 Sara Siskind
Sara Siskind Fangming Zhang1,2
Fangming Zhang1,2 Max Brenner
Max Brenner Ping Wang
Ping Wang- 1Department of Surgery, Zucker School of Medicine at Hofstra/Northwell, Manhasset, NY, United States
- 2Center for Immunology and Inflammation, The Feinstein Institutes for Medical Research, Manhasset, NY, United States
- 3Elmezzi Graduate School of Medicine, Manhasset, NY, United States
Introduction: Acute kidney injury is associated with elevated serum levels of extracellular cold-inducible RNA-binding protein (eCIRP), a damage-associated molecular pattern released during ischemia/reperfusion injury, hemorrhagic shock, and sepsis. It is unknown if circulating eCIRP and eCIRP-induced activation of receptor triggering receptor expressed on myeloid cells-1 (TREM-1), expressed on endothelial cells, play an important role in the pathogenesis of AKI.
Methods: Male B6 wild-type (WT) and TREM-1−/− mice were subjected to intravenous injection of recombinant murine (rm) CIRP. Serum, urine, and renal tissue were collected 6 h later for analysis. Additionally, primary human renal glomerular endothelial cells (HRGEC) were stimulated in vitro with rmCIRP after pretreatment with M3, a novel inhibitory peptide of TREM-1, or vehicle. Supernatants and cells were collected 20 h after stimulation.
Results: After injection with rmCIRP, WT mice had a significant increase in serum levels of BUN, creatinine, and NGAL compared to control. Additionally, NGAL was significantly increased in the urine of rmCIRP-injected mice, suggesting that circulating eCIRP can directly induce AKI. The levels of TREM-1 mRNA in the kidneys, as well as soluble (s) TREM-1 released into the serum and urine, were significantly increased in rmCIRP-injected mice. TREM-1−/− mice injected with rmCIRP had attenuated AKI, indicated by significantly decreased serum BUN, creatinine, and NGAL, and renal mRNA expression of NGAL and KIM-1 compared to WT mice. TREM-1−/− mice also had attenuated endothelial activation, with decreased mRNA and protein expression of ICAM-1 in renal tissue. HRGEC stimulated with rmCIRP in vitro had significant increases in cytokine production and sTREM-1 release, which was attenuated in cells treated with M3.
Conclusion: Activation of renal TREM-1 with circulating eCIRP is sufficient to cause AKI. Elevated levels of eCIRP may be critical for the development of AKI under conditions such as ischemia/reperfusion injury, hemorrhagic shock, and sepsis. Mice deficient in the TREM-1 receptor have attenuated AKI and reduced endothelial cell activation after injection of rmCIRP. TREM-1 inhibition with M3 attenuates HRGEC activation after eCIRP stimulation. Targeting eCIRP activation of TREM-1 may provide a novel and effective treatment for AKI.
Introduction
Acute kidney injury (AKI) is a frequent complication among hospitalized patients, defined as a sudden decrease in kidney function over a period of hours to days (Coca et al., 2009; Rewa and Bagshaw, 2014; Makris and Spanou, 2016). This injury encompasses a wide range of physiologic impairments, from small changes in serum biochemical markers and decreased urine output to kidney failure requiring dialysis, and is associated with substantial short and long-term morbidity and mortality (Coca et al., 2009; Rewa and Bagshaw, 2014; Vijayan, 2021). AKI can result from a variety of insults to the kidney, including hypovolemia secondary to hemorrhage and sepsis, or renal ischemia/reperfusion (RIR) injury (Basile et al., 2012; Makris and Spanou, 2016). These etiologies share a common pathophysiology in that when blood flow to the kidney is decreased or absent, there is an insufficient oxygen and nutrients delivered to the tubular epithelial cells, causing damage and subsequent apoptosis or necrosis. These damaged cells release damage-associated molecular patterns (DAMPs) which activate the innate immune response and amplify cellular injury through the release of pro-inflammatory cytokines (Thurman, 2007; Bonventre and Yang, 2011; Basile et al., 2012; Peerapornratana et al., 2019). Regardless of the etiology, care for AKI remains supportive, with fluid administration and/or diuretics, avoidance of nephrotoxic agents, correcting the underlying etiology, and renal replacement therapy when necessary (Kaushal and Shah, 2014; Godin et al., 2015; Peerapornratana et al., 2019). There is currently no effective treatment aimed at reducing renal injury after an insulting event (Jo et al., 2007).
Cold inducible RNA binding protein (CIRP) is a cold shock protein that under normal conditions is located intracellularly and regulates protein expression (Qiang et al., 2013). During sepsis and hemorrhagic shock, CIRP is released into the extracellular space, acting as a DAMP and aggravating the inflammatory response (Qiang et al., 2013; Aziz et al., 2019). Extracellular CIRP (eCIRP) release has also been implicated in the progression of AKI following RIR injury through activation of triggering receptor expressed on myeloid cells-1 (TREM-1), a pattern recognition receptor (PRR) involved in the innate immune response (Gibot et al., 2004; Cen et al., 2016; Siskind et al., 2022). Both mice genetically deficient in TREM-1 and treated with M3, a novel peptide designed to inhibit the interaction of eCIRP with TREM-1, had attenuated renal injury after RIR (Denning et al., 2020a; Siskind et al., 2022). Recently, we found that eCIRP can independently induce a systemic inflammatory response akin to sepsis through activation of TREM-1, however, the resultant effects on kidney function have not been thoroughly explored (Denning et al., 2020a). Further characterization of the direct effects of eCIRP on the development of AKI via TREM-1 will improve our understanding of the pathophysiology of this disease and target this novel pathway for therapeutic development.
Both eCIRP and TREM-1 are upregulated in AKI, and blockade of TREM-1 attenuates AKI severity (Campanholle et al., 2013; Lo et al., 2014; Cen et al., 2016; Tammaro et al., 2016; Tammaro et al., 2017; Siskind et al., 2022), giving rise to the hypothesis that eCIRP directly induces renal inflammation via activation of TREM-1. In this study, we have demonstrated that circulating eCIRP can independently induce AKI through interaction with TREM-1. We further propose a mechanism of action by providing evidence that eCIRP activates TREM-1 expressed on renal endothelial cells, inducing endothelial cell activation and release of inflammatory and chemoattractant factors. Inhibition of renal glomerular endothelial cells with M3 protects cells from eCIRP-induced activation.
Materials and methods
Experimental animals
C57BL/6 male mice were purchased from Charles River Laboratories. The TREM-1−/− mice were generated by the trans-NIH Knockout Mouse Project (KOMP) and obtained from the KOMP Repository, University of California, Davis, lines maintained by in-house breeding. All experiments used age-matched mice at 8–12 weeks old. Only male mice were used in this study based on evidence that there are sex-specific differences in the immune response to sepsis (Angele et al., 2014). It has been reported that female sex hormones can be protective against the overwhelming inflammatory response in sepsis, and therefore males produce increased levels of inflammatory cytokines (Zellweger et al., 1997; Angele et al., 2014; Kuo, 2016). All experiments involving live animals were evaluated and approved by the Institutional Animal Care and Use Committee (IACUC) at the Feinstein Institutes for Medical Research.
In vivo administration of recombinant murine (rmCIRP)
WT and TREM-1−/− mice were anesthetized with inhaled isoflurane on a surgical plane and restrained in prone position. rmCIRP (produced in our lab as previously described) at a dose of 3 mg/kg BW (100 μL) was administered via retro-orbital injection using a 26G × 1/2″ needle on a 1 ml syringe (Qiang et al., 2013; Denning et al., 2020a). At 6 h after rmCIRP injection, mice were anesthetized, and blood, renal tissue, and urine were collected for analysis. The kidneys were frozen in liquid nitrogen and stored at −80°C for quantitative PCR and protein analysis.
Cell culture, stimulation, and treatment
Primary Human glomerular renal endothelial cells (HRGECs) were purchased from ScienCell Research Laboratories. The cells were maintained in complete endothelial cell medium (ScienCell) in fibronectin coated plates (Invitrogen, Thermo Fischer Scientific) at 37 °C in a 5% CO2 humidified incubator, per culturing instructions. All experiments were performed between passages 2 and 5. Cells were stimulated with 1 μg/ml of rmCIRP 30 min after pretreatment with 10 μg/ml of M3 (RGFFRGG; GenScript) or PBS vehicle. Supernatants from control and stimulated cells were collected at 20 h after stimulation, and cells were lysed for intracellular protein or mRNA analysis.
Determination of BUN and creatinine
Serum levels of BUN and creatinine were determined using specific colorimetric enzymatic assays according to manufacturer instructions (Pointe Scientific).
ELISA and cytokine array
Supernatant, serum, or urine protein was analyzed by ELISA kits specific for IL-6 (BD Biosciences), ICAM-1 (R&D Systems), NGAL, and TREM-1 (Invitrogen, Thermo Fischer Scientific) according to manufacturers’ instructions. HRGEC were dissolved in 100 μL of in-house prepared RIPA lysis buffer. The kidney tissue was crushed in liquid nitrogen and ∼100 mg of powdered tissue was dissolved in 500 μl of in-house prepared RIPA lysis buffer. All samples were sonicated on ice and protein concentration was determined by Bradford protein assay reagent (Bio-Rad). 40 μg of protein from HRGEC was used for assessment of IL-6 and 50 μg of kidney protein was used for assessment of ICAM-1 using respective ELISA kits. Supernatant from HRGEC was analyzed for 105 cytokines using a proteome profiler cytokine array (R&D Systems) per manufacturer’s instructions and resultant pixel density was calculated using ImageJ software.
Isolation of the mRNA and real-time quantitative reverse transcription PCR
Approximately 100 mg of kidney tissue powder were dissolved in lysis buffer from Illustra RNAspin Mini RNA Isolation kit (GE Healthcare) and sonicated on ice. RNA was extracted using the same kit according to manufacturer instructions and subsequently reversed-transcribed into cDNA using MLV reverse transcriptase (Applied Biosystems, Thermo Fisher Scientific). The PCR reaction was performed in a final volume of 24 μL containing 4 μg cDNA, 0.08 μmol of forward and reverse primers, 10 μL SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific), and 11 μL nuclease-free water. cDNA was amplified using a Step One Plus real-time PCR machine (Applied Biosystems, Thermo Fisher Scientific). Mouse β-actin was used for normalization and relative expression of mRNA was calculated using the ΔΔCT method. Results were reported as fold change in comparison with the control mice. Primer sequences are: β-actin: Forward: CGTGAAAAGATGACCCAGATCA, Reverse: TGGTACGACCAGAGGCATACAG, TREM-1: Forward: CTACAACCCGATCCCTACCC, Reverse: AAACCAGGCTCTTGCTGAGA, NGAL: Forward: CTCAGAACTTGATCCCTGCC, Reverse: TCCTTGAGGCCCAGAGACTT, KIM-1: Forward: TGCTGCTACTGCTCCTTGTG, Reverse: GGGCCACTGGTACTCATTCT, ICAM-1: Forward: GGGCTGGCATTGTTCTCTAA, Reverse: CTTCAGAGGCAGGAAACAGG.
Statistical analysis
Data represented in the figures are expressed as mean ± SEM. All data have been tested for normality using the Kolmogorov-Smirnov test of normality. Normally distributed data were analyzed using 1-way ANOVA for comparison among multiple groups, and the significance of differences between individual groups was determined by Tukey’s method. Student’s t-test was utilized for 2-group comparisons. The specific tests used for each graph are identified in the figure legends. All statistical analyses were conducted using Prism (GraphPad by Dotmatics), and the statistical significance threshold was set at p <0.05 between study groups.
Results
Circulating extracellular CIRP induces acute kidney injury and upregulates renal triggering receptor expressed on myeloid cells-1
To determine whether eCIRP is sufficient to induce AKI, we subjected WT mice to i. v. injection of rmCIRP and collected serum, urine, and kidney tissue 6 h later. Serum BUN was increased by 52% and creatinine was increased by 180% in mice injected with rmCIRP compared to control (Figures 1A,B). Additionally, NGAL protein expression, a biomarker for AKI (Soni et al., 2010), was increased by 389% in the serum and 136% in the urine compared to control (Figures 1C,D). We then evaluated if the eCIRP-induced AKI was associated with an increase in TREM-1 expression. After injection with rmCIRP, mice had a 10-fold increase in renal TREM-1 mRNA expression comparted to control (Figure 1E). Next, soluble TREM-1, the extracellular domain of TREM-1 that is cleaved and released into circulation during sepsis and sepsis-associated AKI and serves a marker of TREM-1 activation (Su et al., 2011; Jolly et al., 2021), was evaluated in the serum and urine. Mice with eCIRP-induced AKI had a 9.3-fold increase in serum sTREM-1 and an 11.3-fold increase in sTREM-1 excreted in urine (Figures 1F,G). These data indicate that eCIRP is sufficient to induce AKI, and that eCIRP-induced AKI is associated with upregulation and activation of TREM-1.
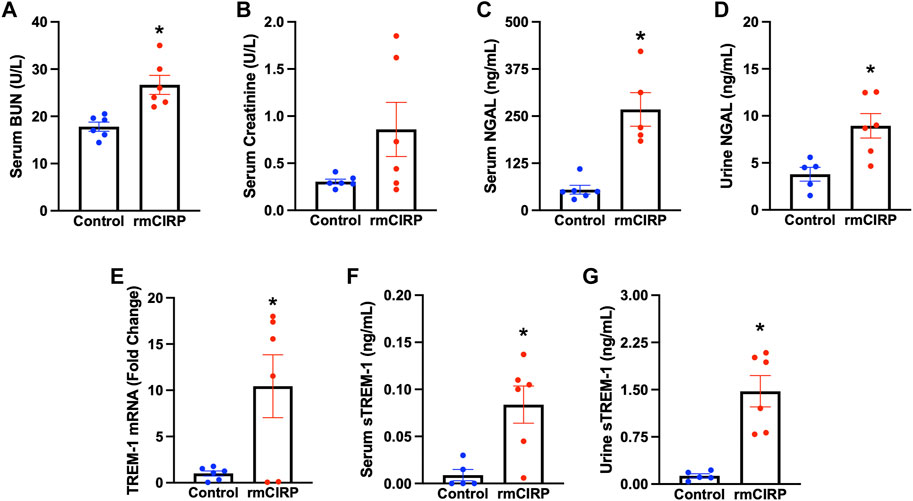
FIGURE 1. eCIRP induces AKI and TREM-1 activation. Adult C57BL/6 mice were randomly assigned to normal control or intravenous injection with recombinant murine (rm) CIRP. At 6 h after the injection, serum levels of (A) BUN and (B) creatinine (p = 0.08) were determined using specific colorimetric enzymatic assays, n = 6 mice per group. (C) Serum and (D) urine levels of NGAL were measured by ELISA, n = 5–6 mice per group. (E) Renal mRNA expression of TREM-1, and serum (F) and (G) urine levels of sTREM-1 were measured by RT-PCR and ELISA, respectively, n = 6 mice per group. All groups were compared using Student’s t-test. *p < 0.05 vs. control. Data are expressed as mean ± SEM.
Triggering receptor expressed on myeloid cells-1 deficiency attenuates extracellular CIRP-induced acute kidney injury
To further verify the role of TREM-1 in AKI, we evaluated if mice deficient in TREM-1 had attenuated AKI after injection with CIRP. TREM-1−/− mice and WT mice received the same dose of IV rmCIRP and collection took place 6 h later. Compared to WT mice that received CIRP, TREM-1−/− mice had a 33.6% reduction in serum BUN and a 55.4% reduction in serum creatinine (Figures 2A,B). Renal mRNA expression of KIM-1, another marker of AKI (Dase et al., 2022), was evaluated in the kidneys of TREM-1−/− mice and was noted to have a 47.1% decrease compared to WT mice (Figure 2C). Additionally, after rmCIRP injection, TREM-1−/− mice had a 41.6% reduction in renal tissue NGAL mRNA expression and a 38.3% decrease in serum NGAL compared to WT mice (Figures 2D,E). eCIRP administration did not result in measurable histological abnormalities (data not shown). These data demonstrate that mice deficient in TREM-1 are protected from eCIRP-induced AKI, and suggest that the eCIRP/TREM-1 pathway likely plays a role in the development of AKI after a proinflammatory insult.
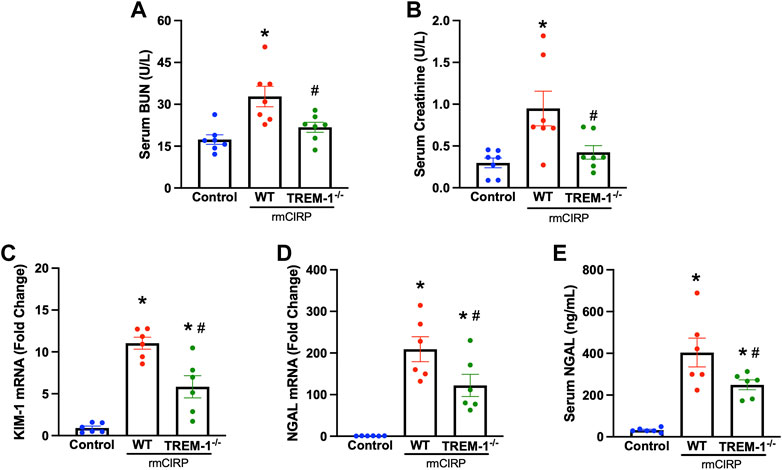
FIGURE 2. TREM-1 mediates eCIRP-induced AKI. Adult C57BL/6 and TREM-1−/− mice were randomly assigned to control or rmCIRP injection. At 6 h after injection, serum levels of (A) BUN and (B) creatinine were determined using specific colorimetric enzymatic assays. Renal mRNA expression of (C) KIM-1 and (D) NGAL were measured by RT-PCR, and (E) serum NGAL was determined using ELISA, n = 6-7 mice per group. All groups were compared by one-way ANOVA and Tukey’s method. *p < 0.05 vs. control, #p < 0.05 vs. WT. Data are expressed as mean ± SEM.
Extracellular CIRP activates renal endothelial cells via triggering receptor expressed on myeloid cells-1
TREM-1 activation on endothelial cells was recently discovered to play a role in the progression of sepsis (Jolly et al., 2018). To determine if endothelial activation within the kidneys during AKI is mediated through the interaction of TREM-1, TREM-1−/− mice and WT mice were injected with rmCIRP to induce AKI and the renal tissue was evaluated for ICAM-1 expression. The renal tissue of WT mice with CIRP-induced AKI had a 100-fold increase in ICAM-1 mRNA expression and a 2-fold increase in ICAM-1 protein compared to that of control. In TREM-1−/− mice, however, ICAM-1 mRNA and protein expression were reduced by 98% and 22%, respectively, compared to WT mice (Figures 3A,B).
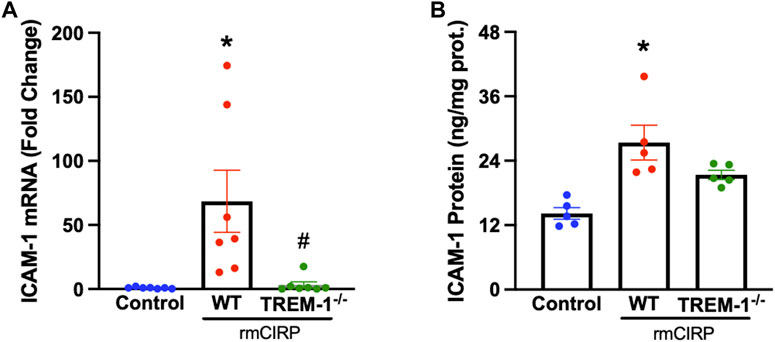
FIGURE 3. rmCIRP activates renal endothelial cells via TREM-1. Adult C57BL/6 and TREM-1−/− mice were randomly assigned to control or rmCIRP injection. At 6 h after injection, renal (A) mRNA and (B) protein levels of ICAM-1 were measured using RT-PCR and ELISA, respectively, n = 5–7 mice per group. All groups were compared by one-way ANOVA and Tukey’s method. *p < 0.05 vs. control, #p < 0.05 vs. WT. Data are expressed as mean ± SEM.
The novel triggering receptor expressed on myeloid cells-1 inhibitor M3 attenuates CIRP-induced renal endothelial cell activation
Activation of renal endothelial cells via the eCIRP/TREM-1 pathway was further studied using a cell line of human renal glomerular endothelial cells (HRGEC). Plated HRGEC were stimulated with rmCIRP, and a subset were pretreated with M3, an inhibitory peptide of TREM-1. Cells and supernatants were collected 20 h after stimulation. HRGEC stimulated with rmCIRP released 65.4% more IL-6 into the supernatant and had a 60.1% increase in intracellular IL-6 protein compared to untreated cells (Figures 4A,B). Additionally, rmCIRP induced a 4.3-fold increase in sTREM-1 release in HRGEC (Figures 4C). Incubation with M3 reduced both IL-6 protein excretion and intracellular expression in HRGEC by 58.7% and 25.6%, respectively (Figures 4A,B). Additionally, HRCEG treated with M3 had decreased activation of TREM-1, represented by a 77.7% reduction in sTREM-1 release compared to cells stimulated with CIRP alone (Figures 4C). CIRP activation of endothelial cells was further demonstrated using a cytokine array to analyze the supernatant of simulated HRGEC. Notably secretion of VCAM-1, proinflammatory cytokines including IL-2 and IL-6, immune cell chemoattractants including CXCL1, CXCL5, MCP-1, IL-8, and PDGF-AA, and proangiogenic factors including osteopontin and angiogenin were increased in cells stimulated with CIRP. Cells treated with M3 had reduced secretion of these factors (Figures 4D,E). These data suggest that eCIRP induces AKI through activation of TREM-1 expressed on renal endothelial cells.
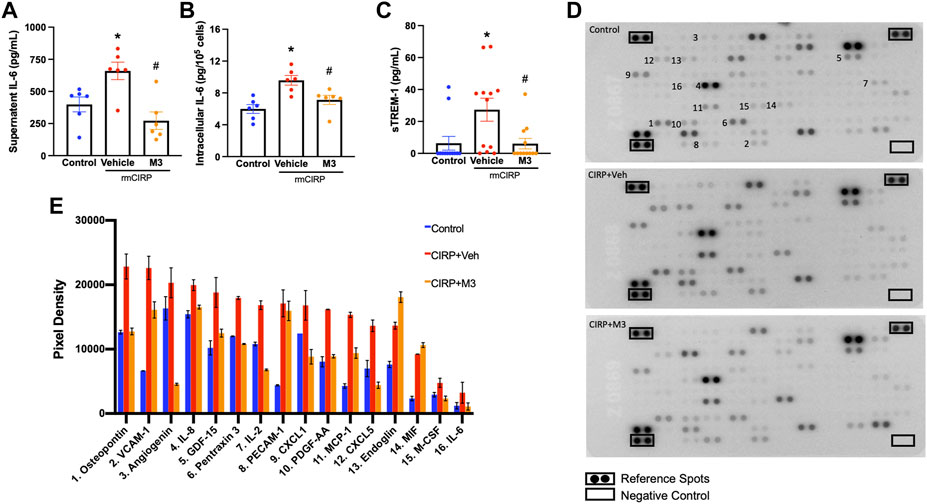
FIGURE 4. M3 attenuates rmCIRP-induced renal endothelial cell activation. HRGEC were treated with 10 μg/ml of M3 or PBS vehicle for 30 min, then stimulated with 1 μg/ml of rmCIRP. After 20 h, IL-6 protein in the (A) supernatant and (B) intracellularly was measured using ELISA. (C) sTREM-1 in the supernatant was measured using ELISA, n = 6–10 samples per group. (D) Relative secretion of 105 cytokines, chemokines, and growth factors in the supernatant were visualized in duplicate using a cytokine array and (E) pixel densities of upregulated factors were measured, n = 1 sample per group. All groups were compared by one-way ANOVA and Tukey’s method. *p < 0.05 vs. control, #p < 0.05 vs. vehicle. Data are expressed as mean ± SEM.
Discussion
Acute kidney injury encompasses a wide range of conditions leading to a sudden decrease in renal function, characterized by a decrease in glomerular filtration rate and urine output, and an increase in serum creatinine (Rewa and Bagshaw, 2014; Makris and Spanou, 2016; Levey and James, 2017; Peerapornratana et al., 2019). Although sepsis is one of the most common precipitators of AKI, its pathophysiology is multifactorial and there are currently no specific interventions for treatment (Makris and Spanou, 2016; Peerapornratana et al., 2019). During sepsis, in addition to decreased blood flow to the kidneys, there is also inflammation, microvascular dysfunction, and tubular cell injury, leading to the release of cytokines and DAMPs, further amplifying the inflammatory response (Makris and Spanou, 2016). eCIRP is a DAMP released during sepsis and is predictive of sepsis severity, associated AKI, and overall mortality (Qiang et al., 2013; Zhou et al., 2015; Zhang et al., 2018). eCIRP was recently explored as an endogenous ligand of TREM-1, capable of inducing a sepsis-like inflammatory response and propagating an inflammatory cascade in murine models of polymicrobial sepsis (Denning et al., 2020a; Denning et al., 2020b). TREM-1 activation has long been studied as a player in the dysregulated immune response in sepsis and, in its soluble form, is also predictive of sepsis severity, mortality, and development of sepsis-induced AKI (SA-AKI) (Derive and Gibot, 2011; Su et al., 2011; Brenner et al., 2017; Jedynak et al., 2018). The interplay of this ligand and receptor had never been evaluated as a potential mechanism for the development of AKI. In this study, we identified that circulating eCIRP can induce AKI via activation of TREM-1 by showing that mice genetically deficient in the TREM-1 receptor have attenuated renal dysfunction after intravenous administration of rmCIRP. We further demonstrate that eCIRP specifically activates renal endothelial cells via TREM-1, and using M3, a peptide that inhibits this interaction, decreased HRGEC activation in vitro. Our overall findings are summarized in Figure 5.
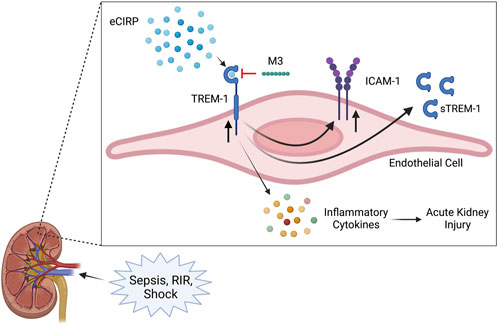
FIGURE 5. Summary of findings. Sepsis and renal ischemia/reperfusion cause an increased release of eCIRP. The circulating eCIRP interacts with TREM-1 on endothelial cells, leading to increased expression of inflammatory cytokines, upregulating TREM-1 and ICAM-1 expression, and releasing sTREM-1. This activation leads to excessive inflammation and renal tissue injury and decreasing renal function, resulting in AKI. M3, a novel inhibitory peptide of the eCIRP-TREM-1 pathway, attenuates the eCIRP-dependent activation of renal endothelial cells, leading to decreased inflammatory cytokine release and improved AKI. Created with BioRender.
We have previously demonstrated that intravenous administration of eCIRP is sufficient to induce systemic inflammation and lung injury (ALI) (Yang et al., 2016). However, mice injected with rmCIRP and treated with LP17, an inhibitory peptide of TREM-1, had significantly lower levels of serum cytokines, and decreased inflammation, lung tissue injury, and endothelial activation in the lungs (Gibot et al., 2004; Denning et al., 2020a). Furthermore, TREM-1−/− mice injected with rmCIRP also had decreased systemic and pulmonary inflammation (Denning et al., 2020a). We have also previously shown that inhibiting eCIRP in mice subjected to the cecal ligation and puncture (CLP) model of polymicrobial sepsis resulted in attenuated SA-AKI, as indicated by decreased serum BUN and creatinine, and NGAL and KIM-1 expression in the kidneys (Zhang et al., 2018). We therefore hypothesized that, analogous to ALI, injecting mice with rmCIRP would be sufficient to cause AKI. Indeed, we found that eCIRP induced AKI.
We also identified that TREM-1 mRNA expression was upregulated in the kidneys and sTREM-1 was increased in the serum and urine of mice with eCIRP-induced AKI, indicating that the eCIRP/TREM-1 pathway may be a novel mechanism for the development of AKI. To date, both serum and urine sTREM-1 have been implicated as diagnostic and predictive markers for SA-AKI in critically ill patients, providing further evidence of eCIRP’s role in the induction of AKI (Su et al., 2011; Dai et al., 2015). However, it remains unknown where the urinary sTREM-1 originates from (Derive and Gibot, 2011). The glomeruli in a functioning kidney freely filter proteins with a molecular weight (MW) less than 15 kDa and rapidly filter proteins up to 45 kDa (Schenk et al., 2008; Jia et al., 2009). Since sTREM-1 is a 27-kDA protein, it is possible that systemic sTREM-1 is rapidly filtered by the glomeruli and excreted by in the urine, reflecting remote production of the protein. It could also be produced locally in the kidneys by the infiltrating inflammatory cells or activated endothelial cells in response to cellular damage in AKI (Derive and Gibot, 2011; Su et al., 2015). Using a human primary cell line of glomerular endothelial cells, we have demonstrated that, when activated with CIRP, endothelial cells release sTREM-1. To our knowledge, this is the first piece of evidence demonstrating that renal endothelial cells release sTREM-1 and, therefore, could be the source of urinary sTREM-1 in AKI.
TREM-1 is expressed in many kidney cells, including renal mesangial cells (Zhao et al., 2018), tubular epithelial cells (Maiese et al., 2019; Pan et al., 2021), and kidney macrophages (Campanholle et al., 2013; Zhang et al., 2019a; Zhang et al., 2019b). TREM-1 is also expressed in endothelial cells (Gibot et al., 2019), although the role of TREM-1 in renal endothelial cells is not very well known. We chose to study renal endothelial cells in particular because during sepsis and hemorrhagic shock the blood levels of eCIRP are increased (Qiang et al., 2013; Zhou et al., 2015; Zhang et al., 2018). Once eCIRP is released into the circulation (which we have modelled by intravenous injection), endothelial cells are the first cells to have direct contact with the eCIRP in the blood. Indeed, we have previously demonstrated that intravenous administration of eCIRP induces systemic inflammation and lung injury (ALI) via endothelial cell activation (Yang et al., 2016).
Endothelial cells are one of the many affected cell types during SA-AKI. Immune cells, tubular epithelial cells (TECs), and endothelial cells all display PRRs, including Toll-like receptors (TLRs) and TREM-1, that are activated by DAMPs and pathogen associated molecular patterns (PAMPs), released from invading pathogens (Jolly et al., 2018; Gibot et al., 2019; Peerapornratana et al., 2019; Tammaro et al., 2019; Denning et al., 2020a). Endothelial cell activation promotes the adhesion of platelets, resulting in increased risk of thrombi formation and decreased flow, and transmigration of leukocytes, causing increased inflammation (Verma and Molitoris, 2015; Peerapornratana et al., 2019). Endothelial activation also leads to increased vascular permeability, causing interstitial edema and increasing the oxygen diffusion distance to the TECs, further aggravating the acute tubular injury (ATI) towards acute tubular necrosis (ATN) (Verma and Molitoris, 2015; Peerapornratana et al., 2019). eCIRP has been demonstrated to activate endothelial cells, increasing expression of cell-surface adhesion molecules of murine lung vascular endothelial cells (Yang et al., 2016). Additionally, septic mice treated with an eCIRP inhibitor had attenuated endothelial activation, demonstrated by decreased E-selectin and ICAM-1 expression in the lungs and kidneys (Zhang et al., 2018). Interestingly, TREM-1, which was originally identified with high expression on neutrophils, monocytes, and macrophages, has recently been identified on endothelial cells (Bouchon et al., 2000; Jolly et al., 2018). Mice deficient in endothelial TREM-1 or treated with a TREM-1 inhibitor were protected against endothelial dysfunction in sepsis (Jolly et al., 2018). Human pulmonary microvascular endothelial cells (HPMEC) stimulated with LPS in vitro had decreased activation and release of inflammatory cytokines after treatment with a TREM-1 inhibitor (Gibot et al., 2019). Further, maortas and mesenteric arteries isolated from TREM-1−/− mice were protected from vascular dysfunction after stimulation with neutrophil extracellular traps (NETs), nuclear DNA expelled from activated neutrophils that propagate the inflammatory response in sepsis (Czaikoski et al., 2016; Boufenzer et al., 2021). Both eCIRP and TREM-1 are upregulated during sepsis, leading to endothelial cell activation and dysfunction, but their interaction has not been studied in this cell type, specifically as a contributory factor to SA-AKI.
Our work continues to explore the role of endothelial cell TREM-1 activation, specifically in the progression of AKI by showing that mice deficient in TREM-1 have attenuated endothelial activation after CIRP-induced AKI. We also show that HRGEC stimulated with CIRP have increased secretion of proinflammatory cytokines including IL-2 and IL-6, immune cell chemoattractants including CXCL1, CXCL5, MCP-1, IL-8, and proangiogenic factors including osteopontin and angiogenin. When TREM-1 was inhibited on these cells using M3, secretion of these factors was reduced. These findings provide insight to a possible novel mechanism for AKI development. Similar to pro-inflammatory cytokines such as TNF-α and IL1-β, circulating eCIRP binds to TREM-1 and stimulates renal endothelial cells, increasing expression of VCAM-1 and ICAM-1 and chemokine release. These work in tandem to recruit and home monocytes and neutrophils to the activated endothelium, and induce chemotaxis (Reitsma et al., 2007; Deshmane et al., 2009; Furuichi et al., 2009; Jourde-Chiche et al., 2019). The influx of immune cells results in further inflammation and release of inflammatory cytokines, in addition from the ones released from the endothelial cells themselves (Verma and Molitoris, 2015; Jourde-Chiche et al., 2019). In response to endothelial cell damage, proangiogenic factors like angiogenin are released and play a critical role in tissue adaptation and recovery from the AKI (Kang et al., 2002; Li and Hu, 2012; Mami et al., 2016).
Although we have focused our investigation on eCIRP/TREM-1 activation of endothelial cells and their potential contribution to AKI, eCIRP may also promote AKI by acting on other renal cells known to express TREM-1 such as renal mesangial cells (Zhao et al., 2018), tubular epithelial cells (Maiese et al., 2019; Pan et al., 2021), and kidney macrophages (Campanholle et al., 2013; Zhang et al., 2019a; Zhang et al., 2019b). However, we have not explored the possible effects of eCIRP/TREM-1 activation of these cells. Neither have we directly injected eCIRP in the renal arteries to exclude the effects of other circulating factors that eCIRP might have induced elsewhere.
In summary, we have demonstrated that systemic eCIRP is sufficient to induce AKI, and that genetic depletion of TREM-1 exhibits protective effects against the development of AKI following CIRP injection. We additionally show that renal endothelial cells are activated in CIRP-induced AKI, and TREM-1 deletion or inhibition decreases this activation. These findings provide insight into a novel pathway of endothelial cell activation involved in AKI development. Future work should focus on further characterizing the effects of CIRP/TREM-1 pathway on renal endothelial cell activation using models for sepsis and exploring M3 as a treatment for SA-AKI.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by Institutional Animal Care and Use Committee (IACUC) at Feinstein Institutes for Medical Research.
Author contributions
SS and MB designed the experiments. SS and FZ performed the experiments. SS analyzed the data, prepared the figures, and wrote the manuscript. PW and MB revised and edited the manuscript. PW conceived the original idea and supervised the project.
Funding
This study was partially supported by the National Institutes of Health (NIH) grants R35GM118337, R01HL076179, and U01AI133655.
Acknowledgments
We thank Ezgi Sari of the Center for Immunology and Inflammation, Feinstein Institutes for Medical Research for her technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Angele M. K., Pratschke S., Hubbard W. J., Chaudry I. H. (2014). Gender differences in sepsis: Cardiovascular and immunological aspects. Virulence 5 (1), 12–19. doi:10.4161/viru.26982
Aziz M., Brenner M., Wang P. (2019). Extracellular CIRP (eCIRP) and inflammation. J. Leukoc. Biol. 106 (1), 133–146. doi:10.1002/JLB.3MIR1118-443R
Basile D. P., Anderson M. D., Sutton T. A. (2012). Pathophysiology of acute kidney injury. Compr. Physiol. 2 (2), 1303–1353. doi:10.1002/cphy.c110041
Bonventre J. V., Yang L. (2011). Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Invest. 121 (11), 4210–4221. doi:10.1172/JCI45161
Bouchon A., Dietrich J., Colonna M. (2000). Cutting edge: Inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J. Immunol. 164 (10), 4991–4995. doi:10.4049/jimmunol.164.10.4991
Boufenzer A., Carrasco K., Jolly L., Brustolin B., Di-Pillo E., Derive M., et al. (2021). Potentiation of NETs release is novel characteristic of TREM-1 activation and the pharmacological inhibition of TREM-1 could prevent from the deleterious consequences of NETs release in sepsis. Cell. Mol. Immunol. 18 (2), 452–460. doi:10.1038/s41423-020-00591-7
Brenner T., Uhle F., Fleming T., Wieland M., Schmoch T., Schmitt F., et al. (2017). Soluble TREM-1 as a diagnostic and prognostic biomarker in patients with septic shock: An observational clinical study. Biomarkers 22 (1), 63–69. doi:10.1080/1354750X.2016.1204005
Campanholle G., Mittelsteadt K., Nakagawa S., Kobayashi A., Lin S. L., Gharib S. A., et al. (2013). TLR-2/TLR-4 TREM-1 signaling pathway is dispensable in inflammatory myeloid cells during sterile kidney injury. PLoS One 8 (7), e68640. doi:10.1371/journal.pone.0068640
Cen C., Yang W. L., Yen H. T., Nicastro J. M., Coppa G. F., Wang P. (2016). Deficiency of cold-inducible ribonucleic acid-binding protein reduces renal injury after ischemia-reperfusion. Surgery 160 (2), 473–483. doi:10.1016/j.surg.2016.04.014
Coca S. G., Yusuf B., Shlipak M. G., Garg A. X., Parikh C. R. (2009). Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am. J. Kidney Dis. 53 (6), 961–973. doi:10.1053/j.ajkd.2008.11.034
Czaikoski P. G., Mota J. M., Nascimento D. C., Sonego F., Castanheira F. V., Melo P. H., et al. (2016). Neutrophil extracellular traps induce organ damage during experimental and clinical sepsis. PLoS One 11 (2), e0148142. doi:10.1371/journal.pone.0148142
Dai X., Zeng Z., Fu C., Zhang S., Cai Y., Chen Z. (2015). Diagnostic value of neutrophil gelatinase-associated lipocalin, cystatin C, and soluble triggering receptor expressed on myeloid cells-1 in critically ill patients with sepsis-associated acute kidney injury. Crit. Care 19, 223. doi:10.1186/s13054-015-0941-6
Dase J., Rasyid H., Masadah R., Cangara M. H., Bukhari A., Dwiyanti R., et al. (2022). Analysis of mRNA and protein kidney injury Molecule-1 (KIM-1) expression in a kidney model during the initiation phase of ischemia reperfusion injury. Ann. Med. Surg. 75, 103373. doi:10.1016/j.amsu.2022.103373
Denning N. L., Aziz M., Murao A., Gurien S. D., Ochani M., Prince J. M., et al. (2020). Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight 5 (5), 134172. doi:10.1172/jci.insight.134172
Denning N. L., Aziz M., Diao L., Prince J. M., Wang P. (2020). Targeting the eCIRP/TREM-1 interaction with a small molecule inhibitor improves cardiac dysfunction in neonatal sepsis. Mol. Med. 26 (1), 121. doi:10.1186/s10020-020-00243-6
Derive M., Gibot S. (2011). Urine sTREM-1 assessment in diagnosing sepsis and sepsis-related acute kidney injury. Crit. Care 15 (6), 1013. doi:10.1186/cc10533
Deshmane S. L., Kremlev S., Amini S., Sawaya B. E. (2009). Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 29 (6), 313–326. doi:10.1089/jir.2008.0027
Furuichi K., Kaneko S., Wada T. (2009). Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin. Exp. Nephrol. 13 (1), 9–14. doi:10.1007/s10157-008-0119-5
Gibot S., Kolopp-Sarda M. N., Bene M. C., Bollaert P. E., Lozniewski A., Mory F., et al. (2004). A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J. Exp. Med. 200 (11), 1419–1426. doi:10.1084/jem.20040708
Gibot S., Jolly L., Lemarie J., Carrasco K., Derive M., Boufenzer A. (2019). Triggering receptor expressed on myeloid cells-1 inhibitor targeted to endothelium decreases cell activation. Front. Immunol. 10, 2314. doi:10.3389/fimmu.2019.02314
Godin M., Murray P., Mehta R. L. (2015). Clinical approach to the patient with AKI and sepsis. Semin. Nephrol. 35 (1), 12–22. doi:10.1016/j.semnephrol.2015.01.003
Jedynak M., Siemiatkowski A., Mroczko B., Groblewska M., Milewski R., Szmitkowski M. (2018). Soluble TREM-1 serum level can early predict mortality of patients with sepsis, severe sepsis and septic shock. Arch. Immunol. Ther. Exp. 66 (4), 299–306. doi:10.1007/s00005-017-0499-x
Jia L., Zhang L., Shao C., Song E., Sun W., Li M., et al. (2009). An attempt to understand kidney's protein handling function by comparing plasma and urine proteomes. PLoS One 4 (4), e5146. doi:10.1371/journal.pone.0005146
Jo S. K., Rosner M. H., Okusa M. D. (2007). Pharmacologic treatment of acute kidney injury: Why drugs haven't worked and what is on the horizon. Clin. J. Am. Soc. Nephrol. 2 (2), 356–365. doi:10.2215/CJN.03280906
Jolly L., Carrasco K., Derive M., Lemarié J., Boufenzer A., Gibot S. (2018). Targeted endothelial gene deletion of triggering receptor expressed on myeloid cells-1 protects mice during septic shock. Cardiovasc. Res. 114 (6), 907–918. doi:10.1093/cvr/cvy018
Jolly L., Carrasco K., Salcedo-Magguilli M., Garaud J. J., Lambden S., van der Poll T., et al. (2021). sTREM-1 is a specific biomarker of TREM-1 pathway activation. Cell. Mol. Immunol. 18 (8), 2054–2056. doi:10.1038/s41423-021-00733-5
Jourde-Chiche N., Fakhouri F., Dou L., Bellien J., Burtey S., Frimat M., et al. (2019). Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 15 (2), 87–108. doi:10.1038/s41581-018-0098-z
Kang D. H., Kanellis J., Hugo C., Truong L., Anderson S., Kerjaschki D., et al. (2002). Role of the microvascular endothelium in progressive renal disease. J. Am. Soc. Nephrol. 13 (3), 806–816. doi:10.1681/ASN.V133806
Kaushal G. P., Shah S. V. (2014). Challenges and advances in the treatment of AKI. J. Am. Soc. Nephrol. 25 (5), 877–883. doi:10.1681/ASN.2013070780
Kuo S. M. (2016). Gender difference in bacteria endotoxin-induced inflammatory and anorexic responses. PLoS One 11 (9), e0162971. doi:10.1371/journal.pone.0162971
Levey A. S., James M. T. (2017). Acute kidney injury. Ann. Intern. Med. 167 (9), ITC66–ITC80. doi:10.7326/AITC201711070
Li S., Hu G. F. (2012). Emerging role of angiogenin in stress response and cell survival under adverse conditions. J. Cell. Physiol. 227 (7), 2822–2826. doi:10.1002/jcp.23051
Lo T. H., Tseng K. Y., Tsao W. S., Yang C. Y., Hsieh S. L., Chiu A. W., et al. (2014). TREM-1 regulates macrophage polarization in ureteral obstruction. Kidney Int. 86 (6), 1174–1186. doi:10.1038/ki.2014.205
Maiese A., Bolino G., Mastracchio A., Frati P., Fineschi V. (2019). An immunohistochemical study of the diagnostic value of TREM-1 as marker for fatal sepsis cases. Biotech. Histochem. 94 (3), 159–166. doi:10.1080/10520295.2018.1535138
Makris K., Spanou L. (2016). Acute kidney injury: Definition, pathophysiology and clinical phenotypes. Clin. Biochem. Rev. 37 (2), 85–98.
Mami I., Bouvier N., El Karoui K., Gallazzini M., Rabant M., Laurent-Puig P., et al. (2016). Angiogenin mediates cell-autonomous translational control under endoplasmic reticulum stress and attenuates kidney injury. J. Am. Soc. Nephrol. 27 (3), 863–876. doi:10.1681/ASN.2015020196
Pan P., Liu X., Wu L., Li X., Wang K., Wang X., et al. (2021). TREM-1 promoted apoptosis and inhibited autophagy in LPS-treated HK-2 cells through the NF-κB pathway. Int. J. Med. Sci. 18 (1), 8–17. doi:10.7150/ijms.50893
Peerapornratana S., Manrique-Caballero C. L., Gómez H., Kellum J. A. (2019). Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 96 (5), 1083–1099. doi:10.1016/j.kint.2019.05.026
Qiang X., Yang W. L., Wu R., Zhou M., Jacob A., Dong W., et al. (2013). Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 19 (11), 1489–1495. doi:10.1038/nm.3368
Reitsma S., Slaaf D. W., Vink H., van Zandvoort M. A., oude Egbrink M. G. (2007). The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 454 (3), 345–359. doi:10.1007/s00424-007-0212-8
Rewa O., Bagshaw S. M. (2014). Acute kidney injury-epidemiology, outcomes and economics. Nat. Rev. Nephrol. 10 (4), 193–207. doi:10.1038/nrneph.2013.282
Schenk S., Schoenhals G. J., de Souza G., Mann M. (2008). A high confidence, manually validated human blood plasma protein reference set. BMC Med. Genomics 1, 41. doi:10.1186/1755-8794-1-41
Siskind S., Royster W., Brenner M., Wang P. (2022). A novel eCIRP/TREM-1 pathway inhibitor attenuates acute kidney injury. Surgery 172, 639–647. doi:10.1016/j.surg.2022.02.003
Soni S. S., Cruz D., Bobek I., Chionh C. Y., Nalesso F., Lentini P., et al. (2010). Ngal: A biomarker of acute kidney injury and other systemic conditions. Int. Urol. Nephrol. 42 (1), 141–150. doi:10.1007/s11255-009-9608-z
Su L. X., Feng L., Zhang J., Xiao Y. J., Jia Y. H., Yan P., et al. (2011). Diagnostic value of urine sTREM-1 for sepsis and relevant acute kidney injuries: A prospective study. Crit. Care 15 (5), R250. doi:10.1186/cc10508
Su L., Xie L., Liu D. (2015). Urine sTREM-1 may be a valuable biomarker in diagnosis and prognosis of sepsis-associated acute kidney injury. Crit. Care 19, 281. doi:10.1186/s13054-015-0998-2
Tammaro A., Kers J., Emal D., Stroo I., Teske G. J. D., Butter L. M., et al. (2016). Effect of TREM-1 blockade and single nucleotide variants in experimental renal injury and kidney transplantation. Sci. Rep. 6, 38275. doi:10.1038/srep38275
Tammaro A., Derive M., Gibot S., Leemans J. C., Florquin S., Dessing M. C. (2017). TREM-1 and its potential ligands in non-infectious diseases: From biology to clinical perspectives. Pharmacol. Ther. 177, 81–95. doi:10.1016/j.pharmthera.2017.02.043
Tammaro A., Scantlebery A. M. L., Rampanelli E., Borrelli C., Claessen N., Butter L. M., et al. (2019). TREM1/3 deficiency impairs tissue repair after acute kidney injury and mitochondrial metabolic flexibility in tubular epithelial cells. Front. Immunol. 10, 1469. doi:10.3389/fimmu.2019.01469
Thurman J. M. (2007). Triggers of inflammation after renal ischemia/reperfusion. Clin. Immunol. 123 (1), 7–13. doi:10.1016/j.clim.2006.09.008
Verma S. K., Molitoris B. A. (2015). Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin. Nephrol. 35 (1), 96–107. doi:10.1016/j.semnephrol.2015.01.010
Vijayan A. (2021). Tackling AKI: Prevention, timing of dialysis and follow-up. Nat. Rev. Nephrol. 17 (2), 87–88. doi:10.1038/s41581-020-00390-3
Yang W. L., Sharma A., Wang Z., Li Z., Fan J., Wang P. (2016). Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci. Rep. 6, 26571. doi:10.1038/srep26571
Zellweger R., Wichmann M. W., Ayala A., Stein S., DeMaso C. M., Chaudry I. H. (1997). Females in proestrus state maintain splenic immune functions and tolerate sepsis better than males. Crit. Care Med. 25 (1), 106–110. doi:10.1097/00003246-199701000-00021
Zhang F., Brenner M., Yang W. L., Wang P. (2018). A cold-inducible RNA-binding protein (CIRP)-derived peptide attenuates inflammation and organ injury in septic mice. Sci. Rep. 8 (1), 3052–3062. doi:10.1038/s41598-017-13139-z
Zhang X., Yang Y., Zhao Y. (2019). Macrophage phenotype and its relationship with renal function in human diabetic nephropathy. PLoS One 14 (9), e0221991. doi:10.1371/journal.pone.0221991
Zhang X., Zhao Y., Zhu X., Guo Y., Yang Y., Jiang Y., et al. (2019). Active vitamin D regulates macrophage M1/M2 phenotypes via the STAT-1-TREM-1 pathway in diabetic nephropathy. J. Cell. Physiol. 234 (5), 6917–6926. doi:10.1002/jcp.27450
Zhao Y. F., Zhu L., Liu L. J., Shi S. F., Lv J. C., Zhang H. (2018). TREM-1 Contributes to inflammation in IgA nephropathy. Kidney Dis. 4 (1), 29–36. doi:10.1159/000485622
Keywords: acute kidney injury, inflammation, TREM-1, eCIRP, DAMP, endothelial cell
Citation: Siskind S, Zhang F, Brenner M and Wang P (2022) Extracellular CIRP induces acute kidney injury via endothelial TREM-1. Front. Physiol. 13:954815. doi: 10.3389/fphys.2022.954815
Received: 31 May 2022; Accepted: 12 September 2022;
Published: 29 September 2022.
Edited by:
Zhanjun Jia, Nanjing Medical University, ChinaCopyright © 2022 Siskind, Zhang, Brenner and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Wang, cHdhbmdAbm9ydGh3ZWxsLmVkdQ==; Max Brenner, bWJyZW5uZXJAbm9ydGh3ZWxsLmVkdQ==
†These authors have contributed equally to this work and share senior authorship