 Ivan Lehman
Ivan Lehman Matija Matošević
Matija Matošević Lovro Lamot
Lovro Lamot Branka Bunoza
Branka Bunoza- 1School of Medicine, University of Zagreb, Zagreb, Croatia
- 2Divison of Pediatric Neurology, Department of Pediatrics, University Hospital Center Zagreb, Zagreb, Croatia
- 3Institute of Emergency Medicine of Zagreb County, Velika Gorica, Croatia
- 4Division of Pediatric Nephrology, Dialysis and Transplantation, Department of Pediatrics, University Hospital Center Zagreb, Zagreb, Croatia
Introduction: The development of novel treatment options and the implementation of newborn screening programs have significantly transformed the landscape of care for patients with spinal muscular atrophy (SMA). In a relatively short span, SMA has evolved from a debilitating and fatal disorder into a treatable condition, primarily due to advancements in gene-targeted therapies. Onasemnogene abeparvovec-xioi, an adeno-associated viral vector-based gene therapy delivering a functional copy of the SMN1 gene, has shown significant efficacy in improving motor function and survival rates. In Croatia, this therapy has been integrated into routine clinical practice for several years, providing valuable real-world data on its long-term outcomes and effectiveness. The presented case study aims to document these clinical experiences, contributing to the growing body of evidence supporting the efficacy and safety of onasemnogene abeparvovec-xioi and highlighting the crucial role of early diagnosis and intervention in SMA management.
Methods: We conducted a retrospective case series analysis of five pediatric patients diagnosed with SMA type 1, treated with onasemnogene abeparvovec-xioi at a tertiary care center in Croatia. Four patients presented with hypotonia and motor developmental delay, and one was identified through newborn screening. All patients had genetically confirmed SMA, underwent CHOP-INTEND (Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders) testing pre- and post-treatment, and were monitored for clinical response and adverse events. In addition, a systematic literature search was conducted using PubMed and Scopus databases to identify reports of pediatric SMA type 1 patients treated with onasemnogene abeparvovec. Keywords used included “onasemnogene abeparvovec” and “spinal muscular atrophy.” A total of 33 articles, describing 408 pediatric patients, were included.
Case report: We describe a series of five patients, four of which initially presented with varying degrees of hypotonia and delay in motor development, while one patient was discovered through newborn screening program. All patients received genetic confirmation of SMA, underwent Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) testing and received onasemnogene abeparvovec treatment. Four out of five patients achieved adequate clinical improvement as show by the increase in CHOP-INTEND score. One patient showed signs of regression and required additional care.
Conclusion: Despite the widespread use of novel treatment modalities that have drastically improved patient outcomes, there remains a paucity of real-world case reports documenting the care of SMA patients. This case series and accompanying literature review reinforce the efficacy and safety of onasemnogene abeparvovec in the treatment of SMA type 1, particularly when initiated early. In addition, our case series emphasize the critical role of newborn screening in identifying affected individuals before the onset of irreversible motor neuron loss as well as prompt start of treatment in all patients. We hope that our findings will contribute meaningfully to the expanding body of literature and knowledge on spinal muscular atrophy, ultimately fostering better patient care and outcomes.
Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disorder caused by mutations in the survival motor neuron 1 (SMN1) gene, which is located in the 5q13 region of chromosome 5, leading predominantly to the loss of lower motor neurons and cranial nerve nuclei in the brainstem, and resulting in progressive muscle weakness and atrophy (1–3). Due to the diverse clinical manifestations and varying age groups affected, SMA is clinically classified into five distinct types, based on the age of onset, the highest motor milestones achieved and life expectancy (Supplementary Table 1) (1–4). The most common type, SMA type I, typically presents during the first 6 months of age with hypotonia, delayed motor milestones, feeding impairment, inability to sit independently, and respiratory muscle involvement. This type affects almost half of SMA patients, usually resulting in death before age of two (2–4). The pathophysiological mechanism of SMA involves biallelic loss-of-function mutations in the SMN1 gene, predominantly due to a homozygous deletion of exon 7, which leads to the production of insufficient and nonfunctional SMN protein. The SMN2 gene, which shares high homology with SMN1, plays a contributory role in the pathogenesis of SMA since it can occasionally retain exon 7 during transcription, resulting in the production of full-length SMN protein that partially compensates for the deficits caused by SMN1 mutations (1–3, 5). Because of that unique property, SMN2 copy number plays a significant role in determining phenotype severity (2, 3, 5).
Until recently, the management of SMA primarily centered on supportive care, which encompassed respiratory and nutritional support in conjunction with physical therapy. In 2016 and 2017, the approval of nusinersen, a modified antisense oligonucleotide, opened new avenues for the management of SMA (6). Subsequently, two additional pharmacotherapies, onasemnogene abeparvovec-xioi and risdiplam, were introduced in 2019 and 2020 respectively, further expanding the therapeutic landscape (7, 8). Onasemnogene abeparvovec (OA), an adeno-associated viral vector-based gene therapy, developed to carry a functional copy of the gene encoding full-length SMN protein directly to motor neuron, was the first gene therapy approved as treatment for SMA in the United States (7). Soon after, Croatian Institute for Health Insurance, following European Medicine Agency approved the use of OA in Croatia, making a step forward in providing advanced treatment options for SMA patients in the country, vast majority of which are treated in a single referral center for pediatric neuromuscular disorders.
The objective of our manuscript is to evaluate the long-term efficacy and tolerability of onasemnogene abeparvovec (OA) in five patients with a genetic diagnosis of spinal muscular atrophy. This evaluation is complemented by a comprehensive systematic literature review, aiming to enhance the robustness and contextualization of our findings within the broader scientific framework.
Methods
Patients
Five pediatric patients (two males and three females) genetically confirmed to have biallelic SMN1 gene mutations with either 2 or 3 SMN2 gene copies and followed at the Division of Pediatric neurology, Department of Pediatrics, University Hospital Center Zagreb were included. All the patients received OA at a dose of 1.1 × 1014 vector genomes per kilogram of body weight between October 2021 and February 2023. Four patients were diagnosed clinically with confirmation through genetic testing. One pre-symptomatic patient was identified via the national newborn screening program in Croatia, which commenced on March 1, 2023.
Ethical approval and consent
Informed written consent was obtained from the parents of all patients following comprehensive counseling regarding the potential risks and benefits of OA treatment and alternative therapeutic options.
Treatment protocol
In alignment with the treatment protocol, all patients were administered prednisone at the equivalent of oral prednisolone dose of 1 mg per kilogram of body weight daily, starting 24 h prior to OA administration and continuing for approximately 30 days. Prednisolone was subsequently tapered over an additional 30-day period.
Clinical and laboratory assessments
Standardized clinical assessments appropriate for the patients’ ages were conducted by physiotherapists using the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND). Comprehensive laboratory evaluations were performed before and after OA therapy. These evaluations included complete blood cell counts, liver function tests (total bilirubin, aspartate aminotransferase [AST], alanine aminotransferase [ALT]), coagulation profiles, and cardiac tests (troponin-I and/or troponin-T levels). Detailed patient data are presented in Table 1.
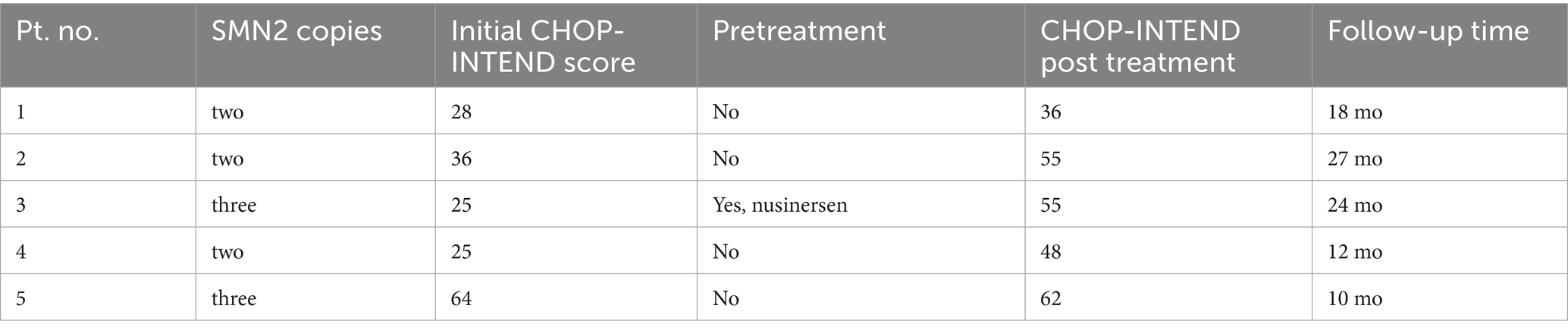
Table 1. Patient characteristics including initial number of SMN2 copies and CHOP-INTEND score compared with CHOP-INTEND score following treatment.
Systemic literature search
A systematic literature search of PudMed and Scopus database was performed to identify pediatric patients with spinal muscular atrophy type 1 that were treated with onasemnogene abeparvovec. The search was conducted by entering the keywords “onasemnogene abeparvovec” and “spinal muscular atrophy” with no time or language constraint, in accordance with the published guidance on narrative reviews. Initial search of both SCOPUS and PubMed/MEDLINE databases yielded 776 articles. In the first round of exclusions, after reviewing all the abstracts, reviews, editorials, comment and pharmacoeconomics articles were excluded as well as articles that were unavailable for retrieval. We assessed 144 full-text articles and further excluded articles that did not describe the use of onasemnogene abeparvovec, articles that contained no patient descriptions, where patients did not have SMA type 1 and were adults. We also excluded articles that contained no data pertaining to the improvement of the patients described. Our search produced 33 papers, describing 408 pediatric patients diagnosed with spinal muscular atrophy type 1 treated with onasemnogene abeparvovec. Details of selected papers can be found in Table 2, while PRISM CHART is depicted in Figure 1.
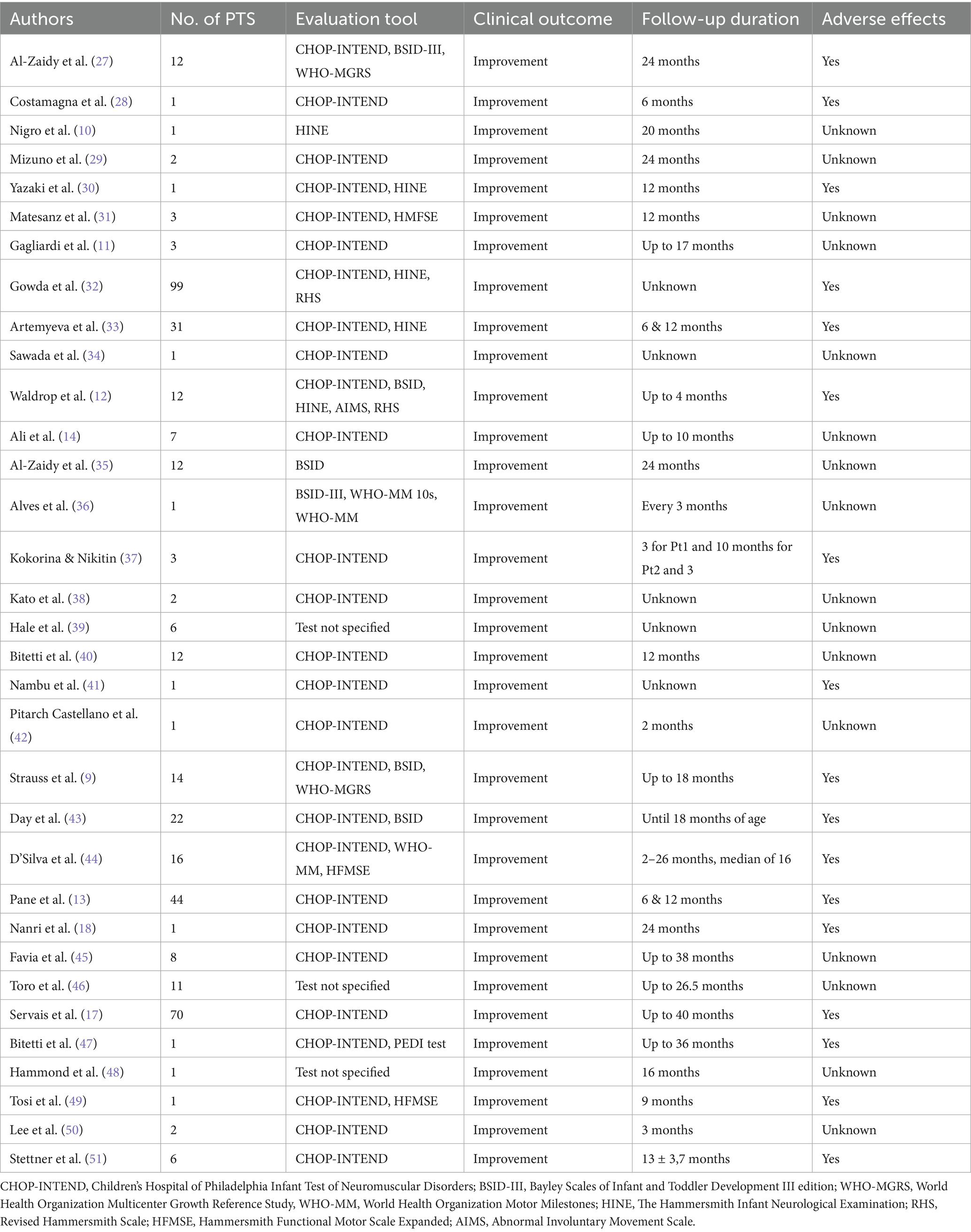
Table 2. Condensed overview of notable findings, outcomes and adverse effects in patients treated with onasemnogene abeparvovec gene replacement therapy for SMA type 1 discovered through literature review.
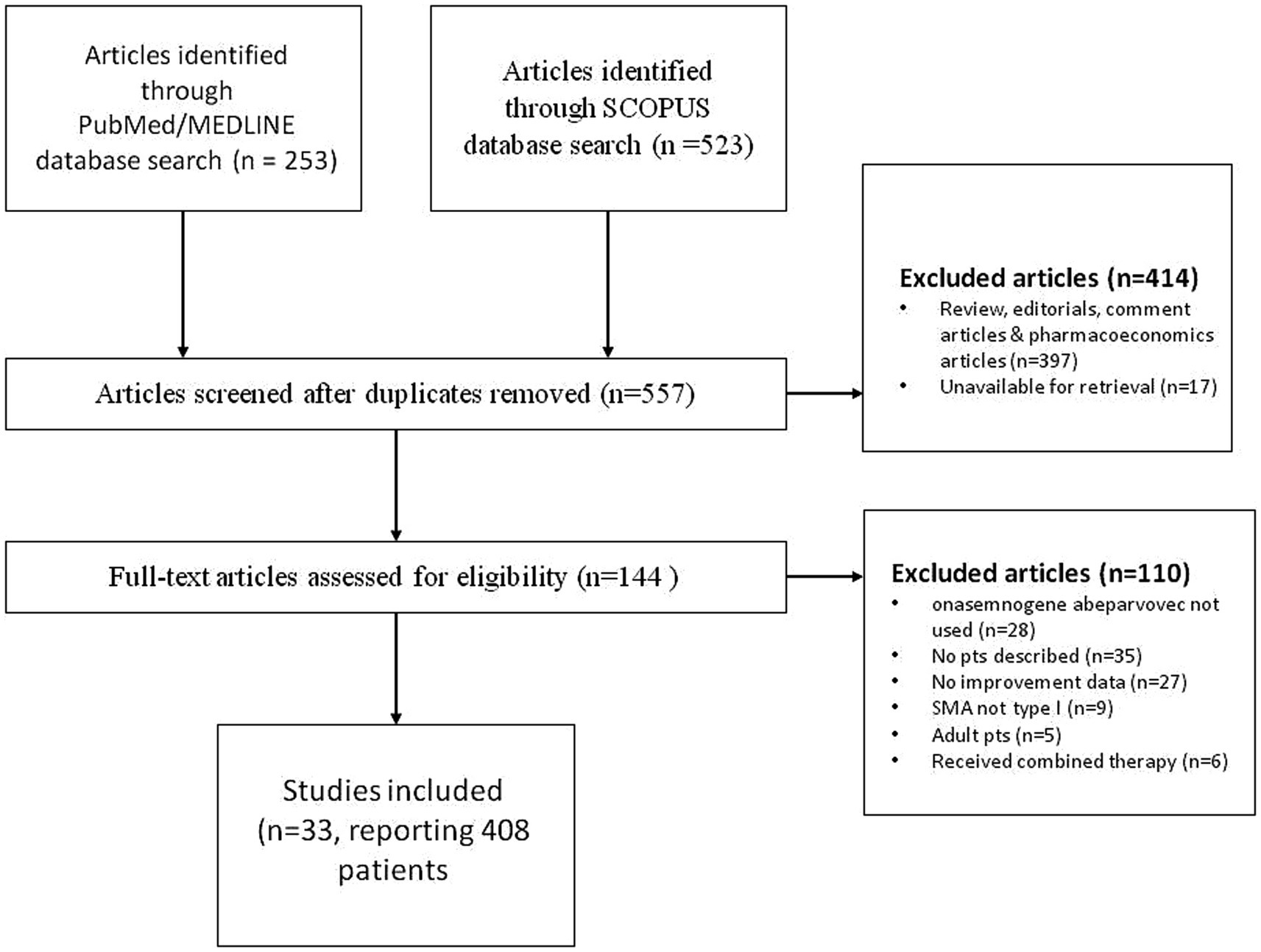
Figure 1. PRISM flowchart of literature review.
Case report
Patient 1
Patient 1 (F) manifested the first signs of hypotonia at the age of 3 weeks and was diagnosed with SMA type 1 with two copies of SMN2 at the age of 3 months. At that age she presented with severe truncal and limb hypotonia and intercostal muscle weakness. Non-invasive ventilation (NIV) was used for 5 h during sleeping. Her initial CHOP-INTEND score was 28/64. She was treated with OA at the age of 116 days. Two days after the treatment, she developed vomiting and diarrhea. Liver enzymes (AST and ALT) were elevated, but not over 2x ULN, at the end of the first week, with preserved liver function. The lowest platelet count was 47 at the end of the first week. The corticosteroid dose was increased to 2 mg/kg of prednisone for 10 days. CHOP-INTEND scores measured after OA treatment were 31, 34, 36 and 38 at 3, 6, 12 and 16 months follow-up, respectively. 11 months after receiving OA, she developed a severe respiratory infection, which required tracheotomy and mechanical ventilation. Following the resolution of infection, only nighttime mechanical ventilation was required. At 18 months, the follow-up CHOP-INTEND score showed a decline, with 36 points. She developed motor deterioration with swallowing difficulties and food aspiration as well as chest deformation. She did not meet motor development milestone of sitting without support. Eventually, add-on therapy with risdiplam was introduced at the age of 28 months. At the age of 42 months she is being fed via percutaneous endoscopic gastrostomy (PEG), is mechanically ventilated during the night and has scoliosis and poor head control.
Patient 2
Patient 2 (F) was diagnosed with SMA type 1 with two copies of SMN2 at the age of 3 months. She initially presented at the age of 2 months with hypotonia, inability to raise upper and lower extremities upward, no head control and inability to raise head in prone position. Her initial CHOP-INTEND score at the age of 3 months was 36/64. She was treated with OA at age of 142 days. The patient was febrile the second day and vomited for 5 days. Liver enzymes were elevated, highest on day 4, AST 3.8xULN and ALT 2.8x ULN. She developed thrombocytopenia with the lowest platelet count 50 on day 7. The corticosteroid dose was increased to 2 mg/kg of prednisone. At her 1-month follow-up, her CHOP-INTEND score was 43. This upward trend continued at subsequent follow-ups at 3, 6, 12 and 24 months, with score being 44, 49, 54 and 55 points, respectively. She started to sit at the age of 16 months. At the last visit at 43 months of age, she attained sitting for 5 s without support, head control in sitting position but no control in traction. She was also able to keep legs flexed at knees, perform abduction and adduction and elevate the hands to the level of the ears. Bulbar movements were normal, but facial muscles showed mild weakness without fasciculations. She developed severe thoracic scoliosis. Parents later started risdiplam treatment at 23 months at their own expense.
Patient 3
Patient 3 (M) was diagnosed with SMA type 1 with three copies of SMN2 at the age of 7 months after initially presenting with signs of hypotonia at 2 months of age. He was primarily treated with 4 doses of nusinersen, with the last dose being at the age of 11 months, after which OA treatment was initiated at 430 days of age. In the moment of OA administration, he was able to sit with support but without head control in traction. He was able to raise his upper extremities but could not raise his lower extremities. He was febrile and vomited in the first week after the drug application. He showed elevation of liver enzymes, with peak being at 5 days after treatment. AST was 4.7xULN and ALT 3.6xULN with preserved liver function. Platelets were lowest on the same day, measuring at 99. After that, laboratory results returned to normal values after 2 weeks. Eventually, corticosteroids were tapered off after 3 months. Four months after application there was a second elevation of liver enzymes (AST 1.6xULN and ALT 2.9xULN), and corticosteroids were introduced again for the next 3 months. CHOP-INTEND score pre-OA treatment was 25 at 8 months of age, before nusinersen treatment and 45 at 14 months after nusinersen treatment. The positive trend of CHOP-INTEND score increase continued, with the scores being 47, 50, 53 and 55 at 3, 6, 12 and 24-month follow-ups. He started to sit independently at the age of 20 months. Neurological exam at 2 years old showcased ability to sit without support for more than a minute and raise upper extremities above shoulder level. He also exhibited thoracic kyphosis and thoracolumbar scoliosis. He was able to stand with support and push wheelchair by himself.
Patient 4
Patient 4 (M) was diagnosed with SMA type 1 with two copies of SMN2 gene at the age of 2 months after presenting with progressive hypotonia and weakness at 1 month of age, tongue fasciculation and weakness of intercostal muscles with areflexia. Initial CHOP-INTEND score was 20. He received OA at the age of 67 days. Granulocytopenia with positive antigranulocyte antibodies was noticed before application OA with ANC of 0.58 that got more severe during the first week after drug application, with the lowest ANC being 0.19. Granulocytopenia resolved after 4 months. During the first week after treatment there was elevation of AST and ALT below 2xULN as well as mild thrombocytopenia of 167 that both resolved afterwards CHOP-INTEND scores were 23, 35, 43 and 48 on follow-up at 1, 3, 6 and 12 months, respectively. He started to sit independently at the age of 15 months and was able to stand with support at the age of 20 months. Language skills were developing adequately, with vocabulary consisting of few words and adequate nonverbal communication.
Patient 5
Patient 5 (F) was diagnosed with SMA type 1 with three copies of SMN2 through newborn screening. Her initial CHOP-INTEND score was 64. She was treated as a pre-symptomatic patient and received OA at age of 26 days. During the first week there was elevation of AST and ALT below 2xULN and mild thrombocytopenia of 114 platelets. She also had an elevation of troponin I level to 128.4 ng/L on day 7 (ref. 0–15.6 ng/L) with a normal heart ultrasound and ECG. Her psychomotor development was normal and without developmental delays. She walks independently at the age of 11 months. Her social development was also normal. She did not show any signs of disease.
The detailed characteristics and outcome of patients is showed in Table 1 while CHOP-INTEND Score Trajectories following onasemnogene abeparvovec treatment are graphically displayed in Figure 2 and Supplementary Table 2.
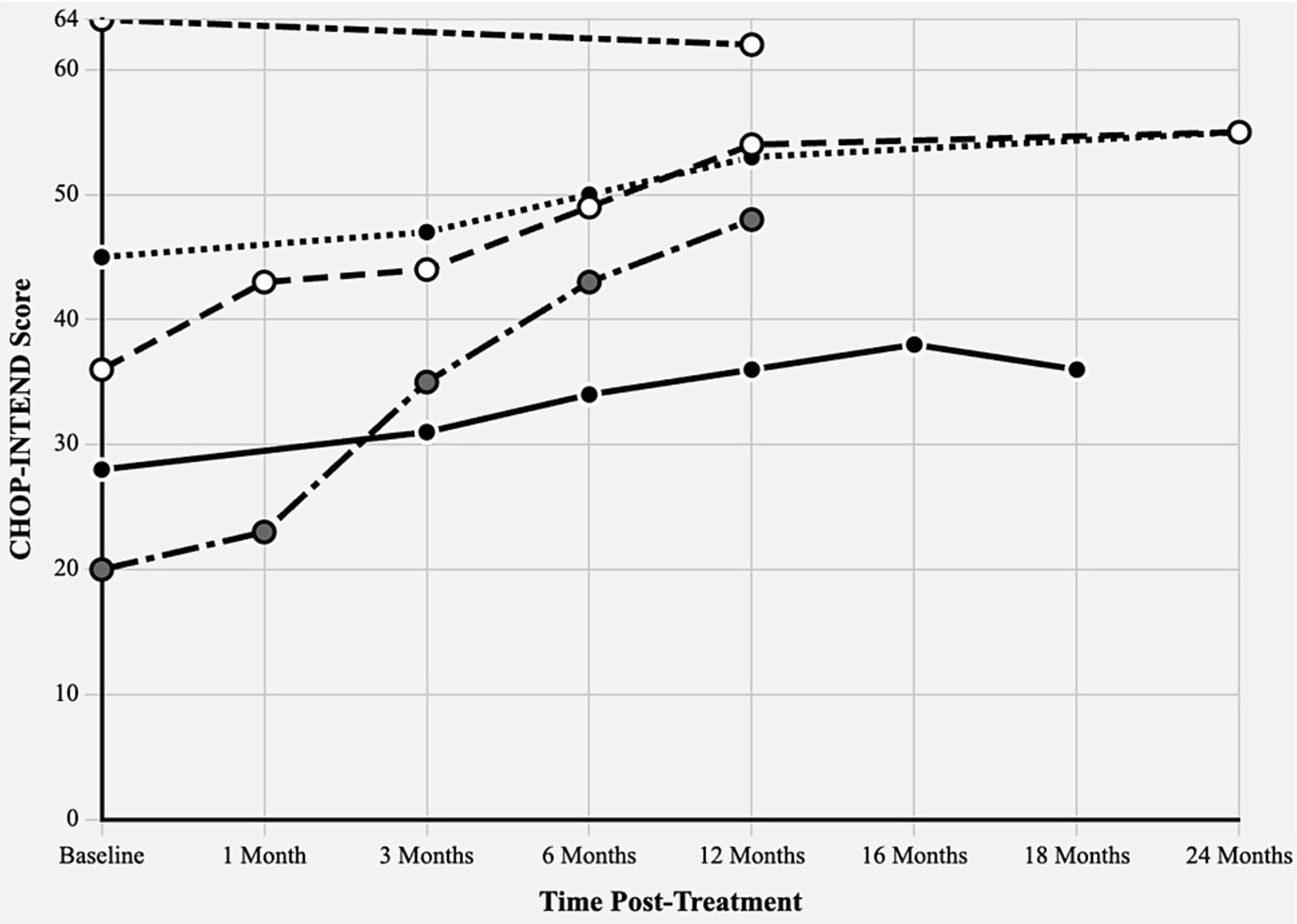

Figure 2. CHOP-INTEND score trajectories following onasemnogene abeparvovec treatment. Children’s hospital of Philadelphia infant test of neuromuscular disorders (CHOP-INTEND) scores from baseline through 24 months post-treatment in five Croatian SMA patients. Maximum possible score: 64 points. Different line patterns represent individual patients with varying SMN2 copy numbers and treatment ages.
Results
Our literature review produced a selection of case reports and studies pertaining to treatment of children with SMA type 1 using onasemnogene abeparvovec for comparison (Supplementary Table 3). The number of patients in every paper noted in our literature review varied greatly, from single case reports to multicenter studies done in university clinical centers. Most commonly used evaluation tool was CHOP-INTEND, often paired with other tests such as The Bayley Scales of Infant and Toddler Development III edition (BSID-III), World Health Organization Multicenter Growth Reference Study (WHO-MGRS), World Health Organization Motor Milestones (WHO-MM) The Hammersmith Infant Neurological Examination (HINE), Revised Hammersmith Scale (RHS), Hammersmith Functional Motor Scale Expanded (HFMSE) and Abnormal Involuntary Movement Scale (AIMS) In two papers, by Hale et al. and Hammond et al., evaluation tools for observing motor milestones and motor development of the patients were not specified. All patients showed clinical improvement upon receiving treatment with OA. The follow-up duration varied greatly, ranging from 2 months up to 40 months. Seventeen out of 33 papers reported some kind of adverse effect with most prevalent being pyrexia and transaminases increase.
Discussion
We presented a single center retrospective cohort analysis of real-world data in treatment of children with SMA type 1 using onasemnogene abeparvovec. In our cohort all children showed clinical improvement, as shown by the increase in CHOP-INTEND score during follow-up visits. Nevertheless, one patient eventually showed deterioration of motor and bulbar functions and a need for mechanical ventilation during sleep. The best outcome of treatment was exhibited by our patient who was diagnosed through a newborn screening program and has three copies of SMN2 gene, which enabled us to treat her at the age of 26 days, before the manifestation of clinical signs of disease. The child reached the milestone of walking independently at the appropriate age. This is comparable with studies showcasing treatment in pre-symptomatic patients within the first weeks of life, which results in favorable or even normal motor function in children with SMA (9–14). Considering that early interventions result in better outcome, a significant step forward in the direction of early treatment was achieved with implementation of newborn screening (15, 16). Even though newborn screening is relatively new in Croatia, our patient greatly benefited from early treatment that was undertaken due to the screening program. Our one patient identified through newborn screening was born in 2023, coinciding with the implementation of SMA newborn screening in Croatia. The remaining four patients were born prior to 2023 and were diagnosed based on clinical presentation. Since the establishment of newborn screening in Croatia in 2023, the program has identified 10 new SMA cases nationally. We deliberately excluded additional newborn screening-identified patients from this case series due to insufficient follow-up duration to assess meaningful clinical outcomes. However, we agree that expanding newborn screening programs internationally represents a critical public health priority, as evidenced by the superior baseline status of our screening-identified patient.
Four out of five children, three of which were diagnosed with SMA type 1, reached the milestone of sitting independently. Similar results of almost 50% or more patients achieving this milestone were presented in studies by Strauss et al. and Servais et al. respectively (9, 17). Patient 3, with three SMN2 copies received four doses of nusinersen prior to beginning treatment with OA at the age of 14 months and continued to improve after the switch. Only he had a second elevation of aminotransferase after discontinuation of corticosteroid treatment. Patient 1 did not achieve a CHOP-INTEND score greater than 40. In addition, she also required tracheostomy 11 months after treatment and mechanical ventilation during the night. It is a child with SMA type 1a who had severe symptoms at the age of 2 months and was severely affected by infection at the age of 11 month. Risdiplam therapy was initiated at the age of 28 months after the deterioration of bulbar motor functions. Combined therapy of nusinersen with OA and risdiplam with OA might have some benefits and is considered safe (18–21). Regarding patient 2, the parent opted for combined therapy of onasemnogene abeparvovec and risdiplam which they procured on their own expense. Due to the popularity of crowdfunding platforms such as GoFundMe or Kickstarter, a viable option for acquiring monetary means for treatment is a crowdfunding campaing (22). However, these types of funding platforms are still relatively unpopular in Croatia, with parents usually depending on their own income or funds raise through various charity organizations and events.
OA is mostly well tolerated in both pre-symptomatic and symptomatic patients, with adverse effect usually consisting of pyrexia, vomiting, loss of appetite and transaminases increase (9, 13, 14, 17). All our patients had an elevation of transaminase levels without liver function deterioration and drop in platelet count without clinical significance. These adverse effects were transitory. One patient had worsening of autoimmune granulocytopenia. In patient 3, who was the oldest at the time of treatment and who was previously treated with nusinersen, there was another elevation of transaminases after discontinuation of corticosteroid therapy that resolved with restarting of corticosteroid therapy. Patient 5 had transitory asymptomatic troponin I elevation. It is important to mention that OA administration can cause some serious adverse events, including hepatic failure and thrombotic microangiopathy, as well as an increase in heart enzymes levels such as troponin-I and troponin-T (23–26).
Conclusion
Spinal muscular atrophy (SMA) poses a significant healthcare challenge due to its progressive nature and the diverse pediatric population it affects. Prior to the introduction of disease-modifying treatments, there was no effective way to combat this debilitating disease. This case series highlights the complexities and urgency involved in decision-making for treating a life-threatening condition like SMA, where timely intervention can critically impact outcomes. The intricate nature of the disease and its treatments necessitates careful consideration of the most appropriate therapy for each patient. Consequently, case reports such as ours are invaluable, providing insights and education for healthcare providers on various treatment and follow-up approaches. These contributions enrich the existing literature and support informed decision-making in the management of SMA.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by University Hospital Center Zagreb Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
IL: Writing – review & editing, Writing – original draft, Data curation, Conceptualization, Validation, Supervision. MM: Visualization, Investigation, Writing – original draft, Data curation. LL: Writing – original draft, Methodology, Resources, Formal analysis, Visualization, Data curation, Conceptualization, Validation, Supervision, Writing – review & editing. BB: Methodology, Conceptualization, Investigation, Validation, Writing – review & editing, Supervision, Visualization, Resources, Formal analysis, Project administration, Writing – original draft, Software.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2025.1609072/full#supplementary-material
References
1. Wirth, B. Spinal muscular atrophy: in the challenge lies a solution. Trends Neurosci. (2021) 44:306–22. doi: 10.1016/j.tins.2020.11.009
2. Nicolau, S, Waldrop, MA, Connolly, AM, and Mendell, JR. Spinal muscular atrophy. Semin Pediatr Neurol. (2021) 37:100878. doi: 10.1016/j.spen.2021.100878
3. Arnold, ES, and Fischbeck, KH. Spinal muscular atrophy In: Handbook of clinical neurology. Handbook of Clinical Neurology, Vol. 148. Neurogenetics, Part II. Eds. D. H. Geschwind, H. L. Paulson, and C. Klein. (Amsterdam, Netherlands: Elsevier) (2018). 591–601.
4. Nishio, H, Niba, ETE, Saito, T, Okamoto, K, Takeshima, Y, and Awano, H. Spinal muscular atrophy: the past, present, and future of diagnosis and treatment. Int J Mol Sci. (2023) 24:1–38. doi: 10.3390/ijms241511939
5. Keinath, MC, Prior, DE, and Prior, TW. Spinal muscular atrophy: mutations, testing, and clinical relevance. Appl Clin Genet. (2021) 14:11–25. doi: 10.2147/TACG.S239603
6. Hoy, SM. Nusinersen: first global approval. Drugs. (2017) 77:473–9. doi: 10.1007/s40265-017-0711-7
7. Hoy, SM. Onasemnogene Abeparvovec: First Global Approval. Drugs. (2019) 79:1255–62. doi: 10.1007/s40265-019-01162-5
9. Strauss, KA, Farrar, MA, Muntoni, F, Saito, K, Mendell, JR, Servais, L, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the phase III SPR1NT trial. Nat Med. (2022) 28:1381–9. doi: 10.1038/s41591-022-01866-4
10. Nigro, E, Grunebaum, E, Kamath, B, Licht, C, Malcolmson, C, Jeewa, A, et al. Case report: a case of spinal muscular atrophy in a preterm infant: risks and benefits of treatment. Front Neurol. (2023) 14:1–7. doi: 10.3389/fneur.2023.1230889
11. Gagliardi, D, Canzio, E, Orsini, P, Conti, P, Sinisi, V, Maggiore, C, et al. Early spinal muscular atrophy treatment following newborn screening: a 20-month review of the first Italian regional experience. Ann Clin Transl Neurol. (2024) 11:1090–6. doi: 10.1002/acn3.52018
12. Waldrop, MA, Karingada, C, Storey, MA, Powers, B, Iammarino, MA, Miller, NF, et al. Gene therapy for spinal muscular atrophy: safety and early outcomes. Pediatrics. (2020) 146:146. doi: 10.1542/PEDS.2020-0729
13. Pane, M, Berti, B, Capasso, A, Coratti, G, Varone, A, D’Amico, A, et al. Onasemnogene abeparvovec in spinal muscular atrophy: predictors of efficacy and safety in naïve patients with spinal muscular atrophy and following switch from other therapies. eClinicalMedicine. (2023) 59:101997. doi: 10.1016/j.eclinm.2023.101997
14. Ali, HG, Ibrahim, K, Elsaid, MF, Mohamed, RB, Abeidah, MIA, Al Rawwas, AO, et al. Gene therapy for spinal muscular atrophy: the Qatari experience. Gene Ther. (2021) 28:676–80. doi: 10.1038/s41434-021-00273-7
15. Butterfield, RJ. Spinal muscular atrophy treatments, newborn screening, and the creation of a Neurogenetics urgency. Semin Pediatr Neurol. (2021) 38:100899. doi: 10.1016/j.spen.2021.100899
16. Aragon-Gawinska, K, Mouraux, C, Dangouloff, T, and Servais, L. Spinal muscular atrophy treatment in patients identified by newborn screening—a. Systematic Rev Genes. (2023) 14:14. doi: 10.3390/genes14071377
17. Servais, L, Day, JW, De Vivo, DC, Kirschner, J, Mercuri, E, Muntoni, F, et al. Real-world outcomes in patients with spinal muscular atrophy treated with Onasemnogene Abeparvovec monotherapy: findings from the RESTORE registry. J Neuromuscul Dis. (2024) 11:425–42. doi: 10.3233/JND-230122
18. Nanri, D, Yuge, K, Goto, K, Kimura, T, Yae, Y, Mizuochi, T, et al. Onasemnogene Abeparvovec treatment after Nusinersen in an infant with spinal muscular atrophy type 1. Kurume Med J. (2024) 69:255–9. doi: 10.2739/kurumemedj.MS6934008
19. Harada, Y, Rao, VK, Arya, K, Kuntz, NL, DiDonato, CJ, Napchan-Pomerantz, G, et al. Combination molecular therapies for type 1 spinal muscular atrophy. Muscle Nerve. (2020) 62:550–4. doi: 10.1002/mus.27034
20. Oechsel, KF, and Cartwright, MS. Combination therapy with onasemnogene and risdiplam in spinal muscular atrophy type 1. Muscle Nerve. (2021) 64:487–90. doi: 10.1002/mus.27375
21. Chiriboga, CA, Bruno, C, Duong, T, Fischer, D, Mercuri, E, Kirschner, J, et al. JEWELFISH: 24-month results from an open-label study in non-treatment-naïve patients with SMA receiving treatment with risdiplam. J Neurol. (2024) 271:4871–84. doi: 10.1007/s00415-024-12318-z
22. Elbashir, A, Nugud, A, and Elbashir, H. Crowdfunding approach for gene therapy: experience from the UAE. Pediatr Neonatol. (2022) 63:567–8. doi: 10.1016/j.pedneo.2022.07.006
23. Feldman, AG, Parsons, JA, Dutmer, CM, Veerapandiyan, A, Hafberg, E, Maloney, N, et al. Subacute liver failure following gene replacement therapy for spinal muscular atrophy type 1. J Pediatr. (2020) 225:252–258.e1. doi: 10.1016/j.jpeds.2020.05.044
24. Chand, D, Mohr, F, McMillan, H, Tukov, FF, Montgomery, K, Kleyn, A, et al. Hepatotoxicity following administration of onasemnogene abeparvovec (AVXS-101) for the treatment of spinal muscular atrophy. J Hepatol. (2021) 74:560–6. doi: 10.1016/j.jhep.2020.11.001
25. Guillou, J, de Pellegars, A, Porcheret, F, Frémeaux-Bacchi, V, Allain-Launay, E, Debord, C, et al. Fatal thrombotic microangiopathy case following adeno-associated viral SMN gene therapy. Blood Adv. (2022) 6:4266–70. doi: 10.1182/bloodadvances.2021006419
26. Chand, DH, Sun, R, Diab, KA, Kenny, D, and Tukov, FF. Review of cardiac safety in onasemnogene abeparvovec gene replacement therapy: translation from preclinical to clinical findings. Gene Ther. (2023) 30:685–97. doi: 10.1038/s41434-023-00401-5
27. Al-Zaidy, SA, Kolb, SJ, Lowes, L, Alfano, LN, Shell, R, Church, KR, et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J Neuromuscul Dis. (2019) 6:307–17. doi: 10.3233/JND-190403
28. Costamagna, G, Govoni, A, Wise, A, and Corti, S. Bridging the gap: gene therapy in a patient with spinal muscular atrophy type 1. Neurology. (2022) 99:952–6. doi: 10.1212/WNL.0000000000201294
29. Mizuno, T, Kanouchi, T, Tamura, Y, Hirata, K, Emoto, R, Suzuki, T, et al. Changes in electrophysiological findings of spinal muscular atrophy type I after the administration of nusinersen and onasemnogene abeparvovec: two case reports. BMC Neurol. (2023) 23:392. doi: 10.1186/s12883-023-03420-2
30. Yazaki, K, Sakuma, S, Hikita, N, Fujimaru, R, and Hamazaki, T. Child neurology: pathologically confirmed thrombotic Microangiopathy caused by Onasemnogene Abeparvovec treatment for SMA. Neurology. (2022) 98:808–13. doi: 10.1212/WNL.0000000000200676
31. Matesanz, SE, Battista, V, Flickinger, J, Jones, JN, and Kichula, EA. Clinical experience with gene therapy in older patients with spinal muscular atrophy. Pediatr Neurol. (2021) 118:1–5. doi: 10.1016/j.pediatrneurol.2021.01.012
32. Gowda, V, Atherton, M, Murugan, A, Servais, L, Sheehan, J, Standing, E, et al. Efficacy and safety of onasemnogene abeparvovec in children with spinal muscular atrophy type 1: real-world evidence from 6 infusion centres in the United Kingdom. Lancet Reg Health - Eur. (2024) 37:100817. doi: 10.1016/j.lanepe.2023.100817
33. Artemyeva, SB, Papina, YO, Shidlovskaya, OA, Monakhova, AV, and Vlodavets, DV. Experience of using gene replacement therapy with Zolgensma® (onasemnogene abeparvovec) in real clinical practice in Russia. Neuromuscul Disord. (2022) 12:29–38. doi: 10.17650/2222-8721-2022-12-1-29-38
34. Sawada, T, Kido, J, Yae, Y, Yuge, K, Nomura, K, Okada, K, et al. Gene therapy for spinal muscular atrophy is considerably effective when administered as early as possible after birth. Mol Genet Metab Rep. (2023) 35:100973. doi: 10.1016/j.ymgmr.2023.100973
35. Al-Zaidy, S, Pickard, AS, Kotha, K, Alfano, LN, Lowes, L, Paul, G, et al. Health outcomes in spinal muscular atrophy type 1 following AVXS-101 gene replacement therapy. Pediatr Pulmonol. (2019) 54:179–85. doi: 10.1002/ppul.24203
36. Alves, CRR, Petrillo, M, Spellman, R, Garner, R, Zhang, R, Kiefer, M, et al. Implications of circulating neurofilamentsfor spinal muscular atrophytreatment early in life: a case series. Mol Ther. (2021) 23:524–38. doi: 10.1016/j.omtm.2021.10.011
37. Kokorina, AA, and Nikitin, SS. Interim analysis of treatment outcomes of young children with 5q spinal muscular atrophy on gene replacement therapy with onasemnogene abeparvovec. Clinical observations. Vopr Sovrem Pediatr - Curr Pediatr. (2022) 21:535–47. doi: 10.15690/vsp.v21i6S.2497
38. Kato, T, Yokomura, M, Urano, M, Sato, Y, Ashihara, Y, Ito, M, et al. Two cases of spinal muscular atrophy type I that received early treatment and achieved improved prognosis by promoting multi-center, multi-professional collaboration after prenatal diagnosis. No To Hattatsu. (2023) 55:443–7. doi: 10.11251/ojjscn.55.443
39. Hale, JE, Darras, BT, Swoboda, KJ, Estrella, E, Chen, JYH, Abbott, M-A, et al. Massachusetts’ findings from statewide newborn screening for spinal muscular atrophy. Int J Neonatal Screen. (2021) 7:1–11. doi: 10.3390/ijns7020026
40. Bitetti, I, Manna, MR, Stella, R, and Varone, A. Motor and neurocognitive profiles of children with symptomatic spinal muscular atrophy type 1 with two copies of SMN2 before and after treatment: a longitudinal observational study. Front Neurol. (2024) 15:1–12. doi: 10.3389/fneur.2024.1326528
41. Nambu, Y, Awano, H, Bo, R, Hong, S, Nishio, H, and Iijima, K. Treatment of a 50-day-old Japanese infant with spinal muscular atrophy type 1 using onasemnogene abeparvovec. No To Hattatsu. (2022) 54:262–5. doi: 10.11251/ojjscn.54.262
42. Pitarch Castellano, I, López Briz, E, Ibáñez Albert, E, Aguado Codina, C, Sevilla, T, and Poveda Andrés, JL. Onasemnogene abeparvovec administration via peripherally inserted central catheter: a case report. Children (Basel). (2024) 11:1–5. doi: 10.3390/children11050590
43. Day, JW, Finkel, RS, Chiriboga, CA, Connolly, AM, Crawford, TO, Darras, BT, et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. (2021) 20:284–93. doi: 10.1016/S1474-4422(21)00001-6
44. D’Silva, AM, Holland, S, Kariyawasam, D, Herbert, K, Barclay, P, Cairns, A, et al. Onasemnogene abeparvovec in spinal muscular atrophy: an Australian experience of safety and efficacy. Ann Clin Transl Neurol. (2022) 9:339–50. doi: 10.1002/acn3.51519
45. Favia, M, Tarantino, D, Cerbo, LD, Sabia, A, Campopiano, R, and Pani, M. Onasemnogene Abeparvovec: post-infusion efficacy and safety in patients with spinal muscular atrophy (SMA)—a Fondazione Policlinico Gemelli IRCCS experience. Hosp Pharm. (2024) 59:39–46. doi: 10.1177/00185787231182562
46. Toro, W, Yang, M, Georgieva, M, Anderson, A, LaMarca, N, Patel, A, et al. Patient and caregiver outcomes after Onasemnogene Abeparvovec treatment: findings from the Cure SMA 2021 membership survey. Adv Ther. (2023) 40:5315–37. doi: 10.1007/s12325-023-02685-w
47. Bitetti, I, Manna, MR, Stella, R, and Varone, A. Sequential treatment with nusinersen, Zolgensma® and risdiplam in a paediatric patient with spinal muscular atrophytype 1: a case report. Acta Myol. (2023) 42:82–5. doi: 10.36185/2532-1900-356
48. Hammond, CK, Oppong, E, Ameyaw, E, and Dogbe, JA. Spinal muscular atrophy in Ghanaian children confirmed by molecular genetic testing: a case series. Pan Afr Med J. (2023) 46:78. doi: 10.11604/pamj.2023.46.78.32240
49. Tosi, M, Catteruccia, M, Cherchi, C, Mizzoni, I, and D’Amico, A. Switching therapies: safety profile of Onasemnogene abeparvovec-xioi in a SMA1 patient previously treated with Risdiplam. Acta Myol. (2022) 41:117–20. doi: 10.36185/2532-1900-077
50. Lee, BH, Waldrop, MA, Connolly, AM, and Ciafaloni, E. Time is muscle: a recommendation for early treatment for preterm infants with spinal muscular atrophy. Muscle Nerve. (2021) 64:153–5. doi: 10.1002/mus.27261
Keywords: onasemnogene abeparvovec, spinal muscle atrophy, CHOP INTEND, SMA type 1, gene therapy
Citation: Lehman I, Matošević M, Lamot L and Bunoza B (2025) Real-world outcomes of spinal muscular atrophy treatment with onasemnogene abeparvovec in Croatia: a comprehensive case series and literature review. Front. Med. 12:1609072. doi: 10.3389/fmed.2025.1609072
Edited by:
Daniele Bottai, University of Milan, ItalyReviewed by:
Ahmed Abd Alwahab Nugud, Sheikh Khalifa Medical City, United Arab EmiratesHuang Jianliang, Zhangjiajie Hospital Affiliated to Hunan Normal University, China
Copyright © 2025 Lehman, Matošević, Lamot and Bunoza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matija Matošević, bWF0aWphLm1hdG9zZXZpY0BnbWFpbC5jb20=