 Jyotsna Mishra1‡
Jyotsna Mishra1‡ Kyle Bevers1†‡
Kyle Bevers1†‡ Keguo Li1Armaan Zare1†James S. Heisner1Ailing Tong1
Keguo Li1Armaan Zare1†James S. Heisner1Ailing Tong1 Wai-Meng Kwok1,2,3,4
Wai-Meng Kwok1,2,3,4 David F. Stowe1,3,5,6
David F. Stowe1,3,5,6 Amadou K. S. Camara1,3,4,5*
Amadou K. S. Camara1,3,4,5*- 1Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, WI, United States
- 2Department of Pharmacology and Toxicology, Medical College of Wisconsin, Milwaukee, WI, United States
- 3Cardiovascular Center, Medical College of Wisconsin, Milwaukee, WI, United States
- 4Cancer Center, Medical College of Wisconsin, Milwaukee, WI, United States
- 5Department of Physiology, Medical College of Wisconsin, Milwaukee, WI, United States
- 6Department of Biomedical Engineering, Medical College of Wisconsin and Marquette University, Milwaukee, WI, United States
Mitochondria regulate intracellular calcium ion (Ca2+) signaling by a fine-tuned process of mitochondrial matrix (m) Ca2+ influx, mCa2+ buffering (sequestration) and mCa2+ release (Ca2+ efflux). This process is critically important in the neurosynaptic terminal, where there is a simultaneous high demand for ATP utilization, cytosolic (c) Ca2+ regulation, and maintenance of ionic gradients across the cell membrane. Brain synaptic and non-synaptic mitochondria display marked differences in Ca2+ retention capacity. We hypothesized that mitochondrial Ca2+ handling in these two mitochondrial populations is determined by the net effects of Ca2+ uptake, buffering or efflux with increasing CaCl2 boluses. We found first that synaptic mitochondria have a more coupled respiration than non-synaptic mitochondria; this may correlate with the higher local energy demand in synapses to support neurotransmission. When both mitochondrial fractions were exposed to increasing mCa2+ loads we observed decreased mCa2+ sequestration in synaptic mitochondria as assessed by a significant increase in the steady-state free extra matrix Ca2+ (ss[Ca2+]e) compared to non-synaptic mitochondria. Since, non-synaptic mitochondria displayed a significantly reduced ss[Ca2+]e, this suggested a larger mCa2+ buffering capacity to maintain [Ca2+]m with increasing mCa2+ loads. There were no differences in the magnitude of the transient depolarizations and repolarizations of the membrane potential (ΔΨm) and both fractions exhibited similar gradual depolarization of the baseline ΔΨm during additional CaCl2 boluses. Adding the mitochondrial Na+/Ca2+ exchanger (mNCE) inhibitor CGP37157 to the mitochondrial suspensions unmasked the mCa2+ sequestration and concomitantly lowered ss[Ca2+]e in synaptic vs. non-synaptic mitochondria. Adding complex V inhibitor oligomycin plus ADP (OMN + ADP) bolstered the matrix Ca2+ buffering capacity in synaptic mitochondria, as did Cyclosporin A (CsA), in non-synaptic. Our results display distinct differences in regulation of the free [Ca2+]m to prevent collapse of ΔΨm during mCa2+ overload in the two populations of mitochondria. Synaptic mitochondria appear to rely mainly on mCa2+ efflux via mNCE, while non-synaptic mitochondria rely mainly on Pi-dependent mCa2+ sequestration. The functional implications of differential mCa2+ handling at neuronal synapses may be adaptations to cope with the higher metabolic activity and larger mCa2+ transients at synaptosomes, reflecting a distinct role they play in brain function.
1 Introduction
Brain mitochondria (m) orchestrate diverse functions including energy transduction (oxidative phosphorylation), neuronal excitability and synaptic neurotransmission and its regulation. A key component in these processes is the interplay of cytosolic (c) and mCa2+ dynamics. Mitochondria contribute to the shaping of cCa2+ transients by regulating mCa2+ uptake, buffering and release. In this process, mitochondria regulate energy metabolism, synaptic activity, vesicular exocytosis, gene expression, fission/fusion, and mitophagy (Pivovarova and Andrews, 2010; Jung et al., 2020). In metabolically active tissues like the brain, mitochondria take up Ca2+ mainly via the outer mitochondrial membrane (OMM) voltage dependent anion channel 1 (VDAC1) into the intermembrane space (IMS) (Camara et al., 2010; Camara et al., 2011; Camara et al., 2017). From the IMS, Ca2+ enters the inner mitochondrial membrane (IMM) via the mCa2+ uniporter (mCU) into the matrix (Camara et al., 2010; Baughman et al., 2011; De Stefani et al., 2011; Camara et al., 2017; Mishra et al., 2017). The large negative electrochemical gradient across the IMM (ΔΨm: −180 to −200 mV) is the main driving force for mCU-mediated Ca2+ uptake.
Mitochondrial Ca2+ release is performed primarily by the mitochondrial Na+/Ca2+/Li+ exchanger (mNCLX) (Palty et al., 2010), a.k.a. mitochondrial Na+/Ca2+ exchanger (mNCE). Another plausible mechanism for mCa2+ release is via the putative Na+-independent Ca2+/H+ exchanger (mCHE) as examined in cardiac myocytes (Haumann et al., 2018; Natarajan et al., 2020; Haumann et al., 2010) even though mCHE is reported to be more active in non-excitable cells (Rottenberg and Marbach, 1990), like hepatocytes. In the matrix, excess free Ca2+ is buffered primarily by inorganic phosphate (Pi), but also by adenine nucleotides (Haumann et al., 2010; Haumann et al., 2018; Mishra et al., 2019) and matrix proteins (Chalmers and Nicholls, 2003; Starkov, 2010; Blomeyer et al., 2013). The buffering system allows mitochondria to accumulate large quantities by mCa2+ uptake, while maintaining free [Ca2+]m in the physiological range (~100 nM) (Chalmers and Nicholls, 2003). Thus, free [Ca2+]m is managed by a balance in mCa2+ influx, mCa2+ buffering, and mCa2+ efflux. Hence, impaired cCa2+ cycling can lead to disruption of [Ca2+]m homeostasis, leading to mCa2+ overload, matrix swelling, collapse of the membrane potential, ΔΨm, and possibly to opening of the mitochondrial permeability transition pore (mPTP), with IMM rupture and total Ca2+ release. This Ca2+ dysregulation in synaptic terminals is implicated in several neurodegenerative diseases (Du et al., 2008; Kalani et al., 2018).
The brain consists of a highly heterogeneous ensemble of cells (e.g., neurons, glia, including astrocytes, and endothelial cells) with distinct anatomical and functional roles. Mitochondrial function, content and morphology vary among cell types, and within cells to account for their different physiological roles in the entire brain (Davis and McLean, 1987; Fecher et al., 2019; Pekkurnaz and Wang, 2022). Mitochondria are abundant in neurons, especially at the synapses (Vos et al., 2010), whereas fewer mitochondria per cell volume are found in glial and endothelial cells (Kannurpatti, 2017) (Figure 1). The high density of mitochondria in synapses are needed to meet the enhanced production of ATP, via oxidative phosphorylation, that is necessary for the high-energy demand to maintain, for example, ionic homeostasis via the Na+/K+ and Ca2+-ATPase pumps during synaptic neurotransmission (Hollenbeck, 2005; Magistretti and Allaman, 2015; Bordone et al., 2019). These processes are necessary for neurons to maintain cellular ionic conditions, i.e., Na+ and K+, and Ca2+ gradients and the cell membrane potential (Meir et al., 1999; Billups and Forsythe, 2002; Verma et al., 2022) to maintain the capability for neurotransmission.
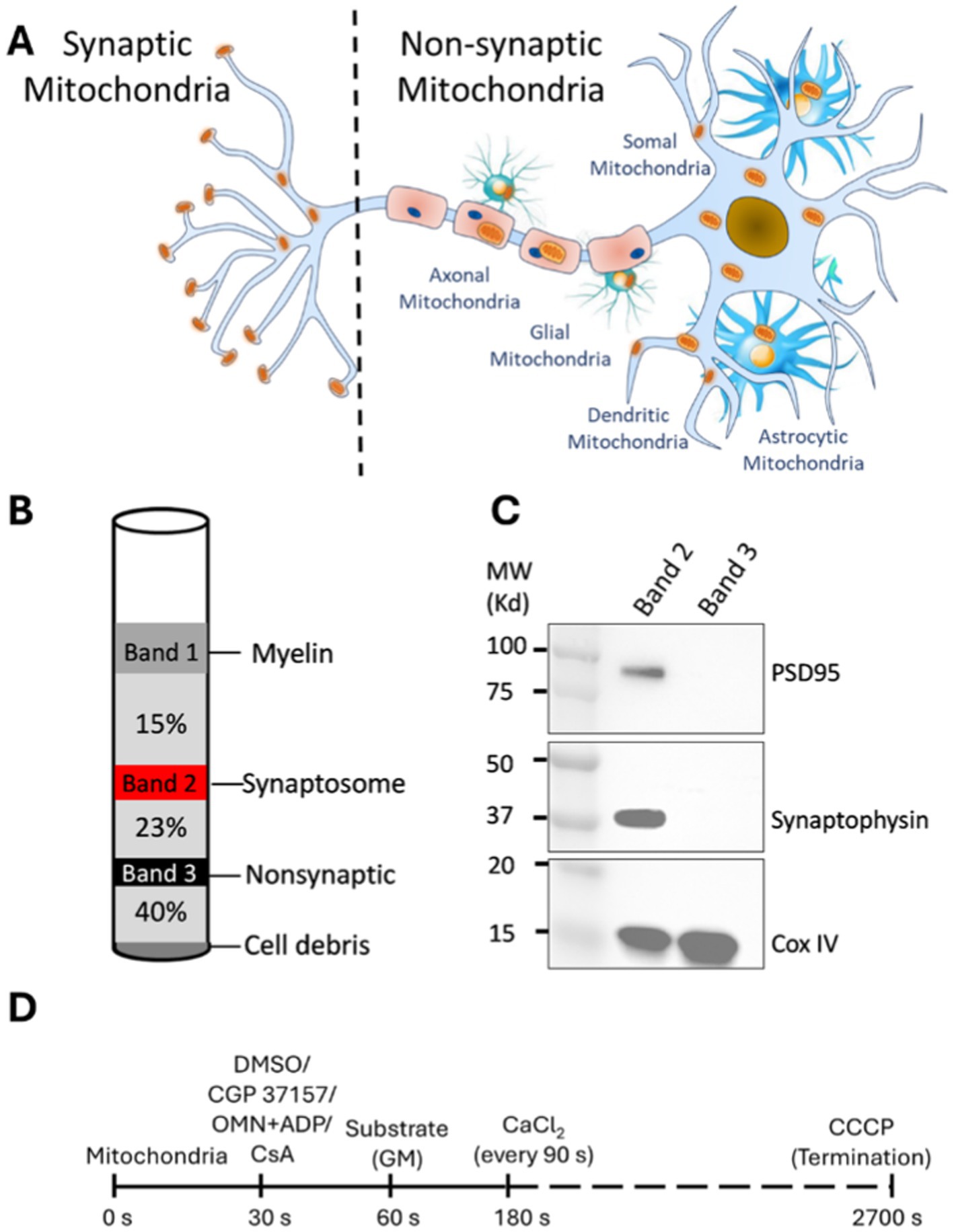
Figure 1. Synaptic and non-synaptic mitochondria isolation and experimental timeline to study Ca2+ handling and bioenergetics in isolated synaptic and non-synaptic mitochondria. (A) Compartmental distribution of mitochondria in subcellular location of neurons and glial cells. Synaptic mitochondria are located within the synaptosome while non-synaptic mitochondria are derived from soma, glial and vascular cells. (B) Schematic diagram of visible bands seen upon ultracentrifugation as described in materials and methods. The synaptosome (band 2; red color) and non-synaptic (band 3; black color) fractions are indicated in their respective gradients following ultracentrifugation. (C) Representative western blot of synaptosome markers in isolated synaptic (band 2) and non-synaptic (band 3) fractions. (D) Timeline of the experimental protocol (in seconds) for isolated synaptic and non-synaptic mitochondria. Different interventions, such as CGP 37157 (2 μM), oligomycin (OMN; 10 μM) + ADP (250 μM) and CsA (0.5 μM) were added at 30 s, followed by the addition of the substrates [Na+-glutamate + Na+-malate (GM)] at 60 s. At 180 s, 40 μM of CaCl2 was added, followed by sequential additions of 40 μM CaCl2 at every 90 s intervals. CCCP (10 μM) was added at the end of each experiment to achieve the maximal dye release from mitochondria to terminate the experiment.
The heterogeneity of the brain mitochondrial population is notable at the neuronal level (Pekkurnaz and Wang, 2022). Synaptic mitochondria are located mainly in the synaptosome, while non-synaptic mitochondria are derived primarily from neuronal soma and axons, and non-neuronal (glia, and endothelial) cells of the brain (Brown et al., 2006; Kulbe et al., 2017; Hill et al., 2020; Olesen et al., 2020). In a previous study, Naga et al. (2007) showed that synaptic mitochondria isolated from rat cerebral cortex are more sensitive to Ca2+ overload than are non-synaptic mitochondria and have higher synaptic mitochondrial levels of cyclophilin-D (Cyp D), a matrix protein implicated in the regulation of the mPTP opening (Naga et al., 2007; Du et al., 2008). In addition, synaptic mitochondria display higher vulnerability to oxidative damage (Du et al., 2010) and exhibit a higher mCa2+ level (Brown et al., 2006; Kulbe et al., 2017) than non-synaptic mitochondria during aging and in neurological disorders (Reddy and Beal, 2008).
These unique features in synaptic mitochondria are likely a result of the high cCa2+ flux demands for instant ATP resupply during neurotransmission (Datta and Jaiswal, 2021). Indeed, studies have demonstrated differential mCa2+ handling in mitochondria isolated from synaptic and non-synaptic brain tissue. As noted above, synaptic mitochondria retained less Ca2+ during CaCl2 pulse challenges and had more Cyp D than non-synaptic mitochondria (Brown et al., 2006; Naga et al., 2007). However, knowledge of the detailed kinetics of synaptic mCa2+ dynamics, particularly mCa2+ uptake, buffering and efflux, and its implications for matrix Ca2+ handling is limited.
In this study, we sought to characterize the dynamics of mCa2+ handling in synaptic and non-synaptic mitochondria isolated from rat cerebral cortex and to assess the physiological and pathophysiological implications of their differential modes of mCa2+ handling. To characterize mCa2+ handling in synaptic and non-synaptic mitochondria we investigated the properties of: (1) mCa2+ extrusion via the mNCE using its inhibitor CGP 37157 (CGP), (2) mCa2+ buffering by changing the matrix ADP, ATP pool using a combination of oligomycin and ADP (OMN + ADP), and (3) mCa2+ buffering by cyclosporin A (CsA), a Cyp D inhibitor (Camara et al., 2010), but which, via a Pi-dependent (Mishra et al., 2019) Ca2+ buffering mechanism, also enhances mCa2+ sequestration during CaCl2 pulse challenges in cardiomyocytes (Haumann et al., 2018; Mishra et al., 2019). Based on these observations, we show that the differences in mCa2+ handling between synaptic and non-synaptic mitochondria is attributed in large part to their differences in mCa2+ efflux and mCa2+ buffering mechanisms.
2 Materials and methods
2.1 Materials
All chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO, United States), unless otherwise stated. CGP37157 (CGP) and Cyclosporin A (CsA) were purchased from Tocris Bioscience (Bristol, United Kingdom). Fluorescent probes Fura-4F penta-K+ salt and tetramethyl-rhodamine methyl ester perchlorate (TMRM) were purchased from Life Technologies (Eugene, OR).
2.2 Animal care
Male Sprague Dawley (SD) rats weighing 300–400 grams were procured from Envigo. Rats were 8 to 12 weeks old at the time of experimentation. All procedures were carried out in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin.
2.3 Synaptic and non-synaptic mitochondria isolation
Synaptic and non-synaptic mitochondria were isolated from SD rats according to the procedure described by Naga et al. (2007), with some modifications. Briefly, the rats were anesthetized with a combination of an intraperitoneal injection of inactin (0.05 mg) for sedation and heparin (500 units) for anticoagulation. After total sedation and decapitation, the brain was rapidly harvested and the cerebral cortex was separated and minced into ice-cold Na+-free isolation buffer containing in mM: 200 mannitol, 50 sucrose, 5 KH2PO4, 5 MOPS, 1 EGTA, and 0.1% bovine serum albumin at pH 7.15 (adjusted with KOH). The minced tissue was suspended in a 5 mL ice-cold isolation buffer containing 0.05% nagarse, a protease, and then homogenized using a Dounce homogenizer. The homogenate was centrifuged at 1,300 g for 5 min. The supernatant was layered on a discontinuous Percoll gradient (2.5 mL each of 15, 23, 40% by volume Percoll in isolation buffer) and centrifuged at 34,000 g for 9 min with a Beckman Optima XPN ultracentrifuge. Band 2 (between 15 and 23% Percoll) and Band 3 (between 23 and 40% Percoll) were carefully removed using a 25-gauge Hamilton syringe; these constitute, respectively, the synaptic and non-synaptic mitochondrial fractions (Figure 1).
Each fraction was resuspended in 5 mL total volume of isolation buffer. A 50 μL aliquot of 2% digitonin was added to both fractions and allowed to incubate on ice for 10 min. Both suspensions were centrifuged at 16,500 g for 15 min and the supernatants were discarded. After resuspension of the pellets in separate aliquots of 5 mL isolation buffer, both fractions were re-centrifuged at 8,000 g for 10 min and the supernatants discarded. Protein concentration was determined by the Bradford method and the final mitochondrial suspensions were adjusted to 6.25 mg protein/mL in the isolation buffer. All centrifugations were performed at 4°C, and all buffers used for the isolation were kept on ice during the procedure. The isolated mitochondria were kept on ice for the duration of the experiments. All experiments on mitochondria function were conducted at room temperature (~25°C), as we have reported previously (Blomeyer et al., 2013; Blomeyer et al., 2016; Haumann et al., 2018; Mishra et al., 2019; Mishra and Camara, 2022; Sun et al., 2022).
2.4 Measurement of mitochondrial O2 consumption rate in synaptic and non-synaptic mitochondria
Respiratory Control Index (RCI) is a method to determine the functional integrity of mitochondria as described previously (Haumann et al., 2010; Mishra et al., 2019). Briefly, mitochondria were suspended in experimental buffer (see 2.5 and 2.6 below) and O2 consumption was measured, as described before (Riess et al., 2004; Riess et al., 2008; Yang et al., 2019; Gerdes et al., 2020; Sun et al., 2022; Kim et al., 2023), using the Clark-type electrode (model 1302, Strathkelvin Instruments) in a water jacketed and air-tight 500 μL chamber (Model MT200A). The mitochondrial suspension in the respiratory chamber was energized with complex I substrate, Na+-glutamate (0.5 mM) and Na+-malate (0.5 mM) (Na+–GM), to determine state 2 respiration, followed by the addition of ADP (250 μM), which results in a faster rate of O2 consumption, i.e., state 3 respiration. The conversion of all the added ADP to ATP, and the concomitant decrease in the O2 consumption rate represents state 4 respiration. The RCI, which determines the magnitude of coupling of oxidative phosphorylation, was defined as the ratio of state 3 to state 4 respiration. The lower RCI (see results) in non-synaptic mitochondria likely represents a degree of uncoupled respiration due to proton leak through the IMM. This process of H+ uptake is independent of H+ influx via the ATP synthase (complex V).
2.5 Measurement of mitochondrial Ca2+ handling in synaptic and non-synaptic mitochondria
Mitochondrial Ca2+ handling (i.e., uptake, sequestration and release), and mitochondrial membrane potential (ΔΨm) experiments were monitored over time using a Photon Technology Instrument fluorescence spectrophotometer (PTI; Qm-8 Horiba, Birmingham NJ, United States) (Haumann et al., 2010; Mishra et al., 2019). The experiments started with 0.5 mg isolated mitochondria added in 1 mL Na+-free experimental buffer containing in mM: 130 KCl, 5 K2 HPO4, 20 MOPS, 0.1% BSA and 40 μM EGTA at pH 7.15 (adjusted with KOH) along with 1 μM of the fluorescent dye Fura-4F K+ salt to measure extra-matrix Ca2+ ([Ca2+]e) transients (Figure 1D). At t = 30 s, vehicle or any drug being tested was added: these were CGP (2 μM) or OMN (10 μM) + ADP (250 μM), OMN alone, ADP alone, or CsA (0.5 μM). At t = 60 s, Na+–GM was added to energize mitochondria and to allow Na+ exchange with Ca2+ when CaCl2 is added. At t = 180 s and for every 90 s after that, 40 μM CaCl2 was added to the mitochondrial suspension in a cuvette placed in the PTI with continuous stirring in the PTI cuvette. At the end of each experiment, when mitochondria could not take up any additional CaCl2, the uncoupler CCCP (10 μM) was added to the suspension to cause maximal release of mCa2+. Changes in [Ca2+]e were monitored at dual-excitation wavelengths λex at 340/380 nm and a single emission wavelength λem at 510 nm.
2.6 Measurement of synaptic and non-synaptic mitochondrial membrane potential (ΔΨm)
As in the CaCl2 pulse experiments above, each experiment started with 0.5 mg isolated mitochondria in 1 mL experimental buffer containing in mM: 130 KCl, 5 K2 HPO4, 20 MOPS, 0.1% BSA and 40 μM EGTA, and at pH 7.15 (adjusted with KOH) along with 1 μM TMRM, a permeant membrane potential (ΔΨm) fluorescence dye. At t = 30 s, any drug being used was added: vehicle, CGP or OMN + ADP, OMN (alone), ADP (alone) or CsA. At t = 60 s, Na+–GM (0.5 M) was added; at t = 180 s and every 90 s after that, 40 μM CaCl2 was added. The TMRM fluorescence changes were measured at two excitations, λex 546 and 573 nm, and a single emission λem 590 nm. CCCP was added as in section 2.6 to induce maximal depolarization of ΔΨm.
2.7 Western blot assay of mitochondrial proteins involved in mCa2+ handling in synaptic and non-synaptic mitochondria
Purified synaptic and non-synaptic mitochondrial fractions were lysed in ice-cold RIPA lysis buffer (Thermo Fisher Scientific) supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF) and 1% protease inhibitor cocktail (Sigma-Aldrich). Lysates were vortexed and incubated for 30 min on ice, followed by centrifugation at 17,000 g for 15 min at 4°C. The supernatant was collected and quantified using the bicinchoninic acid assay (Thermo Fisher Scientific). The protein samples were reduced in 4× Laemmli sample buffer (Bio-Rad) containing 10% β-mercaptoethanol and denatured at 95°C for 5 min. Denatured proteins were resolved on 4–20% SDS-PAGE gels (Bio-Rad) and transferred onto nitrocellulose membranes (0.45 μm, Bio-Rad). The membranes were blotted with the following primary antibodies: mCU, Synaptophysin, PSD95, VDAC, ANT1, (Cell Signaling Technology) and Cyp D (abcam). The IMM protein, COX IV (Cell Signaling Technology), was used as the loading control. Of note, we did not report here the expression levels of mNCE, due to questionable reliability of the commercially available antibodies. The LI-COR infrared fluorescent mouse (925–68,020) and rabbit (925–32,211) secondary antibodies were used for visualization using a LI-COR Odyssey scanner at 680 and 800 nm, respectively. Band quantification (densitometry) was performed using ImageJ software.
2.8 Statistical analyses
All data were imported into a Microsoft Excel 2016 program for analysis. Any statistical difference in RCI was assessed using a paired student’s t-test. The student’s t-test for other experiments was paired when appropriate, i.e., synaptic vs. non-synaptic, synaptic with CGP vs. non-synaptic with CGP, synaptic with OMN + ADP vs. non-synaptic with OMN + ADP, and synaptic with CsA vs. non-synaptic with CsA, but was otherwise performed as a two-sample unequal variance test. All t-tests reported are two-tailed. To assess significance in the Ca2+ handling data (steady state extra-matrix Ca2+ levels), a student’s t-test was performed using the 5 s average of each experiment as a data point just prior to addition of each new CaCl2 bolus. Data are presented as means ± SEM.
3 Results
3.1 Isolation and validation of synaptic and non-synaptic mitochondrial population
Synaptic and non-synaptic mitochondrial fractions were isolated from freshly harvested brain tissue following a well-described protocol (Naga et al., 2007), with some modifications (Figure 1). Western blot analysis showed that the synaptic fractions are highly enriched with well-established protein markers including, postsynaptic density protein 95 (PSD95) and synaptophysin, (Glantz et al., 2007) and the IMM protein COXIV. The non-synaptic fraction showed a COXIV band, but no PSD95 and synaptophysin bands (Figure 1C). These results demonstrate an enriched abundant synaptosomal fraction in the synaptic population and validates our procedure to isolate synaptic and non-synaptic mitochondria from cerebral tissue as also described by others (Villasana et al., 2006; Kamat et al., 2014).
3.2 Differences in oxidative phosphorylation capacity of synaptic and non-synaptic mitochondria
To assess the capacity of oxidative phosphorylation in the two mitochondrial populations we assessed O2 consumption using the Clark-type electrode, as reported previously (Mishra et al., 2019; Mishra and Camara, 2022; Sun et al., 2022). Specifically, we measured the ADP-mediated increase in mitochondrial respiration after energizing with complex I substrate Na+-GM. We quantified the Respiratory Control Indexes, RCIs, i.e., the ratios of state 3 to state 4 respiration. Representative traces of mitochondrial respiration and a summary of RCIs (inset) are displayed in Figure 2. Synaptic and non-synaptic mitochondria exhibited similar state 2 and state 3 respirations (Supplementary Figure 1) but the mean RCI was significantly higher in synaptic mitochondria, 5.92 ± 0.43 vs. non-synaptic mitochondria, 4.17 ± 0.17 (Figure 2; inset). This is because state 4 respiration was faster in non-synaptic mitochondria than in synaptic mitochondria (Supplementary Figure 1), which indicates a degree of uncoupled respiration in non-synaptic mitochondria that is likely due to a mild H+ leak (H+ ions entering the matrix independent of the ATP-synthase).
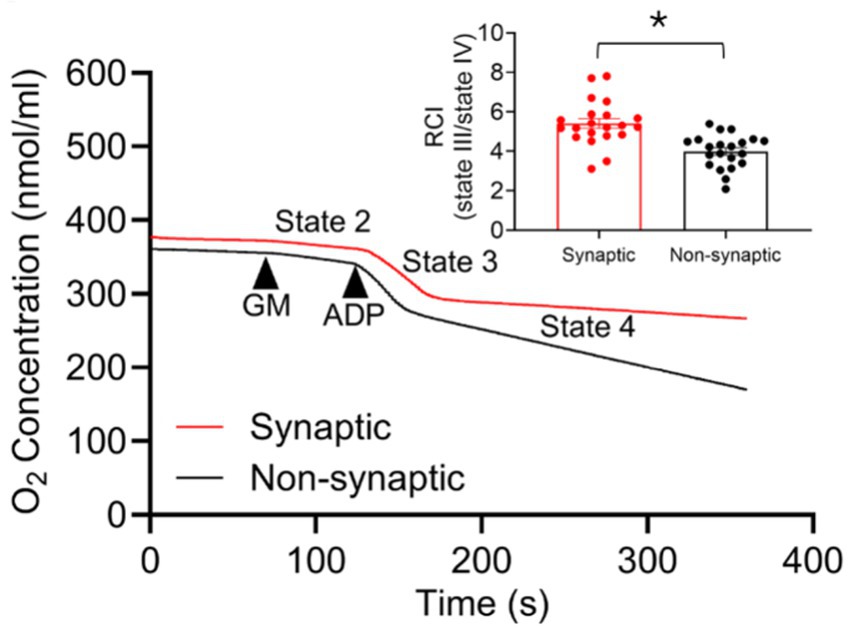
Figure 2. Representative traces of O2 consumption rate (OCR) of isolated synaptic and non-synaptic mitochondria. Complex I substrates [Na+-glutamate + Na+-malate (GM)] and ADP were sequentially injected to assess mitochondrial OCR, the slope, at the different respiratory states. The Respiratory Control Index (RCI) is derived from the ratio of the OCR during state 3 (in the presence of ADP) to the OCR during state 4 (after exhausting the added ADP) respiration. The insets show average RCI of synaptic and non-synaptic mitochondria. Data are expressed as mean ± SEM (*p < 0.0001).
3.3 Differences in [Ca2+]m handling in synaptic and non-synaptic mitochondria
To comprehensively address the [Ca2+]m handling in synaptic and non-synaptic mitochondria, we measured extra-mitochondrial Ca2+ ([Ca2+]e) (expressed as relative fluorescence units, RFU) during repetitive additions of 40 μM CaCl2 boluses, in the presence of 80 μM EGTA, at 90 s intervals until mitochondria stopped taking up the added exogenous CaCl2 (Figure 3). As shown in representative traces, each CaCl2 pulse is marked by a transient peak in the extra-matrix fluorescent signal for Ca2+ followed by a downward slope reaching a baseline steady-state level. For the 90 s inter-pulse interval, the steady-state (ss) [Ca2+]e is defined as the net basal level of the extra-matrix free Ca2+ ([Ca2+]e) following mCa2+ uptake, sequestration, and efflux after each CaCl2 bolus (Blomeyer et al., 2016; Mishra et al., 2019). In this case, Figure 3 shows that non-synaptic mitochondria exhibited a more robust and sustained capacity for mCa2+ uptake and sequestration than the synaptic mitochondria.

Figure 3. Comparison of synaptic and non-synaptic mitochondria Ca2+ handling. (A) Representative traces of extra-matrix Ca2+ ([Ca2+]e) measured with the Ca2+-sensitive ratiometric dye Fura-4FF in synaptic and non-synaptic mitochondria. (B) Change of ΔΨm in synaptic and non-synaptic mitochondria measured using the ΔΨm sensitive dye TMRM (tetramethylrhodamine methyl ester perchlorate). Synaptic (red trace) and non-synaptic (black trace) mitochondria were energized with complex I substrates, [Na+-glutamate + Na+-malate (GM)] at 60 s and 40 μM CaCl2 pulses were added at every 90 s, followed by addition of 10 μM CCCP at the end of the experiment. The insets show quantification of steady-state [Ca2+]e (A) and ΔΨm (B). Error bars represent mean ± SEM (*p < 0.05).
To better elucidate the differences, we quantified ss[Ca2+]e and plotted it against the cumulative additions of CaCl2, i.e., 160, 400, 600, and 840 μM. This corresponds to ss[Ca2+]e at the 4th, 10th, 15th, and 21st CaCl2 pulse, respectively, and are shown as brown arrows in Figure 3. Synaptic mitochondria showed a gradual increase in ss[Ca2+]e, i.e., lesser retained extra-matrix Ca2+ that gradually accumulated in the experimental buffer. We reasoned this could be due to either a lesser mCa2+ uptake or to a greater mCa2+ release over time as additional boluses of CaCl2 were given. As presented below on the impact of the CaCl2 pulses on ΔΨm, lesser Ca2+ uptake is not likely a contributing factor because mCa2+ uptake is dependent on a high ΔΨm. In contrast, in non-synaptic mitochondria, CaCl2 pulses resulted in no change in ss[Ca2+]e until the 21st pulse (Figure 3); this indicated that more mCa2+ was sequestered in mitochondria and so less mCa2+ was released. To assess the potential for differences in Ca2+ uptake rates in the two mitochondrial populations, mCU activity was quantified using a single exponential decay for each CaCl2 pulse to derive the decay constants. There was no significant difference in the Ca2+ decay constants between synaptic and non-synaptic mitochondria (Supplementary Figure 2). These results suggest a differential balance in the mechanism for handling mCa2+ in the two populations, with a likelihood for greater mCa2+ buffering in regulating [Ca2+]m in non-synaptic mitochondria, and the possibility that mCa2+ efflux via mNCE as a potential mechanism for regulating mCa2+ in synaptic mitochondria, as presented later.
A high mitochondrial membrane potential (ΔΨm) is the major determinant for mCa2+ uptake. When ΔΨm is fully charged, it leads to a rapid uptake of mCa2+ via the mCU. Excess free Ca2+ in the matrix causes depolarization and loss of ΔΨm, which diminishes the drive for mCa2+ uptake. Therefore, we next investigated whether the differences in Ca2+ handling between synaptic and non-synaptic mitochondria could be attributed to differences in ΔΨm during CaCl2 pulse challenges. We examined changes in ΔΨm using the similar protocol (Figure 1D) as for measuring [Ca2+]e with CaCl2 boluses. Figure 3B shows small transient depolarizations and repolarizations of ΔΨm with each bolus of CaCl2 in both synaptic and non-synaptic mitochondria. Basal ΔΨm gradually depolarized to similar levels in both populations; this suggest that the driving force for mCa2+ uptake was similar between the two groups. The Ca2+ transients also demonstrate that the differences in Ca2+ flux dynamics in the two mitochondrial populations are not related to differences in ΔΨm in response to repeated mCa2+ loading. In both populations during the latter CaCl2 pulse challenges, mitochondria stopped taking up additional Ca2+ because significant depolarization of ΔΨm terminated the driving force for mCa2+ uptake. Adding the protonophore CCCP after the end of the CaCl2 boluses validates the similar maximal depolarizations with complete collapse of ΔΨm in both mitochondrial fractions.
We have shown that in synaptic mitochondria, each CaCl2 pulse produced a new steady state (ss[Ca2+]e) slightly above the previous one. It is possible that this is due less to reliance on the mCa2+ sequestration system and more to an increase in efflux of mCa2+ via the mNCE. Unlike synaptic mitochondria, non-synaptic mitochondria maintained ss[Ca2+]e at almost the basal level for an extended time during the CaCl2 boluses. We suspected that this is due to a greater capacity for mCa2+ buffering in non-synaptic mitochondria with a minor, or no effect, on mCa2+ efflux via mNCE.
3.4 Synaptic and non-synaptic mitochondria exhibit differences in mNCE function
To further evaluate these differences in mCa2+ handling in synaptic and non-synaptic mitochondria, we assessed the functional differences in mNCE activity. We studied the mCa2+ handling during boluses of CaCl2 in the two mitochondrial populations in the absence or presence of CGP, a specific inhibitor of mNCE (1–10 μM) (Csordas et al., 2012; Palty and Sekler, 2012; Liu et al., 2014; Islam et al., 2020; Natarajan et al., 2020). Any difference in mCa2+ uptake, assessed by ss[Ca2+]e (Figures 4A,B) were attributed to Na+-dependent/CGP-sensitive mCa2+ efflux. Addition of CGP to both mitochondrial fractions energized with Na+-GM before CaCl2 pulses produced a robust mCa2+ uptake with [Ca2+]e transients representing mCa2+ handling that was different in synaptic mitochondria vs. synaptic mitochondria without CGP (Figure 4A). Quantification of ss[Ca2+]e in synaptic mitochondria showed significantly lower ss[Ca2+]e levels, i.e., more mCa2+ retained, in the presence of CGP than in the absence of CGP. The inhibition of mNCE by CGP revealed that the mCa2+ buffering system in synaptic mitochondria is present, but its action may be secondary to the efflux system which may be dominant in mCa2+ handling in this population of mitochondria (Figure 4A, inset). In contrast to synaptic mitochondria, non-synaptic mitochondria did not exhibit significant differences in ss[Ca2+]e with CGP and without CGP for most of the CaCl2 pulses (Figure 4B). Thus, CGP shifted the synaptic handling of excess mCa2+ during CaCl2 pulses to a pattern of mCa2+ dynamics that is like that of non-synaptic mitochondria without CGP.
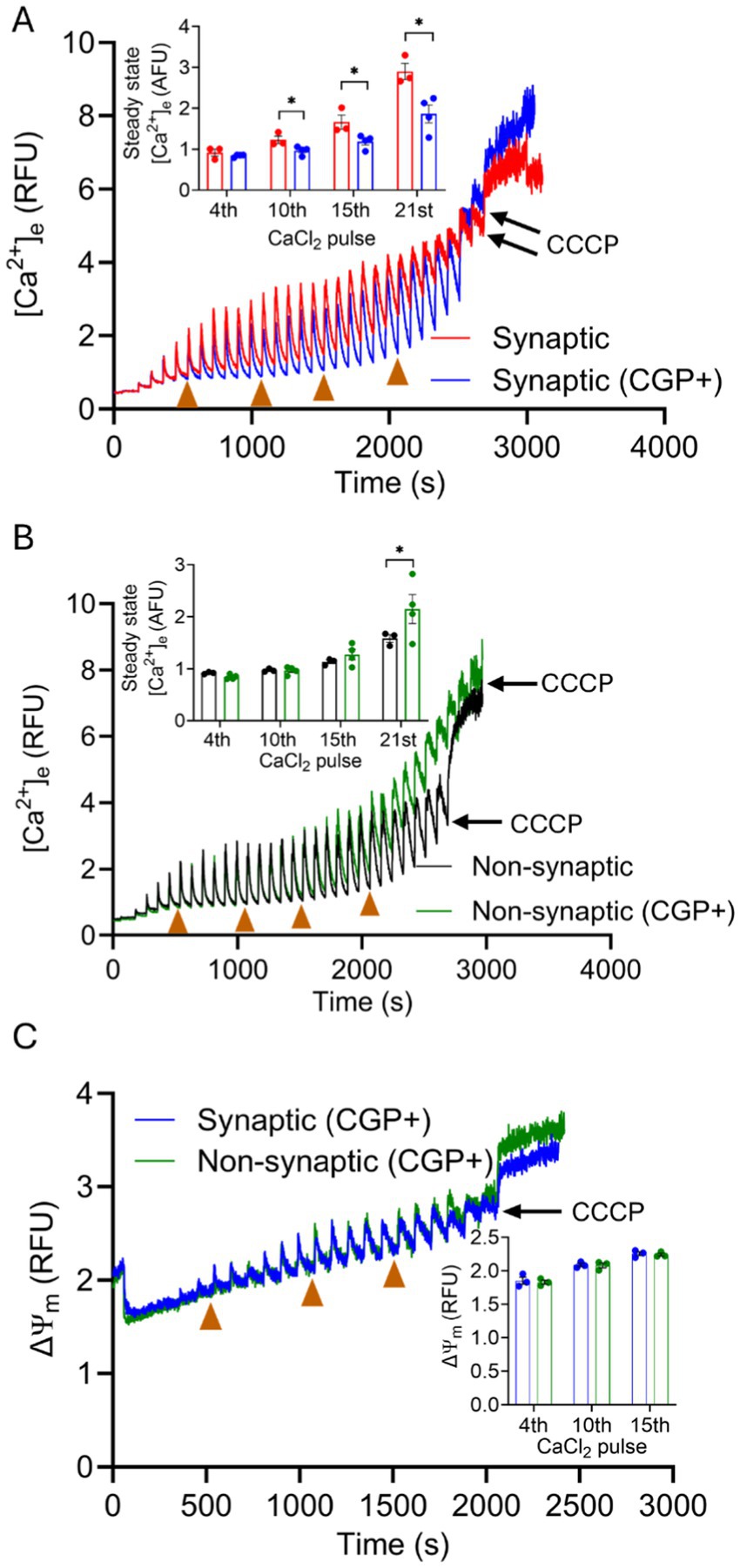
Figure 4. Effect of CGP on extra-mitochondrial calcium ([Ca2+]e) dynamics of synaptic and non-synaptic mitochondria. Representative traces of extra-matrix Ca2+ ([Ca2+]e) measured with the Ca2+-sensitive ratiometric dye Fura-4F in isolated synaptic (A) and non-synaptic (B) mitochondria in the presence and absence of CGP. Changes in ΔΨm in CGP-treated synaptic and non-synaptic mitochondria (C) measured using the ΔΨm sensitive dye TMRM (tetramethylrhodamine methyl ester perchlorate). 2 μM CGP was added in synaptic (blue trace) and non-synaptic (green trace) mitochondria at 30 s, followed by the addition of complex I substrates, [Na+-glutamate + Na+-malate (GM)] at 60 s. 40 μM CaCl2 pulses were added at every 90 s, and 10 μM CCCP was given at the end of the experiment. The insets show quantification of steady-state [Ca2+]e (A,B) and ΔΨm (C) after a cumulative addition of 160, 400, 600, and 840 μM CaCl2. Error bars represent mean ± SEM (*p < 0.05).
Therefore, CGP did not significantly affect mCa2+ dynamics in non-synaptic mitochondria during most of the CaCl2 pulse challenges. Importantly, no significant differences in ΔΨm were noted in mitochondria treated with CGP in either synaptic or non-synaptic mitochondrial fractions (Figure 4C). This suggests that by inhibiting mNCE, and so blocking mCa2+ efflux, the excess mCa2+ becomes more actively buffered and sequestered so that matrix free [Ca2+] remains low enough to preserve ΔΨm. Inhibition of mNCE-mediated Ca2+ extrusion to expose the sequestration of mCa2+ in synaptic mitochondria indicates that mNCE plays a larger role in shaping synaptic [Ca2+]e transients than transients in non-synaptic mitochondria. Differential mCa2+ handling in these two populations might have physiological and pathophysiological implications.
3.5 Effect of altering mitochondrial matrix adenine nucleotide pool with OMN + ADP on mitochondrial Ca2+ handling in synaptic and non-synaptic mitochondria
Since matrix free Ca2+ sequestration plays an important role in shaping mCa2+ influx and efflux transients, we next examined whether some aspects of the mCa2+ buffering system contribute to differences in the Ca2+ handling phenotype between the two mitochondrial fractions. To do this, we again challenged synaptic and non-synaptic mitochondria as above with repeated boluses of CaCl2, but in the prior presence of OMN + ADP, ADP alone, or OMN alone. We reported before (Mishra et al., 2019) in cardiomyocytes that OMN and ADP bolstered the mCa2+ buffering system, purportedly by modulating the matrix adenine nucleotide (AdN) pool (ADP/ATP ratio) (Haumann et al., 2010; Sokolova et al., 2013; Mishra et al., 2019). OMN + ADP given before the CaCl2 pulses showed rapid initial robust mCa2+ uptake and buffering during 7–8 pulses (280 μM–320 μM) by both synaptic and non-synaptic mitochondria, when compared to their respective untreated groups. This robust Ca2+ buffering was manifested by the significant diminution of the [Ca2+]e transients and the markedly reduced ss[Ca2+]e (Figures 5A,B).
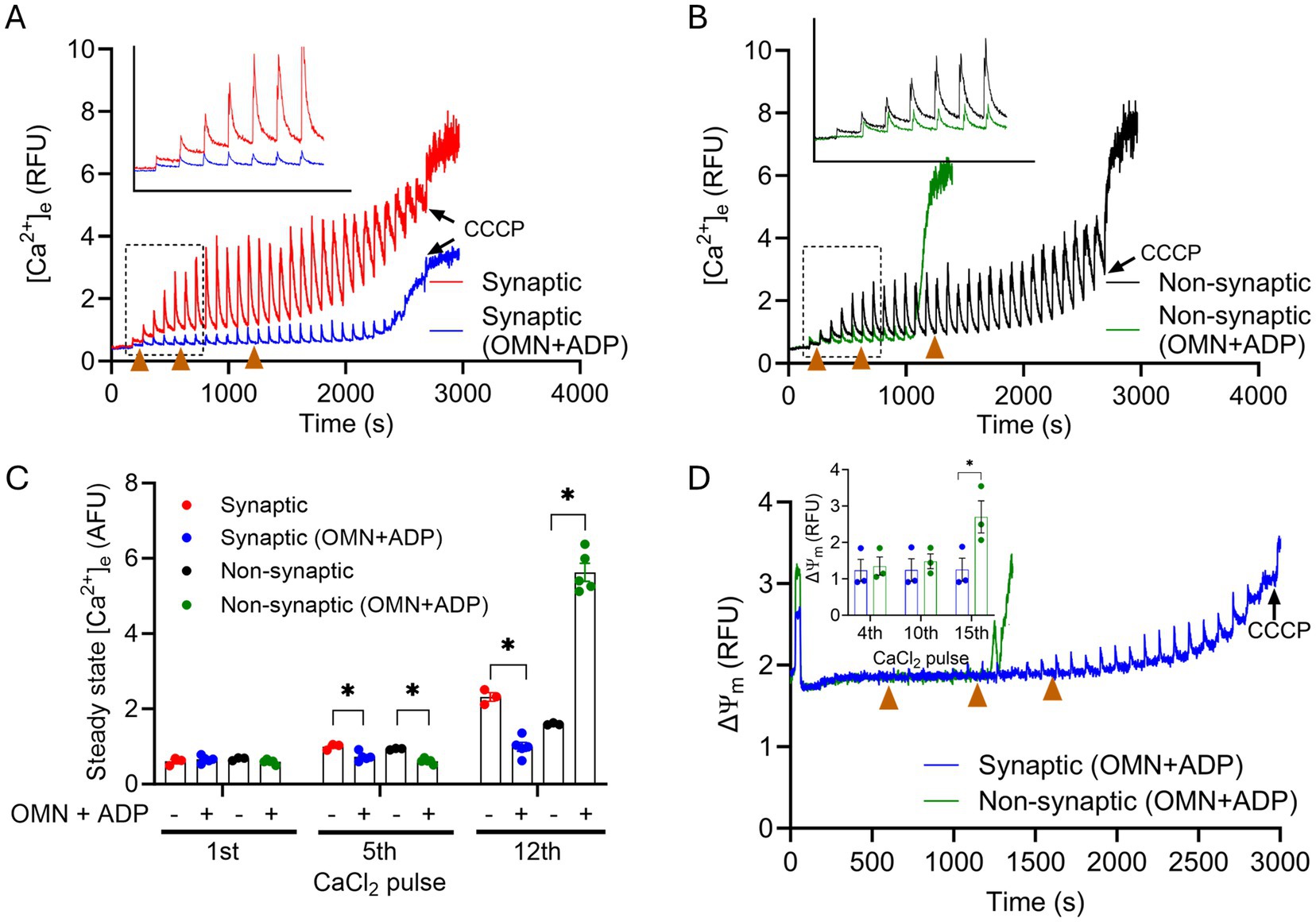
Figure 5. Effect of OMN + ADP on extra-mitochondrial calcium ([Ca2+]e) dynamics of synaptic and non-synaptic mitochondria. Representative traces of extra-matrix Ca2+ ([Ca2+]e) measured with the Ca2+-sensitive ratiometric dye Fura-4F in an isolated synaptic (A) and non-synaptic (B) mitochondria. 10 μM OMN and 250 μM ADP (OMN + ADP) were added in synaptic (blue trace) and non-synaptic (green trace) mitochondria at 30 s followed by the addition of the complex I substrates, [Na+-glutamate + Na+-malate (GM)] at 60 s. 40 μM CaCl2 pulses were added at every 90 s, and 10 μM CCCP was added at the end of each experiment. Quantification of steady-state [Ca2+]e after a cumulative addition of 160, 400, and 600 μM CaCl2 (C). Change in ΔΨm in OMN + ADP-treated synaptic and non-synaptic mitochondria were measured using the ΔΨm sensitive dye TMRM (tetramethylrhodamine methyl ester perchlorate) (D). The insets (A,B) show [Ca2+]m uptake kinetics in detail. The inset (D) shows ΔΨm after a cumulative addition of 160, 400, and 600 μM CaCl2. Error bars represent mean ± SEM (*p < 0.05).
Despite the initial strong mCa2+ uptake and buffering in both mitochondrial fractions, we observed, paradoxically, an early and complete collapse of ΔΨm in OMN + ADP treated non-synaptic mitochondria (Figure 5B). This observation suggests that in the non-synaptic mitochondria, the OMN + ADP-mediated matrix Ca2+ buffering, i.e., AdN pool, may not contribute to the protracted Ca2+ buffering (Figure 5A). Furthermore, if the early collapse of the ΔΨm represents mPTP opening in non-synaptic, but not in synaptic mitochondria, this appears highly unusual because a known activator of mPTP opening in isolated mitochondria is excess matrix free Ca2+ when the mCa2+ buffering system is overwhelmed. In contrast, in synaptic mitochondria, the presence of OMN + ADP resulted in prolonged and robust mCa2+ uptake and sequestration that endured during multiple CaCl2 pulse challenges (Figure 5A). Additionally, the presence of OMN + ADP strongly blunted the increase in ss[Ca2+]e by stimulating faster mCa2+ uptake, sequestration, and delayed collapse of ΔΨm in synaptic mitochondria (Figure 5C). In both synaptic and non-synaptic mitochondria, the addition of ADP or OMN alone (Supplementary Figure 3) elicited a marked difference in the mCa2+ handling profile for both mitochondrial fractions when compared to their respective combined OMN + ADP (Figure 5) treated mitochondria. Interestingly, in non-synaptic mitochondria treated with ADP or OMN alone (Supplementary Figures 3C,D), we did not observe collapse of ΔΨm during the CaCl2 boluses that occurred when non-synaptic mitochondria were treated with combined OMN + ADP (Figure 5B). In addition, the fast Ca2+ uptake in the presence of OMN + ADP was abrogated in both synaptic and non-synaptic mitochondria. These interesting and unexpected observations portend a novel insight into the role of mCa2+ sequestration by the matrix adenine nucleotide pool and the impact it may have on the differential Ca2+ handling in these two populations of mitochondria.
Because we observed significant differences in the OMN + ADP-mediated buffering of Ca2+ between synaptic and non-synaptic mitochondria, we assessed again the differential effects on ΔΨm during similar CaCl2 pulse challenges. As expected, the addition of OMN + ADP in synaptic mitochondria displayed marked preservation of basal ΔΨm during a prolonged exposure to CaCl2 pulse challenges (Figure 5D). However, in the non-synaptic mitochondria, total dissipation of ΔΨm occurred early and was accompanied by maximal mCa2+ release into the extra-matrix space, portending mPTP opening (Figures 5B,D).
The reason for early dissipation of ΔΨm and release of mCa2+ in the presence of OMN + ADP in non-synaptic mitochondria is unclear. However, it is well established that excess Ca2+ and ROS are potent activators of mPTP opening, and they can do so by acting independently or synergistically (Chandra, 1991; Halestrap, 2010; Aldakkak et al., 2013). Thus, a possible explanation can be that the presence of OMN + ADP in non-synaptic mitochondria enhanced ROS generation (Liu and Schubert, 2009; Aklima et al., 2021) that eventually triggers early activation and opening of the mPTP. However, in the classical understanding, we did not observe mPTP opening as evidenced by the abrupt release of matrix Ca2+ in either population of brain mitochondria under control conditions.
In a recent study, we reported that CsA, a potent mPTP opening inhibitor in cardiomyocytes, modulates free [Ca2+]m by enhancing a Pi-dependent mCa2+ buffering (Mishra et al., 2019). In the current study, we tested the buffering effect of CsA on [Ca2+]m handling in synaptic and non-synaptic mitochondria (Figures 6A,B). Interestingly, in the presence of cyclosporin A (CsA), we observed more mCa2+ uptake in non-synaptic vs. synaptic mitochondria. The magnitude of mCa2+ uptake and buffering for the initial CaCl2 pulses, 0–900 s was similar between both groups. However, on additions of subsequent CaCl2 boluses, CsA treated non-synaptic mitochondria continued to show more robust mCa2+ uptake and sequestration with markedly lower ss[Ca2+]e compared to untreated (control) non-synaptic mitochondria (Figures 6B,C) and synaptic mitochondria with CsA. CsA treated synaptic mitochondria, when compared to its control (untreated), exhibited less sequestration as evidenced by the gradual increase in ss[Ca2+]e with additional CaCl2 pulses (Figure 6A) that was not statistically different. Thus, our result demonstrate that in the presence of CsA, non-synaptic mitochondria displayed more robust Ca2+ buffering with a lower ss[Ca2+]e than in synaptic mitochondria (Figure 6B vs. Figure 6A). The magnitude of ΔΨm depolarization in both mitochondrial populations in the presence of CsA (Figure 6D) reflects distinct differences in mCa2+ handling, with the non-synaptic mitochondria exhibiting a more polarized ΔΨm, likely because of less matrix free Ca2+ or greater mCa2+ sequestration. Taken together, the data show that CsA differentially modulates non-synaptic and synaptic mCa2+ handling, which is consistent with a previous observation by Naga et al. (2007), but with the potential for invoking CsA as an enhancer of matrix Ca2+ buffering.
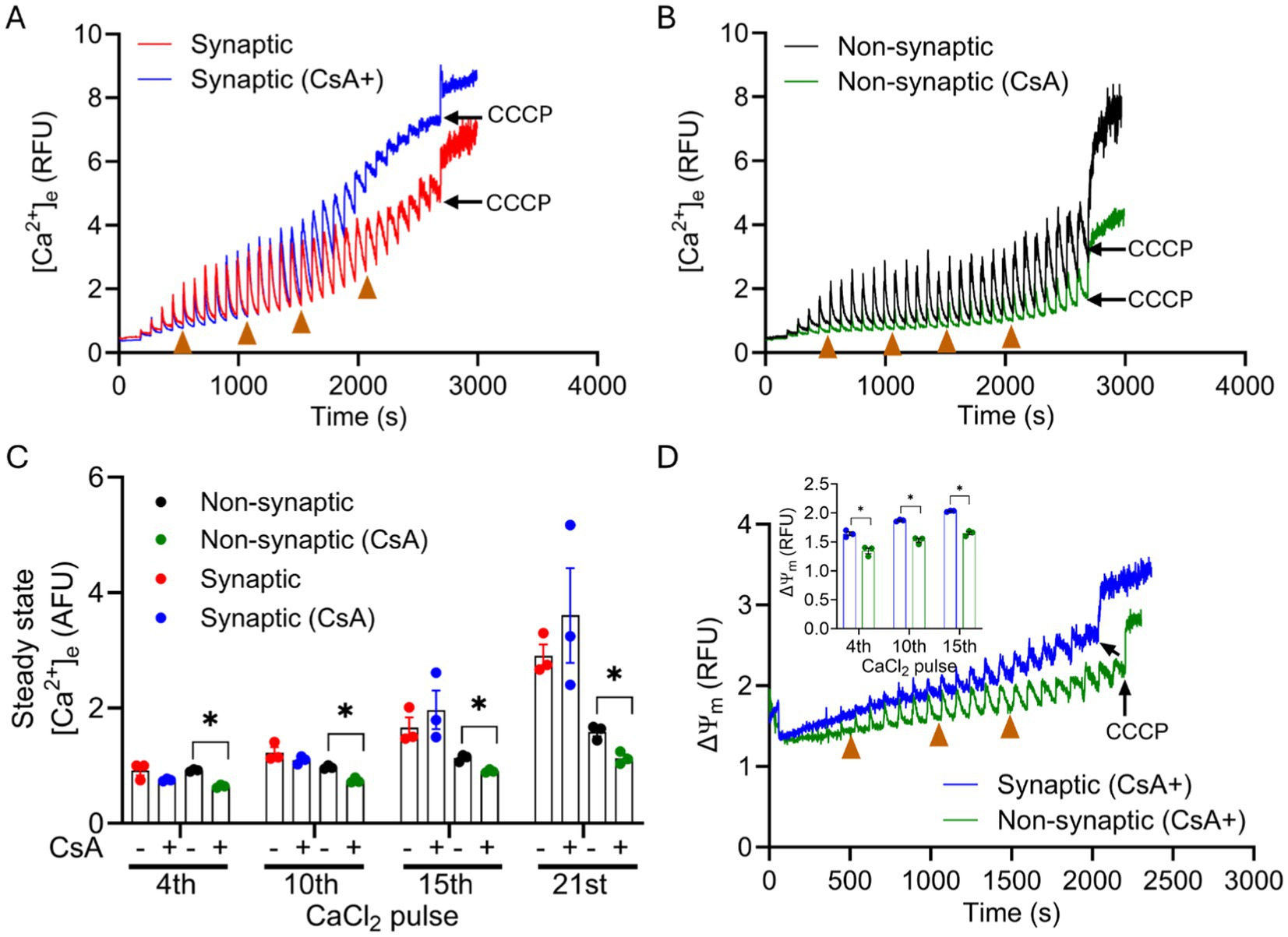
Figure 6. Effect of CsA on extra-mitochondrial calcium ([Ca2+]e) dynamics of synaptic and non-synaptic mitochondria. Representative traces of extra-matrix Ca2+ ([Ca2+]e) were measured with the Ca2+-sensitive ratiometric dye Fura-4FF in an isolated synaptic (A) and non-synaptic (B) mitochondria. 0.5 μM CsA was added in synaptic (blue trace) and non-synaptic (green trace) mitochondria at 30 s followed by the addition of complex I substrates, [Na+-glutamate + Na+-malate (GM)] at 60 s. 40 μM CaCl2 pulses were added at every 90 s, and 10 μM CCCP was added at the end of each experiment. Quantification of steady-state [Ca2+]e after a cumulative addition of 160, 400, 600, and 840 μM CaCl2 (C). Change in ΔΨm of OMN + ADP-treated synaptic and non-synaptic mitochondria were measured using the ΔΨm sensitive dye TMRM (tetramethylrhodamine methyl ester perchlorate) (D). Inset (D) shows ΔΨm after a cumulative addition of 160, 400, and 600 μM CaCl2. Error bars represent mean ± SEM (*p < 0.05).
3.6 Variable expression of mCa2+ handling and bioenergetics proteins in synaptic and non-synaptic mitochondria
Our results show that mitochondria from synaptic and non-synaptic brains display disparity in their bioenergetics and Ca2+ handling. To further elucidate the molecular underpinnings for the differential responses we examined the differences in the expression of key mitochondrial proteins involved in mCa2+ uptake and modulation of bioenergetics. The expression levels of the following crucial proteins were assessed in synaptic and non-synaptic mitochondrial fractions: mCU, voltage dependent anion channel 1 (VDAC1), adenine nucleotide translocase (ANT), and cyclophilin D (Cyp D). Mitochondrial Ca2+ uptake is primarily first through VDAC1 in the OMM, and then via the mCU in the IMM into the matrix. Additionally, VDAC1 is the main conduit for the fluxes of metabolites and nucleotides between the cytoplasm and the IMS (Camara et al., 2017). ANT, abundant in the IMM, mediates the exchange of ATP/ADP between the matrix and the IMS (Brand et al., 2005; Palmieri and Pierri, 2010). Cyp D is a mitochondrial peptidyl-prolyl cis-trans isomerase that promotes mPTP opening in the presence of excess free [Ca2+]m (Du et al., 2008; Camara et al., 2010; Camara et al., 2011).
We show (Figure 7) that mCU expression was higher in the non-synaptic mitochondria vs. synaptic mitochondria. We also show that there is no significant differences in the expression levels of VDAC1, ANT, and Cyp D between the two mitochondrial populations, when normalized to their respective mitochondrial Cox IV levels (Figure 7). We then sought to investigate expression levels of mNCE; however, we were unable to determine the true native levels because of the lack of reliable antibodies. Altogether, these results indicate that the differences in mCa2+ handling in the two mitochondrial populations is attributable to difference in the functional aspects of the mCa2+ uptake, sequestration and release, and less on the molecular levels of these proteins. These differences may have implications in the differential handling of Ca2+ in brain mitochondria during transient and cellular Ca2+ overload.
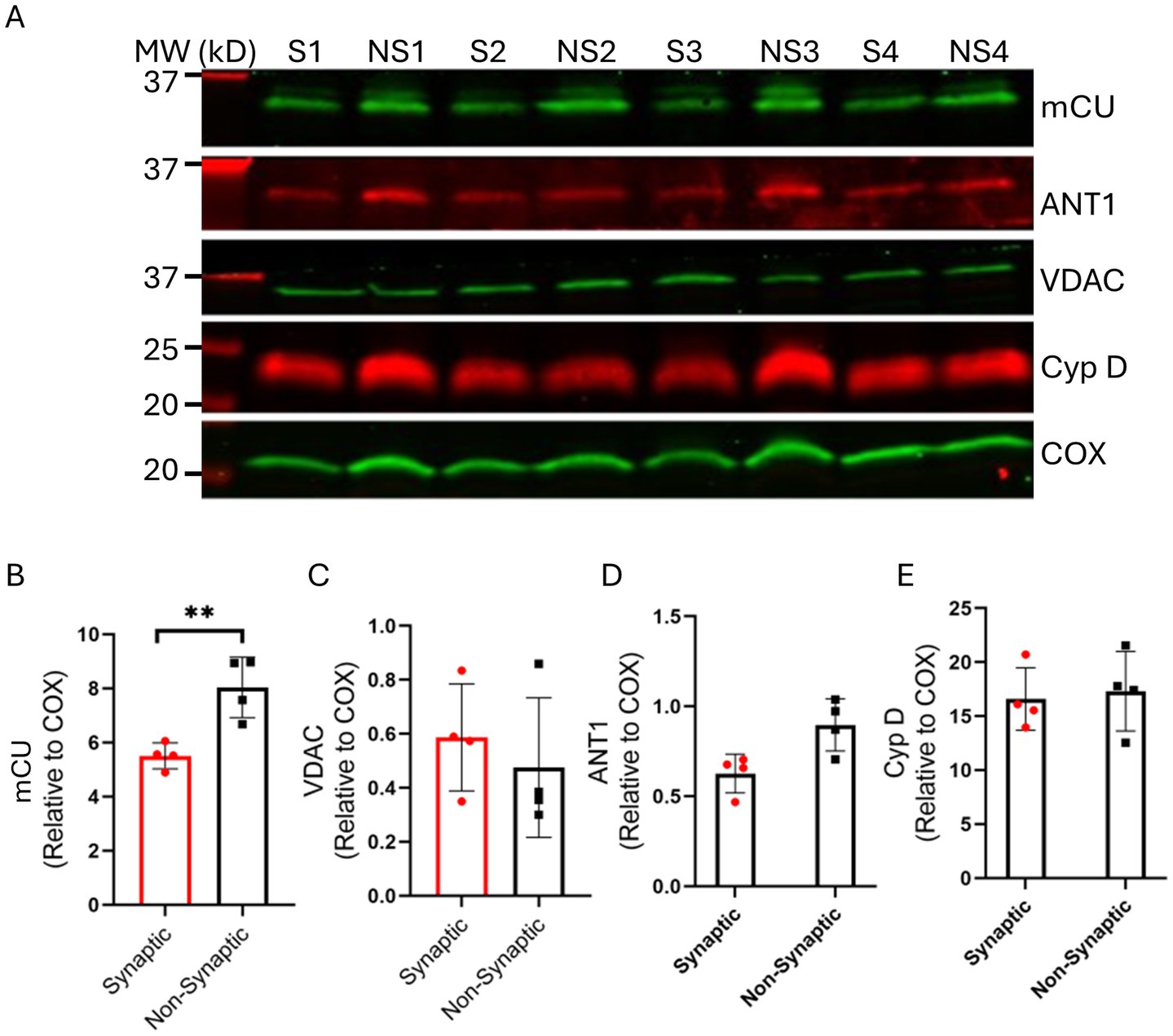
Figure 7. Assessment of synaptic (S) and non-synaptic (NS) mitochondrial proteins associated with mitochondrial bioenergetics and Ca2+ handling. Representative immunoblots (A) and quantification of the relative protein expressions of mCU (B), VDAC (C), ANT1 (D) and CyP D (E) in synaptic and non-synaptic mitochondria normalized to the mitochondrial housekeeping protein, COX IV. Error bars represent mean ± SEM (*p < 0.05 and **p < 0.01).
4 Discussion
In the brain, the large number of different cell types, with distinct metabolic profiles, excitability, and ion channel composition, contribute to the heterogeneity of brain function. Different brain cell mitochondria also exhibit a high degree of functional heterogeneity, depending on the specific cell type (e.g., mitochondria from neurons vs. glial cells) or subcellular locations within the neuron (i.e., soma, dendrites, axons and synapses) (Sonnewald et al., 1998; Pekkurnaz and Wang, 2022; Chen et al., 2023). The regional or local mitochondrial phenotypic differences are found not to consist of more variability in their mitochondrial content, but more variability in their utilization for ATP synthesis and specialization for tasks in different brain cell types (Mosharov et al., 2024). In so far as cCa2+ signaling is a key regulator of these cell functions, this functional heterogeneity may reflect how mitochondria from different brain cells or even within the same cell, display differential sensitivities to metabolism, stress, and trauma based on their differences in mCa2+ handling (Kulbe et al., 2017; Hill et al., 2018; Pekkurnaz and Wang, 2022).
Our goal in this study was to provide a systematic comparison of the dynamics of Ca2+ handling in synaptic and non-synaptic mitochondria from rat brain. Although several studies have examined and reported on the heterogeneity in the Ca2+ handling of synaptic and non-synaptic mitochondria (Brown et al., 2006; Naga et al., 2007), those studies did not delve into a detailed examination of mCa2+ dynamics in these two mitochondrial populations. In this study, we: (1) monitored basal mitochondrial bioenergetics and energetic efficiency, and (2) characterized mCa2+ handling, specifically mCa2+ uptake, buffering (sequestration), and extrusion in synaptic and non-synaptic mitochondria.
Our key functional findings are: (i) the rate of state 3 (ADP-stimulated) respiration is similar between the two populations of mitochondria. However, state 4 (ADP-depleted) respiration was faster in non-synaptic than in synaptic mitochondria, which results in a lower RCI in non-synaptic mitochondria; (ii) basal ΔΨm remains the same before and during CaCl2 boluses between the two populations; (iii) non-synaptic mitochondria take up and sequestered more Ca2+ than synaptic mitochondria consistent with prior reports (Brown et al., 2006; Naga et al., 2007); (iv) synaptic mitochondria exhibit higher mNCE-mediated Ca2+ efflux activity than non-synaptic mitochondria; (v) synaptic and non-synaptic mitochondria exhibit two distinct modes of matrix Ca2+ buffering: (a) enhanced and protracted matrix Ca2+ buffering by AdN pool (OMN + ADP) in synaptic vs. non-synaptic mitochondria, and (b) enhanced and protracted Ca2+ buffering by CsA in non-synaptic vs. synaptic mitochondria. Our quantitative protein assay demonstrate that: (i) there is greater mCU expression in non-synaptic vs. synaptic mitochondria, and (ii) there are no significant differences between the two populations in expression of VDAC1, ANT, and Cyp D proteins. Taken together, our study reveals differential mCa2+ handling features unique to synaptic and non-synaptic mitochondria that might be central to understanding their functional differences in the brain, and that there are potential implications for differential susceptibility to injury (Kulbe et al., 2017; Hill et al., 2018), particularly when this heterogeneity results in differential cCa2+ and mCa2+ overload in different brain areas.
4.1 Basal mitochondrial bioenergetics in synaptic and non-synaptic mitochondria
Synaptic mitochondria are isolated from viable synaptosomes, while non-synaptic mitochondria are isolated from various brain cell types, including glia and soma of neurons (Naga et al., 2007). The two populations of mitochondria display distinct morphological (Lewis et al., 2018; Faitg et al., 2021) and biochemical features (Lai et al., 1977; Graham et al., 2017), as well as differential vulnerabilities to oxidative damage (Banaclocha et al., 1997; Lores-Arnaiz and Bustamante, 2011; Hill et al., 2018) and Ca2+ overload (Brown et al., 2006; Naga et al., 2007; Yarana et al., 2012). To ascertain that the two populations of mitochondria accurately represent synaptic and non-synaptic mitochondria, we conducted western blot studies and showed synaptophysin and PSD95, distinct molecular markers of synaptic mitochondria, were markedly expressed in synaptic, but not in non-synaptic mitochondria (Figure 1). We showed that the isolated mitochondrial fractions were: (1) fractions of comparable purity (Figure 1), (2) analytical grade for key protein markers expression (Figure 7), and (3) functionally competent for bioenergetics and Ca2+ handling studies (Figures 2–6).
Heterogeneity in brain mitochondrial respiratory function has been studied and is due to the different functions and metabolism of different brain cell types (Gilmer et al., 2010a, 2010b; Kulbe et al., 2017). Synaptic mitochondria exhibit either decreased O2 consumption rates compared to non-synaptic mitochondria (Gilmer et al., 2010a, 2010b; Kulbe et al., 2017) or no significant difference in respiratory capacity (Hamberger et al., 1970; Hamilton et al., 2021). We found that synaptic mitochondria display a more coupled respiration, i.e., RCI than non-synaptic mitochondria. This occurred even though both mitochondrial populations exhibited comparable basal (state 2), and ADP stimulated (state 3) respirations, whereas non-synaptic mitochondria exhibited a higher state 4 respiration (after total ADP phosphorylation), likely due to a mild H+ leak, and hence the lower RCI. Mild uncoupling, due to an increase in H+ leak, stimulates respiration to maintain ΔΨm. Oligomycin is a robust and definitive measure of state 4 respiration because it blocks any ATP formation. However, rather than using oligomycin to expose a residual H+ leak in the two mitochondrial populations we used depletion of ADP as a marker of ceased ATP production to assess H+ leak during state 4. Nevertheless, it is unclear why we see the non-synaptic mitochondria display increased state 4 H+ leak that leads to the mild uncoupling.
4.2 Differential mechanisms of mCa2+ handling in synaptic and non-synaptic mitochondria
Ca2+ homeostasis is important for regulating the release of neurotransmitters at synaptic terminals and for the required ATP supply to execute these functions. As excitable cells, neurons, especially at the synaptic terminal, have high fluxes of Ca2+ in and out of their mitochondria that is coupled to their high energetic demand (Kessinger et al., 1991; Li et al., 2004) for neurotransmission. Compared to synaptic mitochondria, non-synaptic mitochondria are reported to be less sensitive to mCa2+ overloading (Brown et al., 2006; Naga et al., 2007). Here, we examined in more detail the changes in mCa2+ dynamics, in synaptic and non-synaptic mitochondria, during transient pulses of CaCl2 in the presence or absence of drugs with known effects to alter mCa2+ uptake, sequestration, or efflux.
Consistent with prior reports (Brown et al., 2006; Naga et al., 2007), we found a marked difference in mCa2+ handling between synaptic and non-synaptic mitochondria (Figure 3). We used measured, intermittent boluses of CaCl2 to have a better appreciation of the detailed mechanisms of mCa2+ handling in the two mitochondrial fractions. In both populations of mitochondria, as CaCl2 is added to the mitochondrial suspension, the extra-matrix Ca2+ signal rapidly and transiently increases, and then decreases as the Ca2+ disappears into the mitochondrial matrix via mCU. The nadir at each pulse interval and after Ca2+ uptake represents the extra-matrix steady-state (ss[Ca2+]e) attained once the bolus of CaCl2 enters the matrix and becomes sequestered. In a recent study (Mishra et al., 2019), we defined ss[Ca2+] as largely attributable to the buffering capacity of mitochondria for Ca2+. This ss[Ca2+] was determined to be inversely proportional to mCa2+ buffering capacity (Mishra et al., 2019). In the present study, while mitochondria were challenged with more CaCl2 pulses, non-synaptic mitochondria continued to markedly take up and buffer the exogenous CaCl2 for an extended period, while synaptic mitochondria buffered less of the added CaCl2. Thus, in synaptic mitochondria, the added CaCl2 gradually accumulated in the external buffer, resulting in a higher ss[Ca2+] than in non-synaptic mitochondria.
We posit that one of two mechanisms could contribute to the increase ss[Ca2+]e: (1) less mCa2+ uptake due to decrease in the ΔΨm, with a corresponding decrease in the driving force for mCU-mediated mCa2+ uptake; or (2) extrusion of the mCa2+ via the mNCE if buffering mechanisms are inadequate or overridden. Although mCU expression is higher in non-synaptic mitochondria (Figure 7), the similarity in ΔΨm (Figure 3) and the Ca2+ decay constant (Supplementary Figure 2) during the CaCl2 pulse challenges in both mitochondrial fractions suggest no differences in mCa2+ uptake contributed to the higher ss[Ca2+]e in synaptic vs. non-synaptic mitochondria. We reasoned that the mCa2+ levels for the two mitochondrial populations were maintained, in part, by different mechanisms that regulate sequestration and/or efflux.
The capacity of mitochondria to sequester mCa2+ depends on the amount of mCa2+ uptake and mCa2+ release. Thus, the matrix-free Ca2+ reflects the Ca2+ fluxes across the IMM and the matrix buffering. We found that the higher ss[Ca2+]e levels in synaptic mitochondria are more likely attributed to increased mNCE activity, resulting in greater mCa2+ release of free mCa2+. This notion of a slow release of Ca2+ from mitochondria leading to a gradual increase in ss[Ca2+]e has also been proposed in brain mitochondria during exposure to multiple boluses of CaCl2 (Hamilton et al., 2021). To test if the increased Ca2+ efflux is mediated by active mNCE during the CaCl2 boluses, we added CGP, a mNCE inhibitor, to both mitochondrial suspensions before adding CaCl2. The presence of CGP markedly increased mCa2+ uptake and mCa2+ sequestration in synaptic mitochondria as evidenced by lowering of the ss[Ca2+]e to levels observed in non-synaptic mitochondria without CGP. The extended robust uptake of mCa2+ and the lower ss[Ca2+]e in synaptic mitochondria in the presence of CGP suggest that mNCE is more active in synaptic mitochondria vs. non-synaptic mitochondria. This differential activity of mNCE could lead to extrusion of some of the added Ca2+ in synaptic mitochondria. In non-synaptic mitochondria, mCa2+ handling with CGP was like that of non-CGP treated non-synaptic mitochondria for most of the duration of the CaCl2 pulse challenges (Figure 4). Furthermore, as there was no significant change in non-synaptic uptake and buffering of mCa2+ with CGP for most of the Ca2+ pulse challenges, this suggests there is a reduced or negligible mNCE role to regulate [Ca2+]m during CaCl2 bolus challenges, compared with synaptic mitochondria. Thus, synaptic mitochondria likely maintain free mCa2+ by an active mNCE-mediated Ca2+ extrusion pathway rather than by mCa2+ buffering and sequestration during CaCl2 pulse challenge.
In comparison, non-synaptic mitochondria did not respond to CGP in regulating their mCa2+ level via a mNCE-mediated Ca2+ extrusion pathway. This heterogeneity in mCa2+ handling may reflect differential mechanisms in intracellular Ca2+ signaling in synaptic and non-synaptic regions of the brain. Impaired mNCE activity has been associated with reduced synaptic activity and mental retardation (Stavsky et al., 2021; Cabral-Costa et al., 2023); in addition, hippocampal neuron-specific deletion of NCLX, aka mNCE, was reported to impair cognitive performance (Jadiya et al., 2023). On the contrary, in vivo genetic deletion of the mNCE in hippocampal astrocytes was associated with improved cognitive performance in behavioral tasks (Cabral-Costa et al., 2023). Other studies report reduced cCa2+ levels following mNCE inhibition (Palty et al., 2010; Cabral-Costa et al., 2023; Ramos et al., 2024). Based on our results, we postulate that mNCE function is the primary regulator at synaptosomes, and that buffering acts only as a secondary regulator of mCa2+ homeostasis when mNCE is inactive during cCa2+ overload. In this case, the mNCE inhibition would contribute to greater cCa2+ dysregulation, unless the mCa2+ buffering system is activated to preserve cCa2+ and mCa2+ homeostasis.
Our premise is that the net ss[Ca2+]e during each bolus of CaCl2 is reflective of the aggregate amount of Ca2+ uptake, buffered and/or released. We have posited that maintenance of a low ss[Ca2+]e is in large part attributable to increased sequestration of mCa2+ by the buffering of Ca2+ with phosphates in the matrix. Indeed, the significance of dynamic mCa2+ buffering and the role of phosphate and other factors in regulating matrix free Ca2+ has been described in detail in our previous studies (Blomeyer et al., 2013; Boelens et al., 2013; Haumann et al., 2018; Mishra et al., 2019). The accumulation of nucleotides ADP + ATP in the matrix has been implicated in delaying mCa2+-induced mPTP opening (Hunter and Haworth, 1979; Halestrap et al., 1997). In an earlier study in cardiomyocytes, we found that adding ADP increased measured free [Ca2+]m transiently, probably by removing matrix [Pi] when ADP + Pi is converted to ATP, which is then removed from the matrix. In the presence of OMN, an ATP synthase (complex V) inhibitor, the transient increase in [Ca2+]m is inhibited because ADP + Pi cannot be converted to ATP (Haumann et al., 2010). Altering the adenine nucleotide (AdN) pool is consistent with our recent findings where we showed OMN + ADP bolstered cardiomyocyte mCa2+ buffering capacity (Mishra et al., 2019). We proceeded to confirm further whether the differences in Ca2+ handling between synaptic and non-synaptic mitochondria are due to differences in their mCa2+ buffering capabilities. To achieve this, we treated mitochondria with OMN + ADP or with CsA, two conditions we have shown enhance matrix Ca2+ buffering in cardiomyocytes (Mishra et al., 2019).
Because our current data show that matrix Ca2+ buffering in the synaptic mitochondria is bolstered by OMN + ADP, this suggests that in the synaptic mitochondria, AdNs may play a role in matrix Ca2+ buffering during exposure to excess Ca2+. Interestingly, the magnitude of mCa2+ uptake and retention are more pronounced in synaptic mitochondria in the presence of OMN + ADP than any other condition (Figure 5A). The matrix Ca2+ buffering in synaptic mitochondria in the presence of OMN + ADP was associated with normal basal ΔΨm (Figure 5D) during the CaCl2 bolus protocol. These observations suggest that the matrix buffering, which is supplemented by the active mNCE, is partly dependent on the AdN pool. In contrast, in non-synaptic mitochondria, the presence of OMN + ADP paradoxically caused early dissipation of the ΔΨm and mCa2+ release (Figures 5B,D), even after initially showing robust mCa2+ uptake and sequestration. This apparent worsening of the mCa2+ handling in non-synaptic mitochondria in the presence of OMN + ADP suggests that the buffering of mCa2+ under control conditions (Figure 2) is not mediated by an AdN-dependent buffering mechanism, but by a different buffering means. This notion is supported by our supplemental data that show in non-synaptic mitochondria, ADP and OMN given alone, led to less mCa2+ retention as evidenced by higher ss[Ca2+]e and unstable mCa2+ dynamics (Supplementary Figures 3C,D). In contrast, in synaptic mitochondria both ADP and OMN given alone before the first CaCl2 pulse showed improved ss[Ca2+]e (Supplementary Figures 3A,B) compared to their untreated controls (Figure 2A). The early collapse of the ΔΨm and concomitant release of mCa2+ despite some initial rapid uptake and buffering of the Ca2+ in the non-synaptic mitochondria is baffling but experiments to understand this are beyond the scope of our study.
4.3 CsA enhances matrix Ca2+ sequestration more in non-synaptic than in synaptic mitochondria
Our results also demonstrate that non-synaptic mitochondria rely on an unidentified matrix buffering system that is not mediated by ADP+ ATP to handle the protracted CaCl2 pulse challenges. So, we explored the possible mCa2+ buffering mechanisms. According to a previous report from our group (Blomeyer et al., 2013), at least two dynamic classes of mCa2+ buffers are known for Ca2+ buffering (Bazil et al., 2013). We propose that one class of buffers bind a single Ca2+ ion at a single binding site, i.e., like classical Ca2+ buffers (Coll et al., 1982; Corkey et al., 1986). Another class of buffer is associated with formation of amorphous Ca2+-phosphates, which have the potential of binding multiple Ca2+ ions at a single site in a cooperative fashion (Bazil et al., 2013). Moreover, we reported recently that CsA maintains low free [Ca2+]m in cardiomyocytes, in part by stimulating and/or potentiating a Pi-dependent matrix Ca2+ buffering system (Mishra et al., 2019).
To investigate further the potential contribution of this buffering system in synaptic vs. non-synaptic mCa2+ handling, CsA was added to the mitochondrial suspensions before the CaCl2 pulses. Interestingly, unlike OMN + ADP, CsA enhanced mCa2+ sequestration as evidenced by the marked reduction in ss[Ca2+]e in non-synaptic mitochondria, but not so well in synaptic mitochondria (Figure 6). The differential responses indicate that the non-synaptic and synaptic mitochondria constitutes different classes of matrix Ca2+ buffering. Furthermore, these differential responses to the buffering factors and their impact on mCa2+ handling have implications for maintaining [Ca2+]m homeostasis in non-synaptic vs. synaptic mitochondria. However, additional studies are needed to delve further into the nature of the Ca2+ buffering systems in these two mitochondrial populations and to delineate the underlying buffering mechanisms in brain mitochondria relevant to understanding Ca2+ regulation at the synapses.
4.4 Atypical or absent mPTP opening in synaptic and non-synaptic mitochondria during CaCl2 pulse challenges
In our study, both synaptic and non-synaptic mitochondria, under control conditions, did not display the characteristic mPTP opening during CaCl2 pulse challenges observed in mitochondria isolated from cardiomyocytes (Aldakkak et al., 2011; Mishra et al., 2019; Mishra and Camara, 2022). This lack of distinct mPTP opening and absence of massive extrusion of mCa2+, as shown by the rapid increase in the extra-matrix fluorescent signal for [Ca2+]e, is consistent with other studies using isolated brain mitochondria (Andreyev et al., 1998; Berman et al., 2000).
Biophysically, classic mPTP opening is characterized by matrix swelling and a dramatic increase in IMM permeability, with the subsequent release of mCa2+, and accompanied by a total collapse of ΔΨm and release of pro-apoptotic factors like cytochrome c (Bernardi et al., 1992; Szabo and Zoratti, 1992; Basso et al., 2005; Mishra et al., 2019; Sun et al., 2022). Unlike in other tissues like heart (Mishra et al., 2019; Sun et al., 2022) and liver (Varanyuwatana and Halestrap, 2012; Kim et al., 2023), we found that repetitive boluses of CaCl2 to both synaptic and non-synaptic mitochondria only led to gradual increase in ss[Ca2+]e, without IMM permeabilization, massive release of matrix Ca2+ or complete loss of ΔΨm. So, we posit that in contrast to heart mitochondria where we observe mPTP opening with lower [Ca2+] (Natarajan et al., 2020), brain mitochondria, with higher [Ca2+], show a much greater capacity to sequester Ca2+, and or to extrude Ca2+ leading to more Ca2+ uptake. This is evidenced by the progressive increase in matrix Ca2+ in non-synaptic mitochondria, and the continuing decrease in ss[Ca2+]e in synaptic mitochondria with similar gradual dissipations of ΔΨm in the two mitochondrial population. This gradual depolarization leads eventually to the diminution of the driving force for further Ca2+ uptake and thereby prevents the matrix from reaching the critical Ca2+ threshold required to open the pore. Another possible explanation is that the mPTP may constitute different entities in brain and heart cells. In concordance with this notion, Bernardi et al. (2023) argued that “remaining issues” in the current debate about the structure and identity of the pore include defining whether there is more than one protein type of mPTP.
In our study, a sudden spike in mCa2+ release or ΔΨm depolarizations at the end of each protocol were caused by the application of CCCP; this was given to establish maximal ΔΨm depolarization and release of some of the matrix Ca2+. An interesting, and unexplainable finding, is that only in the presence of OMN + ADP did both synaptic and non-synaptic mitochondria exhibit collapse of ΔΨm and total release of mCa2+ during the CaCl2 pulse challenges. These events occurred much earlier in non-synaptic mitochondria compared with synaptic mitochondria (Figure 5). As alluded to previously, these unexpected results in the presence of OMN + ADP suggest yet identified features in the constituents of the mPTP complex in different tissues.
4.5 Potential molecular factors contributing to the differential Ca2+ handling and differences in bioenergetics in synaptic vs. non-synaptic mitochondria
Molecular mechanisms could contribute to the differences in mCa2+ handling between synaptic vs. non-synaptic mitochondria. To supplement physiological studies, we examined key mitochondrial proteins involved in regulating [Ca2+]m. Consistent with a previous study (Stauch et al., 2014), western blot showed lower expression of mCU in synaptic vs. non-synaptic mitochondria (Figure 7). Knockdown of endogenous mCU expression leads to reduction of NMDA-mediated increase in mCa2+, lower levels of ΔΨm depolarizations and prevents excitotoxicity (Qiu et al., 2013). Since synaptic mitochondria are reported to be more susceptible to Ca2+ overload (Brown et al., 2006; Naga et al., 2007; Yarana et al., 2012), the lower expression of mCU suggests less Ca2+ uptake, and concomitantly, less [Ca2+]m overload during transient increases in cCa2+. However, whether the differences in the expression of mCU contributed significantly to the differences in mCa2+ handling between the synaptic and non-synaptic mitochondria is unclear, since the pore-forming mCU is regulated by a complex of associated proteins (MacEwen and Sancak, 2023). Furthermore, based on similarities of ΔΨm profiles, mCa2+ uptake (Supplementary Figure 2) and buffering in the presence and absence of CGP in synaptic and non-synaptic mitochondria, respectively, we propose that the uptake phase of mCa2+ may not contribute to the differential regulation of [Ca2+]m in these two fractions.
There was no significant difference in the expressions of VDAC1, ANT and Cyp D between synaptic and non-synaptic mitochondria (Figure 7). The lack of differences in VDAC1 on the OMM indicate that both populations likely have a similar [Ca2+]m in their IMS during the CaCl2 pulse challenges. Similarities in ANT expression suggest no significant differences in the translocation of ADP and ATP across the IMM of the two mitochondrial fractions. A previous study (Naga et al., 2007) showed significant differences in the expression of Cyp D between the two populations, with synaptic mitochondria showing higher Cyp D expression. Increased Cyp D expression sensitizes mitochondria to Ca2+-mediated mPTP opening (Du et al., 2008; Camara et al., 2010). CsA binds to mitochondrial Cyp D and so may desensitize mitochondria to excess Ca2+ (Hamilton et al., 2021) or may enhance matrix Ca2+ sequestration (Mishra et al., 2019). However, in our study, we observed no significant differences in Cyp D expression between the two populations. Moreover, the presence of CsA bolstered the mCa2+ uptake and buffering in non-synaptic vs. synaptic mitochondria. These findings are inconsistent with reports that show more CsA was required in synaptic mitochondria to inhibit mPTP. This inconsistency may be ascribed to the lack of significant differences in CypD between the two fractions in our study. This suggests that in our study the CsA effect on mCa2+ could be attributable to its Pi-dependent buffering effect, as we reported previously (Mishra et al., 2019).
Overall, our result indicate that the mNCE likely plays a prominent role in maintaining free [Ca2+]m in synaptic mitochondria by ejecting much of the Ca2+ during periods of increased extra-matrix CaCl2 challenges. Regarding the role of mNCE activity between the two mitochondrial fractions, we postulate that it could, in part, be attributed to increased expression of the exchanger. Unfortunately, we were unable to confirm this notion because of the lack of reliable mNCE antibodies at the time. Other contributing factors to the high mNCE -mediated Ca2+ efflux could be increased activity through posttranslational modification (PTM) of the exchanger (Kostic et al., 2015). Indeed, there is evidence from a recent report that mNCE activity is increased via a PKA-mediated phosphorylation of the exchanger. This targeted phosphorylation is reported to reverse [Ca2+]m overload by increasing mCa2+ efflux and to promote cell survival (Kostic et al., 2015).
PTM studies are beyond the scope of our study and are also hampered by the lack of reliable commercially available phospho-mNCE antibodies. Nonetheless, delineating these molecular mechanisms could provide novel insights into our understanding of the function of the mNCE in preserving Ca2+ homeostasis during periods of increased synaptic Ca2+ transients during neurotransmission at the synapse. Lastly, mNCE function, which is electrogenic, is dependent on ΔΨm, as we and others have reported (Blomeyer et al., 2013; Blomeyer et al., 2016; Kostic et al., 2018); however, we found no difference in basal ΔΨm in synaptic and non-synaptic mitochondria. In addition, the transient and gradual depolarization of ΔΨm during the CaCl2 pulse challenges were similar for the two populations, suggesting that the mNCE was not hampered by ΔΨm. Altogether, these results indicate that synaptic and non-synaptic mitochondria employ different strategies to cope with the challenges of excess [Ca2+]m to sustain their resting ΔΨm, that is essential to regulate Ca2+ homeostasis.
5 Summary and conclusion
Our study demonstrates that synaptic and non-synaptic mitochondria handle excess mCa2+ by predominately separate mechanisms that likely involve dependencies on mCa2+-induced extrusion via mNCE vs. mCa2+ buffering, respectively. The reliance on Ca2+ efflux in synaptic mitochondria and Ca2+ buffering in non-synaptic mitochondria, combined with no significant differences in ΔΨm between the two populations during the CaCl2 pulse challenges confirm that different mechanisms are involved in regulating [Ca2+]m during mCa2+ overload. In synaptic mitochondria, the quiescent and nominal matrix buffering is dependent, in part, on AdN (OMN + ADP)-mediated mCa2+ sequestration. In contrast, the active matrix buffering in the non-synaptic mitochondria is dependent, in part, on Pi-mediated mCa2+ buffering system that is bolstered by CsA. The implication of these observations may portend differences in the mCa2+ buffering systems and the regulation of ΔΨm in these two populations of mitochondria.
Altogether, our study provides new mechanistic insights into how synaptic and non-synaptic mitochondria differentially handle Ca2+ during exposure to excess extra-matrix Ca2+. Synaptic mitochondria are susceptible to Ca2+ dysregulation during cCa2+ overload (Brown et al., 2006; Naga et al., 2007; Yarana et al., 2012) and oxidative stress (Banaclocha et al., 1997; Lores-Arnaiz and Bustamante, 2011; Hill et al., 2018), as reported in some neurological disorders, like traumatic brain injury (Kulbe et al., 2017; Hill et al., 2018) and other neurodegenerative diseases (Du et al., 2012). Therefore, this in-depth assessment of ss[Ca2+]e on cCa2+ homeostasis in synaptic and non-synaptic mitochondria could contribute to our understanding of the potential roles of these two brain mitochondrial populations in the etiology and progression of neurodegenerative diseases.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. KB: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – review & editing, Writing – original draft. KL: Data curation, Formal analysis, Methodology, Validation, Writing – review & editing. AZ: Formal analysis, Methodology, Validation, Writing – review & editing, Data curation. JH: Data curation, Formal analysis, Methodology, Validation, Writing – review & editing. AT: Investigation, Methodology, Writing – review & editing. W-MK: Writing – review & editing. DS: Funding acquisition, Writing – review & editing. AC: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported in part by funding from the MCW Advancing a Healthier Wisconsin Endowment Project 5520444, NHLBI Training Grant T35 HL072483 and Merit Review BX-002539-01.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnsyn.2025.1562065/full#supplementary-material
SUPPLEMENTARY FIGURE 1 | Respiration in isolated mitochondria from synaptic and non-synaptic fraction. State 2 (A), 3 (B) and 4 (C) respiration with the complex I substrates, [Na+-glutamate + Na+-malate (GM)]. Error bars represent mean ± SEM (*p < 0.05 and **p < 0.01).
SUPPLEMENTARY FIGURE 2 | Representative traces of extra-matrix Ca2+ ([Ca2+]e) measured with the Ca2+-sensitive ratiometric dye Fura-4FF. Inset shows quantification of decay constants. Error bars represent mean ± SEM.
SUPPLEMENTARY FIGURE 3 | Effect of ADP alone (A,C) and OMN alone (B,D) on extra-mitochondrial calcium ([Ca2+]e) dynamics of synaptic (A,B) and non-synaptic (C,D) mitochondria. Representative traces of extra-matrix Ca2+ ([Ca2+]e) measured with the Ca2+-sensitive ratiometric dye Fura-4F in isolated synaptic (A,B) and non-synaptic (C,D) mitochondria. 250 μM ADP (A,C) and 10 μM OMN (B,D) were added to synaptic (pink traces) and non-synaptic (gray traces) mitochondria at 30 s followed by the addition of the complex I substrates, [Na+-glutamate + Na+-malate (GM)] at 60 s. 40 μM CaCl2 pulses were added at every 90 s, and 10 μM CCCP was added at the end of each experiment.
References
Aklima, J., Onojima, T., Kimura, S., Umiuchi, K., Shibata, T., Kuraoka, Y., et al. (2021). Effects of matrix pH on spontaneous transient depolarization and reactive oxygen species production in mitochondria. Front. Cell Dev. Biol. 9:692776. doi: 10.3389/fcell.2021.692776
Aldakkak, M., Camara, A. K., Heisner, J. S., Yang, M., and Stowe, D. F. (2011). Ranolazine reduces Ca2+ overload and oxidative stress and improves mitochondrial integrity to protect against ischemia reperfusion injury in isolated hearts. Pharmacol. Res. 64, 381–392. doi: 10.1016/j.phrs.2011.06.018
Aldakkak, M., Stowe, D. F., Dash, R. K., and Camara, A. K. (2013). Mitochondrial handling of excess Ca2+ is substrate-dependent with implications for reactive oxygen species generation. Free Radic. Biol. Med. 56, 193–203. doi: 10.1016/j.freeradbiomed.2012.09.020
Andreyev, A. Y., Fahy, B., and Fiskum, G. (1998). Cytochrome C release from brain mitochondria is independent of the mitochondrial permeability transition. FEBS Lett. 439, 373–376. doi: 10.1016/s0014-5793(98)01394-5
Banaclocha, M. M., Hernandez, A. I., Martinez, N., and Ferrandiz, M. L. (1997). N-acetylcysteine protects against age-related increase in oxidized proteins in mouse synaptic mitochondria. Brain Res. 762, 256–258. doi: 10.1016/s0006-8993(97)00493-9
Basso, E., Fante, L., Fowlkes, J., Petronilli, V., Forte, M. A., and Bernardi, P. (2005). Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J. Biol. Chem. 280, 18558–18561. doi: 10.1074/jbc.C500089200
Baughman, J. M., Perocchi, F., Girgis, H. S., Plovanich, M., Belcher-Timme, C. A., Sancak, Y., et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. doi: 10.1038/nature10234
Bazil, J. N., Blomeyer, C. A., Pradhan, R. K., Camara, A. K., and Dash, R. K. (2013). Modeling the calcium sequestration system in isolated guinea pig cardiac mitochondria. J. Bioenerg. Biomembr. 45, 177–188. doi: 10.1007/s10863-012-9488-2
Berman, S. B., Watkins, S. C., and Hastings, T. G. (2000). Quantitative biochemical and ultrastructural comparison of mitochondrial permeability transition in isolated brain and liver mitochondria: evidence for reduced sensitivity of brain mitochondria. Exp. Neurol. 164, 415–425. doi: 10.1006/exnr.2000.7438
Bernardi, P., Gerle, C., Halestrap, A. P., Jonas, E. A., Karch, J., Mnatsakanyan, N., et al. (2023). Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions. Cell Death Differ. 30, 1869–1885. doi: 10.1038/s41418-023-01187-0
Bernardi, P., Vassanelli, S., Veronese, P., Colonna, R., Szabo, I., and Zoratti, M. (1992). Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J. Biol. Chem. 267, 2934–2939. doi: 10.1016/S0021-9258(19)50676-7
Billups, B., and Forsythe, I. D. (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 22, 5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002
Blomeyer, C. A., Bazil, J. N., Stowe, D. F., Dash, R. K., and Camara, A. K. (2016). Mg2+ differentially regulates two modes of mitochondrial Ca2+ uptake in isolated cardiac mitochondria: implications for mitochondrial Ca2+ sequestration. J. Bioenerg. Biomembr. 48, 175–188. doi: 10.1007/s10863-016-9644-1
Blomeyer, C. A., Bazil, J. N., Stowe, D. F., Pradhan, R. K., Dash, R. K., and Camara, A. K. (2013). Dynamic buffering of mitochondrial Ca2+ during Ca2+ uptake and Na+ induced Ca2+ release. J. Bioenerg. Biomembr. 45, 189–202. doi: 10.1007/s10863-012-9483-7
Boelens, A. D., Pradhan, R. K., Blomeyer, C. A., Camara, A. K., Dash, R. K., and Stowe, D. F. (2013). Extra-matrix Mg2+ limits Ca2+ uptake and modulates Ca2+ uptake-independent respiration and redox state in cardiac isolated mitochondria. J. Bioenerg. Biomembr. 45, 203–218. doi: 10.1007/s10863-013-9500-5
Bordone, M. P., Salman, M. M., Titus, H. E., Amini, E., Andersen, J. V., Chakraborti, B., et al. (2019). The energetic brain—a review from students to students. J. Neurochem. 151, 139–165. doi: 10.1111/jnc.14829
Brand, M. D., Pakay, J. L., Ocloo, A., Kokoszka, J., Wallace, D. C., Brookes, P. S., et al. (2005). The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem. J. 392, 353–362. doi: 10.1042/BJ20050890
Brown, M. R., Sullivan, P. G., and Geddes, J. W. (2006). Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J. Biol. Chem. 281, 11658–11668. doi: 10.1074/jbc.M510303200
Cabral-Costa, J. V., Vicente-Gutierrez, C., Agulla, J., Lapresa, R., Elrod, J. W., Almeida, A., et al. (2023). Mitochondrial sodium/calcium exchanger NCLX regulates glycolysis in astrocytes, impacting on cognitive performance. J. Neurochem. 165, 521–535. doi: 10.1111/jnc.15745
Camara, A. K., Bienengraeber, M., and Stowe, D. F. (2011). Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2:13. doi: 10.3389/fphys.2011.00013
Camara, A. K., Lesnefsky, E. J., and Stowe, D. F. (2010). Potential therapeutic benefits of strategies directed to mitochondria. Antioxid. Redox Signal. 13, 279–347. doi: 10.1089/ars.2009.2788
Camara, A. K. S., Zhou, Y., Wen, P. C., Tajkhorshid, E., and Kwok, W. M. (2017). Mitochondrial VDAC1: a key gatekeeper as potential therapeutic target. Front. Physiol. 8:460. doi: 10.3389/fphys.2017.00460
Chalmers, S., and Nicholls, D. G. (2003). The relationship between free and total calcium concentrations in the matrix of liver and brain mitochondria. J. Biol. Chem. 278, 19062–19070. doi: 10.1074/jbc.M212661200
Chandra, R. K. (1991). Immunocompetence is a sensitive and functional barometer of nutritional status. Acta Paediatr. Scand. Suppl. 374, 129–132. doi: 10.1111/j.1651-2227.1991.tb12015.x
Chen, L., Zhou, M., Li, H., Liu, D., Liao, P., Zong, Y., et al. (2023). Mitochondrial heterogeneity in diseases. Signal Transduct. Target. Ther. 8:311. doi: 10.1038/s41392-023-01546-w
Coll, K. E., Joseph, S. K., Corkey, B. E., and Williamson, J. R. (1982). Determination of the matrix free Ca2+ concentration and kinetics of Ca2+ efflux in liver and heart mitochondria. J. Biol. Chem. 257, 8696–8704. doi: 10.1016/S0021-9258(18)34184-X
Corkey, B. E., Duszynski, J., Rich, T. L., Matschinsky, B., and Williamson, J. R. (1986). Regulation of free and bound magnesium in rat hepatocytes and isolated mitochondria. J. Biol. Chem. 261, 2567–2574. doi: 10.1016/S0021-9258(17)35825-8
Csordas, G., Varnai, P., Golenar, T., Sheu, S. S., and Hajnoczky, G. (2012). Calcium transport across the inner mitochondrial membrane: molecular mechanisms and pharmacology. Mol. Cell. Endocrinol. 353, 109–113. doi: 10.1016/j.mce.2011.11.011
Datta, S., and Jaiswal, M. (2021). Mitochondrial calcium at the synapse. Mitochondrion 59, 135–153. doi: 10.1016/j.mito.2021.04.006
Davis, S., and McLean, S. (1987). Smoking habits and beliefs, and attitudes to cigarette advertisements, amongst grade 6 children. Community Health Stud. 11, 35s–40s. doi: 10.1111/j.1753-6405.1987.tb00511.x
De Stefani, D., Raffaello, A., Teardo, E., Szabo, I., and Rizzuto, R. (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. doi: 10.1038/nature10230
Du, H., Guo, L., Fang, F., Chen, D., Sosunov, A. A., McKhann, G. M., et al. (2008). Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 14, 1097–1105. doi: 10.1038/nm.1868
Du, H., Guo, L., and Yan, S. S. (2012). Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid. Redox Signal. 16, 1467–1475. doi: 10.1089/ars.2011.4277
Du, H., Guo, L., Yan, S., Sosunov, A. A., McKhann, G. M., and Yan, S. S. (2010). Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 107, 18670–18675. doi: 10.1073/pnas.1006586107
Faitg, J., Lacefield, C., Davey, T., White, K., Laws, R., Kosmidis, S., et al. (2021). 3D neuronal mitochondrial morphology in axons, dendrites, and somata of the aging mouse hippocampus. Cell Rep. 36:109509. doi: 10.1016/j.celrep.2021.109509
Fecher, C., Trovo, L., Muller, S. A., Snaidero, N., Wettmarshausen, J., Heink, S., et al. (2019). Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 22, 1731–1742. doi: 10.1038/s41593-019-0479-z
Gerdes, H. J., Yang, M., Heisner, J. S., Camara, A. K. S., and Stowe, D. F. (2020). Modulation of peroxynitrite produced via mitochondrial nitric oxide synthesis during Ca2+ and succinate-induced oxidative stress in cardiac isolated mitochondria. Biochim. Biophys. Acta Bioenerg. 1861:148290. doi: 10.1016/j.bbabio.2020.148290
Gilmer, L. K., Ansari, M. A., Roberts, K. N., and Scheff, S. W. (2010a). Age-related changes in mitochondrial respiration and oxidative damage in the cerebral cortex of the Fischer 344 rat. Mech. Ageing Dev. 131, 133–143. doi: 10.1016/j.mad.2009.12.011
Gilmer, L. K., Ansari, M. A., Roberts, K. N., and Scheff, S. W. (2010b). Age-related mitochondrial changes after traumatic brain injury. J. Neurotrauma 27, 939–950. doi: 10.1089/neu.2009.1181
Glantz, L. A., Gilmore, J. H., Hamer, R. M., Lieberman, J. A., and Jarskog, L. F. (2007). Synaptophysin and postsynaptic density protein 95 in the human prefrontal cortex from mid-gestation into early adulthood. Neuroscience 149, 582–591. doi: 10.1016/j.neuroscience.2007.06.036
Graham, L. C., Eaton, S. L., Brunton, P. J., Atrih, A., Smith, C., Lamont, D. J., et al. (2017). Proteomic profiling of neuronal mitochondria reveals modulators of synaptic architecture. Mol. Neurodegener. 12:77. doi: 10.1186/s13024-017-0221-9
Halestrap, A. P. (2010). A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 38, 841–860. doi: 10.1042/BST0380841
Halestrap, A. P., Connern, C. P., Griffiths, E. J., and Kerr, P. M. (1997). Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 174, 167–172. doi: 10.1023/A:1006879618176
Hamberger, A., Blomstrand, C., and Lehninger, A. L. (1970). Comparative studies on mitochondria isolated from neuron-enriched and glia-enriched fractions of rabbit and beef brain. J. Cell Biol. 45, 221–234. doi: 10.1083/jcb.45.2.221
Hamilton, J., Brustovetsky, T., and Brustovetsky, N. (2021). The effect of mitochondrial calcium uniporter and cyclophilin D knockout on resistance of brain mitochondria to Ca2+-induced damage. J. Biol. Chem. 296:100669. doi: 10.1016/j.jbc.2021.100669
Haumann, J., Camara, A. K. S., Gadicherla, A. K., Navarro, C. D., Boelens, A. D., Blomeyer, C. A., et al. (2018). Slow Ca2+ efflux by Ca2+/H+ exchange in cardiac mitochondria is modulated by Ca2+ re-uptake via MCU, extra-mitochondrial pH, and H+ pumping by FoF1-ATPase. Front. Physiol. 9:1914. doi: 10.3389/fphys.2018.01914
Haumann, J., Dash, R. K., Stowe, D. F., Boelens, A. D., Beard, D. A., and Camara, A. K. (2010). Mitochondrial free [Ca2+] increases during ATP/ADP antiport and ADP phosphorylation: exploration of mechanisms. Biophys. J. 99, 997–1006. doi: 10.1016/j.bpj.2010.04.069
Hill, R. L., Kulbe, J. R., Singh, I. N., Wang, J. A., and Hall, E. D. (2018). Synaptic mitochondria are more susceptible to traumatic brain injury-induced oxidative damage and respiratory dysfunction than non-synaptic mitochondria. Neuroscience 386, 265–283. doi: 10.1016/j.neuroscience.2018.06.028
Hill, R. L., Singh, I. N., Wang, J. A., Kulbe, J. R., and Hall, E. D. (2020). Protective effects of phenelzine administration on synaptic and non-synaptic cortical mitochondrial function and lipid peroxidation-mediated oxidative damage following TBI in young adult male rats. Exp. Neurol. 330:113322. doi: 10.1016/j.expneurol.2020.113322
Hollenbeck, P. J. (2005). Mitochondria and neurotransmission: evacuating the synapse. Neuron 47, 331–333. doi: 10.1016/j.neuron.2005.07.017
Hunter, D. R., and Haworth, R. A. (1979). The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch. Biochem. Biophys. 195, 453–459. doi: 10.1016/0003-9861(79)90371-0
Islam, M. M., Takeuchi, A., and Matsuoka, S. (2020). Membrane current evoked by mitochondrial Na+-Ca2+ exchange in mouse heart. J. Physiol. Sci. 70:24. doi: 10.1186/s12576-020-00752-3
Jadiya, P., Cohen, H. M., Kolmetzky, D. W., Kadam, A. A., Tomar, D., and Elrod, J. W. (2023). Neuronal loss of NCLX-dependent mitochondrial calcium efflux mediates age-associated cognitive decline. iScience 26:106296. doi: 10.1016/j.isci.2023.106296
Jung, H., Kim, S. Y., Canbakis Cecen, F. S., Cho, Y., and Kwon, S. K. (2020). Dysfunction of mitochondrial Ca2+ regulatory machineries in brain aging and neurodegenerative diseases. Front. Cell Dev. Biol. 8:599792. doi: 10.3389/fcell.2020.599792
Kalani, K., Yan, S. F., and Yan, S. S. (2018). Mitochondrial permeability transition pore: a potential drug target for neurodegeneration. Drug Discov. Today 23, 1983–1989. doi: 10.1016/j.drudis.2018.08.001
Kamat, P. K., Kalani, A., and Tyagi, N. (2014). Method and validation of synaptosomal preparation for isolation of synaptic membrane proteins from rat brain. MethodsX 1, 102–107. doi: 10.1016/j.mex.2014.08.002
Kannurpatti, S. S. (2017). Mitochondrial calcium homeostasis: implications for neurovascular and neurometabolic coupling. J. Cereb. Blood Flow Metab. 37, 381–395. doi: 10.1177/0271678X16680637
Kessinger, A., Vose, J. M., Bierman, P. J., and Armitage, J. O. (1991). High-dose therapy and autologous peripheral stem cell transplantation for patients with bone marrow metastases and relapsed lymphoma: an alternative to bone marrow purging. Exp. Hematol. 19, 1013–1016
Kim, J., Zimmerman, M. A., Shin, W. Y., Boettcher, B. T., Lee, J. S., Park, J. I., et al. (2023). Effects of subnormothermic regulated hepatic reperfusion on mitochondrial and transcriptomic profiles in a porcine model. Ann. Surg. 277, e366–e375. doi: 10.1097/SLA.0000000000005156
Kostic, M., Katoshevski, T., and Sekler, I. (2018). Allosteric regulation of NCLX by mitochondrial membrane potential links the metabolic state and Ca2+ signaling in mitochondria. Cell Rep. 25, 3465–3475.e4. doi: 10.1016/j.celrep.2018.11.084
Kostic, M., Ludtmann, M. H., Bading, H., Hershfinkel, M., Steer, E., Chu, C. T., et al. (2015). PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 13, 376–386. doi: 10.1016/j.celrep.2015.08.079
Kulbe, J. R., Hill, R. L., Singh, I. N., Wang, J. A., and Hall, E. D. (2017). Synaptic mitochondria sustain more damage than non-synaptic mitochondria after traumatic brain injury and are protected by cyclosporine A. J. Neurotrauma 34, 1291–1301. doi: 10.1089/neu.2016.4628
Lai, J. C., Walsh, J. M., Dennis, S. C., and Clark, J. B. (1977). Synaptic and non-synaptic mitochondria from rat brain: isolation and characterization. J. Neurochem. 28, 625–631. doi: 10.1111/j.1471-4159.1977.tb10434.x
Lewis, T. L. Jr., Kwon, S. K., Lee, A., Shaw, R., and Polleux, F. (2018). MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 9:5008. doi: 10.1038/s41467-018-07416-2
Li, Z., Okamoto, K., Hayashi, Y., and Sheng, M. (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. doi: 10.1016/j.cell.2004.11.003
Liu, Y., and Schubert, D. R. (2009). The specificity of neuroprotection by antioxidants. J. Biomed. Sci. 16:98. doi: 10.1186/1423-0127-16-98
Liu, T., Takimoto, E., Dimaano, V. L., DeMazumder, D., Kettlewell, S., Smith, G., et al. (2014). Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 115, 44–54. doi: 10.1161/CIRCRESAHA.115.303062
Lores-Arnaiz, S., and Bustamante, J. (2011). Age-related alterations in mitochondrial physiological parameters and nitric oxide production in synaptic and non-synaptic brain cortex mitochondria. Neuroscience 188, 117–124. doi: 10.1016/j.neuroscience.2011.04.060
MacEwen, M. J. S., and Sancak, Y. (2023). Beyond the matrix: structural and physiological advancements in mitochondrial calcium signaling. Biochem. Soc. Trans. 51, 665–673. doi: 10.1042/BST20220317
Magistretti, P. J., and Allaman, I. (2015). A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901. doi: 10.1016/j.neuron.2015.03.035
Meir, A., Ginsburg, S., Butkevich, A., Kachalsky, S. G., Kaiserman, I., Ahdut, R., et al. (1999). Ion channels in presynaptic nerve terminals and control of transmitter release. Physiol. Rev. 79, 1019–1088. doi: 10.1152/physrev.1999.79.3.1019
Mishra, J., and Camara, A. K. S. (2022). Mitochondrial calcium handling in isolated mitochondria from a Guinea pig heart. Methods Mol. Biol. 2497, 97–106. doi: 10.1007/978-1-0716-2309-1_6
Mishra, J., Davani, A. J., Natarajan, G. K., Kwok, W. M., Stowe, D. F., and Camara, A. K. S. (2019). Cyclosporin A increases mitochondrial buffering of calcium: an additional mechanism in delaying mitochondrial permeability transition pore opening. Cells 8:1052. doi: 10.3390/cells8091052
Mishra, J., Jhun, B. S., Hurst, S., Csordas, G., and Sheu, S. S. (2017). The mitochondrial Ca2+ uniporter: structure, function, and pharmacology. Handb. Exp. Pharmacol. 240, 129–156. doi: 10.1007/164_2017_1
Mosharov, E. V., Rosenberg, A. M., Monzel, A. S., Osto, C. A., Stiles, L., Rosoklija, G. B., et al. (2024). A human brain map of mitochondrial respiratory capacity and diversity. Res Sq. doi: 10.21203/rs.3.rs-4047706/v1
Naga, K. K., Sullivan, P. G., and Geddes, J. W. (2007). High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J. Neurosci. 27, 7469–7475. doi: 10.1523/JNEUROSCI.0646-07.2007
Natarajan, G. K., Glait, L., Mishra, J., Stowe, D. F., Camara, A. K. S., and Kwok, W. M. (2020). Total matrix Ca2+ modulates Ca2+ efflux via the Ca2+/H+ exchanger in cardiac mitochondria. Front. Physiol. 11:510600. doi: 10.3389/fphys.2020.510600
Olesen, M. A., Torres, A. K., Jara, C., Murphy, M. P., and Tapia-Rojas, C. (2020). Premature synaptic mitochondrial dysfunction in the hippocampus during aging contributes to memory loss. Redox Biol. 34:101558. doi: 10.1016/j.redox.2020.101558
Palmieri, F., and Pierri, C. L. (2010). Mitochondrial metabolite transport. Essays Biochem. 47, 37–52. doi: 10.1042/bse0470037
Palty, R., and Sekler, I. (2012). The mitochondrial Na+/Ca2+ exchanger. Cell Calcium 52, 9–15. doi: 10.1016/j.ceca.2012.02.010
Palty, R., Silverman, W. F., Hershfinkel, M., Caporale, T., Sensi, S. L., Parnis, J., et al. (2010). NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U.S.A. 107, 436–441. doi: 10.1073/pnas.0908099107
Pekkurnaz, G., and Wang, X. (2022). Mitochondrial heterogeneity and homeostasis through the lens of a neuron. Nat. Metab. 4, 802–812. doi: 10.1038/s42255-022-00594-w
Pivovarova, N. B., and Andrews, S. B. (2010). Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 277, 3622–3636. doi: 10.1111/j.1742-4658.2010.07754.x
Qiu, J., Tan, Y. W., Hagenston, A. M., Martel, M. A., Kneisel, N., Skehel, P. A., et al. (2013). Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 4:2034. doi: 10.1038/ncomms3034
Ramos, V. M., Serna, J. D. C., Vilas-Boas, E. A., Cabral-Costa, J. V., Cunha, F. M., Kataura, T., et al. (2024). Mitochondrial sodium/calcium exchanger (NCLX) regulates basal and starvation-induced autophagy through calcium signaling. FASEB J. 38:e23454. doi: 10.1096/fj.202301368RR
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53. doi: 10.1016/j.molmed.2007.12.002
Riess, M. L., Camara, A. K., Heinen, A., Eells, J. T., Henry, M. M., and Stowe, D. F. (2008). KATP channel openers have opposite effects on mitochondrial respiration under different energetic conditions. J. Cardiovasc. Pharmacol. 51, 483–491. doi: 10.1097/FJC.0b013e31816bf4a4
Riess, M. L., Eells, J. T., Kevin, L. G., Camara, A. K., Henry, M. M., and Stowe, D. F. (2004). Attenuation of mitochondrial respiration by sevoflurane in isolated cardiac mitochondria is mediated in part by reactive oxygen species. Anesthesiology 100, 498–505. doi: 10.1097/00000542-200403000-00007
Rottenberg, H., and Marbach, M. (1990). The Na+-independent Ca2+ efflux system in mitochondria is a Ca2+/2H+ exchange system. FEBS Lett. 274, 65–68. doi: 10.1016/0014-5793(90)81330-q
Sokolova, N., Pan, S., Provazza, S., Beutner, G., Vendelin, M., Birkedal, R., et al. (2013). ADP protects cardiac mitochondria under severe oxidative stress. PLoS One 8:e83214. doi: 10.1371/journal.pone.0083214
Sonnewald, U., Hertz, L., and Schousboe, A. (1998). Mitochondrial heterogeneity in the brain at the cellular level. J. Cereb. Blood Flow Metab. 18, 231–237. doi: 10.1097/00004647-199803000-00001
Starkov, A. A. (2010). The molecular identity of the mitochondrial Ca2+ sequestration system. FEBS J. 277, 3652–3663. doi: 10.1111/j.1742-4658.2010.07756.x
Stauch, K. L., Purnell, P. R., and Fox, H. S. (2014). Quantitative proteomics of synaptic and nonsynaptic mitochondria: insights for synaptic mitochondrial vulnerability. J. Proteome Res. 13, 2620–2636. doi: 10.1021/pr500295n
Stavsky, A., Stoler, O., Kostic, M., Katoshevsky, T., Assali, E. A., Savic, I., et al. (2021). Author correction: aberrant activity of mitochondrial NCLX is linked to impaired synaptic transmission and is associated with mental retardation. Commun. Biol. 4:755. doi: 10.1038/s42003-021-02312-w
Sun, J., Mishra, J., Yang, M., Stowe, D. F., Heisner, J. S., An, J., et al. (2022). Hypothermia prevents cardiac dysfunction during acute ischemia reperfusion by maintaining mitochondrial bioenergetics and by promoting hexokinase II binding to mitochondria. Oxid. Med. Cell. Longev. 2022, 4476448–4476419. doi: 10.1155/2022/4476448
Szabo, I., and Zoratti, M. (1992). The mitochondrial megachannel is the permeability transition pore. J. Bioenerg. Biomembr. 24, 111–117. doi: 10.1007/BF00769537
Varanyuwatana, P., and Halestrap, A. P. (2012). The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 12, 120–125. doi: 10.1016/j.mito.2011.04.006
Verma, M., Lizama, B. N., and Chu, C. T. (2022). Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl. Neurodegener. 11:3. doi: 10.1186/s40035-021-00278-7
Villasana, L. E., Klann, E., and Tejada-Simon, M. V. (2006). Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J. Neurosci. Methods 158, 30–36. doi: 10.1016/j.jneumeth.2006.05.008
Vos, M., Lauwers, E., and Verstreken, P. (2010). Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2:139. doi: 10.3389/fnsyn.2010.00139
Yang, M., Xu, Y., Heisner, J. S., Sun, J., Stowe, D. F., Kwok, W. M., et al. (2019). Peroxynitrite nitrates adenine nucleotide translocase and voltage-dependent anion channel 1 and alters their interactions and association with hexokinase II in mitochondria. Mitochondrion 46, 380–392. doi: 10.1016/j.mito.2018.10.002
Keywords: synaptic mitochondria, non-synaptic mitochondria, Ca2+ buffering, Ca2+ efflux, bioenergetics
Citation: Mishra J, Bevers K, Li K, Zare A, Heisner JS, Tong A, Kwok W-M, Stowe DF and Camara AKS (2025) Differential Ca2+ handling by isolated synaptic and non-synaptic mitochondria: roles of Ca2+ buffering and efflux. Front. Synaptic Neurosci. 17:1562065. doi: 10.3389/fnsyn.2025.1562065
Edited by:
Shelley Halpain, University of California, San Diego, United StatesReviewed by:
Timothy Gomez, University of Wisconsin-Madison, United StatesWilliam Brad Hubbard, University of Kentucky, United States
Copyright © 2025 Mishra, Bevers, Li, Zare, Heisner, Tong, Kwok, Stowe and Camara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amadou K. S. Camara, YWtzY0BtY3cuZWR1
†Present addresses: Kyle Bevers, Department of Anesthesiology and Perioperative Medicine, University of Pittsburgh, Pittsburgh, PA, United States
Armaan Zare, Department of Psychiatry, University of California, Riverside, Riverside, CA, United States
‡These authors have contributed equally to this work