead to reentrant VT or VF, with the driving rotors preferentially occurring in the RVOT [reused with permission from the American Journal of Physiology (Warren and Zaitsev, 2017) and Zaitsev et al. (2019)].
PKA is a cAMP sensitive protein kinase which responds to beta-adrenergic receptor activation (Taylor et al., 2013; Soni et al., 2014), and is inhibited by muscarinic receptor activation (Harvey and Belevych, 2003). PKA activation drives the physiological response to stress by targeting molecular components of cardiac function and excitability (Soni et al., 2014). Interestingly, CaMKII and PKA work hand in hand upregulating excitation–contraction coupling in response to stress (Grimm and Brown, 2010). Consequently, and given that Nav1.5 phosphorylation sites targeted by CaMKII or PKA cluster mostly in the first intracellular linker loop of the channel (Marionneau and Abriel, 2015), including a potentially shared site at Ser571 (Hund et al., 2010; Marionneau et al., 2012), we hypothesized that PKA regulates ventricular conduction similarly to CaMKII. This was subsequently confirmed (Zaitsev et al., 2019). Figure 1A (rightmost panel) illustrates how PKA blockade with H89 caused a highly non-uniform, RVOT-centric, depression of conduction, which initiated arrhythmia (Figure 1B).
Our organ-level findings are generally consistent with single cell patch clamp studies indicating that PKA and/or its upstream signals (beta-agonists and cAMP) upregulate INa (Matsuda et al., 1992; Frohnwieser et al., 1997; Lu et al., 1999; Aiba et al., 2010). Although some early studies suggested opposite effects (Ono et al., 1989; Schubert et al., 1989), slow background shifts in INa kinetics common in isolated myocytes may have confounded those results (Ono et al., 1993). Of note, diverse mechanisms of PKA-mediated INa upregulation were proposed, including PKA-induced increase in Na+ channel trafficking to the sarcolemma (Zhou et al., 2000, 2002). To date, there is no cellular counterpart to our whole heart study (Zaitsev et al., 2019) that would clarify how inhibition of PKA modulates INa gating. However, similarities in the conduction depression patterns elicited by CaMKII and PKA inhibition (Figure 1) suggest that the underlying ionic mechanisms are also similar.
Role of Phosphatases – A “Known Unknown”
In the 1980s the notion emerged that opposing actions of endogenous phosphatases and associated kinases set the basal level of membrane currents, such as the L-type inward Ca2+ current (Hescheler et al., 1988), IK1 (Koumi et al., 1995a), and connexins (Moreno et al., 1994). It is suggested that type 1 and type 2A phosphatases (PP1 and PP2A) are key to regulating ion channel phosphorylation (Luss et al., 2000; Terentyev and Hamilton, 2016), but improved understanding is pending. A recent study by El Refaey et al. (2019) using a PP2A phosphatase-defective transgenic mouse showed that adrenergic stimulation of myocytes induced aberrant action potentials attributed to a deficient dephosphorylation of Nav1.5 affecting INaL. This study reported no significant difference in the fast component of INa (El Refaey et al., 2019).
In our experiments, administration of broad-acting phosphatase inhibitor calyculin to isolated rabbit hearts led to a small yet significant acceleration of ventricular conduction, the effect being greater in the RV than in the LV (Zaitsev et al., 2019). This supports a phosphatase-mediated negative regulation of excitability, which differs from El Refaey et al. (2019). Given the broad action of calyculin, our data might indicate that a phosphatase other than PP2A (e.g., PP1) regulates the fast component of INa.
Additional indirect evidence of the phosphatase-mediated negative regulation of excitability is the progressive nature of CaMKII/PKA-inhibition-mediated conduction defects (steady state after >1 h of kinase blockade) (Zaitsev et al., 2019). The progressive conduction deterioration is most easily explained in terms of continuing phosphatase activity amid decreased total kinase activity. Because the RV is revealed as the most vulnerable region, it is plausible that phosphorylation/dephosphorylation imbalance affects it more prominently.
Stress Kinases Regulate Ventricular Conduction in a Rate-Dependent Manner
Besides the progressive conduction deterioration caused by CaMKII and/or PKA blockade, the effect of each blocker was strongly rate dependent. Combined, these two effects resulted in a frequency-dependence which progressed with time. At short durations of drug exposure, the detrimental effect of either CaMKII or PKA blockade was noticeable only at relatively high pacing rates (short pacing intervals), but with increased exposure to drug conduction was affected even at physiological pacing rates. The detrimental effects of kinases’ blockade were always largest in the basal RV (approximately corresponding to the RVOT), culminating in 2:1 conduction block, turbulence, and initiation of VF in that region at pacing intervals as long as 400 ms (Figure 1B; Zaitsev et al., 2019). Remarkably, severe conduction depression induced by either CaMKII or PKA inhibition was almost fully abolished just by prolongation of the pacing interval to >6000 ms (Zaitsev et al., 2019).
This result is revealing. First, it suggests that IK1 or Cx43 channels, the two major factors of ventricular syncytial conduction besides INa, do not play a significant role in mediating adverse effects caused by stress kinases’ inhibition. Even though current evidence points to CaMKII and PKA targeting both connexins (Burt and Spray, 1988; Moreno et al., 1992; Procida et al., 2009; Huang et al., 2011) and IK1 (Fakler et al., 1994; Koumi et al., 1995a,b,c; Wagner et al., 2009), neither is known to possess time-dependent gating properties. Second, given Yoon et al.’s (2009) data, enhancement of intermediate/slow inactivation of INa emerges as the likely mechanism causing conduction abnormalities in the presence of CaMKII inhibition. Furthermore, by similarity of the CaMKII and PKA inhibition effects, we predict that PKA also regulates intermediate/slow inactivation of INa, an effect not reported thus far. Lastly, the fact that long period of quiescence abolishes the effect of PKA and/or CaMKII inhibition disfavors the notion that our observed upregulation of INa by PKA and/or CaMKII is due to Na+ channel trafficking to the plasma membrane (Zhou et al., 2000). From the perspective of Nav1.5 gating mechanisms, it remains to be understood how insufficient phosphorylation of the protein, likely at locations identified in the 1st intracellular loop (Marionneau and Abriel, 2015), favors stabilization of the channel in the inactivated state.
Constitutive Activity of CaMKII and PKA in the Heart – How Much of It?
De Koninck and Schulman (1998) demonstrated that CaMKII can act as an intracellular Ca2+ ([Ca2+]i) transducer and activation frequency sensor in vitro. In line with this, using FRET-based biosensor Camui, Erickson et al. (2011) showed that increased activation rates (range 0–1 Hz) significantly increased CaMKII activity in isolated rabbit ventricular myocytes. Since the tested rates are much below the resting heart rate, one should expect a much higher level of CaMKII activation at the physiological baseline. In addition, even in quiescent cells, a significant activation of CaMKII produced by various neurohumoral ligands acting upon GPCR was reported (Erickson et al., 2011). Thus, the normal physiological level of CaMKII activity in the heart is a complex integral of various signals. Whereas it has not yet been quantitatively assessed, the degree at which CaMKII blockade affects conduction in hearts paced at normal physiological rate and in the absence of GPCR stimulation (Zaitsev et al., 2019), clearly indicates that the basal level of CaMKII activity is far from zero. To which extent this level is regulated by acute variations in heart rate and [Ca2+]i, or by phasic changes in autonomic and endocrine regulation, remains unknown and needs to be further investigated.
PKA constitutive activity is largely determined by the relationship between the half-maximal cAMP concentration required for PKA activation and the basal level of cAMP in cells. Measures of cAMP concentration yielded values around 1 μM (Terasaki and Brooker, 1977; Iancu et al., 2008). According to several in vitro studies, the cAMP concentration required for half-maximal activation of PKA is in the range 90–300 nM (Adams et al., 1991; Mongillo et al., 2004). Based on these estimates, PKA should be fully activated under resting conditions, precluding the possibility of a dynamic response to upstream signals acting via cAMP, which clearly contradicts fundamental physiology. Various schemes were proposed to resolve this controversy, including cAMP sequestration that renders it inactive (Exton et al., 1971), or the presence of intracellular domains in which cAMP concentration is much lower than the bulk cytosol concentration (Iancu et al., 2008). Recently, Koschinski and Zaccolo (2017) challenged the prior in vitro estimates of PKA sensitivity, reporting significantly higher half-maximal concentrations for the enzyme (5.2 μM cAMP) measured in intact Chinese hamster ovary cells. Upon inhibition of PKA (10 μM H89) in unstimulated cells, they found no detectable change in FRET-based PKA activity reporter signal, suggesting a negligible level of PKA activity at baseline cAMP levels. However, 10 μM H89 induced a dramatic slowing of ventricular conduction in perfused rabbit hearts, supporting the presence of robust endogenous PKA activity, even in the absence of beta-adrenergic and other neural and hormonal signals (Zaitsev et al., 2019). Perhaps, PKA signaling is very different between ovary cells and cardiac myocytes. Since isolated hearts respond to both beta-adrenergic stimulation and stimulation of cAMP synthesis with vigorous increase in PKA activity (Zhang and MacLeod, 1996), the constitutive level of PKA activity in the intact heart must be far from both inactive and fully activated state, but where it stands exactly, remains to be established.
Conduction Vulnerability in the RV: The Achilles’ Heel of the Heart
The spatial patterns of ventricular conduction depression induced by “stress” kinase inhibitors are unique insomuch that the RV is affected much more prominently than the LV, and within the RV the most affected region is RVOT. This pattern is remarkably similar among the kinase inhibitors KN93 and H89, as well as the calmodulin inhibitor W7 (Figure 1A), suggesting a common mechanism of action.
Figures 2A–C illustrate how the specific RVOT vulnerability can be explained in terms of its intrinsically reduced excitability (reflected in reduced dV/dtmax, see Figure 2C inset), compared to other ventricular regions such as the anterior–apical left ventricle (AALV) (Warren et al., 2017). The reduced excitability presumably results from the locally reduced expression levels of Nav1.5 (Veeraraghavan and Poelzing, 2008; Boukens et al., 2013). The fact that, despite regional differences in excitability, the impulse conduction is uniform through the ventricular chambers (see Figure 1A, upper panels; see also Figures 3D–E in Boukens et al., 2013) requires explanation. We believe that two factors are involved. One is that cardiac excitability operates within a wide margin of safety across which impulse conduction is robust (Figure 2D). The presence of such a safety margin stems from the asymptotic relationship between action potential upstroke (dV/dtmax) and conduction (Buchanan et al., 1985; Shaw and Rudy, 1997), whereby dV/dtmax needs to cross a critical threshold before conduction is noticeably affected (Figure 2D).
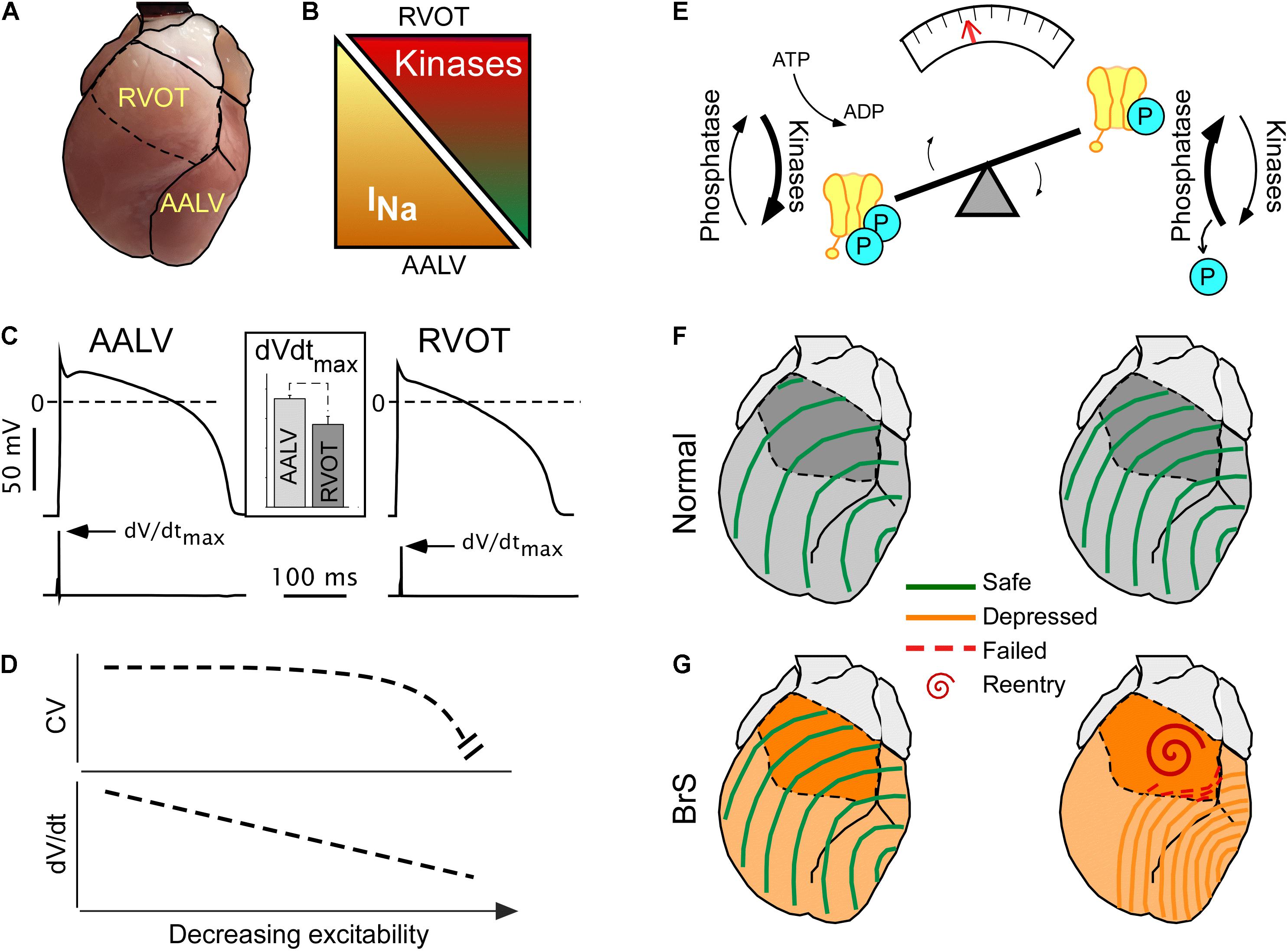
Figure 2. Concealed conduction vulnerability in the right ventricular outflow tract (RVOT). The left-hand part (A–D) illustrates the currently established static component of the phenomenon, whereas the right-hand part (E–G) illustrates a largely hypothetical dynamic component. (A) The anterior view of the heart showing RVOT and the anterior–apical left ventricle (AALV), the two regions with demonstrated significant differences in properties affecting conduction. (B) Compared to AALV (and possibly other ventricular regions), RVOT has a lesser expression of Nav 1.5, the pore-forming subunit of INa (Boukens et al., 2013), but a larger expression of “stress” kinases PKA and CaMKII (Zaitsev et al., 2019). (C) Cardiac myocytes from RVOT have reduced excitability (assessed by the maximum slope of the action potential upstroke slope, dV/dtmax) than cardiac myocytes from AALV [reused with permission from the American Journal of Physiology (Warren et al., 2017)]. (D) The theoretical non-linear relationship between excitability and conduction velocity (Shaw and Rudy, 1997) which may explain the relatively uniform conduction velocities across the ventricles (Zaitsev et al., 2019) despite the regional differences in the expression of Nav 1.5 and excitability [modified Shaw and Rudy (1997), with permission]. (E) A diagram highlighting the dynamic nature of the phosphorylated state of Nav 1.5, which depends on the dynamic equilibrium between the sum of all kinase and phosphatase activity. Overall, little is known about the actual temporal fluctuations of Nav 1.5 phosphorylation, but such can be inferred from the fact that both PKA and CaMKII are downstream of highly dynamic autonomic and endocrine signals. (F,G) Physiological fluctuations of Nav 1.5 phosphorylation may lead to variations in excitability and conduction velocity [slower conduction with less phosphorylation (Zaitsev et al., 2019)]. While such variations are benign under normal conditions (F), they may lead to acute conduction crisis and reentrant arrhythmias when INa expression is globally reduced (G) (Remme and Wilde, 2014), or when the phosphorylation balance of Nav 1.5 is compromised (Aiba et al., 2014) in patients with Brugada syndrome (BrS).
The second factor is the presence of spatial heterogeneity in stress kinase-related signaling (Figures 2A,B). We found that the protein expression levels of CaMKII-δ (Maier and Bers, 2002) and of the catalytic subunit of PKA (PKA-Cα) (Yin et al., 2008) are significantly higher in the RVOT than in the AALV (Zaitsev et al., 2019), suggesting increased local activities of these enzymes. Others showed that the RV hemodynamic response was more sensitive to β-adrenergic stimulation than the LV (Irlbeck et al., 1996). Additionally, RV myocytes subject to isoproterenol exhibited increased sarcomere shortening, Ca2+ transient amplitude, cytoplasmic cAMP accumulation, and PKA activity compared to the LV counterparts (Molina et al., 2014). These findings suggest the existence of an organ-wide program which controls local cellular signaling to maintain a specific functional profile. Whereas the physiological advantage of such an adaptation remains unclear, an apparent physiological role of stress kinases is to upregulate functional INa (Aiba et al., 2010) to maintain RV excitability well within the margins of safety. The source of this heterogeneity may be linked to the distinct development origin of the RVOT (Boukens et al., 2009), which has been associated to a more persistent RVOT-regional slow impulse conduction during cardiac development (de Jong et al., 1992; Boukens et al., 2013). Alternatively, we speculate that higher expression levels of CaMKII and PKA in RVOT could develop in response to a comparatively higher local mechanical stress caused by the distinct hemodynamic context in which RVOT operates (Geva et al., 1998). However, the physiological advantage or necessity of intrinsically reduced RVOT excitability (Warren et al., 2017) remains elusive.
Finally, the role of regional differences in the myocardial organization properties such as connexin distribution (Ou et al., 2005), fiber orientation (Burgess et al., 1988), or cleft geometry (Kelly et al., 2018) might be important. We should note, however, that isolated cells subject to kinase inhibitors developed depressed excitability characteristics akin to that observed in intact tissue (Warren et al., 2017), indicating that tissue architecture is not required for the phenomenon to develop. Additionally, the dynamic nature of the phenomenon cannot be readily ascribed to tissue architecture components which are functionally time-independent. Thus, it seems that the tissue architecture is not directly responsible for the core phenomenon of kinase inhibition-induced conduction depression. However, it may contribute to increased conduction vulnerability in the RVOT by conferring the region a narrower safety margin for conduction.
Stress Kinase Activity and the Brugada Syndrome
The BrS is a hereditary lethal cardiac condition associated with conduction abnormalities in the RVOT (Kasanuki et al., 1997; Postema et al., 2010; Zhang et al., 2015). Abnormal INa function likely underlies the BrS phenotype, since the majority of known mutations causing the disease affect this current (Antzelevitch et al., 2017). The ECG signature of BrS, an abnormal ST segment elevation in precordial leads, manifests intermittently (Veltmann et al., 2006). Circadian biases in the development of abnormal ST segment elevation (Gray et al., 2017), as well as in the initiation of VF (Matsuo et al., 1999), underscore the dynamic nature of BrS. Increased parasympathetic tone can also underlie the dynamic unmasking of the disease phenotype, which can be reverted by interventions that increase the sympathetic signals (Kasanuki et al., 1997).
Conduction patterns induced by “stress” kinase inhibition (Figure 1) are strikingly similar to RVOT-centric conduction defects described in BrS patients (Zhang et al., 2015). A prevailing paradox is that the permanent nature of dysfunctional Nav1.5 mutations afflicting BrS patients is typically associated to intermittent or lacking phenotypes. We speculate that the intermittent display of BrS phenotype may in part depend on dynamic fluctuations in stress kinases’ activity. Supporting this, half-maximal blockade of INa with TTX [which is similar to the degree of INa loss-of-function in some BrS models (Papadatos et al., 2002; Remme et al., 2009)] caused only a uniform slowing of conduction, while a subsequent short exposure to CaMKII inhibitor KN93 disproportionally slowed RV conduction, bringing about BrS-like phenotype (Zaitsev et al., 2019). We submit that following kinase activity fluctuations (Figure 2E), ventricular excitability is dynamically shifted back-and-forth within the limits of safe conduction, which are wide in healthy hearts (Figure 2F), but narrower in hearts affected by the BrS (Figure 2G). When this limit is breached, conduction in the RVOT fails first, and VF initiates in a manner similar to that observed during kinase inhibition (Figure 2G, right panel). Whereas there are no known cases of BrS involving CaMKII signaling, a family with BrS bore a SCN5A mutation in a PKA consensus phosphorylation site, which effectively disrupted positive regulation of INa by PKA (Aiba et al., 2014). Overall, the role of stress kinase signaling in BrS remains poorly understood and definitely merits further investigation.
Conclusion
Because of an intrinsically reduced safety margin for conduction in the RV/RVOT, the constitutive activity of both CaMKII and PKA is required for normal ventricular conduction. Consequently, any intervention decreasing activity of these kinases is potentially pro-arrhythmic and life-threatening. Further, any condition leading to additional reduction in the RV excitability (BrS, ischemia, and electrolyte imbalance) bears increased risk. Normal ventricular conduction hinges on the delicate balance of phosphorylation/dephosphorylation, which is a result of a very complex and highly dynamic summation of upstream signals mediated through nervous and endocrine regulation, as well as circadian rhythms. Whereas a wealth of knowledge has been accumulated at the level of molecular mechanisms involved in regulation of cardiac ionic channels by phosphorylation, there is a deficiency in translating these mechanisms to the level of whole-heart physiology and pathophysiology. We hope that this mini-review will stimulate investigations to bridge this gap.
Author Contributions
Both authors contributed equally to the preparation of this review.
Funding
This work was supported by the Nora Eccles Treadwell Foundation Research Grant (AZ) and the National Institutes of Health (NIH) grants 1RO1HL103877 (AZ) and RO1HL128752 (MW, Dosdall PI).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Junco (Shibayama) Warren for interesting discussions on the reviewed topics.
References
Adams, S. R., Harootunian, A. T., Buechler, Y. J., Taylor, S. S., and Tsien, R. Y. (1991). Fluorescence ratio imaging of cyclic AMP in single cells. Nature 349, 694–697. doi: 10.1038/349694a0
Aiba, T., Farinelli, F., Kostecki, G., Hesketh, G. G., Edwards, D., Biswas, S., et al. (2014). A mutation causing Brugada syndrome identifies a mechanism for altered autonomic and oxidant regulation of cardiac sodium currents. Circ. Cardiovasc. Genet. 7, 249–256. doi: 10.1161/CIRCGENETICS.113.000480
Aiba, T., Hesketh, G. G., Liu, T., Carlisle, R., Villa-Abrille, M. C., O’Rourke, B., et al. (2010). Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc. Res. 85, 454–463. doi: 10.1093/cvr/cvp324
Ajijola, O. A., Lux, R. L., Khahera, A., Kwon, O., Aliotta, E., Ennis, D. B., et al. (2017). Sympathetic modulation of electrical activation in normal and infarcted myocardium: implications for arrhythmogenesis. Am. J. Physiol. Heart Circ. Physiol. 312, H608–H621. doi: 10.1152/ajpheart.00575.2016
Anderson, M. E. (2005). Calmodulin kinase signaling in heart: an intriguing candidate target for therapy of myocardial dysfunction and arrhythmias. Pharmacol. Ther. 106, 39–55. doi: 10.1016/j.pharmthera.2004.11.002
Anderson, M. E., Braun, A. P., Schulman, H., and Premack, B. A. (1994). Multifunctional Ca2+/calmodulin-dependent protein kinase mediates Ca(2+)-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ. Res. 75, 854–861. doi: 10.1161/01.res.75.5.854
Antzelevitch, C., Yan, G. X., Ackerman, M. J., Borggrefe, M., Corrado, D., Guo, J., et al. (2017). J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Europace 19, 665–694. doi: 10.1093/europace/euw235
Ashpole, N. M., Herren, A. W., Ginsburg, K. S., Brogan, J. D., Johnson, D. E., Cummins, T. R., et al. (2012). Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 287, 19856–19869. doi: 10.1074/jbc.M111.322537
Bers, D. M., and Grandi, E. (2009). Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J. Cardiovasc. Pharmacol. 54, 180–187. doi: 10.1097/FJC.0b013e3181a25078
Boukens, B. J., Christoffels, V. M., Coronel, R., and Moorman, A. F. (2009). Developmental basis for electrophysiological heterogeneity in the ventricular and outflow tract myocardium as a substrate for life-threatening ventricular arrhythmias. Circ. Res. 104, 19–31. doi: 10.1161/CIRCRESAHA.108.188698
Boukens, B. J., Sylva, M., de Gier-de Vries, C., Remme, C. A., Bezzina, C. R., Christoffels, V. M., et al. (2013). Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ. Res. 113, 137–141. doi: 10.1161/CIRCRESAHA.113.301565
Buchanan, J. W., Saito, T., and Gettes, L. S. (1985). The effects of antiarrhythmic drugs, stimulation frequency, and potassium-induced resting membrane potential changes on conduction velocity and dV/dtmax in guinea pig myocardium. Circ. Res. 56, 696–703. doi: 10.1161/01.res.56.5.696
Burel, S., Coyan, F. C., Lorenzini, M., Meyer, M. R., Lichti, C. F., Brown, J. H., et al. (2017). C-terminal phosphorylation of NaV1.5 impairs FGF13-dependent regulation of channel inactivation. J. Biol. Chem. 292, 17431–17448. doi: 10.1074/jbc.M117.787788
Burgess, M. J., Steinhaus, B. M., Spitzer, K. W., and Ershler, P. R. (1988). Nonuniform epicardial activation and repolarization properties of in vivo canine pulmonary conus. Circ. Res. 62, 233–246. doi: 10.1161/01.res.62.2.233
Burt, J. M., and Spray, D. C. (1988). Inotropic agents modulate gap junctional conductance between cardiac myocytes. Am. J. Physiol. 254(6 Pt 2), H1206–H1210. doi: 10.1152/ajpheart.1988.254.6.H1206
de Boer, T. P., van Rijen, H. V., Van der Heyden, M. A., Kok, B., Opthof, T., Vos, M. A., et al. (2007). Beta-, not alpha-adrenergic stimulation enhances conduction velocity in cultures of neonatal cardiomyocytes. Circ. J. 71, 973–981. doi: 10.1253/circj.71.973
de Jong, F., Opthof, T., Wilde, A. A., Janse, M. J., Charles, R., Lamers, W. H., et al. (1992). Persisting zones of slow impulse conduction in developing chicken hearts. Circ. Res. 71, 240–250. doi: 10.1161/01.res.71.2.240
De Koninck, P., and Schulman, H. (1998). Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 279, 227–230. doi: 10.1126/science.279.5348.227
Deschenes, I., Neyroud, N., DiSilvestre, D., Marban, E., Yue, D. T., and Tomaselli, G. F. (2002). Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ. Res. 90, E49–E57. doi: 10.1161/01.res.0000012502.92751.e6
El Refaey, M., Musa, H., Murphy, N. P., Lubbers, E. R., Skaf, M., Han, M., et al. (2019). Protein phosphatase 2A regulates cardiac Na(+) channels. Circ. Res. 124, 737–746. doi: 10.1161/CIRCRESAHA.118.314350
Erickson, J. R. (2014). Mechanisms of CaMKII activation in the heart. Front. Pharmacol. 5:59. doi: 10.3389/fphar.2014.00059
Erickson, J. R., Patel, R., Ferguson, A., Bossuyt, J., and Bers, D. M. (2011). Fluorescence resonance energy transfer-based sensor Camui provides new insight into mechanisms of calcium/calmodulin-dependent protein kinase II activation in intact cardiomyocytes. Circ. Res. 109, 729–738. doi: 10.1161/CIRCRESAHA.111.247148
Exton, J. H., Lewis, S. B., Ho, R. J., Robison, G. A., and Park, C. R. (1971). The role of cyclic AMP in the interaction of glucagon and insulin in the control of liver metabolism. Ann. N. Y. Acad. Sci. 185, 85–100. doi: 10.1111/j.1749-6632.1971.tb45239.x
Fakler, B., Brandle, U., Glowatzki, E., Zenner, H. P., and Ruppersberg, J. P. (1994). Kir2.1 inward rectifier K+ channels are regulated independently by protein kinases and ATP hydrolysis. Neuron 13, 1413–1420. doi: 10.1016/0896-6273(94)90426-x
Frohnwieser, B., Chen, L. Q., Schreibmayer, W., and Kallen, R. G. (1997). Modulation of the human cardiac sodium channel alpha-subunit by cAMP-dependent protein kinase and the responsible sequence domain. J. Physiol. 498(Pt 2), 309–318. doi: 10.1113/jphysiol.1997.sp021859
Geva, T., Powell, A. J., Crawford, E. C., Chung, T., and Colan, S. D. (1998). Evaluation of regional differences in right ventricular systolic function by acoustic quantification echocardiography and cine magnetic resonance imaging. Circulation 98, 339–345. doi: 10.1161/01.cir.98.4.339
Glynn, P., Musa, H., Wu, X., Unudurthi, S. D., Little, S., Qian, L., et al. (2015). Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132, 567–577. doi: 10.1161/CIRCULATIONAHA.114.015218
Gray, B., Kirby, A., Kabunga, P., Freedman, S. B., Yeates, L., Kanthan, A., et al. (2017). Twelve-lead ambulatory electrocardiographic monitoring in Brugada syndrome: potential diagnostic and prognostic implications. Heart Rhythm 14, 866–874. doi: 10.1016/j.hrthm.2017.02.026
Grimm, M., and Brown, J. H. (2010). Beta-adrenergic receptor signaling in the heart: role of CaMKII. J. Mol. Cell. Cardiol. 48, 322–330. doi: 10.1016/j.yjmcc.2009.10.016
Harvey, R. D., and Belevych, A. E. (2003). Muscarinic regulation of cardiac ion channels. Br. J. Pharmacol. 139, 1074–1084. doi: 10.1038/sj.bjp.0705338
Hescheler, J., Mieskes, G., Ruegg, J. C., Takai, A., and Trautwein, W. (1988). Effects of a protein phosphatase inhibitor, okadaic acid, on membrane currents of isolated guinea-pig cardiac myocytes. Pflugers Arch. 412, 248–252. doi: 10.1007/bf00582504
Huang, R. Y., Laing, J. G., Kanter, E. M., Berthoud, V. M., Bao, M., Rohrs, H. W., et al. (2011). Identification of CaMKII phosphorylation sites in Connexin43 by high-resolution mass spectrometry. J. Proteome Res. 10, 1098–1109. doi: 10.1021/pr1008702
Hund, T. J., Decker, K. F., Kanter, E., Mohler, P. J., Boyden, P. A., Schuessler, R. B., et al. (2008). Role of activated CaMKII in abnormal calcium homeostasis and I(Na) remodeling after myocardial infarction: insights from mathematical modeling. J. Mol. Cell. Cardiol. 45, 420–428. doi: 10.1016/j.yjmcc.2008.06.007
Hund, T. J., Koval, O. M., Li, J., Wright, P. J., Qian, L., Snyder, J. S., et al. (2010). A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519. doi: 10.1172/JCI43621
Iancu, R. V., Ramamurthy, G., Warrier, S., Nikolaev, V. O., Lohse, M. J., Jones, S. W., et al. (2008). Cytoplasmic cAMP concentrations in intact cardiac myocytes. Am. J. Physiol. Cell. Physiol. 295, C414–C422. doi: 10.1152/ajpcell.00038.2008
Irlbeck, M., Muhling, O., Iwai, T., and Zimmer, H. G. (1996). Different response of the rat left and right heart to norepinephrine. Cardiovasc. Res. 31, 157–162. doi: 10.1016/0008-6363(95)00188-3
Kasanuki, H., Ohnishi, S., Ohtuka, M., Matsuda, N., Nirei, T., Isogai, R., et al. (1997). Idiopathic ventricular fibrillation induced with vagal activity in patients without obvious heart disease. Circulation 95, 2277–2285. doi: 10.1161/01.cir.95.9.2277
Kelly, A., Salerno, S., Connolly, A., Bishop, M., Charpentier, F., Stolen, T., et al. (2018). Normal interventricular differences in tissue architecture underlie right ventricular susceptibility to conduction abnormalities in a mouse model of Brugada syndrome. Cardiovasc. Res. 114, 724–736. doi: 10.1093/cvr/cvx244
Koschinski, A., and Zaccolo, M. (2017). Activation of PKA in cell requires higher concentration of cAMP than in vitro: implications for compartmentalization of cAMP signalling. Sci. Rep. 7:14090. doi: 10.1038/s41598-017-13021-y
Koumi, S., Backer, C. L., Arentzen, C. E., and Sato, R. (1995a). beta-Adrenergic modulation of the inwardly rectifying potassium channel in isolated human ventricular myocytes. Alteration in channel response to beta-adrenergic stimulation in failing human hearts. J. Clin. Invest. 96, 2870–2881. doi: 10.1172/JCI118358
Koumi, S., Wasserstrom, J. A., and Ten Eick, R. E. (1995b). Beta-adrenergic and cholinergic modulation of inward rectifier K+ channel function and phosphorylation in guinea-pig ventricle. J. Physiol. 486(Pt 3), 661–678. doi: 10.1113/jphysiol.1995.sp020842
Koumi, S., Wasserstrom, J. A., and Ten Eick, R. E. (1995c). beta-adrenergic and cholinergic modulation of the inwardly rectifying K+ current in guinea-pig ventricular myocytes. J. Physiol. 486(Pt 3), 647–659. doi: 10.1113/jphysiol.1995.sp020841
Lang, D., Holzem, K., Kang, C., Xiao, M., Hwang, H. J., Ewald, G. A., et al. (2015). Arrhythmogenic remodeling of beta2 versus beta1 adrenergic signaling in the human failing heart. Circ. Arrhythm. Electrophysiol. 8, 409–419. doi: 10.1161/CIRCEP.114.002065
Lu, T., Lee, H. C., Kabat, J. A., and Shibata, E. F. (1999). Modulation of rat cardiac sodium channel by the stimulatory G protein alpha subunit. J. Physiol. 518(Pt 2), 371–384. doi: 10.1111/j.1469-7793.1999.0371p.x
Luss, H., Klein-Wiele, O., Boknik, P., Herzig, S., Knapp, J., Linck, B., et al. (2000). Regional expression of protein phosphatase type 1 and 2A catalytic subunit isoforms in the human heart. J. Mol. Cell. Cardiol. 32, 2349–2359. doi: 10.1006/jmcc.2000.1265
Macfarlane, P. W., McLaughlin, S. C., Devine, B., and Yang, T. F. (1994). Effects of age, sex, and race on ECG interval measurements. J. Electrocardiol. 27(Suppl.), 14–19. doi: 10.1016/s0022-0736(94)80039-1
Maier, L. S., and Bers, D. M. (2002). Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J. Mol. Cell. Cardiol. 34, 919–939. doi: 10.1006/jmcc.2002.2038
Maltsev, V. A., Reznikov, V., Undrovinas, N. A., Sabbah, H. N., and Undrovinas, A. (2008). Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 294, H1597–H1608. doi: 10.1152/ajpheart.00484.2007
Marionneau, C., and Abriel, H. (2015). Regulation of the cardiac Na+ channel NaV1.5 by post-translational modifications. J. Mol. Cell. Cardiol. 82, 36–47. doi: 10.1016/j.yjmcc.2015.02.013
Marionneau, C., Lichti, C. F., Lindenbaum, P., Charpentier, F., Nerbonne, J. M., Townsend, R. R., et al. (2012). Mass spectrometry-based identification of native cardiac Nav1.5 channel alpha subunit phosphorylation sites. J. Proteome Res. 11, 5994–6007. doi: 10.1021/pr300702c
Matsuda, J. J., Lee, H., and Shibata, E. F. (1992). Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circ. Res. 70, 199–207. doi: 10.1161/01.res.70.1.199
Matsuo, K., Kurita, T., Inagaki, M., Kakishita, M., Aihara, N., Shimizu, W., et al. (1999). The circadian pattern of the development of ventricular fibrillation in patients with Brugada syndrome. Eur. Heart J. 20, 465–470. doi: 10.1053/euhj.1998.1332
Mattiazzi, A., and Kranias, E. G. (2014). The role of CaMKII regulation of phospholamban activity in heart disease. Front. Pharmacol. 5:5. doi: 10.3389/fphar.2014.00005
Molina, C. E., Johnson, D. M., Mehel, H., Spatjens, R. L., Mika, D., Algalarrondo, V., et al. (2014). Interventricular differences in beta-adrenergic responses in the canine heart: role of phosphodiesterases. J. Am. Heart Assoc. 3:e000858. doi: 10.1161/JAHA.114.000858
Mongillo, M., McSorley, T., Evellin, S., Sood, A., Lissandron, V., Terrin, A., et al. (2004). Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ. Res. 95, 67–75. doi: 10.1161/01.RES.0000134629.84732.11
Moreno, A. P., Fishman, G. I., and Spray, D. C. (1992). Phosphorylation shifts unitary conductance and modifies voltage dependent kinetics of human connexin43 gap junction channels. Biophys. J. 62, 51–53. doi: 10.1016/S0006-3495(92)81775-7
Moreno, A. P., Saez, J. C., Fishman, G. I., and Spray, D. C. (1994). Human connexin43 gap junction channels. Regulation of unitary conductances by phosphorylation. Circ. Res. 74, 1050–1057. doi: 10.1161/01.res.74.6.1050
Munger, T. M., Johnson, S. B., and Packer, D. L. (1994). Voltage dependence of beta-adrenergic modulation of conduction in the canine Purkinje fiber. Circ. Res. 75, 511–519. doi: 10.1161/01.res.75.3.511
Murphy, B. J., Rogers, J., Perdichizzi, A. P., Colvin, A. A., and Catterall, W. A. (1996). cAMP-dependent phosphorylation of two sites in the alpha subunit of the cardiac sodium channel. J. Biol. Chem. 271, 28837–28843. doi: 10.1074/jbc.271.46.28837
Nakagawa, M., Iwao, T., Ishida, S., Yonemochi, H., Fujino, T., Saikawa, T., et al. (1998). Circadian rhythm of the signal averaged electrocardiogram and its relation to heart rate variability in healthy subjects. Heart 79, 493–496. doi: 10.1136/hrt.79.5.493
Nattel, S., and Jing, W. (1989). Rate-dependent changes in intraventricular conduction produced by procainamide in anesthetized dogs. A quantitative analysis based on the relation between phase 0 inward current and conduction velocity. Circ. Res. 65, 1485–1498. doi: 10.1161/01.res.65.6.1485
Okin, P. M., Roman, M. J., Devereux, R. B., and Kligfield, P. (1995). Gender differences and the electrocardiogram in left ventricular hypertrophy. Hypertension 25, 242–249. doi: 10.1161/01.hyp.25.2.242
Ono, K., Fozzard, H. A., and Hanck, D. A. (1993). Mechanism of cAMP-dependent modulation of cardiac sodium channel current kinetics. Circ. Res. 72, 807–815. doi: 10.1161/01.res.72.4.807
Ono, K., Kiyosue, T., and Arita, M. (1989). Isoproterenol, DBcAMP, and forskolin inhibit cardiac sodium current. Am. J. Physiol. 256(6 Pt 1), C1131–C1137. doi: 10.1152/ajpcell.1989.256.6.C1131
Ou, B., Nakagawa, M., Kajimoto, M., Nobe, S., Ooie, T., Ichinose, M., et al. (2005). Heterogeneous expression of connexin 43 in the myocardium of rabbit right ventricular outflow tract. Life Sci. 77, 52–59. doi: 10.1016/j.lfs.2004.12.030
Papadatos, G. A., Wallerstein, P. M., Head, C. E., Ratcliff, R., Brady, P. A., Benndorf, K., et al. (2002). Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc. Natl. Acad. Sci. U.S.A. 99, 6210–6215. doi: 10.1073/pnas.082121299
Pilhall, M., Riha, M., and Jern, S. (1992). Exercise-induced QRS changes in healthy men and women: a multivariate analysis on their relation to background data and exercise performance. Eur. Heart J. 13, 1316–1324. doi: 10.1093/oxfordjournals.eurheartj.a060060
Postema, P. G., van Dessel, P. F., Kors, J. A., Linnenbank, A. C., van Herpen, G., Ritsema van Eck, H. J., et al. (2010). Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in brugada syndrome a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J. Am. Coll. Cardiol. 55, 789–797. doi: 10.1016/j.jacc.2009.11.033
Procida, K., Jorgensen, L., Schmitt, N., Delmar, M., Taffet, S. M., Holstein-Rathlou, N. H., et al. (2009). Phosphorylation of connexin43 on serine 306 regulates electrical coupling. Heart Rhythm 6, 1632–1638. doi: 10.1016/j.hrthm.2009.07.043
Remme, C. A., Scicluna, B. P., Verkerk, A. O., Amin, A. S., van Brunschot, S., Beekman, L., et al. (2009). Genetically determined differences in sodium current characteristics modulate conduction disease severity in mice with cardiac sodium channelopathy. Circ. Res. 104, 1283–1292. doi: 10.1161/CIRCRESAHA.109.194423
Remme, C. A., and Wilde, A. A. (2014). Targeting sodium channels in cardiac arrhythmia. Curr. Opin. Pharmacol. 15, 53–60. doi: 10.1016/j.coph.2013.11.014
Sato, P. Y., Chuprun, J. K., Schwartz, M., and Koch, W. J. (2015). The evolving impact of g protein-coupled receptor kinases in cardiac health and disease. Physiol. Rev. 95, 377–404. doi: 10.1152/physrev.00015.2014
Schubert, B., VanDongen, A. M., Kirsch, G. E., and Brown, A. M. (1989). Beta-adrenergic inhibition of cardiac sodium channels by dual G-protein pathways. Science 245, 516–519. doi: 10.1126/science.2547248
Shaw, R. M., and Rudy, Y. (1997). Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ. Res. 81, 727–741. doi: 10.1161/01.res.81.5.727
Siebens, A. A., Hoffman, B. F., Enson, Y., Farrell, J. E., and Brooks, C. M. (1953). Effects of l-epinephrine and l-nor-epinephrine on cardiac excitability. Am. J. Physiol. 175, 1–7. doi: 10.1152/ajplegacy.1953.175.1.1
Soni, S., Scholten, A., Vos, M. A., and van Veen, T. A. (2014). Anchored protein kinase A signalling in cardiac cellular electrophysiology. J. Cell. Mol. Med. 18, 2135–2146. doi: 10.1111/jcmm.12365
Swaminathan, P. D., Purohit, A., Hund, T. J., and Anderson, M. E. (2012). Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ. Res. 110, 1661–1677. doi: 10.1161/CIRCRESAHA.111.243956
Takanari, H., Bourgonje, V. J., Fontes, M. S., Raaijmakers, A. J., Driessen, H., Jansen, J. A., et al. (2016). Calmodulin/CaMKII inhibition improves intercellular communication and impulse propagation in the heart and is antiarrhythmic under conditions when fibrosis is absent. Cardiovasc. Res. 111, 410–421. doi: 10.1093/cvr/cvw173
Taylor, S. S., Zhang, P., Steichen, J. M., Keshwani, M. M., and Kornev, A. P. (2013). PKA: lessons learned after twenty years. Biochim. Biophys. Acta 1834, 1271–1278. doi: 10.1016/j.bbapap.2013.03.007
Terasaki, W. L., and Brooker, G. (1977). Cardiac adenosine 3′:5′-monophosphate. Free and bound forms in the isolated rat atrium. J. Biol. Chem. 252, 1041–1050.
Terentyev, D., and Hamilton, S. (2016). Regulation of sarcoplasmic reticulum Ca(2+) release by serine-threonine phosphatases in the heart. J. Mol. Cell. Cardiol. 101, 156–164. doi: 10.1016/j.yjmcc.2016.08.020
Veeraraghavan, R., Gourdie, R. G., and Poelzing, S. (2014). Mechanisms of cardiac conduction: a history of revisions. Am. J. Physiol. Heart Circ. Physiol. 306, H619–H627. doi: 10.1152/ajpheart.00760.2013
Veeraraghavan, R., and Poelzing, S. (2008). Mechanisms underlying increased right ventricular conduction sensitivity to flecainide challenge. Cardiovasc. Res. 77, 749–756. doi: 10.1093/cvr/cvm090
Veltmann, C., Schimpf, R., Echternach, C., Eckardt, L., Kuschyk, J., Streitner, F., et al. (2006). A prospective study on spontaneous fluctuations between diagnostic and non-diagnostic ECGs in Brugada syndrome: implications for correct phenotyping and risk stratification. Eur. Heart J. 27, 2544–2552. doi: 10.1093/eurheartj/ehl205
Vincent, K. P., McCulloch, A. D., and Edwards, A. G. (2014). Toward a hierarchy of mechanisms in CaMKII-mediated arrhythmia. Front. Pharmacol. 5:110. doi: 10.3389/fphar.2014.00110
Wagner, S., Dybkova, N., Rasenack, E. C., Jacobshagen, C., Fabritz, L., Kirchhof, P., et al. (2006). Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138. doi: 10.1172/JCI26620
Wagner, S., Hacker, E., Grandi, E., Weber, S. L., Dybkova, N., Sossalla, S., et al. (2009). Ca/calmodulin kinase II differentially modulates potassium currents. Circ. Arrhythm. Electrophysiol. 2, 285–294. doi: 10.1161/CIRCEP.108.842799
Warren, M., Sciuto, K. J., Taylor, T. G., Garg, V., Torres, N. S., Shibayama, J., et al. (2017). Blockade of CaMKII depresses conduction preferentially in the right ventricular outflow tract and promotes ischemic ventricular fibrillation in the rabbit heart. Am. J. Physiol. Heart Circ. Physiol. 312, H752–H767. doi: 10.1152/ajpheart.00347.2016
Warren, M., and Zaitsev, A. V. (2017). CaMKII blockade, cardiac conduction, and arrhythmia. Cardiovasc. Res. 113, 1798–1799. doi: 10.1093/cvr/cvx199
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004). Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2
Wu, Y., Valdivia, H. H., Wehrens, X. H., and Anderson, M. E. (2016). A single protein kinase a or calmodulin kinase II site does not control the cardiac pacemaker Ca2+ Clock. Circ. Arrhythm. Electrophysiol. 9:e003180. doi: 10.1161/CIRCEP.115.003180
Yin, Z., Jones, G. N., Towns, W. H. II, Zhang, X., Abel, E. D., Binkley, P. F., et al. (2008). Heart-specific ablation of Prkar1a causes failure of heart development and myxomagenesis. Circulation 117, 1414–1422. doi: 10.1161/CIRCULATIONAHA.107.759233
Yoon, J. Y., Ho, W. K., Kim, S. T., and Cho, H. (2009). Constitutive CaMKII activity regulates Na+ channel in rat ventricular myocytes. J. Mol. Cell. Cardiol. 47, 475–484. doi: 10.1016/j.yjmcc.2009.06.020
Zaitsev, A. V., Torres, N. S., Cawley, K. M., Sabry, A. D., Warren, J. S., and Warren, M. (2019). Conduction in the right and left ventricle is differentially regulated by protein kinases and phosphatases: implications for arrhythmogenesis. Am. J. Physiol. Heart Circ. Physiol. 316, H1507–H1527. doi: 10.1152/ajpheart.00660.2018
Zhang, J., Sacher, F., Hoffmayer, K., O’Hara, T., Strom, M., Cuculich, P., et al. (2015). Cardiac electrophysiological substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 131, 1950–1959. doi: 10.1161/CIRCULATIONAHA.114.013698
Zhang, J. Z., and MacLeod, K. M. (1996). Dissociation of negative inotropic effect of carbachol from changes in cAMP and PKA in perfused rat hearts. Am. J. Physiol. 271(2 Pt 2), H404–H409. doi: 10.1152/ajpheart.1996.271.2.H404
Zhang, T., Johnson, E. N., Gu, Y., Morissette, M. R., Sah, V. P., Gigena, M. S., et al. (2002). The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J. Biol. Chem. 277, 1261–1267. doi: 10.1074/jbc.M108525200
Zhang, T., Maier, L. S., Dalton, N. D., Miyamoto, S., Ross, J., Bers, D. M., et al. (2003). The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919. doi: 10.1161/01.RES.0000069686.31472.C5
Zhou, J., Shin, H. G., Yi, J., Shen, W., Williams, C. P., and Murray, K. T. (2002). Phosphorylation and putative ER retention signals are required for protein kinase A-mediated potentiation of cardiac sodium current. Circ. Res. 91, 540–546. doi: 10.1161/01.res.0000033598.00903.27
Keywords: right ventricle, right ventricle outflow tract, PKA, CaMKII, phosphatase, conduction velocity, ventricular fibrillation, Brugada syndrome
Citation: Zaitsev AV and Warren M (2020) “Heart Oddity”: Intrinsically Reduced Excitability in the Right Ventricle Requires Compensation by Regionally Specific Stress Kinase Function. Front. Physiol. 11:86. doi: 10.3389/fphys.2020.00086
Received: 14 October 2019; Accepted: 27 January 2020;
Published: 18 February 2020.
Edited by:
Carol Ann Remme, University of Amsterdam, NetherlandsReviewed by:
Bas J. Boukens, University of Amsterdam, NetherlandsSteve Poelzing, Virginia Tech, United States
Copyright © 2020 Zaitsev and Warren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Warren, bWFyay53YXJyZW5AdXRhaC5lZHU=