 Richard L. Momparler
Richard L. Momparler Sylvie Côté2
Sylvie Côté2 Youssef Idaghdour
Youssef Idaghdour- 1Département de Pharmacologie, Université de Montréal, Montreal, QC, Canada
- 2Centre de recherche, Service d’hématologie/oncologie, CHU-Saint-Justine, Montréal, QC, Canada
- 3Department of Biology, New York University Abu Dhabi, Abu Dhabi, United Arab Emirates
Epigenetic alterations play an important role in the development of acute myeloid leukemia (AML) by silencing of genes that suppress leukemogenesis and differentiation. One of the key epigenetic changes in AML is gene silencing by DNA methylation. The importance of this alteration is illustrated by the induction of remissions in AML by 5-aza-2′-deoxycytidine (5-AZA-CdR, decitabine), a potent inhibitor of DNA methylation. However, most patients induced into remission by 5-AZA-CdR will relapse, suggesting that a second agent should be sought to increase the efficacy of this epigenetic therapy. An interesting candidate for this purpose is 3-deazaneplanocin A (DZNep). This analog inhibits EZH2, a histone methyltransferase that trimethylates lysine 27 histone H3 (H3K27me3), a marker for gene silencing. This second epigenetic silencing mechanism also plays an important role in leukemogenesis as shown in preclinical studies where DZNep exhibits potent inhibition of colony formation by AML cells. We reported previously that 5-AZA-CdR in combination with DZNep exhibits a synergistic antineoplastic action against human HL-60 AML cells and the synergistic activation of several tumor suppressor genes. In this report, we showed that this combination also induced a synergistic activation of apoptosis in HL-60 cells. The synergistic antineoplastic action of 5-AZA-CdR plus DZNep was also observed on a second human myeloid leukemia cell line, AML-3. In addition, 5-AZA-CdR in combination with the specific inhibitors of EZH2, GSK-126, or GSK-343, also exhibited a synergistic antineoplastic action on both HL-60 and AML-3. The combined action of 5-AZA-CdR and DZNep on global gene expression in HL-60 cells was investigated in greater depth using RNA sequencing analysis. We observed that this combination of epigenetic agents exhibited a synergistic activation of hundreds of genes. The synergistic activation of so many genes that suppress malignancy by 5-AZA-CdR plus DZNep suggests that epigenetic gene silencing by DNA and histone methylation plays a major role in leukemogenesis. Targeting DNA and histone methylation is a promising approach that merits clinical investigation for the treatment of AML.
Introduction
Acute myeloid leukemia (AML) is the most common type of acute leukemia in adults (1). Despite the use of intensive chemotherapy and stem cell transplantation, AML is still fatal in approximately half of younger patients and in about 80% of elderly patients as a result of primary drug refractoriness, relapse, or treatment-related mortality (2). Consequently, there is an urgent need to develop more specific and effective drugs to target leukemia-specific alterations. Both genetic and epigenetic alterations play an important role in leukemogenesis (3). Epigenetics is defined as heritable changes in gene expression that is not caused by changes in the DNA sequence. These changes involve primarily DNA methylation, histone modifications, chromatin remodeling, and microRNAs.
A frequent epigenetic alteration that is observed in AML is aberrant DNA methylation (4–7). This event occurs by the conversion of cytosine to 5-methylcytosine by DNA methyltransferase in the promoter region of genes. The genes silenced by DNA methylation include tumor suppressors, transcription factors, apoptosis-related proteins, and regulators of myeloid differentiation (7). The importance of this epigenetic alteration is shown by the successful use of inhibitor of DNA methylation (5-aza-2′-deoxycytidine, decitabine, 5-AZA-CdR) to induce complete remissions in patients with AML (8–10). However, therapy of AML with 5-AZA-CdR is not curative (5, 11, 12).
It may be possible to increase the effectiveness of epigenetic therapy by using combinations of epigenetic agents. Chemical modifications of histones can also produce epigenetic changes in gene expression. The initial discovery that a histone deacetylase (HDAC) inhibitor enhanced gene expression of 5-AZA-CdR on leukemic cells (13) provided the rationale to investigate combinations of these two classes of epigenetic agents in clinical trials. This approach was supported by preclinical studies that showed the synergistic interaction of 5-AZA-CdR and the HDAC inhibitor, sodium phenylbutyrate, on leukemic cells and in an animal model of leukemia (14). Interesting clinical responses were observed in AML patients treated with 5-AZA-CdR and the HDAC inhibitor, valproic acid (5). However, these combinational chemotherapies did not improve the long-term outcome of AML (5), indicating that other approaches should be investigated.
An interesting target for epigenetic therapy is the histone methyltransferases EZH2 (15, 16). EZH2 is a subunit of the polycomb repressive complex 2. It plays a key role in embryonic development by suppression of the genes for differentiation so that embryonic stem cells maintain their “self-renewal” capacity and pluripotency (17). EZH2 trimethylates lysine 27 on histone 3 (H3K27me3), a repressive marker for gene expression. Several studies have shown that EZH2 can function as an oncogene. Knockdown of EZH2 protein by RNA interference results in growth inhibition in several tumor models (18). EZH2 overexpression has been reported in several types of cancer and is associated with adverse outcomes (19). The overexpression of EZH2 can be due to gene amplification (20) or a gain-in-function mutation (21).
In this report, we have selected 3-deazaneplanocin-A (DZNep) to target EZH2 and investigated its mechanism and capacity to enhance the antineoplastic action of 5-AZA-CdR. DZNep inhibits S-adenosylhomocysteine hydrolase, resulting in the cellular accumulation of S-adenosylhomocysteine, which acts as a competitive inhibitor of S-adenosyl-l-methionine, the methyl donor for methyltransferases (22). DZNep is a global inhibitor of methyltransferases (23). The preferential target of DZNep is EZH2 as shown by the reduction in the level of this enzyme and H3K27me3 after treatment of tumor cells with this analog (24). DZNep also induces apoptosis of tumor cells. DZNep activates developmental genes that are not silenced by DNA methylation (23, 24). Other EZH2 inhibitors that target the catalytic site of this enzyme are under investigation and show promising clinical activity against lymphoma (15, 16).
The rationale to use DZNep in combination with 5-AZA-CdR for the therapy of AML is as follows: first, DZNep as a single agent shows activity against AML cells as indicated by the reduction in colony formation, induction of differentiation and increase survival of mice with AML (25). Second, some genes silenced by DNA methylation can escape gene activation by 5-AZA-CdR if they contain the repressive marker H3K27me3 (26). Third, genes that program differentiation and marked with H3K27me3 in embryonic stem cells have the highest probability to undergo DNA methylation in cancers (27). Fourth, a “cross-talk” exists between histone and DNA methylation. The trimethylation of H3K27 by EZH2 pre-marks genes for DNA methylation in cancer (28–31). These observations suggest that the interaction between DNA and histone methylation (H3K27me3) can promote leukemogenesis by silencing the genes that program differentiation. Targeting these two gene epigenetic silencing mechanisms using 5-AZA-CdR and DZNep has the potential to reverse this process and lead to effective therapy of AML (32).
We have reported previously that 5-AZA-CdR in combination with DZNep exhibits a synergistic antineoplastic action against AML cells and the reactivation of several genes that suppress leukemogenesis (33). In this report, in order to fully understand the chemotherapeutic potential of DZNep in combination with 5-AZA-CdR for the treatment of AML, we have investigated in depth their activation of a large number of genes that can suppress leukemogenesis using RNA sequence analysis. In addition, we demonstrated that their antineoplastic action was related to the induction of apoptosis and their synergistic antileukemic action was also observed with a second human myeloid leukemic cell line, AML-3 (34).
Materials and Methods
Cell Lines and Materials
Human HL-60 myeloid leukemic cells were obtained from ATCC. The HL-60 cells were maintained in RPMI-1640-HEPES containing 10% fetal bovine serum (Wisent). Human OCI-AML-3 (AML-3) myeloid leukemic cells were kindly provided by Mark Minden, Princess Margaret Hospital, Toronto. The AML-3 cells were maintained in alpha-MEM containing 10% fetal bovine serum (Wisent). 5-AZA-CdR was obtained from Dr. Alois Piskala, Institute of Organic Chemistry, Czechoslovak Academy of Sciences, Prague. DZNep was kindly provided by Dr. Victor E. Marquez, Chemical Biology Laboratory, National Cancer Institute, Frederick, MD, USA. 5-AZA-CdR and DZNep were dissolved in sterile phosphate buffer saline (PBS) pH 6.8 solution. GSK-126 and GSK-343 were dissolved in DMSO and obtained from Xcess Biosciences Inc. or Structural Genomics Consortium (Toronto, ON, Canada), respectively.
Antineoplastic Assay
The in vitro antineoplastic activity of the drugs was evaluated by reduction of colony formation after drug treatment. The HL-60 and AML-3 cells were treated with the indicated concentrations of drugs. At the end of drug treatment, a cell count was performed using the Beckmann Model Z Coulter Counter. For colony assays, the cells were placed in 0.3% soft agar medium containing 20% serum. The number of colonies (>500 cells) was counted after 16–18 days of incubation. The cloning efficiency was in the range of 60%.
Apoptosis Analysis
Annexin V and propidium iodide (PI) staining were used to assess apoptosis and was determined using flow cytometry. The cells were treated as indicated. Twenty-four hours after the end of drug treatment, the cells were washed twice with cold PBS and resuspended in 1× Annexin V binding buffer (BD Biosciences Pharmingen). Then, 2 × 105 cells were mixed gently with Annexin V-FITC (BD Biosciences Pharmingen) and PI solution (Sigma-Aldrich) and incubated for 15 min in the dark at room temperature. The cells were suspended in 1× Annexin V binding buffer, and staining was immediately quantified using a BD LSR Fortessa flow cytometer (San Jose, CA, USA) and analyzed with the BD DIVA (San Jose, CA, USA) software program. A minimum of 10,000 cells within the gated region was analyzed per measurement.
Analysis of Gene Expression
Drug treatment of the HL-60 leukemic cells was performed as described previously (33). At 24 h after the end of drug treatment, total RNA was isolated from HL-60 cells using the RNeasy Plus Mini kit (Qiagen). Quantity and integrity of total RNA were checked with a 2100 Bioanalyzer instrument (Agilent). All samples had an RNA integrity number >8. Paired-end RNASeq libraries were constructed using the TruSeq RNA Sample Prep kit v2 (Illumina). Quantification and quality control of RNASeq libraries were performed prior to sequencing using Illumina’s recommended protocols. Hundred base pairs of paired-end RNA sequencing were performed using eight samples per sequencing lane on the Illumina HiSeq 2000 platform at the Genome Quebec Innovation Centre, Montreal, Canada. Reads were assembled to a reference genome [hg19, European Hapmap (CEU) Major Allele release] using TopHat v1.3.2. The number of mismatches allowed per read was set to 2. PCR duplicates were removed using Picard-tools1 and non-properly paired and non-uniquely mapped reads were filtered out with SAMtools.2 Recalibration and local realignment was performed with GATK tools. BAM files were processed with Cufflinks to estimate isoform-level relative abundances and to perform differential expression analysis. Unsupervised analysis and hierarchical clustering was performed using JMP Genomics v6.0 (SAS Institute). The data are deposited in GEO (Gene Expression Omnibus) database under accession number GSE94344.
For comparative analysis of the expression of the transcripts, the genes were divided into different groups with similar biological functions (e.g., differentiation, senescence, apoptosis, etc.) using the gene lists of pathway-focused arrays from SA Biosciences.3 Gene function and comparison of expression of transcripts in normal human tissues was done using BioGPS (35) and Gene Card (36). The list of tumor suppressor genes (TSGs) and oncogenes was obtained from Zhao et al. (37) and UniProt Consortium (38).
Ingenuity Pathway Analysis
To elucidate the probable downstream effects on biological functions of the observed gene expression changes, we used the downstream effects analysis approach implemented in Ingenuity Pathway Analysis. The algorithm uses the Ingenuity Knowledge Base and is based on a collection of experimental observations manually curated from the literature or other databases. Nodes and network edges from these data are used in combination with the observed gene expression changes to infer cause–effect relationships with a direction of the causal effect, i.e., either activating or inhibiting (39).
Ethical Considerations
This study was exempted from ethical approval by the Research Ethics Board of CHU Sainte-Justine since no patient samples were used.
Results
Antineoplastic Action of 5-AZA-CdR Plus EZH2 Inhibitors on Myeloid Leukemic Cells
We reported previously that the combination of 5-AZA-CdR and DZNep exhibited a synergistic antineoplastic effect on human HL-60 myeloid leukemic cells (33). In this report, we confirm that this synergy also takes place in human AML-3 myeloid leukemic cells as determined by a colony assay (Figures 1A,C). The AML-3 cells are more sensitive to 5-AZA-CdR than the HL-60 cells. For both cell lines, the combination of 5-AZA-CdR and DZNep showed a similar synergistic antileukemic effect. The antineoplastic action of DZNep on EZH2 is indirect and due to its potent inhibition of adenosyl homocysteine hydrolase (22). This action results in the accumulation of adenosyl homocysteine, a competitive inhibitor of S-adenosyl-l-methionine-dependent methylations. EZH2 uses S-adenosyl-l-methionine as the methyl donor to methylate H3K27. In support that the principle target for DZNep is EZH2, we compared its antileukemic action with the specific inhibitors of EZH2, GSK-126, and GSK-343 (40). We observed a similar synergy with all the combinations with these agents, suggesting that EZH2 is the primary target of DZNep (Figures 1B,D). However, DZNep was more active than GSK-343 or GSK-126 (Figures 1A,C). We also investigated the induction of apoptosis by 5-AZA-CdR and DZNep on HL-60 and AML-3 leukemic cells (Figures 1E,F). The combination showed a synergistic induction of apoptosis for both cell lines.
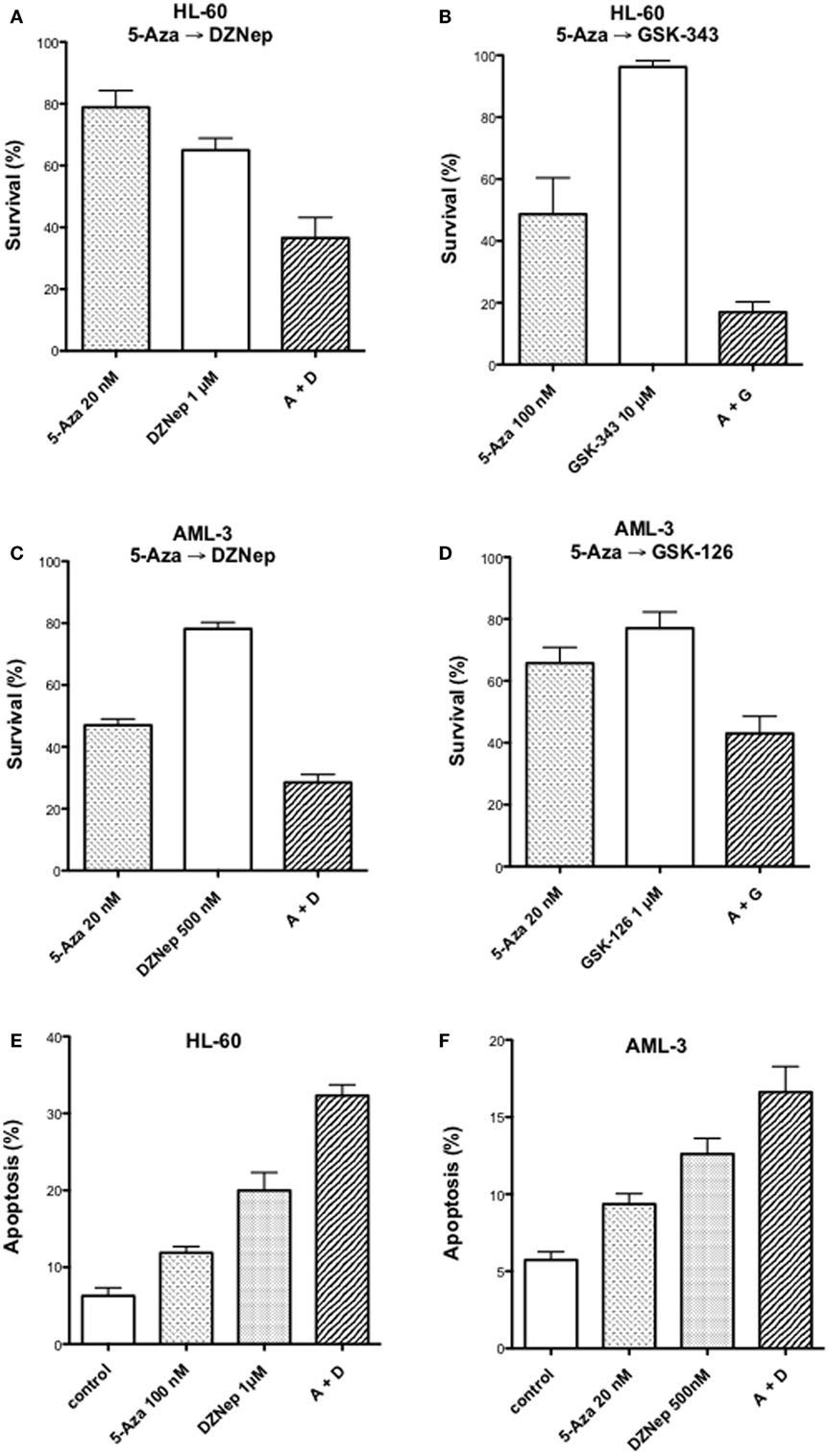
Figure 1. Antineoplastic activity on myeloid leukemic cells after treatment with inhibitors of DNA methylation and EZH2. Survival of human HL-60 (A,B) and AML-3 (C,D) leukemic cells after treatment with 5-Aza-CdR alone or in combination with inhibitors of EZH2: DZNep, GSK-343, or GSK-126. The cells were treated with the indicated concentration of 5-AZA-CdR, and, at 24 h, the different EZH2 inhibitors were added. At 48 h, the leukemic cells were placed in soft agar medium for colony assay to measure survival. Analysis of induction of apoptosis at 72 h after drug treatment in HL-60 (E) and AML-3 (F) leukemic cells. The results are expressed as mean ± SEM, n = 3. 5-AZA-CdR (5-Aza), DZNep (D), GSK-343 (G), GSK-126 (G).
Effect of 5-AZA-CdR and DZNep on Global Gene Expression in HL-60 Leukemic Cells
The effect of 5-AZA-CdR and DZNep on global gene expression in HL-60 leukemic cells was determined by RNA sequence analysis (Figure 2). This figure shows the heat map displaying relative transcript abundances of 12,866 transcripts, each of which is differentially expressed (FDR 5%) at least in one of the pairwise comparisons of the four conditions. Relative to the control sample, 5-AZA-CdR alone showed a greater increase in the expression of transcripts as compared to DZNep. DZNep increased the activation of many cohorts of genes, but also showed a reduction in the expression of some genes. The combination of 5-AZA-CdR plus DZNep showed a synergistic increase in expression of hundreds of genes (Figure 2). Specific data on all of the genes are shown in Table S1 in Supplementary Material. In order to evaluate this very large data set, we used Ingenuity Pathway Analysis, an application for interpreting biological data and modeling complex networks (39). This analysis can identify the most relevant signaling pathways and has the potential to predict the downstream effects of the experimental chemotherapy. We performed a comparison analysis of the three pairwise contrasts (control versus 5-AZA-CdR, control versus DZNep, and control versus 5-AZA-CdR + DZNep) from the database generated from HL-60 leukemic cells treated with these epigenetic agents. The analysis revealed the clusters of biological functions that show increased or decreased activity across the different contrasts and enabled visualization across the three contrasts (Figure 3A). In particular, this analysis revealed the top most synergistically affected biological functions are apoptosis (activation z-score = 0.22 in 5-AZA-CdR + DZNep) and cell death (activation z-score = 0.18 in 5-AZA-CdR + DZNep) of leukemia cell lines. Based on expression levels of all genes implicated in these processes, both functions are predicted as highly activated under the synergistic treatment. The network of the top activated biological functions in 5-AZA-CdR + DZNep treatment (apoptosis of leukemia cell lines) is shown in Figure 3B. This conclusion was confirmed by our data on specific genes related to these phenomena as discussed in the section on apoptosis.
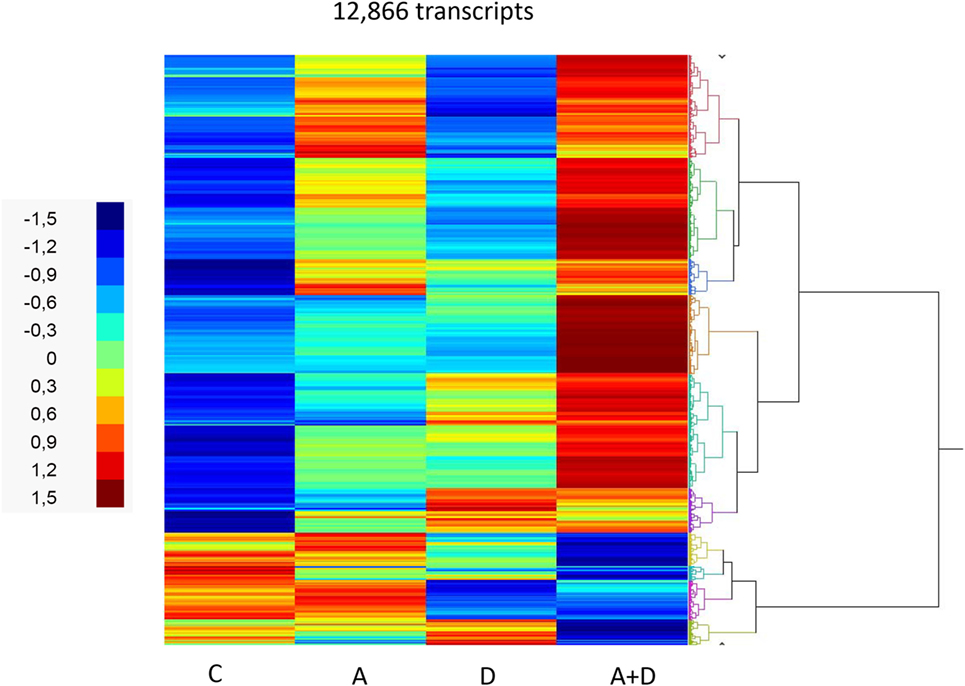
Figure 2. RNA sequence analysis of global gene expression in HL-60 leukemic cells after treatment with 5-AZA-CdR and/or DZNep. Heat map of expression of transcripts. HL-60 leukemic cells were treated with 100 nM 5-AZA-CdR (A). At 24 h 1,000 nM DZNep (D) was added to the medium. Drug removal at 48 h. RNA isolation at 72 h as described in Section “Materials and Methods.” The pairwise comparisons were control (C) versus A; C versus D; and C versus A + D.
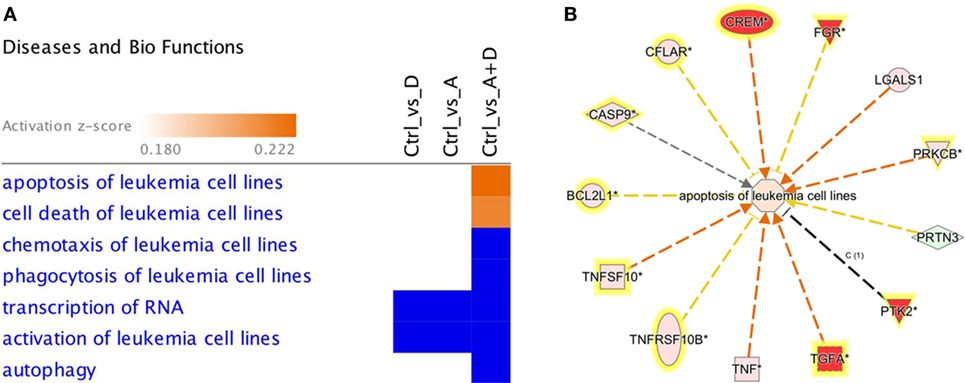
Figure 3. Downstream effects analysis enables prediction of the effect of gene expression changes in each treatment. (A) The heat map visualizes the downstream effects analysis results across the multiple treatments. The top clusters of diseases or biological functions that are predicted to increase or decrease in at least one treatment are shown. The disease or function scores are displayed using a gradient from dark blue to orange for activation z-scores. (B) The network of the top activated biological function in 5-AZA-CdR + DZNep treatment (apoptosis of leukemia cell lines) is shown. The edges are colored orange when leading to activation of the downstream node, blue when leading to its inhibition, and yellow if the findings underlying the relationship are inconsistent with the state of the downstream node. The predicted downstream effect of the observed expression changes on apoptosis is stronger pathway activation under the joint effect of 5-AZA-CdR and DZNep (activation z-score = 0.22).
Below, we focused on the effect of treatment with 5-AZA-CdR and DZNep on gene expression levels of individual genes from several cohorts of genes based on their functional ontologies and in particular genes implicated in leukemogenesis, apoptosis and differentiation. Due to the relatively large number of genes whose expression was modulated by treatment with these epigenetic agents, we limited our focus to a subset of genes that are highly related to leukemogenesis (35–38). Nonetheless, Table S1 in Supplementary Material contains the extended list of specific genes affected by the action of 5-AZA-CdR plus DZNep.
Effect of 5-AZA-CdR and DZNep on Expression of Differentiation Genes
Since a block in differentiation is one of the hallmarks of AML, we investigated the action of this epigenetic therapy on the expression of genes that program differentiation. The transcriptional responses of genes related to differentiation after treatment with 5-AZA-CdR plus DZNep in HL-60 leukemic cells are shown in Table 1 and Figure 4. Many of the genes involved in differentiation were activated, some of which showed a remarkable synergistic activation by this combination of epigenetic agents. The expression of these genes is observed primarily in differentiated tissues.4 NEFH gene codes for neurofilaments that are normally expressed in neuronal cells. MYH11 gene codes for myosin and exhibits high expression in smooth muscle. PMEL, a premelanosome gene, is expressed predominantly in the retina. These genes are important markers for cellular differentiation. These data indicate that 5-AZA-CdR plus DZNep turn on several differentiation genes that are not related to the phenotype of myeloid leukemic cells and may contribute to the antileukemic action of these epigenetic agents. 5-AZA-CdR plus DZNep also exhibited a synergistic activation of many genes involved in development (Table S2 in Supplementary Material).
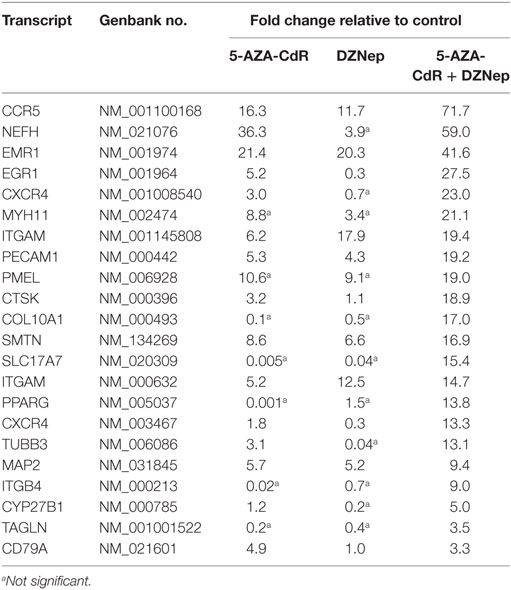
Table 1. Relative expression for differentiation-related genes in HL-60 leukemic cells after treatment with 5-AZA-CdR and DZNep.
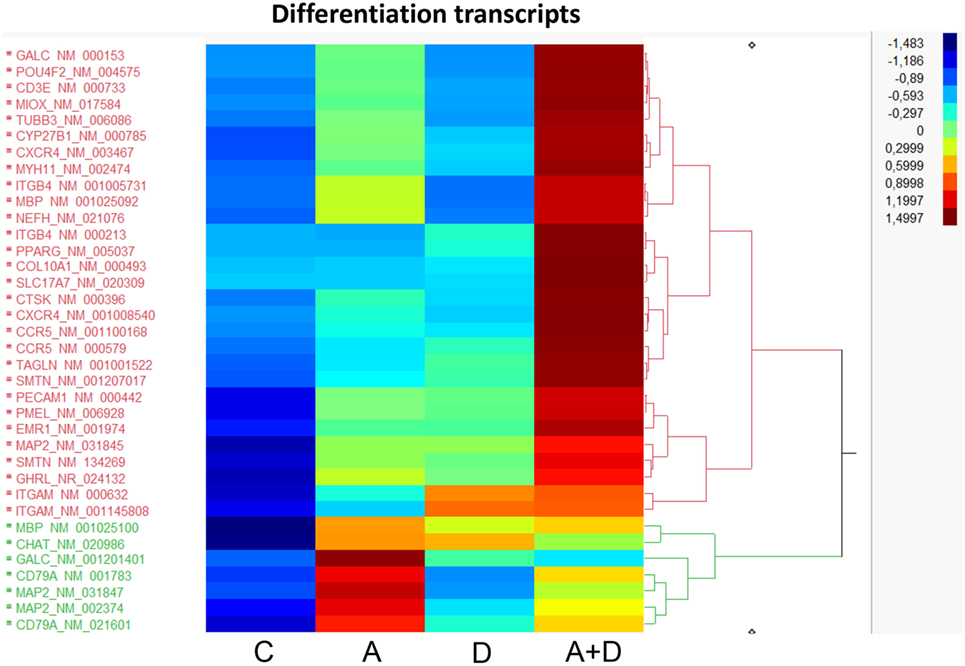
Figure 4. Heat map expression of genes related to differentiation after 5-AZA-CdR and DZNep treatment of HL-60 myeloid leukemic cells. Drug treatment of the leukemic cells is described in Figure 2. C, control; A, 5-AZA-CdR; D, DZNep; A + D, 5-AZA-CdR plus DZNep.
Effect of 5-AZA-CdR and DZNep on Expression of Genes Related to Apoptosis and Senescence
The individual gene data on the synergistic activation of apoptosis-related genes in HL-60 leukemic cells by 5-AZA-CdR and/or DZNep are shown in Table 2 and Figure 5. The combination of 5-AZA-CdR and DZNep exhibited an increase of the expression of many genes that are positive regulators of apoptosis, such as TNFSF10, CASP6, CASP10, BNIP3L, and GADD45A (41). However, the combination also exhibited the activation of expression of several antiapoptotic genes, such as BCL2A1, BCL2L1, and XIAP (41). The action of 5-Aza-CdR and DZNep on the induction of apoptosis in HL-60 and AML-3 leukemic cells (Figures 1E,F) indicates activation of proapoptotic genes is the predominant action of these epigenetic agents and is clearly supported by our Ingenuity Pathway Analysis described above (Figure 3).
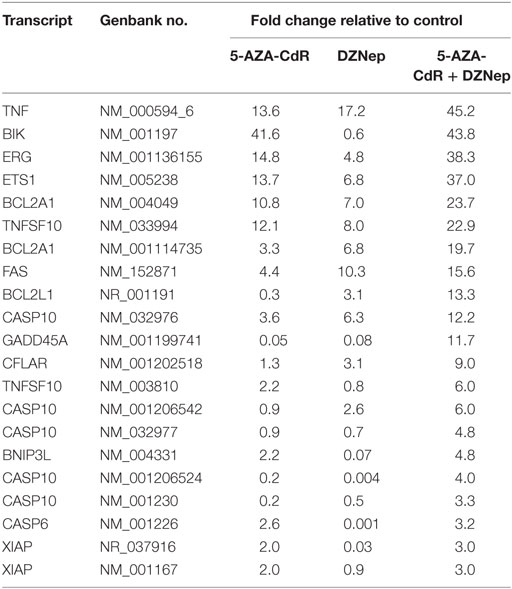
Table 2. Relative expression for apoptosis-related genes in HL-60 leukemic cells after treatment with 5-AZA-CdR and DZNep.
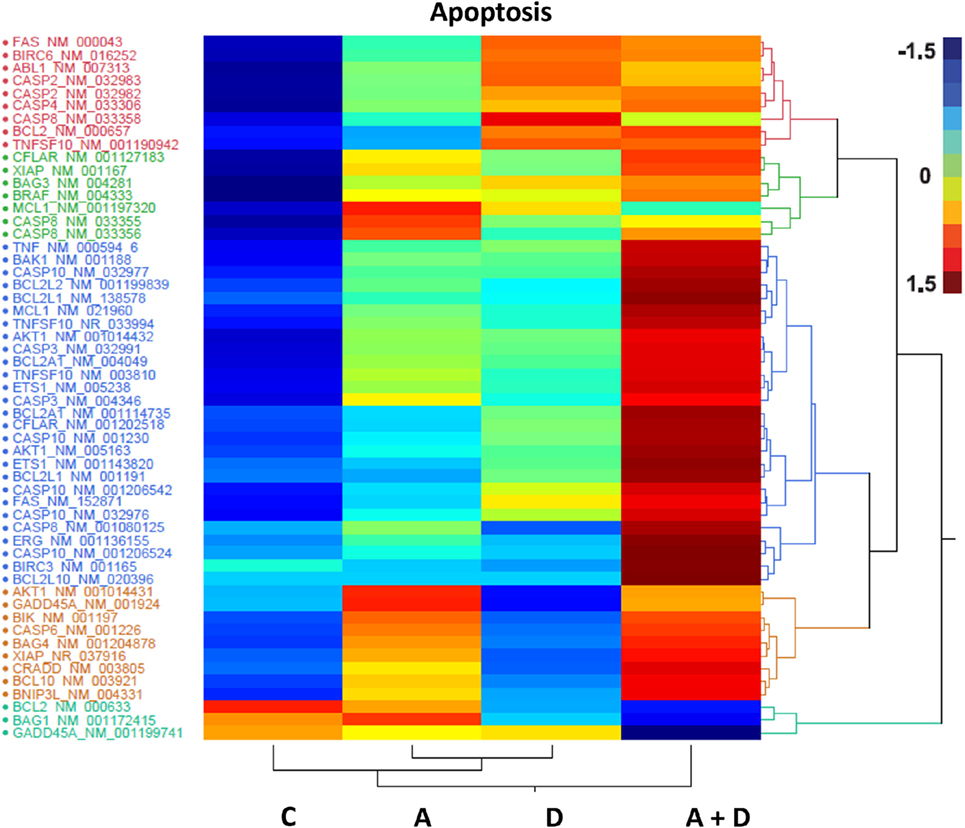
Figure 5. Heat map expression of genes related to apoptosis after 5-AZA-CdR and DZNep treatment of HL-60 myeloid leukemic cells. Drug treatment of the leukemic cells is described in Figure 2. C, control; A, 5-AZA-CdR; D, DZNep; A + D, 5-AZA-CdR plus DZNep.
Cellular senescence is the phenomenon by which normal cells cease to divide after threshold limit of cell divisions. This phenomenon is known as “replicative senescence,” or the Hayflick limit (42). Suppression of senescence can favor the development of malignancy (43). The treatment of HL-60 leukemic cells with 5-AZA-CdR and/or DZNep leads to the synergistic activation of many genes related to senescence (Table 3; Figure 6). Several of the activated genes are initiators of senescence, including SPARC, EGR1, SERPINE1, and TP53BP1. The activation of these genes by 5-AZA-CdR and/or DZNep may play an important role with respect to the antineoplastic action on HL-60 leukemic cells.
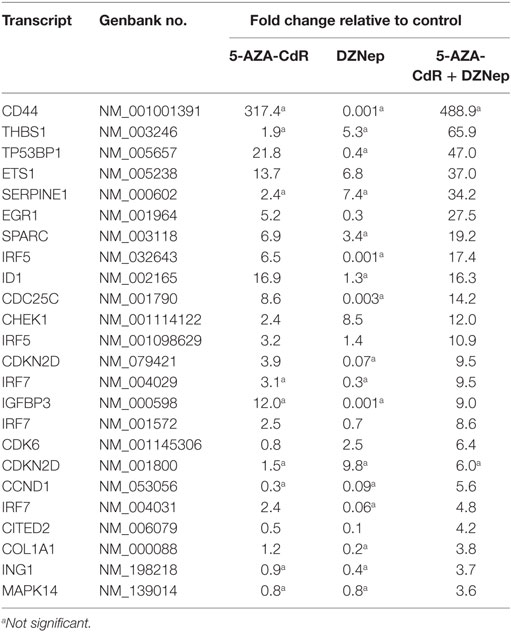
Table 3. Relative expression for senescence related genes in HL-60 leukemic cells after treatment with 5-AZA-CdR and DZNep.
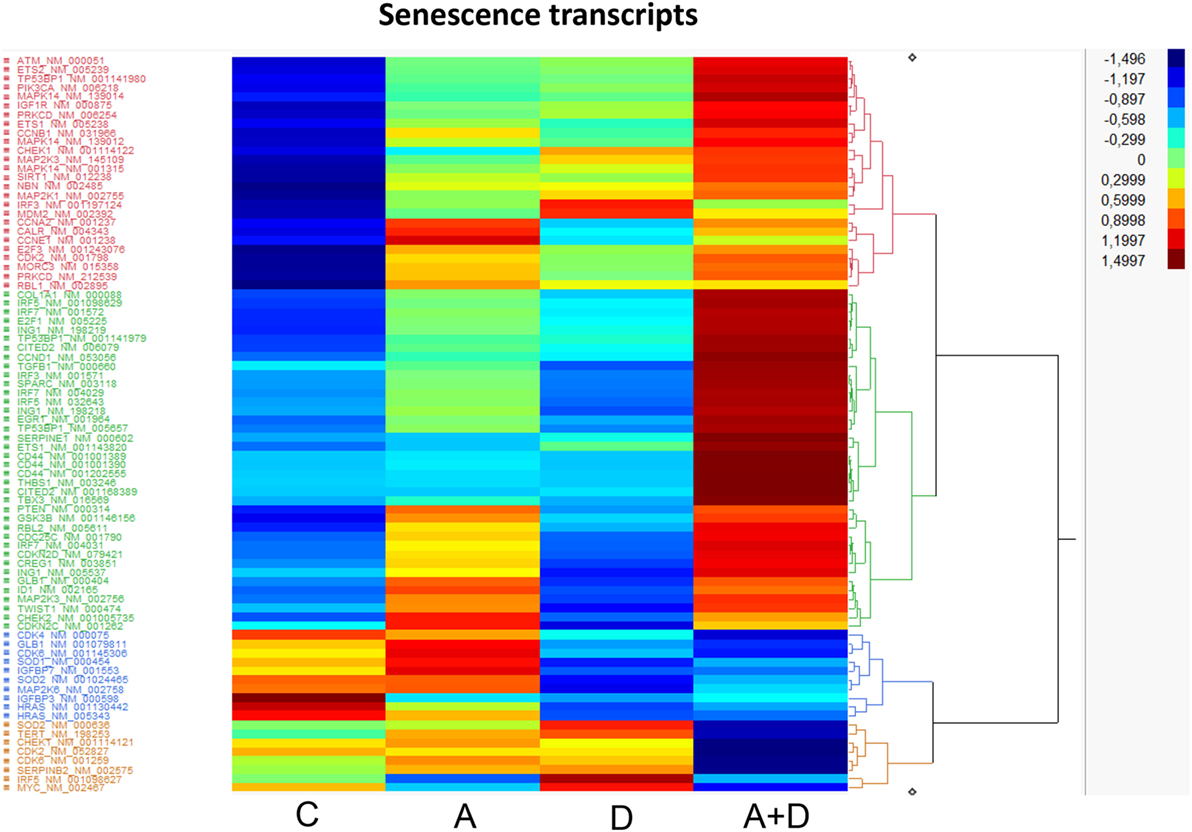
Figure 6. Heat map expression of genes related to senescence after 5-AZA-CdR and DZNep treatment of HL-60 myeloid leukemic cells. Drug treatment of the leukemic cells is described in Figure 2. C, control; A, 5-AZA-CdR; D, DZNep; A + D, 5-AZA-CdR plus DZNep.
Effect of 5-AZA-CdR and DZNep on Expression of TSGs
5-AZA-CdR and DZNep exhibited a synergistic activation of the expression of several important TSGs in HL-60 leukemic cells (Table 4). For example, CDKN2B (p15) is a cyclin-dependent kinase (CDK) inhibitor that inhibits cell growth by blocking G1 progression into S phase. CDKN2B function as a TSG in AML is well established (44). Hypermethylation of CpG islands in its promoter region was shown to occur frequently (45). This epigenetic state is associated with reduced expression (46) and is an important molecular predictor of outcome. Loss of expression of CDKN2B accelerates the development of myeloid leukemia in transgenic mice (47). Another important TSG is CDKN1A (p21), the expression of which is correlated with cell-cycle arrest that precedes terminal differentiation in a variety of tissues (48). CDKN1A is also implicated in regulation of cell growth and cell response to DNA damage. In response to DNA damage, p53 induces CDKN1A expression, which is responsible for the cell-cycle arrest at the G1 checkpoint (48). Other TSGs show similar patterns. We observed massive overexpression of EGR1, a zinc finger protein that functions as a transcriptional regulator. Overexpression of EGR1, one of the derepressed genes in EZH2-deficient leukemic cells, promoted profoundly the differentiation of AML cells (49). EXT1 (Exostosin-1), a glycosyltransferase, is another highly upregulated gene whose expression is reduced in human cancer cells by DNA methylation (50). Introduction of a functional EXT1 gene into cancer cell lines induces tumor-suppressor-like features, such as reduced colony formation and tumor growth in nude mouse xenografts. DNA methylation of EXT1 is common in leukemia (50). HES1, a helix-loop transcriptional repressor, is frequently downregulated in AML (51). HES1 activation suppresses proliferation of leukemia cells in AML. These results demonstrate the TSG role of HES1 in AML. Finally, we observe upregulation of RUNX3 a runt-domain transcriptional factor. The hypermethylation of this gene is frequent in AML cell lines (52). Relapse-free survival of AML patients without RUNX3 methylation was significantly better than that of methylated cases. These results support the idea that RUNX3 silencing is an important event in AML.
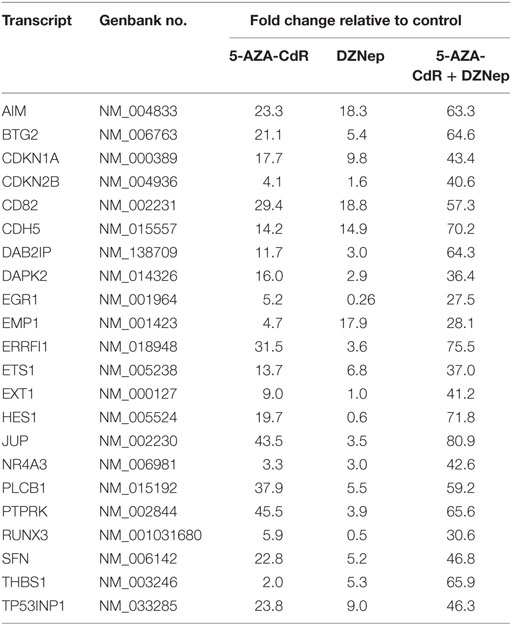
Table 4. Expression of tumor suppressor genes in HL-60 leukemic cells after treatment with 5-AZA-CdR and DZNep.
Discussion
Epigenetic alterations that silence gene expression play an important role in the development of AML. Aberrant DNA methylation that silences genes which suppress leukemogenesis occurs frequently in AML (45). The importance of this epigenetic modification is illustrated by the clinical activity of the potent inhibitor of DNA methylation, 5-AZA-CdR, for the treatment of AML (9). However, 5-AZA-CdR therapy of AML for most patients is not curative, and new approaches are needed to optimize the treatment of AML with this epigenetic drug. One factor that may limit the clinical effectiveness of 5-AZA-CdR for the treatment of AML is that this cytosine nucleoside analog has difficulty to activate DNA-methylated genes that contain the histone repressive marker, H3K27me3 (26). This latter epigenetic alteration is catalyzed by EZH2 methyltransferase (53). DZNep, a potent inhibitor of EZH2, shows remarkable antineoplastic activity against AML cells (25). This observation provided a good rationale to use DZNep in combination with 5-AZA-CdR. We reported previously that inhibition of DNA methylation and EZH2 by 5-AZA-CdR and DZNep, respectively, exhibits a synergistic antineoplastic effect on HL-60 AML cells (33). We confirm in this study that the synergistic antineoplastic action of this combination is also observed with a second human myeloid leukemic cell line, AML-3. DZNep is a global inhibitor of histone methylation (23). GSK-126 and GSK-343, specific inhibitors of EZH2, exhibit a similar synergy in combination with 5-AZA-CdR as observed with DZNep. This observation supports that EZH2 is the preferential target for DZNep.
In a previous study using real-time PCR and microarray analysis, we observed that 5-AZA-CdR plus DZNep exhibited a synergistic activation of several TSGs in HL-60 leukemic cells (33). In this report, we investigated the action of this combination on gene expression in greater depth using RNA sequence analysis. Each methodology has particular methodologically related detection biases and limitations, which could explain why some differences were observed between our previous microarray and RT-PCR results and the present RNA sequencing with respect to the expression of several of genes that were detected. Ideally for genes of interest, our RNASeq data would have been confirmed using a second methodology, but there was insufficient mRNA available to do this. However, we nevertheless observed a synergistic increase in expression of hundreds of different transcripts by this combination of epigenetic agents. This synergistic effect of 5-AZA-CdR plus DZNep is highly consistent with the antineoplastic action of this combination. Epigenetic activation of specific genes that program apoptosis are likely one of the major contributors to the antileukemic action of 5-AZA-CdR and DZNep (Figure 5). This is shown by a synergistic activation of several genes that induce apoptosis, such as CASP6 (Table 2). These gene expression data are also supported by the synergistic induction of apoptosis in the HL-60 leukemic cells by 5-AZA-CdR plus DZNep (Figures 1E,F).
The genes that program differentiation also merit serious consideration (Table 1; Figure 4). These genes show a high frequency of DNA methylation in many different types of cancer and contain high levels of the H3K27me3 marker in adult stem/progenitor cells (27). The synergistic activation of so many genes that program differentiation by 5-AZA-CdR plus DZNep support the hypothesis that gene silencing by DNA methylation and H3K27me3 plays a very important role in the leukemogenesis of AML. Since these epigenetic alterations in the leukemic cells resemble that of embryonic stem cells (54, 55), it is possible that this epigenetic therapy has the potential to target leukemic stem cells (32). This interesting hypothesis merits further investigation. This may be a one of the key mechanisms that explains the block in differentiation that is a hallmark of AML. The synergistic activation of genes that promote senescence may also contribute to its antileukemic action of this combination of these epigenetic agents.
It is of interest to investigate if the antineoplastic synergy that we observed between 5-AZA-CdR and DZNep on myeloid leukemic cells also occurs in tumors. In this regard, we reported recently that 5-AZA-CdR and DZNep show a synergistic antineoplastic action against human lung carcinoma cells (56). The synergistic interaction between these two epigenetic agents was also observed against rhabdoid tumor cells (57). 5-AZA-CdR in combination with the EZH2 inhibitor, GSK-126, inhibited the in vivo growth of a prostate tumor xenograft more effectively than treatment with 5-AZA-CdR alone (58). These observations suggest that the reversal of gene silencing by DNA and histone methylation using epigenetic agents may also be an effective therapy for tumors.
Conclusion
Our data support the hypothesis that the antileukemic activity of 5-AZA-CdR plus DZNep results from the summation of the activation of different cohorts of genes that suppress leukemogenesis by different mechanisms, including the induction of apoptosis, differentiation, development, and senescence. However, since so many genes in these different categories are activated, it is not possible to define the precise mechanisms involved. The genes that program differentiation may play a prominent role in this process since the block in differentiation is one of the hallmarks of AML. In order to accomplish this, leukemic stem cells have to “turn off” the genes that program differentiation by DNA and H3K27 methylation. The synergistic activation of so many genes that suppress malignancy by 5-AZA-CdR plus DZNep suggests that epigenetic gene silencing by DNA and histone methylation plays a major role in leukemogenesis. 5-AZA-CdR plus DZNep have the potential to reverse this “double lock” gene silencing mechanism and thus target leukemic stem cells (32). This interesting epigenetic therapy merits clinical investigation in patients with advanced myeloid leukemia.
Supporting Information
The dataset supporting the conclusions of this article is available in the BioProject database of the National Center for Biotechnology Information (NCBI) (BioProject ID: PRJNA316529; http://www.ncbi.nlm.nih.gov/bioproject/316529).
Author Contributions
RM, YI, and SC conceived and designed the experiments; RM, SC, and LM performed the experimental work. RM, SC, and YI analyzed the data. All the authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AC and handling Editor declared their shared affiliation, and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
Acknowledgments
The authors would like to thank Genome Québec, McGill University, for RNA sequence analysis and Christian Young, Lady Davis Institute Flow Cytometry and Cell Imaging Facility, Montreal, for assistance for analysis of cell apoptosis.
Funding
This research was funded by the Canadian Cancer Society (grant #700795). Youssef Idaghdour is supported by New York University Abu Dhabi Research Grant AD105.
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fonc.2017.00019/full#supplementary-material.
Table S1. RNA sequence analysis of global gene expression in HL-60 leukemic cells after 5-AZA-CdR (A) and/or DZNep (D).
Table S2. Developmental genes that show frequent DNA hypermethylation in cancer: relative expression after treatment of HL-60 leukemic cells with 5-AZA-CdR and DZNep.
Footnotes
References
1. Ferrara F, Schiffer CA. Acute myeloid leukaemia in adults. Lancet (2013) 381(9865):484–95. doi: 10.1016/S0140-6736(12)61727-9
2. Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol (2011) 29(5):487–94. doi:10.1200/JCO.2010.30.1820
3. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet (2016) 17(10):630–41. doi:10.1038/nrg.2016.93
4. Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell (2010) 17(1):13–27. doi:10.1016/j.ccr.2009.11.020
5. Kim TK, Gore SD, Zeidan AM. Epigenetic therapy in acute myeloid leukemia: current and future directions. Semin Hematol (2015) 52(3):172–83. doi:10.1053/j.seminhematol.2015.04.003
6. Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood (2016) 127(1):42–52. doi:10.1182/blood-2015-07-604512
7. Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol (2016) 8(9). doi:10.1101/cshperspect.a019505
8. Cashen AF, Schiller GJ, O’Donnell MR, DiPersio JF. Multicenter, phase II study of decitabine for the first-line treatment of older patients with acute myeloid leukemia. J Clin Oncol (2010) 28(4):556–61. doi:10.1200/JCO.2009.23.9178
9. Lubbert M, Ruter BH, Claus R, Schmoor C, Schmid M, Germing U, et al. A multicenter phase II trial of decitabine as first-line treatment for older patients with acute myeloid leukemia judged unfit for induction chemotherapy. Haematologica (2012) 97(3):393–401. doi:10.3324/haematol.2011.048231
10. Kantarjian HM, Thomas XG, Dmoszynska A, Wierzbowska A, Mazur G, Mayer J, et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol (2012) 30(21):2670–7. doi:10.1200/JCO.2011.38.9429
11. Diesch J, Zwick A, Garz AK, Palau A, Buschbeck M, Gotze KS. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin Epigenetics (2016) 8:71. doi:10.1186/s13148-016-0237-y
12. Yun S, Vincelette ND, Abraham I, Robertson KD, Fernandez-Zapico ME, Patnaik MM. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: a systematic review of hypomethylating agents trials. Clin Epigenetics (2016) 8:68. doi:10.1186/s13148-016-0233-2
13. Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet (1999) 21(1):103–7. doi:10.1038/5047
14. Lemaire M, Momparler LF, Farinha NJ, Bernstein M, Momparler RL. Enhancement of antineoplastic action of 5-aza-2’-deoxycytidine by phenylbutyrate on L1210 leukemic cells. Leuk Lymphoma (2004) 45(1):147–54. doi:10.1080/1042819031000149304
15. Italiano A. Role of the EZH2 histone methyltransferase as a therapeutic target in cancer. Pharmacol Ther (2016) 165:26–31. doi:10.1016/j.pharmthera.2016.05.003
16. Morera L, Lubbert M, Jung M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin Epigenetics (2016) 8:57. doi:10.1186/s13148-016-0223-4
17. Bracken AP, Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat Rev Cancer (2009) 9(11):773–84. doi:10.1038/nrc2736
18. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A (2003) 100(20):11606–11. doi:10.1073/pnas.1933744100
19. Jiang T, Wang Y, Zhou F, Gao G, Ren S, Zhou C. Prognostic value of high EZH2 expression in patients with different types of cancer: a systematic review with meta-analysis. Oncotarget (2016) 7(4):4584–97. doi:10.18632/oncotarget.6612
20. Saramaki OR, Tammela TL, Martikainen PM, Vessella RL, Visakorpi T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer (2006) 45(7):639–45. doi:10.1002/gcc.20327
21. Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood (2011) 117(8):2451–9. doi:10.1182/blood-2010-11-321208
22. Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, et al. 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem Biophys Res Commun (1986) 135(2):688–94. doi:10.1016/0006-291X(86)90048-3
23. Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther (2009) 8(6):1579–88. doi:10.1158/1535-7163.MCT-09-0013
24. Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev (2007) 21(9):1050–63. doi:10.1101/gad.1524107
25. Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, et al. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood (2009) 114(13):2733–43. doi:10.1182/blood-2009-03-213496
26. Si J, Boumber YA, Shu J, Qin T, Ahmed S, He R, et al. Chromatin remodeling is required for gene reactivation after decitabine-mediated DNA hypomethylation. Cancer Res (2010) 70(17):6968–77. doi:10.1158/0008-5472.CAN-09-4474
27. Easwaran H, Johnstone SE, Van Neste L, Ohm J, Mosbruger T, Wang Q, et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res (2012) 22(5):837–49. doi:10.1101/gr.131169.111
28. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature (2006) 439(7078):871–4. doi:10.1038/nature04431
29. Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet (2007) 39(2):237–42. doi:10.1038/ng1972
30. Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet (2007) 39(2):232–6. doi:10.1038/ng1950
31. Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, et al. Epigenetic stem cell signature in cancer. Nat Genet (2007) 39(2):157–8. doi:10.1038/ng1941
32. Momparler RL, Cote S. Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin Investig Drugs (2015) 24(8):1031–43. doi:10.1517/13543784.2015.1051220
33. Momparler RL, Idaghdour Y, Marquez VE, Momparler LF. Synergistic antileukemic action of a combination of inhibitors of DNA methylation and histone methylation. Leuk Res (2012) 36(8):1049–54. doi:10.1016/j.leukres.2012.03.001
34. Momparler RL, Cote S, Momparler LF, Idaghdour Y. Epigenetic therapy of acute myeloid leukemia using 5-aza-2’-deoxycytidine (decitabine) in combination with inhibitors of histone methylation and deacetylation. Clin Epigenetics (2014) 6(1):19. doi:10.1186/1868-7083-6-19
35. Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, et al. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol (2009) 10(11):R130. doi:10.1186/gb-2009-10-11-r130
36. Harel A, Inger A, Stelzer G, Strichman-Almashanu L, Dalah I, Safran M. GIFtS: annotation landscape analysis with GeneCards. BMC Bioinformatics (2009) 10:348. doi:10.1186/1471-2105-10-348
37. Zhao M, Sun J, Zhao Z. TSGene: a web resource for tumor suppressor genes. Nucleic Acids Res (2013) 41(Database issue):D970–6. doi:10.1093/nar/gks937
38. UniProt C. UniProt: a hub for protein information. Nucleic Acids Research (2015) 43(Database issue):D204–12. doi:10.1093/nar/gku989
39. Kramer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics (2014) 30(4):523–30. doi:10.1093/bioinformatics/btt703
40. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature (2012) 492(7427):108–12. doi:10.1038/nature11606
41. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35(4):495–516. doi:10.1080/01926230701320337
42. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol (2013) 75:685–705. doi:10.1146/annurev-physiol-030212-183653
43. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell (2007) 130(2):223–33. doi:10.1016/j.cell.2007.07.003
44. Issa JP, Baylin SB, Herman JG. DNA methylation changes in hematologic malignancies: biologic and clinical implications. Leukemia (1997) 11(Suppl 1):S7–11.
45. Deneberg S, Grövdal M, Karimi M, Jansson M, Nahi H, Corbacioglu A, et al. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia (2010) 24(5):932–41. doi:10.1038/leu.2010.41
46. Markus J, Garin MT, Bies J, Galili N, Raza A, Thirman MJ, et al. Methylation-independent silencing of the tumor suppressor INK4b (p15) by CBFbeta-SMMHC in acute myelogenous leukemia with inv(16). Cancer Res (2007) 67(3):992–1000. doi:10.1158/0008-5472.CAN-06-2964
47. Humeniuk R, Koller R, Bies J, Aplan P, Wolff L. Brief report: loss of p15Ink4b accelerates development of myeloid neoplasms in Nup98-HoxD13 transgenic mice. Stem Cells (2014) 32(5):1361–6. doi:10.1002/stem.1635
48. Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res (2005) 65(10):3980–5. doi:10.1158/0008-5472.CAN-04-3995
49. Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood (2012) 120(5):1107–17. doi:10.1182/blood-2011-11-394932
50. Ropero S, Setien F, Espada J, Fraga MF, Herranz M, Asp J, et al. Epigenetic loss of the familial tumor-suppressor gene exostosin-1 (EXT1) disrupts heparan sulfate synthesis in cancer cells. Hum Mol Genet (2004) 13(22):2753–65. doi:10.1093/hmg/ddh298
51. Tian C, Yu Y, Jia Y, Zhu L, Zhang Y. HES1 activation suppresses proliferation of leukemia cells in acute myeloid leukemia. Ann Hematol (2015) 94(9):1477–83. doi:10.1007/s00277-015-2413-0
52. Estecio MR, Maddipoti S, Bueso-Ramos C, DiNardo CD, Yang H, Wei Y, et al. RUNX3 promoter hypermethylation is frequent in leukaemia cell lines and associated with acute myeloid leukaemia inv(16) subtype. Br J Haematol (2015) 169(3):344–51. doi:10.1111/bjh.13299
53. Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer (2006) 6(11):846–56. doi:10.1038/nrc1991
54. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet (2008) 40(5):499–507. doi:10.1038/ng.127
55. Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell (2010) 143(2):313–24. doi:10.1016/j.cell.2010.09.010
56. Fernandes do Nascimento AS, Côté S, Jeong LS, Yu J, Momparler RL. Synergistic antineoplastic action of 5-aza-2’deoxycytidine (decitabine) in combination with different inhibitors of enhancer of zeste homolog 2 (EZH2) on human lung carcinoma cells. J Cancer Res Ther (2016) 4(5):42–9. doi:10.14312/2052-4994.2016-8
57. Unland R, Borchardt C, Clemens D, Kool M, Dirksen U, Fruhwald MC. Analysis of the antiproliferative effects of 3-deazaneoplanocin A in combination with standard anticancer agents in rhabdoid tumor cell lines. Anticancer Drugs (2015) 26(3):301–11. doi:10.1097/CAD.0000000000000181
Keywords: 5-aza-2-deoxycytidine, 3-deazaneplanocin A, myeloid leukemia, epigenetics, gene expression, chemotherapy
Citation: Momparler RL, Côté S, Momparler LF and Idaghdour Y (2017) Inhibition of DNA and Histone Methylation by 5-Aza-2′-Deoxycytidine (Decitabine) and 3-Deazaneplanocin-A on Antineoplastic Action and Gene Expression in Myeloid Leukemic Cells. Front. Oncol. 7:19. doi: 10.3389/fonc.2017.00019
Received: 24 October 2016; Accepted: 31 January 2017;
Published: 15 February 2017
Edited by:
Michael Eccles, University of Otago, New ZealandReviewed by:
Qiang Yu, Genome Institute of Singapore, SingaporeAniruddha Chatterjee, University of Otago, New Zealand
Copyright: © 2017 Momparler, Côté, Momparler and Idaghdour. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard L. Momparler, richard.l.momparler@umontreal.ca;
Youssef Idaghdour, youssef.idaghdour@nyu.edu