 Baojian Xue
Baojian Xue Alan Kim Johnson
Alan Kim Johnson Alexander G. Bassuk1,3,4*
Alexander G. Bassuk1,3,4*- 1Stead Family Department of Pediatrics, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa, IA, United States
- 2Department of Psychological and Brain Sciences, College of Liberal Arts and Sciences, University of Iowa, Iowa, IA, United States
- 3The Iowa Neuroscience Institute, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa, IA, United States
- 4The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa, IA, United States
Studies have demonstrated that there are sex differences in the timing of onset and severity of prenatally programmed hypertension, with consistently milder phenotypes observed in females relative to male offspring. However, the root cause(s) for these sex-specific effects is unknown. Activation of the renin-angiotensin system (RAS), elevated oxidative stress and inflammation, and sympathetic hyperactivity in the cardiovascular organs and cardiovascular regulatory systems, are all involved in the pathogenesis of hypertension. Sex hormones interact with these prohypertensive systems to modulate blood pressure, and this interaction may lead to a sex-specific development of programmed hypertension. A more complete understanding of the functional capabilities of the sex hormones and their interactions with prohypertensive factors in offspring, from early life to aging, would likely lead to new insights into the basis of sex differences in programmed hypertension. Recently, we have discovered that sex differences also occur in the sensitization of offspring hypertension as programmed by maternal gestational hypertension and that this requires the brain RAS and proinflammatory factors. In this review, we will discuss the possible mechanisms underlying sex differences in sensitization to hypertension in the offspring of mothers exposed to various prenatal insults. These mechanisms operate at various levels from the periphery to the central nervous system (e.g., blood vessel, heart, kidney, and brain). Understanding the sex-specific mechanisms responsible for the sensitized state in offspring can help to develop therapeutic strategies for interrupting the vicious cycle of transgenerational hypertension and for treating hypertension in men and women differentially to maximize efficacy.
Introduction
The Developmental Origin of Health and Disease (DOHaD) theory (i.e., Barker hypothesis) was first proposed by David Barker, who had discovered the association between lower birth weight and higher mortality from ischemic heart disease (Barker et al., 1989). Subsequently, mounting human epidemiological evidence and animal studies indicated a link between in utero adverse stimuli during gestation and an increased risk of hypertension later in life (Arima and Fukuoka, 2020; Falkner et al., 2024; Warrington et al., 2024). The sex differences of prenatally programmed hypertension in offspring have been reported in many different animal models inducing hypertension through a variety of drivers, such as hypertensive disorders during pregnancy, nutritional deficits/excess, uteroplacental perfusion insufficiency, glucocorticoids, nicotine, testosterone treatment or hypoxia during pregnancy (Tomat and Salazar, 2014; Dasinger and Alexander, 2016; Dasinger et al., 2016; Xue et al., 2018; Xue et al., 2021; Xue et al., 2022a; Drury et al., 2024; Darlas et al., 2025). Most of these studies observed that male offspring develop hypertension, whereas female offspring seem to be protected, suggesting a role for the sex hormones such as testosterone and estrogen in modulating the long-term changes in blood pressure (BP) in response to these prenatal insults (Tomat and Salazar, 2014; Dasinger and Alexander, 2016; Drury et al., 2024). The etiology of prenatally programmed hypertension is multifactorial and may include reduced nephron number, endothelial dysfunction, activation of the sympathetic nervous system (SNS) and the renin-angiotensin system (RAS), and elevated oxidative stress and inflammation as well as epigenetic mechanisms (Tomat and Salazar, 2014; Dasinger and Alexander, 2016; Dasinger et al., 2016; Johnson and Xue, 2018; Liu and Liang, 2019; Xue and Johnson, 2023; Drury et al., 2024; Tain and Hsu, 2024). Sex hormones interacting with these prohypertensive systems in male and female offspring from early life to aging may reinforce the formation of the sex differences seen in programmed hypertension (Tomat and Salazar, 2014; Dasinger and Alexander, 2016; Dasinger et al., 2016; Drury et al., 2024).
By using an Induction-Delay-Expression paradigm, we demonstrated that the pretreatment with the non-pressor dose of angiotensin (ANG) II given during Induction (1 week) produced a significantly greater pressor response to the subsequent infusion of slow-pressor doses of ANG II during Expression (2 weeks) when compared to rats pretreated with saline during Induction. This differential pressor response is referred to as hypertensive response sensitization (HTRS) (Xue et al., 2012). Thereafter, we have used a prenatal insult model to investigate the pathogenesis of hypertension in offspring; we demonstrated similar sensitizing effects of maternal gestational hypertension on the development of hypertension in offspring when they are challenged as adults (10 weeks of age) with ANG II or a high fat diet (HFD), as “second hits” (Xue et al., 2017; Xue et al., 2021; Xue et al., 2022a). Sex differences in the effects of prenatal insult to the mother on sensitized hypertension expressed in adult offspring were evident (Xue et al., 2018; Xue et al., 2021; Xue et al., 2022a).
In this review by way of background of our and other studies, we will review sex differences in the sensitizing effects of various prenatal insults to development of hypertension in the offspring. In particular, we will discuss the possible mechanisms of action underlying these sex differences at several levels, ranging from peripheral tissues to the central nervous system (CNS), including blood vessel, kidney, heart, and brain.
Central nervous system mechanisms
Excessive SNS activation, autonomic dysfunction, activation of the RAS and increased inflammation in the CNS have been associated with development of hypertension (DiBona, 2013; Johnson and Xue, 2018; Xue et al., 2020). Numerous clinical and experimental studies have shown that various prenatal insults can enhance the impact of these central prohypertensive systems. In other words, such prenatal insults can create a sensitized state in the CNS that facilitates the development of prenatally programmed hypertension in offspring later in life (Jones et al., 2007; Tomat and Salazar, 2014; Johnson and Xue, 2018; Zhang et al., 2018; Lim et al., 2021).
Adult obesity is a major risk factor for hypertension, with increased SNS activity serving as the link between the increased adiposity and the elevated BP (Da Silva et al., 2013). For example, leptin is an adipokine that elevates BP via central activation of the SNS, serving as a key mediator for obesity-related hypertension (Da Silva et al., 2013; Taylor et al., 2014). Likewise, it has been established that HFD-induced maternal obesity leads to hypertension in the offspring that is also of sympathetic origin. This was demonstrated by the elevated cardiovascular stress responses to restraint, the enhanced pressor effects of a leptin challenge, the increased sympathetic component of heart rate variability and reduced baroreflex sensitivity in offspring (Prior et al., 2014; Taylor et al., 2014; Gemici et al., 2021).
Perhaps not surprisingly, in animal models, the hypertension in the adult arises from a direct influence of maternal obesity/HFD on the development of BP regulatory pathways in the fetus (Samuelsson et al., 2010). Samuelsson and colleagues support this notion by demonstrating not only that HFD-induced maternal obesity results in a significant increase in BP accompanied by elevated SNS activity in rat offspring as adults, but also that these two effects are already established in the juvenile offspring of obese dams (at 30 days of age). This indicates that the sympathetic overactivation and hypertension arise as a direct consequence of in utero exposure to maternal obesity (Samuelsson et al., 2010). Furthermore, there are marked changes in anxiety and spatial learning in offspring from obese dams, and these effects are all observed in adulthood, even after the pups are placed on standard chow at weaning, confirming that these outcomes are programmed early in life (Bilbo and Tsang, 2010). Finally, offspring exposed to maternal obesity/HFD show sex-specific changes in metabolic, behavioral, and BP regulation involving activation of SNS and RAS, hyperleptinemia and increased neuroinflammation that persist into adulthood (Samuelsson et al., 2016; Gemici et al., 2021; do Carmo et al., 2023).
Leptin, acting on melanocortin four receptors (MC4R) in the paraventricular nucleus of hypothalamus (PVN), appears to be required for early-life programming of hypertension arising from either maternal obesity or neonatal hyperleptinemia (Samuelsson et al., 2016). Using transgenic technology to restore MC4R in the PVN of MC4R knockout mice, Samuelsson and colleagues reported that neonatal hyperleptinemia due to maternal obesity induces persistent changes in the central melanocortin system resulting in sympathetic hyperactivity, thereby contributing to offspring hypertension (Samuelsson et al., 2016). Furthermore, do Carmo et al. (2023) found that the male and female offspring of dams with maternal obesity were a greater risk for developing hypertension when the offspring were also kept on the same obesogenic diet, and crucially, found that offspring showed greater reduction in BP during MC4R blockade. However, only the male offspring from obese dams exhibited elevated BP when fed a normal diet and they showed significant BP reduction after adrenergic and ANG II type I receptor (AT1R) blockade. These results confirmed that brain leptin-mediated SNS hyperactivity contributes to the sensitized BP response in obese offspring from obese dams and indicated a sex difference in the mechanisms of BP sensitization in male and female offspring of obese dams (do Carmo et al., 2023).
Obesity is also characterized as a systemic inflammatory condition. Studies showed that microglial activation markers and expression of proinflammatory genes were basally increased in the brain nuclei of neonates from obese dams (Bilbo and Tsang, 2010; Zhang et al., 2018; Wang et al., 2020; Wijenayake et al., 2020). The PVN, amygdala, hippocampus, and prefrontal cortex were among the affected nuclei. Dudele et al. demonstrated that offspring exposed solely to maternal inflammation resemble those born to obese dams. The similarities between offspring subjected to maternal lipopolysaccharide (LPS) or a HFD provide experimental evidence that inflammation is likely to be a key programming factor in pregnancies produced by obesity/HFD (Dudele et al., 2017; Deng et al., 2018). Our studies also confirmed that maternal HFD modulates the brain RAS, oxidative stress, and proinflammatory cytokines (PICs) that alter the actions of ANG II and TNF-α and sensitize the ANG II-elicited hypertensive response in adult offspring. Systemic inhibition of the RAS and PICs can block maternal HFD-induced sensitization of ANG II hypertension, which is associated with attenuation of brain RAS and PIC expression in offspring (Zhang et al., 2018; Wang et al., 2020). Notably, male HFD offspring showed greater proinflammatory gene expression, whereas female HFD offspring exhibited increased anti-inflammatory gene expression in response to simultaneous cortisol and LPS administration. These findings suggest that exposure to maternal HFD leads to sex-specific changes that alter inflammatory responses in the brain (Dudele et al., 2017; Wijenayake et al., 2020). Blood-borne PICs induce a pressor response and sympathetic activation, operating on the brain cardiovascular nuclei to increase RAS activity and inflammation (Wei et al., 2013; Wei et al., 2015; Wei et al., 2018). Estrogen is anti-inflammatory at several levels, including immune cells, adipose tissue, and the brain (Shi et al., 2020). Considering these observations, it is likely that increased PICs in the brains of both male and female offspring induced by prenatal insults including maternal HFD/diabetes may be one of the origins of SNS activation. Since the enhanced brain proinflammatory response and activation of SNS programmed by prenatal insults are sex-specific, they eventually lead to a sex difference in the development of hypertension.
Age may exert a secondary impact on the development of programmed hypertension (Virdis et al., 2011). Intapad et al. showed that growth-restricted male offspring exhibit an increase in BP after puberty, whereas BP is normalized after puberty in growth-restricted female offspring (Alexander, 2003). However, BP was significantly elevated in growth-restricted female offspring by 12 months of age, which was accompanied by increased circulating leptin. This effect could be abolished by bilateral renal denervation. These data indicate that age induces increases in visceral fat and circulating leptin that result in a significant increase in BP in growth-restricted female offspring, with the renal nerves serving as an underlying mechanism (Intapad et al., 2013). Renal denervation also abolishes hypertension in growth-restricted male rats at 3 months of age, further suggesting an important role for activation of the SNS in the etiology of programmed hypertension and it age-dependent and sex-specific manifestations (Alexander et al., 2005). Consistent with the effect of aging on BP in the growth-restricted female offspring, maternal HFD renders the offspring metabolically imbalanced and impairs their ability to cope with a HFD when challenged during aging. The metabolic effects of HFD challenge were more profound in female offspring (Sadagurski et al., 2019) that exhibited more significant changes in blood-brain barrier (BBB) permeability and hypothalamic inflammation compared to male animals (Contu et al., 2019). Moreover, sex differences in hypothalamic estrogen receptor α (ER-α) expression levels were lost in female offspring upon HFD challenge, supporting a link between ER-α levels and hypothalamic inflammation in offspring. These studies highlight the programming potential of hypothalamic inflammatory responses, loss of BBB integrity and maternal obesity, especially in aging females (Contu et al., 2019; Sadagurski et al., 2019).
The RAS is a systemic hormonal regulator of vasoconstriction, aldosterone production, and SNS activity that directly regulates BP. The RAS exhibits autocrine and paracrine capabilities in numerous organs including the brain (Nakagawa et al., 2020). It has been demonstrated that the RAS is involved in prenatal programming of sympathetic overactivity and hypertension. For example, studies show that hypertension induced by maternal protein restriction is associated with an enhanced AT1R expression in several brain nuclei and that the intracerebroventricular administration of an AT1R blocker results in a significant reduction in BP (Pladys et al., 2004; Mizuno et al., 2014). Blockade of the RAS also abolishes hypertension in growth-restricted adult rats exposed to placental insufficiency (Ojeda et al., 2007; Ojeda et al., 2010). In our studies with the ANG II-induced maternal hypertension model, we found that adult male offspring exhibited upregulated expression of both RAS components as well as upregulation of PICs in the lamina terminalis (LT) and PVN. The LT and PVN are forebrain structures with key roles in sodium and water homeostasis and regulation of the cardiovascular system. These adult male offspring also displayed HTRS to a slow-pressor dose of ANG II or to a switch to a HFD post-weaning, when compared with the offspring of normotensive dams (Xue et al., 2017; Xue et al., 2021). A compromised BBB, elevated brain reactivity to pressor stimuli and augmented sympathetic drive to the cardiovascular system likely contributed to the HTRS (Xue et al., 2021). However, this maternal hypertension-induced HTRS was sex-specific, as intact female offspring exhibited an attenuated increase in BP when compared to male offspring (Xue et al., 2018; Xue et al., 2022a). Importantly, different central mechanisms were responsible for the sex differences in the HTRS. Leptin was involved in the expression of HTRS induced by both maternal hypertension and post-weaning HFD feeding in male offspring, but not in females (Xue et al., 2021; Xue et al., 2022a), while antihypertensive components of the RAS, such as angiotensin converting enzyme 2 and AT2R, play a protective role in antagonizing the expression of the HTRS in females (Xue et al., 2014; Xue et al., 2021; Xue and Johnson, 2023).
It is well established that sex hormones such as estrogen and testosterone, are involved in sex differences in the development of hypertension. In our previous study, we demonstrated that relative to males, females were protected against the induction of sensitized hypertension, whereas sensitized hypertensive response was enhanced in male and ovariectomized (OVX) female rats. Central administration of estrogen in either male or OVX female rats during induction blocked ANG II-induced sensitization of hypertension (Xue et al., 2014). Similarly, central Cytochrome P450 1B1 (CYP1B1)-estradiol metabolite, 2-Methoxyestradiol, protects from neuroinflammation and hypertension in female mice (Singh et al., 2020b). In contrast to the protective effects of female sex hormones against the development of hypertension, chronic dihydrotestosterone (DHT) treatment in female rats induced an increase in BP through activation of the SNS and hypothalamic MC4R (Maranon et al., 2015). Testosterone-CYP1B1-generated metabolite 6β-hydroxytestosterone, most likely in the PVN via androgen receptor and G protein-coupled receptor C6A, elicited an increased reactive oxygen species (ROS) production, activation of microglia and astrocyte, and elevated neuroinflammation, contributing to ANG II-induced hypertension in male mice (Singh et al., 2020a). There results indicate that sex hormones and their metabolites act through CNS to contribute to sex differences in the development of hypertension in adult animals. However, few studies have explored pathways and mechanisms through which these sex hormones and their metabolites regulate BP in offspring exposed to various prenatal insults, and further investigations are warranted in the future.
Collectively, the studies highlight the pathogenesis of elevated CNS activity programmed by prenatal insults, which involve the leptin/MC4R pathway, a compromised BBB, central activation of the RAS and increased inflammation, either alone or synergistically. These developmental origins of SNS hyperactivity eventually alter the offspring’s phenotype so as to sensitize their response to prohypertensive agents and/or HFD challenge (i.e., second hits), thereby developing hypertension in adulthood. Sex hormones and aging play an important role in this sensitization process resulting in sex-specific development of programmed hypertension.
Kidney mechanisms
Epidemiological and animal studies have shown that various prenatal insults all lead to low-birth-weight neonates with increased risk for chronic kidney disease and hypertension (Baum, 2010; Baum, 2018). The kidney dysfunction and elevation of BP that are caused by prenatal insults involve a low nephron endowment, dysregulation of the systemic and intrarenal RAS, increased renal sympathetic nerve activity, and increased tubular sodium transport (Moritz et al., 2009; Lankadeva et al., 2014; Singh and Denton, 2015). Thus, kidney dysfunction plays an important role in generating and maintaining prenatally programmed hypertension in humans and in animal models. Sex difference in the timing of onset and severity of hypertension after prenatal programming also may reflect, in part, sex-specific differences in kidney development and/or the different effects of sex hormones on renal function (Moritz et al., 2010; Dasinger and Alexander, 2016; Drury et al., 2024).
Since nephrogenesis in rodents is not completed until postnatal day 10, the kidney can be programmed not only by various intrauterine perturbations but also by neonatal insults (Baum, 2010; Yim and Yoo, 2015; Baum, 2018). Woods et al. reported that nephron number is reduced in male, but not in female, offspring of low protein dams, and that the formation of a low nephron endowment results in impaired renal function, which in turn contributes to the development of hypertension only in male offspring (Woods et al., 2010). Similarly, both male and female growth-restricted rat offspring have nephron deficits but only the males develop kidney dysfunction and hypertension (Doan et al., 2021). Maternal low-protein diet, neonatal overnutrition and glucocorticoid exposure have similar effects on the reduction of nephron number and cause similar alterations of glomerular morphology and renal cortical oxidative stress in offspring (McMullen and Langley-Evans, 2005; Pedroza et al., 2019; Alhamoud et al., 2021).
do Carmo et al. (2024) examined the impact of maternal obesity on offspring kidney function, morphology, and markers of kidney damage after acute kidney injury (AKI) induced by ischemia-reperfusion at 24–26 weeks of age. They found an increased mortality rate and worse kidney injury scores after AKI in male offspring from obese dams. Female offspring were protected from major kidney injury after AKI. These results indicate that maternal obesity predisposes offspring to kidney dysfunction that sensitizes responses to ischemia-reperfusion injury in a sex-dependent manner (do Carmo et al., 2024). Similarly, hypoxia during late pregnancy disrupted growth of the kidney, particularly the collecting duct network, in male neonates as early as postnatal day 7. By mid-late adulthood (4–12 months of age), these offspring developed early signs of kidney disease, notably a compromised response to water deprivation. Female offspring showed no obvious signs of impaired kidney development and did not develop kidney disease, suggesting an underlying protective mechanism against the hypoxia insult-induced kidney injury in females (Walton et al., 2018).
The RAS is critical for normal renal development (Moritz et al., 2010). In this regard, administration of an angiotensin-converting enzyme inhibitor or an AT1 receptor antagonist during renal development induces significant decreases in body weight and nephron endowment that lead to deterioration of renal function and a significant increase in BP (Saez et al., 2007; Gilbert and Nijland, 2008; Tomat and Salazar, 2014). McMullen and Langley-Evans found that prenatal low-protein and glucocorticoid exposure induce a similar reduction of nephron number. They also found that there are age- and sex-specific differences in the enhancing effects of these two prenatal conditions on postnatal angiotensin receptor expression (AT1R and AT2R) in the kidney, suggesting that upregulation of the RAS plays an important role in the pathogenesis of programmed hypertension through receptor-mediated changes in ANG II activity (McMullen and Langley-Evans, 2005). In a rat model of intrauterine growth restriction (IUGR) induced by placental insufficiency, BP at 4 months of age is increased in male but not female offspring with a normal glomerular filtration rate (GFR). However, in response to a second hit such as acute ANG II, the GFR is reduced in male, but not in female offspring with IUGR (Ojeda et al., 2010; Ojeda et al., 2013; Intapad et al., 2019). Similarly, administration of a postnatal HFD, as a second hit to prenatal dexamethasone, sex-specifically alters protein profiles in offspring kidneys and increases the vulnerability to prenatal-dexamethasone-exposure-induced programmed hypertension, but only in male offspring (Hsu et al., 2018). These data revealed a role for a sensitized renal response to second hits, such as postnatal acute ANG II or HFD, in the pathogenesis of prenatally programmed hypertension in male, but not female, offspring.
Sex hormones such as estrogen and testosterone interact with the RAS to modulate BP (Tomat and Salazar, 2014). In young male and aging female offspring programmed for hypertension by placental insufficiency, the enhanced responsiveness to acute ANG II is testosterone-dependent, and testosterone plays an important role in maintaining the hypertension through enhancement of intrarenal angiotensinogen (Ojeda et al., 2010; Davis et al., 2019a). In contrast, growth-restricted female offspring are normotensive in adulthood. OVX induces a marked increase in BP that is abolished by RAS blockade, and renal hemodynamic responses to acute ANG II are significantly enhanced in growth-restricted female offspring that had undergone OVX, suggesting that sensitivity to acute ANG II is modulated by ovarian hormones in growth-restricted female offspring (Ojeda et al., 2011; Davis et al., 2019b). In our ANG II-induced maternal hypertensive rat model, we also found sex differences in the maternal hypertension-induced sensitization of ANG II hypertension in offspring. Castration did not alter the hypertensive response to ANG II in male offspring, whereas OVX induced a greater increase in the pressor response to ANG II in female offspring of hypertensive dams compared with female offspring of normotensive dams. Furthermore, either RAS blockade or renal denervation abolished the maternal hypertension-induced sensitization of hypertension in offspring (Xue et al., 2017; Xue et al., 2018). It has been shown that estrogens promote the anti-hypertensive effects of the RAS by enhancing the Ang-(1–7)/AT2R pathway while diminishing the ACE/AT1R pathway (Colafella and Denton, 2018). Therefore, modulation of the renal RAS by estrogen or testosterone serves as a potential mechanism in mediating the sex-specific differences in hypertension in offspring programmed by prenatal insults.
In summary, various prenatal insults lead to a reduced number of nephrons, impaired renal development and function, and disturbance of the RAS activity in the kidney, which may fundamentally lead to a sensitized response to second hits (e.g., ANG II or HFD) that participate in both the development and the maintenance of prenatally programmed hypertension in offspring. Sex hormone interactions with the renal RAS play important roles in the sex-specific processes during the development of programmed hypertension.
Cardiovascular mechanisms
Alterations in the structure and function of vascular smooth muscle, endothelium, and the heart are involved in the development of prenatally programmed hypertension and of myocardial injury, in which nitric oxide (NO), ROS, RAS activity and β-adrenergic receptors play a mediating role (Tomat and Salazar, 2014). Sex differences in vascular and heart dysfunction in different animal models of prenatal programming are also observed, and sex hormones play a modulatory role in cardiovascular responses to an adverse fetal environment (Chen et al., 2019; Drury et al., 2024).
After maternal malnutrition or fetal glucocorticoid exposure, it is only the male offspring who suffer an increase in BP. Vascular NO and ROS contribute to this sex-specific programmed hypertension. While maternal malnutrition increased both superoxide-mediated vasoconstriction and NO mediated vasodilation, the balance of these factors favored the development of hypertension in males and hypotension in females (Roghair et al., 2009). Moreover, females counteract the adverse effects of maternal malnutrition through the development of a better antioxidant status during the critical developmental window of prenatal life. This early female advantage, together with the ability of estrogen to scavenge free radicals during pregnancy (Reyes et al., 2006), contribute to the milder consequences of programmed hypertension in females (Rodriguez-Rodriguez et al., 2015).
Cardiovascular changes are also involved in other forms of prenatally programmed hypertension. Prenatal nicotine or hypoxia had no effect on baseline BP but caused a heightened vascular response to ANG II and increased the BP in adult male, but not in female, rat offspring. This increase in BP was associated with increased arterial media thickness and the ratio of AT1R/AT2R in the aorta, but not with endothelial NO synthase activity in males. These results suggest that prenatal nicotine or hypoxia exposure alters vascular function via changes in ANG II receptor-mediated signaling pathways in a sex-specific manner (Xiao et al., 2008; Xiao et al., 2014). Estrogen counteracts heightened ROS production, leading to protection of females from prenatal programming of a hypertensive phenotype in adulthood (Xiao et al., 2013; Xiao et al., 2014). In mouse offspring of dams with maternal hypertension induced by vasopressin infusion throughout gestation, a sensitized vascular response to a low dose of ANG II was also evident. Moreover, genetic interference with peroxisome proliferator-activated receptor-γ (PPARγ) specifically in the vascular endothelium of these offspring augmented ANG II-induced endothelial dysfunction. This impairment in endothelial function was attenuated by scavengers of ROS, an effect more prominent in male offspring than female offspring (Nair et al., 2019). In addition, the finding that prenatal nicotine or hypoxia had no significant effect on BP under resting conditions in adult offspring, but enhanced the BP response to ANG II treatment in adult male offspring are consistent with findings in several different animal models including our maternal hypertension model (Peyronnet et al., 2002; Xiao et al., 2009; Intapad et al., 2015; Xue et al., 2018). These data suggest a sensitizing effect of prenatal insults on vascular responses, thereby resulting in the development of hypertension later in life when the subjects encounter second hits.
Similarly, a maternal HFD causes a sex-specific regulation of vascular AT1R and AT2R gene expression through epigenetic DNA methylation, which leads to heightened vascular contraction in adult male, but not female, offspring (Chen et al., 2021). Estrogen plays a key role in this sex difference, as it normalizes vascular dysfunction induced by maternal HFD in female offspring by regulating ATRs, thereby leading to a reduced development of a hypertensive phenotype in adulthood (Chen et al., 2022).
In the rat model using prenatal dietary protein restriction, BP is significantly higher in male than in female offspring. However, OVX induces a significant increase in BP in such females, while estrogen replacement partially reduces the increased BP (Sathishkumar et al., 2009, 2012). It has been shown that prenatal protein restriction leads to compromised ovarian function, thereby contributing to reduced levels of vascular ERα receptors and plasma estrogen, and increased testosterone in female offspring (Leonhardt et al., 2003; Guzman et al., 2006). Therefore, it is likely that prenatal insults could have adversely impacted developing organs (e.g., ovarian dysfunction, and reduced ERα in blood vessels) such that they became less responsive to estrogen. As a result, estrogen replacement reversed only the OVX-induced increase in BP but not that induced by maternal protein restriction (i.e., BP was not recovered to control level) (Sathishkumar et al., 2012). Similar to the aforementioned studies, in our maternal hypertension rat model, for female offspring challenged with postnatal ANG II, OVX increased BP and estrogen replacement partially rescued this effect (Xue et al., 2018).
The sympathetic branch of the autonomic nervous system exerts its predominant impact on cardiomyocytes via β-adrenergic receptors, which can be affected by prenatal programming (Giussani et al., 2012; Harvey et al., 2015). The inotropic and chronotropic responses to the β-adrenergic receptor agonist, isoproterenol, following prenatal protein restriction, were unaltered in female offspring, but significantly higher in male offspring. Estrogen plays a key role in regulation of the expression of β-adrenergic receptors in the heart. Expression of β-adrenergic receptors is upregulated in the OVX rat heart but quickly reversed following estrogen replacement (Elmes et al., 2009). Moreover, male offspring exposed to prenatal hypoxia had an increased susceptibility to ischemic myocardial injury (similar to heart failure involving diastolic dysfunction in adult life) compared with both offspring from healthy pregnancies and with their female counterparts (Shah et al., 2017). These results suggest that prenatal insults such as prenatal low protein or hypoxia, sex-specifically program the sensitivity of β-adrenergic receptors to stimulation and alter AT1R/AT2R expression patterns in the offspring’s heart, which may explain the higher sympathovagal balance and increased susceptibility to ischemia-reperfusion injury in male offspring subject to prenatal insult when compared to female offspring (Elmes et al., 2009; Giussani et al., 2012; Harvey et al., 2015; Xue et al., 2015).
In summary, the prenatal insults induce an increased ratio of AT1R/AT2R, elevated ROS production, and decreased NO bioavailability in conduit and resistance arteries, and these changes act collectively to elicit a sensitized vascular responsiveness, thereby resulting in elevation of the BP. The prenatal insults also program the expression and sensitivity of β-adrenergic receptor in cardiomyocytes to increase susceptibility to ischemic myocardial injury. Female sex hormones, especially estrogen, play a protective role in the prenatal insult-induced sensitization processes, which manifest as cardiovascular dysfunction in a sex-specific manner.
Alterations and effects of maternal sex hormones in prenatal insults
Sex hormones, including estrogen, progesterone, and testosterone, are essential for the physiological regulation of pregnancy such as vascular adaptations, pregnancy maintenance and labor process. However, low levels of estrogen or high levels of testosterone during pregnancy have been shown to initiate various prenatal insults, such as preeclampsia (Shu et al., 2021; Darlas et al., 2025).
Several clinical studies have documented a significant decrease in the levels of estradiol during preeclampsia (Berkane et al., 2017; Cantonwine et al., 2019). The decrease in estrogen levels is due to alterations in enzyme activities including a decrease in Hydroxysteroid (17-β) dehydrogenase 1 (17β-HSD1, an enzyme converting estrone to estradiol), aromatase (an enzyme converting androgens to estrogens), catechol-O-methyltransferase (COMT, an enzyme for the synthesis of 2-methoxyestradiol) (Taravati et al., 2017; Berkane et al., 2018). The exogenous administration of estrogen normalized the BP and other associated symptoms of preeclampsia in both animal models and preeclampsia patients (Djordjevic et al., 2010; Babic et al., 2018; Lin et al., 2020). These beneficial effects of estrogen involve the activation of GPR30, endothelial NOS and PI3K-Akt signaling pathway, reduced release of inflammatory cytokines and oxidative stress, leading to improved placental perfusion (Shu et al., 2021).
A meta-analysis reveals that expectant mothers with hyperandrogenic polycystic ovary syndrome (PCOS) had increased odds ratios for gestational diabetes mellitus and preeclampsia compared to those with a non-hyperandrogenic PCOS (Guo et al., 2024). Indeed, maternal androgen levels are already elevated in the early second trimester among women who eventually develop preeclampsia. Thus, hyperandrogenism is involved in the pathogenesis of preeclampsia, and may be considered an early risk marker of preeclampsia (Carlsen et al., 2005). Chinnathambi et al. confirmed that increased maternal testosterone in female rats, at concentrations relevant to abnormal clinical conditions, induces blunting of NO-mediated vasodilation and increased vascular resistance, leading to maternal gestational hypertension (Chinnathambi et al., 2013a).
Conditions of excess androgen in women, such as PCOS, often exhibit intergenerational transmission. In a human study, women born to mothers with the highest levels of maternal bioactive androgens demonstrated a 4.84-fold increased odds for having hypertension and were associated with an increased risk for incident metabolic syndrome (Huang et al., 2018). In animal studies, Sherman et al. demonstrated that prenatal exposure to excess androgen negatively impacted cardiovascular function by increasing BP and decreasing heart rate in adult female offspring. Prenatal androgen was also associated with gut microbial dysbiosis. These results suggest that prenatal exposure to hyperandrogenemia in daughters of women with PCOS may lead to long-term alterations in gut microbiota and cardiometabolic function (Sherman et al., 2018). Similarly, male offspring of hyperandrogenemic dams had a normal baseline BP, but an exaggerated pressor response to ANG II infusion, suggesting that adult sons of PCOS mothers may also be at increased risk of cardiometabolic disease (Zuchowski et al., 2021). Further, prenatal testosterone leads to an increase in BP in both male and female offspring but involves a sex-specific mechanism responsible for blunting of endothelial cell-associated relaxation: endothelium-derived hyperpolarizing factor (EDHF)-related in males and NO-related in females (Chinnathambi et al., 2013b). The ACE inhibitor enalapril has a positive influence on endothelial function with improvement in EDHF relaxation (More et al., 2015).
In summary, both decreased estrogen and increased testosterone during pregnancy contribute to the initiation of prenatal insults, particularly preeclampsia, which is associated with detrimental consequences for both mothers and offspring, exhibiting sex-specific cardiovascular dysfunctions.
Conclusion
In this review, we have described the sex differences in the pathogenesis of prenatally programmed hypertension and the associated peripheral and central mechanisms. The prenatal insults program the cardiovascular system and cardiovascular-regulating organs, such as the blood vessels, heart, kidney, and the CNS, to adversely alter their structures and functions in a way that increases the risk for hypertension in adults. These prenatal and/or early life alterations include reduced nephron endowment, changes in factors that affect endothelial and arterial compliance, and alterations in RAS and SNS activity in the periphery and the CNS. These changes increased the sensitivity of the offspring of mothers exposed to various prenatal insults to a second hit (e.g., stress, HFD or prohypertensive agents), increasing the likelihood that the offspring will develop hypertension later in life. Sex hormone interactions with the prohypertensive systems in the cardiovascular organs play a pivotal role in the pathogenesis of sex-specific and age-dependent hypertension programmed by various prenatal insults (Figure 1). However, despite intense investigation into the mechanisms underlying fetal programming of adult hypertension, there is still no consensus on how these different mechanisms and pathways are programmed in utero and how they interact with sex hormones to ultimately lead to a sex-specific increase in BP later in life. Future studies are warranted to investigate the mechanisms involved in initiating programming events in utero, and the subsequent sensitization to postnatal challenges (i.e., stresses). Understanding these mechanisms is important for the development of novel, sex-specific strategies for prevention and treatment of hypertension in men and women.
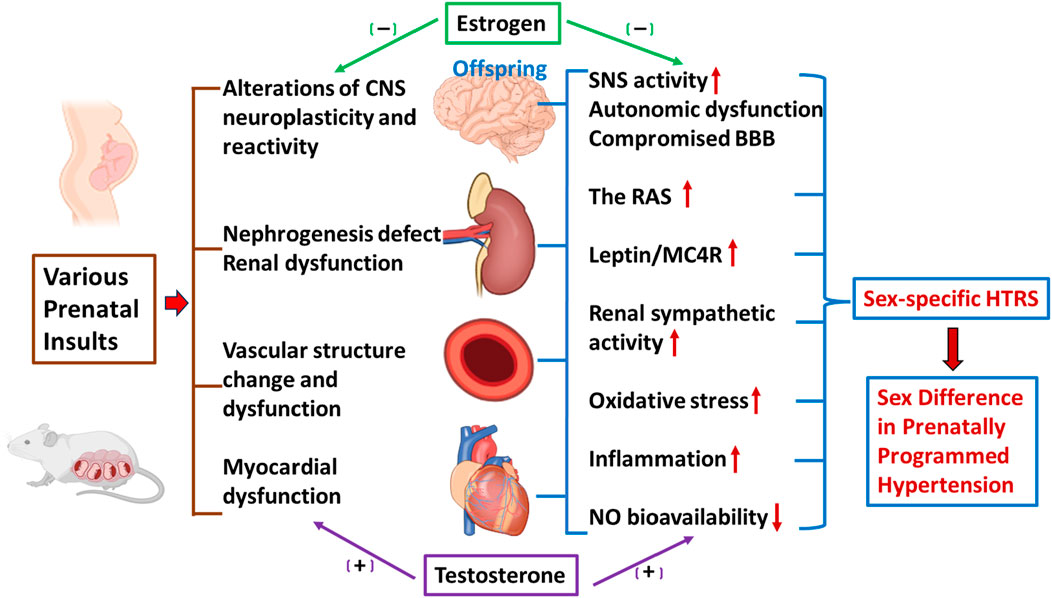
Figure 1. Schematic representation of sex differences in prenatal insult-elicited hypertensive response sensitization (HTRS) and associated peripheral and central mechanisms. The figure shows prenatal insults elicit a sex-specific HTRS (i.e., prenatally programmed hypertension) through cardiovascular dysfunction, nephrogenesis defects, and alterations of central nervous system (CNS) neuroplasticity and reactivity. Decreased nitric oxide (NO) bioavailability, increased oxidative stress and inflammation, activation of the renin-angiotensin system (RAS), disruption of the blood-brain barrier (BBB), and elevation of leptin/melanocortin 4 receptors (MC4R) and sympathetic nervous system (SNS) activity in the peripheral and CNS networks controlling blood pressure (BP) mediate these sensitization processes, in which sex hormones such as estrogen and testosterone, are involved.
Since the current evidence shows that fetal and early postnatal life are critical developmental windows, one opportunity for interventions to ameliorate prenatally programmed hypertension is around these developmental periods. We and others have demonstrated the beneficial effects of anti-RAS or anti-inflammatory agents administered to mothers or offspring on improving fetal programming outcomes (Mizuno et al., 2014; Xue et al., 2017; Deng et al., 2018; Li et al., 2020). However, these therapies cannot be used during pregnancy or postnatally in human because these treatments are likely to interfere with renal and CNS development in the offspring. Thus, there is great need for developing therapeutic strategies for women exposed to prenatal insults which do not involve the risk of teratological toxicity. Exercise in the mother during pregnancy and in the offspring may be a promising strategy to prevent or reprogram latent HTRS (Kusuyama et al., 2020). Our recent studies showed that voluntary exercise in offspring plays a beneficial role in preventing maternal hypertension-induced HTRS elicited by postweaning HFD (Xue et al., 2022b). Further studies are needed to investigate the underlying mechanisms involved in the positive reprogramming effects of “good” therapeutic strategies, not limited to exercise, that will improve maternal and offspring health and prevent the vicious cycle of transgenerational hypertension.
Author contributions
BX: Writing – original draft, Writing – review and editing, Conceptualization. AKJ: Writing – review and editing, Conceptualization, Funding acquisition. AGB: Writing – review and editing, Conceptualization, Funding acquisition.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by The Tross Family Epilepsy Fund (AGB) and the NIH grants 5P50HD103556-03 (AGB), 5R01EY030151-05 (AGB), 5R01EY031952-04 (AGB), 5R01NS127428-02 (AGB), 1P01HL158500-01 (AKJ), R01HL-98207 (AKJ and BX) and R01HL-139575 (AKJ and BX).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Alexander B. T. (2003). Placental insufficiency leads to development of hypertension in growth-restricted offspring. Hypertension 41, 457–462. doi:10.1161/01.HYP.0000053448.95913.3D
Alexander B. T., Hendon A. E., Ferril G., Dwyer T. M. (2005). Renal denervation abolishes hypertension in low-birth-weight offspring from pregnant rats with reduced uterine perfusion. Hypertension 45, 754–758. doi:10.1161/01.HYP.0000153319.20340.2a
Alhamoud I., Legan S. K., Gattineni J., Baum M. (2021). Sex differences in prenatal programming of hypertension by dexamethasone. Exp. Biol. Med. (Maywood) 246, 1554–1562. doi:10.1177/15353702211003294
Arima Y., Fukuoka H. (2020). Developmental origins of health and disease theory in cardiology. J. Cardiol. 76, 14–17. doi:10.1016/j.jjcc.2020.02.003
Babic G. M., Markovic S. D., Varjacic M., Djordjevic N. Z., Nikolic T., Stojic I., et al. (2018). Estradiol decreases blood pressure in association with redox regulation in preeclampsia. Clin. Exp. Hypertens. 40, 281–286. doi:10.1080/10641963.2017.1368538
Barker D. J., Winter P. D., Osmond C., Margetts B., Simmonds S. J. (1989). Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580. doi:10.1016/s0140-6736(89)90710-1
Baum M. (2010). Role of the kidney in the prenatal and early postnatal programming of hypertension. Am. J. Physiol. Ren. Physiol. 298, F235–F247. doi:10.1152/ajprenal.00288.2009
Baum M. (2018). Role of renal sympathetic nerve activity in prenatal programming of hypertension. Pediatr. Nephrol. 33, 409–419. doi:10.1007/s00467-016-3359-8
Berkane N., Liere P., Lefevre G., Alfaidy N., Nahed R. A., Vincent J., et al. (2018). Abnormal steroidogenesis and aromatase activity in preeclampsia. Placenta 69, 40–49. doi:10.1016/j.placenta.2018.07.004
Berkane N., Liere P., Oudinet J. P., Hertig A., Lefevre G., Pluchino N., et al. (2017). From pregnancy to preeclampsia: a key role for estrogens. Endocr. Rev. 38, 123–144. doi:10.1210/er.2016-1065
Bilbo S. D., Tsang V. (2010). Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J. 24, 2104–2115. doi:10.1096/fj.09-144014
Cantonwine D. E., Mcelrath T. F., Trabert B., Xu X., Sampson J., Roberts J. M., et al. (2019). Estrogen metabolism pathways in preeclampsia and normal pregnancy. Steroids 144, 8–14. doi:10.1016/j.steroids.2019.01.005
Carlsen S. M., Romundstad P., Jacobsen G. (2005). Early second-trimester maternal hyperandrogenemia and subsequent preeclampsia: a prospective study. Acta Obstet. Gynecol. Scand. 84, 117–121. doi:10.1111/j.0001-6349.2005.00493.x
Chen F., Cao K., Zhang H., Yu H., Liu Y., Xue Q. (2021). Maternal high-fat diet increases vascular contractility in adult offspring in a sex-dependent manner. Hypertens. Res. 44, 36–46. doi:10.1038/s41440-020-0519-9
Chen F., Zhao R., Zhang H., Huang C., Liu Y., Xue Q. (2022). Estrogen normalizes maternal HFD-induced vascular dysfunction in offspring by regulating ATR. Hypertens. Res. 45, 1743–1753. doi:10.1038/s41440-022-01002-2
Chen Z., Wang L., Ke J., Xiao D. (2019). Effects of estrogen in gender-dependent fetal programming of adult cardiovascular dysfunction. Curr. Vasc. Pharmacol. 17, 147–152. doi:10.2174/1570161116666180301142453
Chinnathambi V., Balakrishnan M., Ramadoss J., Yallampalli C., Sathishkumar K. (2013). Testosterone alters maternal vascular adaptations: role of the endothelial NO system. Hypertension 61, 647–654. doi:10.1161/HYPERTENSIONAHA.111.00486
Chinnathambi V., Yallampalli C., Sathishkumar K. (2013). Prenatal testosterone induces sex-specific dysfunction in endothelium-dependent relaxation pathways in adult male and female rats. Biol. Reprod. 89, 97. doi:10.1095/biolreprod.113.111542
Colafella K. M. M., Denton K. M. (2018). Sex-specific differences in hypertension and associated cardiovascular disease. Nat. Rev. Nephrol. 14, 185–201. doi:10.1038/nrneph.2017.189
Contu L., Nizari S., Heath C. J., Hawkes C. A. (2019). Pre- and post-natal high fat feeding differentially affects the structure and integrity of the neurovascular unit of 16-month old male and female mice. Front. Neurosci. 13, 1045. doi:10.3389/fnins.2019.01045
Darlas M., Kalantaridou S., Valsamakis G. (2025). Maternal hyperandrogenemia and the long-term neuropsychological, sex developmental, and metabolic effects on offspring. Int. J. Mol. Sci. 26, 2199. doi:10.3390/ijms26052199
Da Silva A. A., Do Carmo J. M., Hall J. E. (2013). Role of leptin and central nervous system melanocortins in obesity hypertension. Curr. Opin. Nephrol. Hypertens. 22, 135–140. doi:10.1097/MNH.0b013e32835d0c05
Dasinger J. H., Alexander B. T. (2016). Gender differences in developmental programming of cardiovascular diseases. Clin. Sci. (Lond) 130, 337–348. doi:10.1042/CS20150611
Dasinger J. H., Davis G. K., Newsome A. D., Alexander B. T. (2016). Developmental programming of hypertension: physiological mechanisms. Hypertension 68, 826–831. doi:10.1161/HYPERTENSIONAHA.116.06603
Davis G. K., Intapad S., Newsome A. D., Coats L. E., Bamrick D. R., Ojeda N. B., et al. (2019). Androgen receptor blockade differentially regulates blood pressure in growth-restricted versus ovarian deficient rats. Hypertension 74, 975–982. doi:10.1161/HYPERTENSIONAHA.119.13257
Davis G. K., Newsome A. D., Cole A. B., Ojeda N. B., Alexander B. T. (2019). Chronic estrogen supplementation prevents the increase in blood pressure in female intrauterine growth-restricted offspring at 12 months of age. Hypertension 73, 1128–1136. doi:10.1161/HYPERTENSIONAHA.118.12379
Deng Y., Song L., Nie X., Shou W., Li X. (2018). Prenatal inflammation exposure-programmed cardiovascular diseases and potential prevention. Pharmacol. Ther. 190, 159–172. doi:10.1016/j.pharmthera.2018.05.009
Dibona G. F. (2013). Sympathetic nervous system and hypertension. Hypertension 61, 556–560. doi:10.1161/HYPERTENSIONAHA.111.00633
Djordjevic N. Z., Babic G. M., Markovic S. D., Ognjanovic B. I., Stajn A. S., Saicic Z. S. (2010). The antioxidative effect of estradiol therapy on erythrocytes in women with preeclampsia. Reprod. Toxicol. 29, 231–236. doi:10.1016/j.reprotox.2009.11.004
Doan T. N. A., Briffa J. F., Phillips A. L., Leemaqz S. Y., Burton R. A., Romano T., et al. (2021). Epigenetic mechanisms involved in intrauterine growth restriction and aberrant kidney development and function. J. Dev. Orig. Health Dis. 12, 952–962. doi:10.1017/S2040174420001257
Do Carmo J. M., Dai X., Aitken N., Larson K. M., Omoto A. C. M., Gulke R. R., et al. (2023). Sex differences in weight gain, blood pressure control, and responses to melanocortin-4 receptor antagonism in offspring from lean and obese parents. Am. J. Physiol. Regul. Integr. Comp. Physiol. 325, R401–R410. doi:10.1152/ajpregu.00106.2023
do Carmo J. M., Hall J. E., Dai X., Aitkens N., Larson K., Luna-Suarez E. M., et al. (2024). Parental obesity predisposes offspring to kidney dysfunction and increased susceptibility to ischemia-reperfusion injury in a sex-dependent manner. Am. J. Physiol. Ren. Physiol. 326, F727–F736. doi:10.1152/ajprenal.00294.2023
Drury E. R., Wu J., Gigliotti J. C., Le T. H. (2024). Sex differences in blood pressure regulation and hypertension: renal, hemodynamic, and hormonal mechanisms. Physiol. Rev. 104, 199–251. doi:10.1152/physrev.00041.2022
Dudele A., Hougaard K. S., Kjolby M., Hokland M., Winther G., Elfving B., et al. (2017). Chronic maternal inflammation or high-fat-feeding programs offspring obesity in a sex-dependent manner. Int. J. Obes. (Lond) 41, 1420–1426. doi:10.1038/ijo.2017.136
Elmes M. J., Haase A., Gardner D. S., Langley-Evans S. C. (2009). Sex differences in sensitivity to beta-adrenergic agonist isoproterenol in the isolated adult rat heart following prenatal protein restriction. Br. J. Nutr. 101, 725–734. doi:10.1017/S0007114508025075
Falkner B., Alexander B. T., Nuyt A. M., South A. M., Ingelfinger J. (2024). Cardiovascular health starts in the womb. Hypertension 81, 2016–2026. doi:10.1161/HYPERTENSIONAHA.124.21359
Gemici A., Sinen O., Bulbul M. (2021). Sexual dimorphism in rats exposed to maternal high fat diet: alterations in medullary sympathetic network. Metab. Brain Dis. 36, 1305–1314. doi:10.1007/s11011-021-00736-1
Gilbert J. S., Nijland M. J. (2008). Sex differences in the developmental origins of hypertension and cardiorenal disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 295, R1941–R1952. doi:10.1152/ajpregu.90724.2008
Giussani D. A., Camm E. J., Niu Y., Richter H. G., Blanco C. E., Gottschalk R., et al. (2012). Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS One 7, e31017. doi:10.1371/journal.pone.0031017
Guo X., Yao Y., Wang T., Wu J., Jiang R. (2024). The impact of hyperandrogenemia on pregnancy complications and outcomes in patients with PCOS: a systematic review and meta-analysis. Hypertens. Pregnancy 43, 2379389. doi:10.1080/10641955.2024.2379389
Guzman C., Cabrera R., Cardenas M., Larrea F., Nathanielsz P. W., Zambrano E. (2006). Protein restriction during fetal and neonatal development in the rat alters reproductive function and accelerates reproductive ageing in female progeny. J. Physiol. 572, 97–108. doi:10.1113/jphysiol.2005.103903
Harvey T. J., Murphy R. M., Morrison J. L., Posterino G. S. (2015). Maternal nutrient restriction alters Ca2+ handling properties and contractile function of isolated left ventricle bundles in male but not female juvenile rats. PLoS One 10, e0138388. doi:10.1371/journal.pone.0138388
Hsu C. N., Lai W. T., Lin Y. J., Tain Y. L. (2018). Postnatal high-fat diet sex-specifically exacerbates prenatal dexamethasone-induced hypertension: mass spectrometry-based quantitative proteomic approach. J. Nutr. Biochem. 57, 268–275. doi:10.1016/j.jnutbio.2018.04.006
Huang G., Cherkerzian S., Loucks E. B., Buka S. L., Handa R. J., Lasley B. L., et al. (2018). Sex differences in the prenatal programming of adult metabolic syndrome by maternal androgens. J. Clin. Endocrinol. Metab. 103, 3945–3953. doi:10.1210/jc.2018-01243
Intapad S., Dasinger J. H., Johnson J. M., Brown A. D., Ojeda N. B., Alexander B. T. (2019). Male and female intrauterine growth-restricted offspring differ in blood pressure, renal function, and glucose homeostasis responses to a postnatal diet high in fat and sugar. Hypertension 73, 620–629. doi:10.1161/HYPERTENSIONAHA.118.12134
Intapad S., Ojeda N. B., Varney E., Royals T. P., Alexander B. T. (2015). Sex-specific effect of endothelin in the blood pressure response to acute angiotensin II in growth-restricted rats. Hypertension 66, 1260–1266. doi:10.1161/HYPERTENSIONAHA.115.06257
Intapad S., Tull F. L., Brown A. D., Dasinger J. H., Ojeda N. B., Fahling J. M., et al. (2013). Renal denervation abolishes the age-dependent increase in blood pressure in female intrauterine growth-restricted rats at 12 months of age. Hypertension 61, 828–834. doi:10.1161/HYPERTENSIONAHA.111.00645
Johnson A. K., Xue B. (2018). Central nervous system neuroplasticity and the sensitization of hypertension. Nat. Rev. Nephrol. 14, 750–766. doi:10.1038/s41581-018-0068-5
Jones A., Beda A., Ward A. M., Osmond C., Phillips D. I., Moore V. M., et al. (2007). Size at birth and autonomic function during psychological stress. Hypertension 49, 548–555. doi:10.1161/01.HYP.0000257196.13485.9b
Kusuyama J., Alves-Wagner A. B., Makarewicz N. S., Goodyear L. J. (2020). Effects of maternal and paternal exercise on offspring metabolism. Nat. Metab. 2, 858–872. doi:10.1038/s42255-020-00274-7
Lankadeva Y. R., Singh R. R., Tare M., Moritz K. M., Denton K. M. (2014). Loss of a kidney during fetal life: long-term consequences and lessons learned. Am. J. Physiol. Ren. Physiol. 306, F791–F800. doi:10.1152/ajprenal.00666.2013
Leonhardt M., Lesage J., Croix D., Dutriez-Casteloot I., Beauvillain J. C., Dupouy J. P. (2003). Effects of perinatal maternal food restriction on pituitary-gonadal axis and plasma leptin level in rat pup at birth and weaning and on timing of puberty. Biol. Reprod. 68, 390–400. doi:10.1095/biolreprod.102.003269
Li H. B., Yang T., Richards E. M., Pepine C. J., Raizada M. K. (2020). Maternal treatment with captopril persistently alters gut-brain communication and attenuates hypertension of male offspring. Hypertension 75, 1315–1324. doi:10.1161/HYPERTENSIONAHA.120.14736
Lim K., Burke S. L., Marques F. Z., Jackson K. L., Gueguen C., Sata Y., et al. (2021). Leptin and melanocortin signaling mediates hypertension in offspring from female rabbits fed a high-fat diet during gestation and lactation. Front. Physiol. 12, 693157. doi:10.3389/fphys.2021.693157
Lin Z. H., Jin J., Shan X. Y. (2020). The effects of estradiol on inflammatory and endothelial dysfunction in rats with preeclampsia. Int. J. Mol. Med. 45, 825–835. doi:10.3892/ijmm.2020.4465
Liu Y., Liang M. (2019). Functional role of epigenetic regulation in the development of prenatal programmed hypertension. Kidney Int. 96, 10–12. doi:10.1016/j.kint.2019.03.003
Maranon R., Lima R., Spradley F. T., Do Carmo J. M., Zhang H., Smith A. D., et al. (2015). Roles for the sympathetic nervous system, renal nerves, and CNS melanocortin-4 receptor in the elevated blood pressure in hyperandrogenemic female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R708–R713. doi:10.1152/ajpregu.00411.2014
Mcmullen S., Langley-Evans S. C. (2005). Sex-specific effects of prenatal low-protein and carbenoxolone exposure on renal angiotensin receptor expression in rats. Hypertension 46, 1374–1380. doi:10.1161/01.HYP.0000188702.96256.46
Mizuno M., Lozano G., Siddique K., Baum M., Smith S. A. (2014). Enalapril attenuates the exaggerated sympathetic response to physical stress in prenatally programmed hypertensive rats. Hypertension 63, 324–329. doi:10.1161/HYPERTENSIONAHA.113.02330
More A. S., Mishra J. S., Hankins G. D., Yallampalli C., Sathishkumar K. (2015). Enalapril normalizes endothelium-derived hyperpolarizing factor-mediated relaxation in mesenteric artery of adult hypertensive rats prenatally exposed to testosterone. Biol. Reprod. 92, 155. doi:10.1095/biolreprod.115.130468
Moritz K. M., Cuffe J. S., Wilson L. B., Dickinson H., Wlodek M. E., Simmons D. G., et al. (2010). Review: sex specific programming: a critical role for the renal renin-angiotensin system. Placenta 31, S40–S46. doi:10.1016/j.placenta.2010.01.006
Moritz K. M., Singh R. R., Probyn M. E., Denton K. M. (2009). Developmental programming of a reduced nephron endowment: more than just a baby's birth weight. Am. J. Physiol. Ren. Physiol. 296, F1–F9. doi:10.1152/ajprenal.00049.2008
Nair A. R., Silva S. D., Agbor L. N., Wu J., Nakagawa P., Mukohda M., et al. (2019). Endothelial PPARγ (peroxisome proliferator-activated receptor-γ) protects from angiotensin II-induced endothelial dysfunction in adult offspring born from pregnancies complicated by hypertension. Hypertension 74, 173–183. doi:10.1161/HYPERTENSIONAHA.119.13101
Nakagawa P., Gomez J., Grobe J. L., Sigmund C. D. (2020). The renin-angiotensin system in the central nervous system and its role in blood pressure regulation. Curr. Hypertens. Rep. 22, 7. doi:10.1007/s11906-019-1011-2
Ojeda N. B., Grigore D., Yanes L. L., Iliescu R., Robertson E. B., Zhang H., et al. (2007). Testosterone contributes to marked elevations in mean arterial pressure in adult male intrauterine growth restricted offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R758–R763. doi:10.1152/ajpregu.00311.2006
Ojeda N. B., Intapad S., Royals T. P., Black J. T., Dasinger J. H., Tull F. L., et al. (2011). Hypersensitivity to acute ANG II in female growth-restricted offspring is exacerbated by ovariectomy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 301, R1199–R1205. doi:10.1152/ajpregu.00219.2011
Ojeda N. B., Royals T. P., Alexander B. T. (2013). Sex differences in the enhanced responsiveness to acute angiotensin II in growth-restricted rats: role of fasudil, a Rho kinase inhibitor. Am. J. Physiol. Ren. Physiol. 304, F900–F907. doi:10.1152/ajprenal.00687.2012
Ojeda N. B., Royals T. P., Black J. T., Dasinger J. H., Johnson J. M., Alexander B. T. (2010). Enhanced sensitivity to acute angiotensin II is testosterone dependent in adult male growth-restricted offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1421–R1427. doi:10.1152/ajpregu.00096.2010
Pedroza A., Ferreira D. S., Santana D. F., Da Silva P. T., De Aguiar Junior F. C. A., Sellitti D. F., et al. (2019). A maternal low-protein diet and neonatal overnutrition result in similar changes to glomerular morphology and renal cortical oxidative stress measures in male Wistar rats. Appl. Physiol. Nutr. Metab. 44, 164–171. doi:10.1139/apnm-2018-0288
Peyronnet J., Dalmaz Y., Ehrstrom M., Mamet J., Roux J. C., Pequignot J. M., et al. (2002). Long-lasting adverse effects of prenatal hypoxia on developing autonomic nervous system and cardiovascular parameters in rats. Pflugers Arch. 443, 858–865. doi:10.1007/s00424-001-0766-9
Pladys P., Lahaie I., Cambonie G., Thibault G., Le N. L., Abran D., et al. (2004). Role of brain and peripheral angiotensin II in hypertension and altered arterial baroreflex programmed during fetal life in rat. Pediatr. Res. 55, 1042–1049. doi:10.1203/01.PDR.0000127012.37315.36
Prior L. J., Davern P. J., Burke S. L., Lim K., Armitage J. A., Head G. A. (2014). Exposure to a high-fat diet during development alters leptin and ghrelin sensitivity and elevates renal sympathetic nerve activity and arterial pressure in rabbits. Hypertension 63, 338–345. doi:10.1161/HYPERTENSIONAHA.113.02498
Reyes M. R., Sifuentes-Alvarez A., Lazalde B. (2006). Estrogens are potentially the only steroids with an antioxidant role in pregnancy: in vitro evidence. Acta Obstet. Gynecol. Scand. 85, 1090–1093. doi:10.1080/00016340500453685
Rodriguez-Rodriguez P., De Pablo A. L., Condezo-Hoyos L., Martin-Cabrejas M. A., Aguilera Y., Ruiz-Hurtado G., et al. (2015). Fetal undernutrition is associated with perinatal sex-dependent alterations in oxidative status. J. Nutr. Biochem. 26, 1650–1659. doi:10.1016/j.jnutbio.2015.08.004
Roghair R. D., Segar J. L., Volk K. A., Chapleau M. W., Dallas L. M., Sorenson A. R., et al. (2009). Vascular nitric oxide and superoxide anion contribute to sex-specific programmed cardiovascular physiology in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R651–R662. doi:10.1152/ajpregu.90756.2008
Sadagurski M., Debarba L. K., Werneck-De-Castro J. P., Ali Awada A., Baker T. A., Bernal-Mizrachi E. (2019). Sexual dimorphism in hypothalamic inflammation in the offspring of dams exposed to a diet rich in high fat and branched-chain amino acids. Am. J. Physiol. Endocrinol. Metab. 317, E526–E534. doi:10.1152/ajpendo.00183.2019
Saez F., Castells M. T., Zuasti A., Salazar F., Reverte V., Loria A., et al. (2007). Sex differences in the renal changes elicited by angiotensin II blockade during the nephrogenic period. Hypertension 49, 1429–1435. doi:10.1161/HYPERTENSIONAHA.107.087957
Samuelsson A. M., Morris A., Igosheva N., Kirk S. L., Pombo J. M., Coen C. W., et al. (2010). Evidence for sympathetic origins of hypertension in juvenile offspring of obese rats. Hypertension 55, 76–82. doi:10.1161/HYPERTENSIONAHA.109.139402
Samuelsson A. S., Mullier A., Maicas N., Oosterhuis N. R., Eun Bae S., Novoselova T. V., et al. (2016). Central role for melanocortin-4 receptors in offspring hypertension arising from maternal obesity. Proc. Natl. Acad. Sci. U. S. A. 113, 12298–12303. doi:10.1073/pnas.1607464113
Sathishkumar K., Elkins R., Yallampalli U., Yallampalli C. (2009). Protein restriction during pregnancy induces hypertension and impairs endothelium-dependent vascular function in adult female offspring. J. Vasc. Res. 46, 229–239. doi:10.1159/000166390
Sathishkumar K., Elkins R., Yallampalli U., Yallampalli C. (2012). Protein restriction during pregnancy induces hypertension in adult female rat offspring--influence of oestradiol. Br. J. Nutr. 107, 665–673. doi:10.1017/S0007114511003448
Shah A., Matsumura N., Quon A., Morton J. S., Dyck J. R. B., Davidge S. T. (2017). Cardiovascular susceptibility to in vivo ischemic myocardial injury in male and female rat offspring exposed to prenatal hypoxia. Clin. Sci. (Lond) 131, 2303–2317. doi:10.1042/CS20171122
Sherman S. B., Sarsour N., Salehi M., Schroering A., Mell B., Joe B., et al. (2018). Prenatal androgen exposure causes hypertension and gut microbiota dysbiosis. Gut Microbes 9, 400–421. doi:10.1080/19490976.2018.1441664
Shi Z., Wong J., Brooks V. L. (2020). Obesity: sex and sympathetics. Biol. Sex. Differ. 11, 10. doi:10.1186/s13293-020-00286-8
Shu C., Han S., Xu P., Wang Y., Cheng T., Hu C. (2021). Estrogen and preeclampsia: potential of estrogens as therapeutic agents in preeclampsia. Drug Des. Devel Ther. 15, 2543–2550. doi:10.2147/DDDT.S304316
Singh P., Dutta S. R., Song C. Y., Oh S., Gonzalez F. J., Malik K. U. (2020). Brain testosterone-CYP1B1 (Cytochrome P450 1B1) generated metabolite 6β-hydroxytestosterone promotes neurogenic hypertension and inflammation. Hypertension 76, 1006–1018. doi:10.1161/HYPERTENSIONAHA.120.15567
Singh P., Song C. Y., Dutta S. R., Gonzalez F. J., Malik K. U. (2020). Central CYP1B1 (Cytochrome P450 1B1)-estradiol metabolite 2-methoxyestradiol protects from hypertension and neuroinflammation in female mice. Hypertension 75, 1054–1062. doi:10.1161/HYPERTENSIONAHA.119.14548
Singh R. R., Denton K. M. (2015). Role of the kidney in the fetal programming of adult cardiovascular disease: an update. Curr. Opin. Pharmacol. 21, 53–59. doi:10.1016/j.coph.2014.12.010
Tain Y. L., Hsu C. N. (2024). Interplay between maternal nutrition and epigenetic programming on offspring hypertension. J. Nutr. Biochem. 127, 109604. doi:10.1016/j.jnutbio.2024.109604
Taravati A., Tohidi F., Moniri M., Kamali K. (2017). Catechol-O-methyltransferase gene polymorphism (Val158Met) and development of pre-eclampsia. Arch. Med. Res. 48, 180–186. doi:10.1016/j.arcmed.2017.03.006
Taylor P. D., Samuelsson A. M., Poston L. (2014). Maternal obesity and the developmental programming of hypertension: a role for leptin. Acta Physiol. (Oxf) 210, 508–523. doi:10.1111/apha.12223
Tomat A. L., Salazar F. J. (2014). Mechanisms involved in developmental programming of hypertension and renal diseases. Gender differences. Horm. Mol. Biol. Clin. Investig. 18, 63–77. doi:10.1515/hmbci-2013-0054
Virdis A., Bruno R. M., Neves M. F., Bernini G., Taddei S., Ghiadoni L. (2011). Hypertension in the elderly: an evidence-based review. Curr. Pharm. Des. 17, 3020–3031. doi:10.2174/138161211798157711
Walton S. L., Singh R. R., Little M. H., Bowles J., Li J., Moritz K. M. (2018). Prolonged prenatal hypoxia selectively disrupts collecting duct patterning and postnatal function in male mouse offspring. J. Physiol. 596, 5873–5889. doi:10.1113/JP275918
Wang X. F., Li J. D., Huo Y. L., Zhang Y. P., Fang Z. Q., Wang H. P., et al. (2020). Blockade of angiotensin-converting enzyme or tumor necrosis factor-alpha reverses maternal high-fat diet-induced sensitization of angiotensin II hypertension in male rat offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 318, R351–R359. doi:10.1152/ajpregu.00200.2019
Warrington J. P., Collins H. E., Davidge S. T., Do Carmo J. M., Goulopoulou S., Intapad S., et al. (2024). Guidelines for in vivo models of developmental programming of cardiovascular disease risk. Am. J. Physiol. Heart Circ. Physiol. 327, H221–H241. doi:10.1152/ajpheart.00060.2024
Wei S. G., Yu Y., Felder R. B. (2018). Blood-borne interleukin-1β acts on the subfornical organ to upregulate the sympathoexcitatory milieu of the hypothalamic paraventricular nucleus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 314, R447–R458. doi:10.1152/ajpregu.00211.2017
Wei S. G., Yu Y., Zhang Z. H., Felder R. B. (2015). Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension 65, 1126–1133. doi:10.1161/HYPERTENSIONAHA.114.05112
Wei S. G., Zhang Z. H., Beltz T. G., Yu Y., Johnson A. K., Felder R. B. (2013). Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 62, 118–125. doi:10.1161/HYPERTENSIONAHA.113.01404
Wijenayake S., Rahman M. F., Lum C. M. W., De Vega W. C., Sasaki A., Mcgowan P. O. (2020). Maternal high-fat diet induces sex-specific changes to glucocorticoid and inflammatory signaling in response to corticosterone and lipopolysaccharide challenge in adult rat offspring. J. Neuroinflammation 17, 116. doi:10.1186/s12974-020-01798-1
Woods L. L., Morgan T. K., Resko J. A. (2010). Castration fails to prevent prenatally programmed hypertension in male rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1111–R1116. doi:10.1152/ajpregu.00803.2009
Xiao D., Huang X., Xu Z., Yang S., Zhang L. (2009). Prenatal cocaine exposure differentially causes vascular dysfunction in adult offspring. Hypertension 53, 937–943. doi:10.1161/HYPERTENSIONAHA.108.121830
Xiao D., Huang X., Xue Q., Zhang L. (2014). Antenatal hypoxia induces programming of reduced arterial blood pressure response in female rat offspring: role of ovarian function. PLoS One 9, e98743. doi:10.1371/journal.pone.0098743
Xiao D., Huang X., Yang S., Zhang L. (2013). Estrogen normalizes perinatal nicotine-induced hypertensive responses in adult female rat offspring. Hypertension 61, 1246–1254. doi:10.1161/HYPERTENSIONAHA.113.01152
Xiao D., Xu Z., Huang X., Longo L. D., Yang S., Zhang L. (2008). Prenatal gender-related nicotine exposure increases blood pressure response to angiotensin II in adult offspring. Hypertension 51, 1239–1247. doi:10.1161/HYPERTENSIONAHA.107.106203
Xue B., Beltz T. G., Guo F., Johnson A. K. (2018). Sex differences in maternal gestational hypertension-induced sensitization of angiotensin II hypertension in rat offspring: the protective effect of estrogen. Am. J. Physiol. Regul. Integr. Comp. Physiol. 314, R274–R281. doi:10.1152/ajpregu.00216.2017
Xue B., Johnson A. K. (2023). Sensitization of hypertension: the impact of earlier life challenges: excellence award for hypertension research 2021. Hypertension 80, 1–12. doi:10.1161/HYPERTENSIONAHA.122.18550
Xue B., Yin H., Guo F., Beltz T. G., Thunhorst R. L., Johnson A. K. (2017). Maternal gestational hypertension-induced sensitization of angiotensin II hypertension is reversed by renal denervation or angiotensin-converting enzyme inhibition in rat offspring. Hypertension 69, 669–677. doi:10.1161/HYPERTENSIONAHA.116.08597
Xue B., Yu Y., Beltz T. G., Guo F., Felder R. B., Wei S. G., et al. (2021). Maternal angiotensin II-induced hypertension sensitizes postweaning high-fat diet-elicited hypertensive response through increased brain reactivity in rat offspring. J. Am. Heart Assoc. 10, e022170. doi:10.1161/JAHA.121.022170
Xue B., Yu Y., Beltz T. G., Guo F., Wei S. G., Johnson A. K. (2022). Loss of the protective effect of estrogen contributes to maternal gestational hypertension-induced hypertensive response sensitization elicited by postweaning high-fat diet in female offspring. J. Am. Heart Assoc. 11, e023685. doi:10.1161/JAHA.121.023685
Xue B., Yu Y., Beltz T. G., Guo F., Wei S. G., Johnson A. K. (2022). Voluntary exercise eliminates maternal gestational hypertension-induced hypertensive response sensitization to postweaning high-fat diet in male adult offspring. Hypertension 79, 2016–2027. doi:10.1161/HYPERTENSIONAHA.122.19608
Xue B., Zhang Y., Johnson A. K. (2020). Interactions of the brain renin-angiotensin-system (RAS) and inflammation in the sensitization of hypertension. Front. Neurosci. 14, 650. doi:10.3389/fnins.2020.00650
Xue B., Zhang Z., Beltz T. G., Guo F., Hay M., Johnson A. K. (2014). Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am. J. Physiol. Heart Circ. Physiol. 307, H191–H198. doi:10.1152/ajpheart.01012.2013
Xue B., Zhang Z., Johnson R. F., Johnson A. K. (2012). Sensitization of slow pressor angiotensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment. Hypertension 59, 459–466. doi:10.1161/HYPERTENSIONAHA.111.185116
Xue Q., Xiao D., Zhang L. (2015). Estrogen regulates angiotensin II receptor expression patterns and protects the heart from ischemic injury in female rats. Biol. Reprod. 93, 6. doi:10.1095/biolreprod.115.129619
Yim H. E., Yoo K. H. (2015). Early life obesity and chronic kidney disease in later life. Pediatr. Nephrol. 30, 1255–1263. doi:10.1007/s00467-014-2922-4
Zhang Y. P., Huo Y. L., Fang Z. Q., Wang X. F., Li J. D., Wang H. P., et al. (2018). Maternal high-fat diet acts on the brain to induce baroreflex dysfunction and sensitization of angiotensin II-induced hypertension in adult offspring. Am. J. Physiol. Heart Circ. Physiol. 314, H1061–H1069. doi:10.1152/ajpheart.00698.2017
Keywords: sex differences, sensitization, blood pressure, prenatal programming, renin-angiotensin system, inflammation
Citation: Xue B, Johnson AK and Bassuk AG (2025) Sex differences in the sensitization of prenatally programmed hypertension. Front. Physiol. 16:1589615. doi: 10.3389/fphys.2025.1589615
Received: 07 March 2025; Accepted: 18 April 2025;
Published: 28 April 2025.
Edited by:
Carlos Fernandez-Patron, University of Alberta, CanadaReviewed by:
Fernando A. C. Seara, Federal University of Rio de Janeiro, BrazilPedro Lopez-Sanchez, Escuela Supeior de Medicina del Instituto Politecnico Nacional, Mexico
Copyright © 2025 Xue, Johnson and Bassuk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexander G. Bassuk, YWxleGFuZGVyLWJhc3N1a0B1aW93YS5lZHU=