 Simone Patergnani
Simone Patergnani Veronica A.M. Vitto
Veronica A.M. Vitto Paolo Pinton
Paolo Pinton Alessandro Rimessi
Alessandro Rimessi- 1Department of Medical Sciences and Laboratory for Technologies of Advanced Therapies (LTTA), University of Ferrara, Ferrara, Italy
- 2Center of Research for Innovative Therapies in Cystic Fibrosis, University of Ferrara, Ferrara, Italy
Cystic fibrosis (CF) is a genetic disease associated to mutations in the cystic fibrosis transmembrane conductance regulator gene, which results in the alteration of biological fluid and electrolyte homeostasis. The characteristic pathological manifestation is represented by exaggerated proinflammatory response in lung of CF patients, driven by recurrent infections and worsen by hypersecretion of proinflammatory mediators and progressive tissue destruction. Treating inflammation remains a priority in CF. However, current anti-inflammatory treatments, including non-steroidal agents, are poorly effective and present dramatic side effects in CF patients. Different studies suggest an intimate relationship between mitochondria and CF lung disease, supporting the hypothesis that a decline in mitochondrial function endorses the development of the hyperinflammatory phenotype observed in CF lung. This allowed the implementation of a new concept: the “mito-inflammation,” a compartmentalization of inflammatory process, related to the role of mitochondria in engage and sustain the inflammatory responses, resulting a druggable target to counteract the amplification of inflammatory signals in CF. Here, we will offer an overview of the contribution of mitochondria in the pathogenesis of CF lung disease, delving into mitochondrial quality control responses, which concur significantly to exacerbation of CF lung inflammatory responses. Finally, we will discuss the new therapeutic avenues that aim to target the mito-inflammation, an alternative therapeutic advantage for mitochondrial quality control that improves CF patient’s inflammatory state.
Introduction
Cystic fibrosis (CF) is a genetic and multi-organ disease, caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, resulting in lacked or reduced expression and function of protein (Elborn, 2016). ΔF508 is the most common mutation that displays a frequency of ∼66% worldwide, which determine the inadequate processing of protein with consequent trapping in the endoplasmic reticulum (ER) (Marson et al., 2016). CFTR protein functions as cyclic adenosine monophosphate-regulated chloride channel and regulator of epithelial transport proteins, including epithelial sodium channel (Csanady et al., 2019). Defective CFTR alters fluid and electrolyte homeostasis, resulting in abnormally viscous and sticky secretions that at pulmonary level facilitates the adhesion and proliferation of pathogens, especially by Pseudomonas aeruginosa (P. aeruginosa), triggering exaggerated pro-inflammatory responses characterized by elevated secretion of pro-inflammatory mediators (such as the interleukin (IL)-8, IL-1β, and IL-18) (Malhotra et al., 2019). The intrinsic defects associated with CFTR deficiency and the repeated pulmonary infections induce hyperinflammation, leading to reduced bacterial clearance capacity and tissue damage in airways, compromising the respiration (Rafeeq and Murad, 2017).
While the underlying mechanisms that promote CF lung disease progression are not fully delineated, more studies highlighted the connection between mitochondrial health and CF pathogenesis, shading light on this organelle’s role in regulating host response and inflammation.
Mitochondria are central signaling hub that communicate with the cell to regulate several functions, such as metabolism, regulation of calcium (Ca2+) homeostasis, inflammation, stress responses, and cell death. Differently from other organelles, mitochondria present two membranes constituted by low levels of sphingolipids and cholesterol with exclusively components, such as posphosphatidylglycerol and cardiolipin (CL). The outer mitochondrial membrane (OMM) fully surrounds the inner mitochondrial membrane, separating the intermembrane space (IMS) from the matrix (Mejia and Hatch, 2016). They create a dynamic tubular and organized network, where the shape is continuously controlled by opposing fission and fusion events. These dynamic events are relevant during cell cycle in mitochondrial movement and in the interaction with other intracellular organelles (Nunnari and Suomalainen, 2012).
The interorganelle communication plays a key role in the mitochondrial functions, in particular with ER. Approximately, 5–20% of mitochondria surface is juxtaposed to specialized ER regions, linked by specific physical tethers at distance of 10–30 nm distance. Functional ER-mitochondria tethering is due by inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs), glucose-regulated protein 75, and voltage-dependent anion channels (VDAC) to transfer Ca2+ from ER to mitochondria (Giorgi et al., 2018a). While among structural tethering: the ER-located mitofusin 2 (MFN2) that builds a bridge with a heterodimer complex with MFN1/2 on OMM, and the integral ER protein vesicle-associated membrane protein-associated protein B (VAPB), which interacts with protein tyrosine phosphatase-interacting protein-51 (PTPIP51) (Lee and Min, 2018). Disruption or remodeling of ER-mitochondria tethering lead to a mismatch between the two organelles, causing dysregulation of lipid trafficking, Ca2+ signaling, autophagosome formation and apoptosis progression (Rowland and Voeltz, 2012; Rimessi et al., 2020a).
The mitochondria’s innermost compartment is the matrix, where resides the mitochondrial DNA (mtDNA) that contains genetic coding information for 13 proteins, core constituents of the mitochondrial respiratory complexes I-IV. The matrix is also site of replication and transcription of mtDNA, protein biosynthesis, and ATP synthesis. During this last process, electrons move along the respiratory complexes of the mitochondrial electron transport chain (mETC). This flow is coupled to the pumping of protons from matrix into IMS, creating an electromotive force used to produce ATP (Turrens, 2003). During this process, some electrons leak out of ETC, interact with oxygen to produce reactive oxygen species (ROS), in particular the superoxide anion (O2-). Complex-I (NADH dehydrogenase) and complex-III (CoQH2-cytochrome c reductase) result the primary sites where electron leak occurs and ROS are produced.
The mitochondrial membrane potential (Δψ) is the driving force necessary for protein import via translocase of outer membrane/translocase of inner membrane complex and for mitochondrial Ca2+-uptake into the matrix through the mitochondrial Ca2+ uniporter (MCU). The Ca2+-released from ER, via IP3Rs, may enter into matrix through VDAC and MCU (Giorgi et al., 2018b). Mitochondrial Ca2+ may stimulate OXPHOS, but upon cell stress, excessive mitochondrial Ca2+ accumulation induces ROS production, autophagic flux reduction, and mitochondrial permeability transition pore (mPTP) opening, which lead to an irreversible collapse of Δψ, swelling of mitochondria, and release of pro-apoptotic factors, such as cytochrome c (Pedriali et al., 2017).
In this review, we highlight the role of mitochondria in the pathogenesis of CF lung disease. In particular, we will discuss of role of mitochondrial quality control responses, which are modulated by defective CFTR and persistent infections, contributing significantly to CF lung hyperinflammation. The complete understanding of these alterations, their molecular mechanisms, and the importance of each compensatory pathways engaged will help us to find new CF therapy strategies.
Mitochondrial Dysfunction in Cystic Fibrosis
Endogenous and/or environmental stresses may perturb the mitochondrial homeostasis and interfere with the steady-state activity of mitochondrial functions, promoting a state of mitochondrial stress characterized by the inability to maintain basal Δψ and mitochondrial ROS production, with repercussions on mitochondrial protein import, mitochondrial Ca2+ signaling, and oxidation state. All this could harm airway epithelial and immune cells, contributing to the development and exacerbation of CF lung disease. Studies revealed that the mitochondrial impairments in the CF lung and the mitochondrial quality control responses are associated with CFTR deficiency and inflammatory environment.
Mitochondrial Defects Associated to CFTR Deficiency
The first evidence of mitochondrial defects related to CFTR deficiency was obtained in the 80s, showing that the oxygen consumption rate of isolated mitochondria from CF patients was affected due to complex-I and Na+/K+ ATPase alterations (Feigal and Shapiro, 1979; Shapiro et al., 1979). Consistently, the gene MT-ND4 and CISD1, fundamentals for a proper functioning of mETC, resulted downregulated in CF patient-derived tracheal cells (Valdivieso et al., 2007). Indeed, case reports of CF patients reported impairments in cytochrome c oxidase and in 6-phosphate dehydrogenase (Congdon et al., 1981; Battino et al., 1986). Oxygen consumption, complex-I activity, Δψ, and OXPHOS were also found dysregulated in CFTR-silencing intestinal epithelial and in F508del-CFTR airway cells (Kleme et al., 2018). However, treatment with “CFTR correctors,” including VX809 and 4,6,4’-trimethylangelicin, improved all the mitochondrial parameters, indicating that the CFTR rescue is linked to recovery of mitochondrial function (Atlante et al., 2016).
Other mitochondrial defects, such as the mitochondrial protein pattern, the intracellular pH and mitochondrial Ca2+ signaling have been described in CF to be sufficient to promote ROS production and membrane lipid peroxidation (Turrens et al., 1982; Picci et al., 1991; de Meer et al., 1995; Rimessi et al., 2015). Mitochondrial alteration due to oxidative stress has been also reported in CFTR-knockout mice, where oxidative mtDNA damage and reduced aconitase activity were described (Velsor et al., 2006). Also the ROS detoxification capacity in CF appears compromised. Low levels of mitochondrial reduced glutathione (mtGSH) and defects in GSH transport have been found in CF patient-derived airway cells and in CFTR-knockout, resulting in an altered extracellular ratio between reduced and oxidized GSH (Gao et al., 1999; Velsor et al., 2006; Kelly-Aubert et al., 2011). Accordingly, functional CFTR reintroduction restored mtGSH levels, attenuating the Δψ depolarization and IL-8 secretion28.
Mitochondrial Defects Associated to Persistence Infections
Pathogens affect mitochondria, generally causing Δψ loss and mitochondrial fragmentation, to influence their intracellular survival or to evade host immunity (Tiku et al., 2020). Airway epithelial and immune cells are sensitive to pathogen upon infection, activating mitochondrial stress responses to preserve the mitochondrial homeostasis.
In uninfected conditions, no significant differences in mitochondrial physiology and in inflammatory profile were detected between human CF and non-CF airway cells. Both mitochondrial networks exhibited the classical ultrastructure integrity of a well-defined OMM with numerous pleomorphic cristae. Contrariwise, the exposition to P. aeruginosa strains displayed extensive mitochondrial swollen and fragmentation with derangement of cristae in CF airway epithelial cells, resulting in Δψ loss, excessive O2- production, and nod-like receptor 3 (NLRP3) inflammasome activation (Rimessi et al., 2015). Using non-motile P. aeruginosa mutants, it has been demonstrated that the mitochondrial dysfunction in CF airway epithelial cells is triggered by the bacterial constituent flagellin through a Toll-like receptor 5 (TLR5)-dependent pathway (Rimessi et al., 2015). The recovery of mitochondrial integrity in CF airway epithelial cells during infection was obtained silencing or pharmacologically inhibiting MCU, with KB-R7943, indicating that mitochondrial Ca2+ signaling has a role in P. aeruginosa-dependent mitochondrial impairments in CF (Rimessi et al., 2015; Rimessi et al., 2020b).
These data show that a functional CFTR channel may prevent the P. aeruginosa-triggered mitochondrial dysfunction, regulating the susceptibility of airway cells to infection and thus conditioning the degree of innate immune response.
Mitochondrial Stress Responses in Cystic Fibrosis
It is not yet entirely clear whether mitochondrial dysfunction is a trigger for, a consequence, or both for CF lung disease. In resting conditions, compensatory mitochondrial stress responses are transiently activated to restore the mitochondrial homeostasis, while during the recurrent infections, the chronic mitochondrial stress condition leads to amplification and persistence of these responses. Functional fusion complementation, mitophagy, mitochondrial unfolded protein response (UPRmt), and apoptosis are recruited to recover and preserve the mitochondrial homeostasis to regulate metabolism and innate immune response and cell viability. In CF, the persistent infections and the defects associated with CFTR deficiency alter the mitochondria quality control machinery, acquiring potential relevance to the disease state.
Functional Fusion Complementation and Mitophagy
The morphology of mitochondrial network is regulated by fusion/fission events and mitophagy to sustain an adequate supply of healthy mitochondria. Mitochondrial fusion compensation optimize the functional efficiency of organelle under stressful conditions, allowing the exchange of materials among partially damaged mitochondria (Campello and Scorrano, 2010). The MFN1/2 are pivotal mediators of this process (Song et al., 2009). A sustained stress condition favors the segregation of damaged/depolarized parts of mitochondria in autophagosomes that then will be eliminated through lysosomal degradation by mitophagy. This catabolic process minimizes the quote of dysfunctional mitochondria, removing excess of ROS, oxidized mtDNA, and other mitochondrial dangerous factors relevant to disease state (Eisner et al., 2018) (Figure 1, mitophagy). Mitochondrial-targeted kinase PINK1 and E3 ubiquitin ligase Parkin have a central role for mitophagy. Parkin is recruited to OMM by perturbed mitochondrial import of PINK1 in stressed mitochondria, where catalyzes the ubiquitination of MFNs and other OMM proteins to sequester the organelle in autophagosome. At the same time, PINK1 contributes to strengthen the mitophagic response, phosphorylating both parkin and ubiquitin and recruiting the mitophagic receptors NDP-52 and optineurin to mitochondria (Koyano et al., 2014; Lazarou et al., 2015).

Figure 1 Mitochondrial stress responses in cystic fibrosis. Schematic representation of mitochondrial stress response pathways in healthy and in CF. In healthy condition, compensatory mitochondrial stress responses are transiently triggered to restore the mitochondrial homeostasis during P. aeruginosa infection. Damaged mitochondrial portions are removed by mitophagy, where the PINK1-Parkin signaling pathway promotes ubiquitination of OMM proteins while the mitophagic receptors, optineurin (OPN) and NDP52, act as adaptors to recruit autophagosomal membranes to mitochondria, interacting with LC3. In CF, defective mitophagy leads to dysfunctional mitochondria accumulation with consequent release of mitochondrial DAMPs, which contribute to hyper-inflammatory responses in CF airway cells during the persistent P. aeruginosa infection. The mammalian UPRmt is regulated by the transcription factors ATF5, which in healthy condition is imported into mitochondria to be degraded. In CF, the persistent mitochondrial stress in airway cells induces abnormal UPRmt activation with consequent nuclear translocation of ATF5, which supports the regulation of innate immunity response during pathogen infection. At front of the mitochondrial Ca2+-overload, a higher susceptibility to irreversible damages in response to pathogen infection occur in CF airway cells, contributing to promote organelle dysfunction and cell death. In CF, the exacerbation of inflammatory environment is due also by mtDAMPs release and by accumulation of phagocyte cells that intervene to scavenge the dying cells and pathogens. PTEN induced kinase 1 (PINK-1); Microtubule-associated proteins 1A/1B light chain 3B (LC3); Nuclear domain 10 protein 52 (NDP52); Optineurin (OPN); Translocase of the outer membrane (TOM); Translocase of the inner membrane (TIM); Activating Transcription Factor 5 (ATF5); Lon protease homolog 1 (LON-1); heat shock protein 60 (HSP60); heat shock protein 10 (HSP10); Caseinolytic Mitochondrial Matrix Peptidase Proteolytic Subunit (CLPP); mitochondrial deoxyribonucleic acid (mtDNA); cytochrome c (cyt c); B-cell lymphoma 2 protein (Bcl2); BCL2 Associated X protein (BAX); Bcl-2 homologous antagonist/killer protein (BAK); reactive oxygen species (ROS); calcium (Ca2+); Pseudomonas aeruginosa (P. aeruginosa); apoptotic protease activating factor-1 (APAF-1). This figure has been created with “BioRender.com”.
In airway epithelial cells the accumulation of NDP-52, optineurin and autophagic form of microtubule-associated protein light chain 3 (LC3-II) to mitochondria triggers mitophagy during P. aeruginosa infection. The minor mitochondrial redistribution of LC3-II indicated a slower kinetic of mitochondrial sequestration into autophagosome in CF cells respect to non-CF cells, confirmed also by a reduced recruitment of Parkin to stressed mitochondria (Rimessi et al., 2020b) (Figure 1, mitophagy). Defective mitophagy led to excessive O2- production and NLRP3 inflammasome activation in CF airway cells during P. aeruginosa infection (Rimessi et al., 2015; Rimessi et al., 2020b). Similar defect was emerged also in xenophagy, the selective autophagic response involved to sequester into the cell invading pathogens (Gatica et al., 2018). The higher number of colony-forming unit/ml and of interactions between xenophagic receptors and invading P. aeruginosa in CF airway cells compared with non-CF cells, indicated a reduced bacterial clearance capacity of CF airway cells, resulting in further cell stress and pyroptosis induction (Rimessi et al., 2020b). The worsening of mitophagy and xenophagy in CF cells during infection is consequence of an enhanced ER-mitochondria juxtaposition, which making the organelles more prone to interorganelle Ca2+-exchange. CF airway cells exposed to P. aeruginosa showed increased expression and interaction between ER protein VAPB and OMM protein PTPIP51, which induced tightening of tethers and concomitant impairment of selective autophagic responses (Rimessi et al., 2020b). Pharmacologically control of mitochondria Ca2+-uptake, by MCU inhibitor KB-R7943, abrogated the inhibitory effects of VAPB and PTPIP51 on the selective autophagic responses, promoting cellular and mitochondrial resistance to infection and inflammatory reduction (Rimessi et al., 2020b). This highlights a central role for mitochondrial stress in progression of CF lung inflammatory state, with detrimental repercussions on the autophagic responses, which may further affect the expression, trafficking, and function of CFTR channel (Luciani et al., 2010; Villella et al., 2013). Defective macroautophagy in CF has been also associated to upregulation of transglutaminase (TG2), which led to ROS production and decrease of aggresomes clearance (Luciani et al., 2010). The rescue of autophagy, mediating antioxidants, cystamine (TG2 inhibitor), or modulators of Ca2+-dependent signaling (KB-R7943), resulted in improved CFTR transport to PM, reduced oxidative stress, and cytokines release in CF airway cells30,37.
Mitochondrial Unfolded Protein Response
Perturbed mitochondrial protein import and the accumulation misfolded proteins within of organelle induces UPRmt activation, a transcriptional program that results in a number of cellular- and mito-protective outcomes (Melber and Haynes, 2018). In C. elegans, the P. aeruginosa exposure induced mitochondrial stress and recruitment of stress-activated transcription factor 1, which induced the transcription of mitochondrial chaperones [such as heat shock protein (HSP) 10 and HSP 60], proteases (including the caseinolytic mitochondrial matrix peptidase proteolytic subunit), ROS detoxification, and innate immune genes, resulting a key regulator of UPRmt (Nargund et al., 2012; Pellegrino et al., 2014). A similar transcriptional response has been described in mammals, identifying ATF5, which upon mitochondrial stress, fails to be imported into mitochondria for its degradation by mitochondrial protease lon protease homolog 1 but moves to the nucleus inducing gene transcription (Figure 1, UPRmt) (Fiorese et al., 2016).
Rimessi et al. demonstrated that CFTR deficiency reduced the mitophagic clearance during infection with detrimental repercussion on mitochondrial homeostasis, triggering an abnormal UPRmt activation in CF airway epithelial cells (Rimessi et al., 2020b). An extensive nuclear ATF5 redistribution, associated to an increased expression levels of UPRmt reporters, was measured in CF airway cells during P. aeruginosa infection. The persistent UPRmt activation also increased the inflammatory-sensitivity of CF cells to pathogen, as shown by higher levels of NLRP3 inflammasome-dependent IL-1β and IL-18 released during infection (Rimessi et al., 2020b). The amplitude of inflammasome responses were correlated to amount of dysfunctional mitochondria accumulated and by the levels of nuclear ATF5. UPRmt and NLRP3 inflammasome activation in turn led to worsening of mitophagic and xenophagic defects in CF cells, favoring a vicious cycle that contributed to exacerbate the P. aeruginosa-dependent cellular and mitochondrial stress (Rimessi et al., 2020b). These findings showed as the impairments in selective autophagy in CF are sustained by abnormal UPRmt and NLRP3 activation, contributing to persistent damaged mitochondria and invading bacteria accumulation in CF cells.
Apoptosis
During infection, in front of irreversible damages in response to prolonged stress conditions, mitochondria may activate the intrinsic apoptotic pathway (Green and Kroemer, 2004). Many pathogens activate the intrinsic apoptotic pathway, causing Δψ depolarization and mitochondrial permeabilization followed by enhanced levels of ROS and pro-apoptotic Bcl2 family proteins, such as Bax and Bid (Green and Kroemer, 2004; Wood et al., 2015). In turn, the excessive oxidative stress may oxidize mtDNA and active redox-sensitive kinases and transcription factors, which exacerbates the inflammatory environment contributing significantly to promote apoptosis in CF cells (Figure 1, apoptosis) (Jendrossek et al., 2001; Rottner et al., 2007; Kamdar et al., 2008; Galli et al., 2012; Rimessi et al., 2016).
Other studies suggest that the increased apoptotic susceptibility in CF cells is dependent by CFTR deficiency; e.g., I) due by abnormal intracellular Ca2+ signaling, which upon stress sensitizes the cell to organelle dysfunction and death (Antigny et al., 2011; Rimessi et al., 2020b); II) due to a reduced antioxidant activity, CFTR deficiency induced dysregulation of GSH concentration and transport, while reducing the expression of superoxide dismutase in F508del-CFTR mutant pancreatic cells (Gao et al., 1999; L’hoste et al., 2010; Kelly-Aubert et al., 2011; Rottner et al., 2011); III) due to a reduced expression of anti-apoptotic proteins, such as Bcl2 in CFTR-silencing intestinal cells20; IV) due to altered intracellular pH (de Meer et al., 1995; Barriere et al., 2001).
In any case, the enhanced apoptosis in CF cells contributes to hasten the tissue damage and functional loss in CF, worsening the disease state.
Mito-Inflammation in Cystic Fibrosis
The extensive studies of these years revealed a supplementary role of mitochondria as drivers of inflammatory responses, leading us to reflect on new concept: the mito-inflammation. A compartmentalization of inflammatory process related to the role of mitochondria in engage and sustain the inflammatory responses and thus a druggable target to counteract the exacerbations of responses. In CF, mitochondria function as central regulator of danger signals, arbitrators with a double role in the pathogenesis of hyperinflammatory state.
Firstly, they act as checkpoints of intracellular downstream signal cascades to pathogen recognition receptor responses, induced by exogenous pathogen-associated molecular patterns. Bacterial ligands induce macrophage bactericidal-activity binding TLR 1, 2 and 4, which modulate the mitochondrial respiratory chain assembly factor, ECSIT, to increase mitochondrial ROS production (West et al., 2011). Again, the mitochondrial antiviral signaling protein (MAVS), located on the OMM regulates the transduction of interferon-dependent signaling pathway to amplify antiviral innate immune responses (Banoth and Cassel, 2018).
Second, mitochondria act as a key source of mitochondrial danger-associated molecular patterns (DAMPs), where ROS, mtDNA, ATP, CL and Ca2+ are released as danger signals into the cytosol or in the extracellular milieu, which recognized by TLRs and/or the cytosolic nucleotide-binding oligomerization domain-like receptors (NLRs) trigger the inflammatory responses inducing the expression and secretion of numerous pro-inflammatory mediators (Figure 2) (Lavelle et al., 2010).
Upon stress, the mitochondrial ROS production results excessive in CF cells, determining (Figure 2, ROS): I) oxidative damage to intracellular macromolecules, including mtDNA; II) activation of redox-sensitive transcription factors, inducing the up-regulation of cytokines and inflammasomes. The transcription factors NF-kB, AP-1 and HIF-1 result hyperactivated in murine and human CF airway cells, due to also by intrinsic defects associated to defective CFTR (like abnormal intracellular Ca2+ signaling), causing production of chemokine IL-8 and priming of NLRP3 and pro-IL-1β expression (McNamara et al., 2006; Tabary et al., 2006; Levy et al., 2009; Legendre et al., 2011; Saadane et al., 2011; Nichols and Chmiel, 2015); III) NLRP3 inflammasome activation; and IV) additional mitochondrial impairments in a feed-back stimulatory manner that exacerbates the inflammatory response (Brookes et al., 2004; Rimessi et al., 2016).
mtDNA may be oxidized by ROS and its damages may alter the OXPHOS activity, producing additional ROS (Figure 2, mtDNA). mtDNA may escape from matrix, due to impaired mitophagy or Ca2+- and oxidative stress-dependent mPTP opening, and binds directly the NLR component of inflammasomes to trigger theirs activation (Escames et al., 2012). CL is accidentally released from damaged mitochondria and acts as direct activator of NLRP3 inflammasome in the cytosol (Iyer et al., 2013).
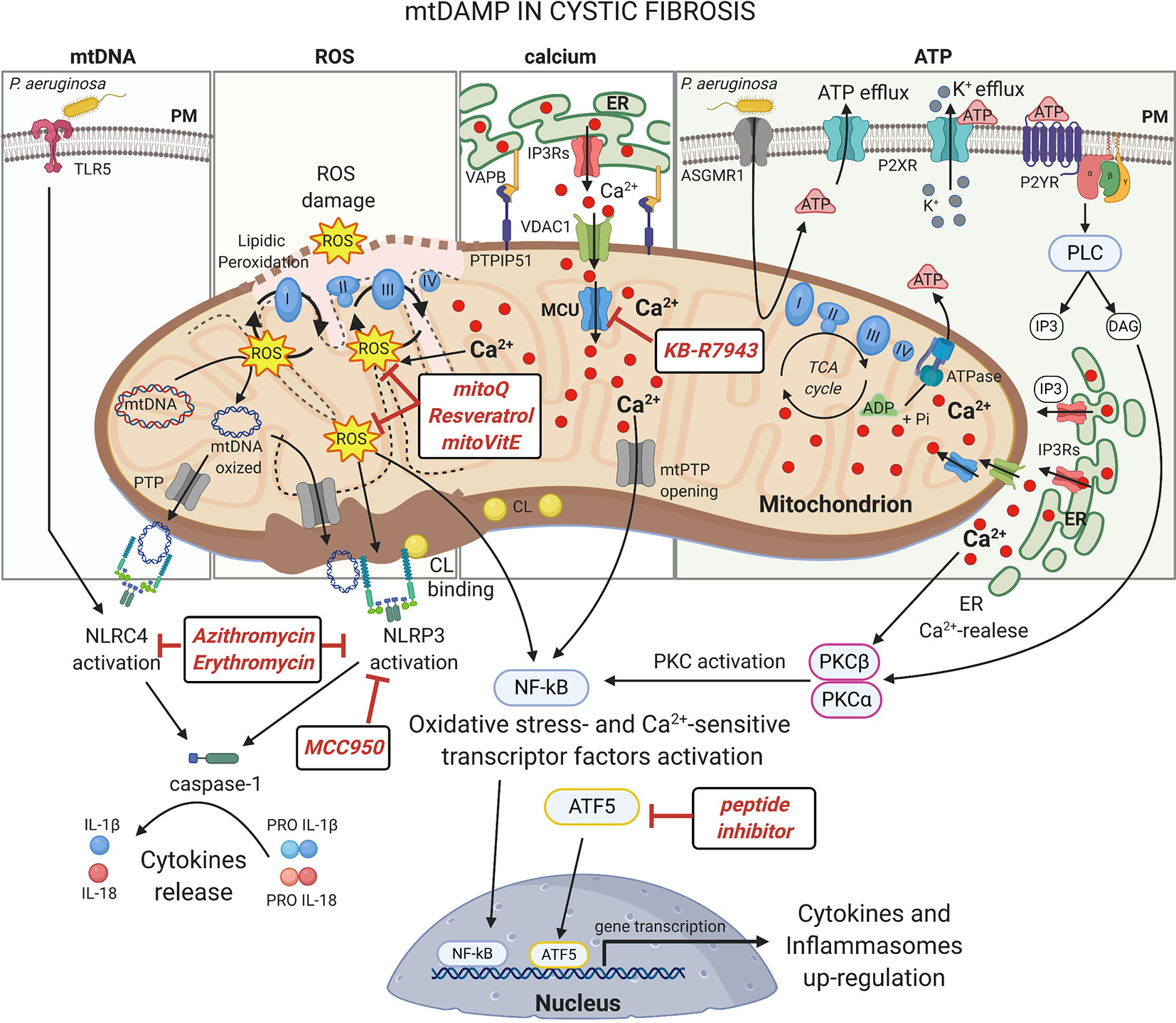
Figure 2 mtDAMPs in cystic fibrosis Under stress, mitochondria generate and/or release immunogenic molecules, named mitochondrial damage associated molecular patters (mtDAMPs), that influence significantly the inflammatory response in CF lung disease. The accumulation of dysfunctional mitochondria leads to excessive ROS production in CF airway cells with detrimental effects on mitochondrial constituents, inducing oxidative damages on mtDNA and mitochondrial respiratory complexes or the lipidic peroxidation of mitochondrial membranes. The ROS may also active: I) oxidative-stress sensitive transcriptor factors, which after nuclear translocation, up-regulate the expression of cytokines and inflammasomes; II) or the NLRP3 inflammasome to promote the release of interleukine-1β (IL-1β) and IL-18. NLRP3 and NLRC4 inflammasomes are also activated by direct interaction with mtDNA and cardiolipin (CL), released following mitochondrial damage or PTP opening, with consequent activation of caspase-1. The abnormal ER-mitochondria Ca2+ transfer, due by increased interorganelle crosstalk mediated by VAPB-PTPIP51 tethering during pathogen infection in CF, contributes to mitochondrial stress inducing ROS production, PTP opening and UPRmt activation, which facilitate the activation of NF-kB and NLRP3 inflammasome. Increased levels of extracellular ATP, induced by P. aeruginosa-ASGMR1 signaling pathway, mediate ionic flux and intracellular potassium (K+) depletion through the ATP-binding to ligand-gated ion channels P2X receptors, sensitizing the CF airway cells to NLRP3 inflammasome activation. Indeed, mediating P2Y receptors, the extracellular ATP induces the Ca2+-dependent activation of classical Protein Kinase C (PKC) isoforms through the IP3-triggering ER Ca2+-release, resulting in NF-kB activation (Pinton et al., 2004). Several classes of molecules may exert anti-inflammatory activity targeting mitochondria at different levels. In this figure are shown the discussed drugs that may counteract the mito-inflammation in CF lung disease. Plasma membrane (PM); Toll-like receptor 5 (TLR5); NLR Family CARD Domain Containing 4 (NLRC4); permeability transition pore (PTP); NLR Family Pyrin Domain Containing 3 (NLRP3); Mitochondrial Calcium Uniporter (MCU); Voltage-dependent anion-selective channel 1 (VDAC1); Vesicle-associated membrane protein-associated protein B (VAPB); Protein tyrosine phosphatase interacting protein 51 (PTPIP51); Inositol 1,4,5-Triphosphate (IP3); diacylglycerol (DAG); Inositol trisphosphate receptors (IP3Rs); Endoplasmic Reticulum (ER); nuclear factor-κB (NF-kB); adenosine triphosphate (ATP); adenosine diphosphate (ADP); Asialo GM1 receptor (ASGMR1); P2X purinoceptor (P2XR); Purinergic Receptor (P2YR); Phospholipase C (PLC). This figure has been created with “BioRender.com”.
ATP released from mitochondria acts in the extracellular milieu as ligand of purinergic receptors, resulting in increased ROS production and cytokines release, influencing also NLRP3 activation (Bonora et al., 2012). Finally, dysfunctional mitochondria present altered mitochondrial Ca2+ signals, critical for inflammation, which influence the mitochondrial stress responses with consequent repercussions on inflammasome activation and on up-regulation and release of cytokines (Rimessi et al., 2015).
The mitochondrial DAMPs are preferentially bound by different NLRs to modulate the amplitude of immune responses activating the inflammasomes, multiprotein complexes that lead to caspase-1 activation and subsequent maturation of the pro-inflammatory cytokines IL-1β, IL-18, and IL-33 (Figure 2) (Broz and Dixit, 2016). Elevated concentrations of IL-1β and IL-18 were detected in human airway epithelial cells, monocytes and serum of CF patients (Levy et al., 2009; Rimessi et al., 2015; Iannitti et al., 2016; Scambler et al., 2019). Mitochondrial DAMPs are the principal initiator of NLRP3 inflammasome activation. NLRP3 is a cytosolic receptor that, once activated, interacts with mitochondria where oligomerizes recruiting the adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and the procaspase-1 (Zhou et al., 2011). Its mitochondrial localization and association depends by MAVS, which promotes also its activation, and by mitochondrial-anchored protein ligase (Park et al., 2013; Subramanian et al., 2013). Rimessi et al. showed the NLRP3 mitochondrial recruitment and activation in CF airway cells during P. aeruginosa infection, since defective CFTR induced mitochondrial Ca2+-overload and ROS production, which in turn driven the NLRP3 inflammasome activation and release of IL-1β and IL-18 (Rimessi et al., 2015). The increased susceptibility to pathogen-dependent mitochondrial dysfunction and the mitochondrial Ca2+-overload in human CF airway cells resulted in the recruitment of both NLRP3 and NLRC4 inflammasome with consequent worsen of inflammation (Rimessi et al., 2015). NLRC4/IPAF inflammasome activation has been associated to mitochondrial DAMPs during P. aeruginosa infection also in macrophages, due to ROS and direct binding with oxidized mtDNA (Jabir et al., 2015). Typically, it activated by TLR5-recognition on PM via bacterial flagellin and in the cytosol via the microbial type III secretion system (Sutterwala et al., 2007; Tolle et al., 2015). The interplay between NLRP3 and NLRC4 inflammasome has been also reported in CFTR-null mouse model and in alveolar CF macrophages and neutrophils, where NLRP3 greatly contributed (Iannitti et al., 2016). In CF airway cells, NLRP3 inflammasome activation induces also the downregulation of mitophagy and xenophagy, promoting the accumulation of damaged mitochondria and invading bacteria into the cells (Rimessi et al., 2020b). In turn, released IL-1β induces mitochondrial ROS production, down-regulating the complex-I activity and Δψ, and NF-kB activation, generating a loop that sustain and exacerbate the inflammatory response (Lopez-Armada et al., 2006; Escames et al., 2012).
The hyperinflammation observed in CF lung is in part conditioned by altered phenotype of airway immune cells associated to defective CFTR. CF neutrophils exhibit altered chlorination of phagocytosed bacteria while the macrophages showed reduced selective autophagic activity, becoming a replicative niche for bacteria (Painter et al., 2006; Lamothe and Valvano, 2008; Ratner and Mueller, 2012; Assani et al., 2014). Mitochondrial metabolism and the metabolic state of the cell drive the pro- or anti-inflammatory responses, regulating the polarization and activation of different immune cells, including neutrophils and macrophages (Michalek et al., 2011; Tarique et al., 2015). Accordingly, a compromised metabolism was found in CF neutrophils in response to LPS, shifting to a state of increased aerobic glycolysis with consequent exacerbation of IL-1β production (McElvaney et al., 2019). CF macrophages did not respond to IL-13/IL-4 and failed to polarize into M2, contributing to excessive production of cytokines (Tarique et al., 2017). The pro-inflammatory Th17 cells are highly glycolytic; meanwhile the immunosuppressive Treg cells present an elevated rate of lipid oxidation. In CF patients, the differentiation of T lymphocytes to Th17 phenotype was increased (Kushwah et al., 2013), indicating that the maintenance of mitochondrial homeostasis and of a functional metabolic reprogramming are critical conditions to regulate the immune responses also in CF.
Targeting Mito-Inflammation as Therapeutic Approach in Cystic Fibrosis
In CF, the ideal drug should be one that interrupts the vicious CF cycle that sustains lung hyperinflammation, but also generates favorable conditions to rescue or potentiate the residue functionality of defective CFTR. In this regard, several modulators of mitochondrial behaviors have shown anti-inflammatory activity, such as mitochondrial antioxidants, modulators of mitochondrial Ca2+-exchange, selective autophagic-inducing compounds and inflammasome and IL-1β inhibitors.
Strategies that limit mitochondrial ROS production may be useful to control the hyperactivation of redox-sensitive inflammatory transcription factors and inflammasomes (Figure 2, ROS). An example may be found in the natural phenolic antioxidant resveratrol (3,4′,5-trihydroxystilbene), which efficiently reduces oxidative stress and subsequent inflammation, preserving mitochondrial Δψ and mtDNA in vitro and in vivo (Manna et al., 2000; Xu et al., 2012). At high concentration, in preclinical studies resveratrol rescues the expression of F508del-CFTR, the chloride secretion and the intracellular transport in human primary airway epithelial cells and CF mouse models (Hamdaoui et al., 2011; Dhooghe et al., 2015; Lu et al., 2020). Indeed, in presence of the modulator, ivacaftor, resveratrol augmented the G155D-CFTR activation in human primary sinonasal cells (Cho et al., 2019). Unfortunately, resveratrol exhibits poor bioavailability with maximal achievable plasma concentration of about 2 μM (a concentration no effective to improve CFTR function), that could limit its clinical usefulness (Walle, 2011; Jai et al., 2015). However, different therapies in CF involve topical application of drugs to lung mediating aerosol, a strategy that may overcome the limit of bioavailability.
Other antioxidants, such NAC, have been locally administrated to CF adults and children in different clinical trials. Acute administration of NAC was found to be well tolerated and free of adverse effects. Despite this, it was only found beneficial variations in sputum rheology and hydration, two factors that may be predictive of improved airway mucus clearance. Any significant change in pulmonary function and in clinical indicator were registered (App et al., 2002). Of more interest were the results obtained in a phase II randomized placebo-controlled trial, where it was performed long-term treatment with oral NAC (Conrad et al., 2015). Indeed, authors found improvements in lung functions, reduction in the incidence of pulmonary exacerbations. Unfortunately, they found no change in sputum human neutrophil elastase activity and other biomarkers of inflammation, suggesting to consider more specialized antioxidants as therapeutic strategy in CF (Cantin, 2004). Antioxidants that have shown promising results in oxidative stress-related diseases are mitovitamin E (MitoVit-E) and mitoquinone (Mito-Q), mitochondrial antioxidants that contain the triphenylphosphonium cation moiety that facilitates theirs accumulation into the organelle (Smith et al., 2003). MitoVit-E suppressed the sepsi-induced peripheral and myocardial production of cytokines, including IL-1β, improving mitochondrial function and heart activity, while Mito-Q restored mitochondrial function in chronic obstructive pulmonary disease (COPD) patients, reducing inflammation and IL-8 release in preclinical studies (Zang et al., 2012; Wiegman et al., 2015). In CF patients, the low level of vitamin-E in serum is normally correct with a dietary supplement, this implementation should be enriched also with MitoVit-E to safeguard the mitochondrial function (Sokol et al., 1989).
The hyperinflammation in CF lung is due also by altered mitochondrial Ca2+ exchange, in which MCU is involved (Rimessi et al., 2015; Rimessi et al., 2020b). Pharmacological MCU inhibition, mediating KB-R7943, attenuated in vitro and in vivo the P. aeruginosa-dependent mitochondrial dysfunction and hyperinflammation in CF lung, controlling UPRmt and NLRP3 inflammasome activation (Figure 2, calcium) (Rimessi et al., 2020b). KB-R7943 is the first freely PM permeable and only one available dose-selective MCU inhibitor, in opposing to more used MCU inhibitors, Ruthenium Red and Ru360 that are cell impermeable (Santo-Domingo et al., 2007). A new class of cell-permeable selective MCU inhibitors are now available, and Ru265 and DS16570511 are minimally toxic in vitro and could have important implications in CF in the future (Kon et al., 2017; Woods et al., 2019). KB-R7943 also restored mitophagy and xenophagy in CF, removing stressed mitochondria and pathogens able to induce mtDAMPs release, it attenuated the inflammatory state (Rimessi et al., 2020b). Thus, selective autophagic inducers may mitigate the hyperinflammation in CF. Rapamycin and MK-2206, both inhibitors of PI3K/Akt/mTOR pathway, reduced the severity of CF inflammation and improved the CFTR stability in airway epithelial cells in preclinical studies (Abdulrahman et al., 2011; Reilly et al., 2017). In a perspective of mitochondrial quality control–based therapy, the synthetic cell-penetrating dominant negative ATF5 peptide (Cs Bio®) should be useful to dampen the persistent UPRmt activation in CF, restoring the autophagy process (Figure 2) (Sheng et al., 2011).
Inflammasome and IL-1β inhibitors may rescue selective autophagy and attenuate the hyperinflammation in CF. MCC950 inhibits NLRP3 and AIM2 inflammasome, but not NLRC4, blocking the formation of ASC complexes (Coll et al., 2011). In vivo, MCC950 improved the clearance of P. aeruginosa and reduced IL-1β release in the lung (McElvaney et al., 2019). The pathogenic NLRP3 activity in CF may be reduced using the IL-1 antagonist receptor Anakinra, that by blocking the biological IL-1α and IL-1β activity ameliorated tissue damage and inflammation against P. aeruginosa in CF, with positive repercussion also on autophagy and neutrophils infiltration (Iannitti et al., 2016). The questionable limit of this approach is the indiscriminate block of IL-1 activity, which may compromise the global inflammatory responses important in fighting the persistent infections, in particular in those body areas where the microbe-host interactions are strategic. Despite this consideration, a phase 2 study (ClinicalTrials.gov Identifier: NCT03925194) to evaluate safety and efficacy on lung function of Anakinra subcutaneous administration in CF patients is ongoing. Recently, it has been demonstrated that also azithromycin and erythromycin (macrolides used in antibiotic CF therapies) inhibit the activation of inflammasome NLRP3 and NLRC4, which attenuated lung injury and inflammation enhancing the P. aeruginosa clearance in mice and in bronchiectasis patients (Figure 2) (Fan et al., 2017). Considering that the beneficial effects of these antibiotics is difficult to maintain over the years, treatments engaging specific inflammasome inhibitors may be used to improve the effects of these drugs in long-term treatment (Samson et al., 2016).
In conclusion, all these studies reveal the pathogenic role of mito-inflammation in CF and support mitochondria as new pharmacological targets. Emerging concepts of mitochondrial quality control provide opportunities to develop mitochondrial therapies, which aim to preserve the mitochondrial function as an alternative anti-inflammatory approach. The therapeutically restoring of mitochondrial homeostasis will be useful to improve the clinical state of CF lung disease, avoiding the overstimulation of inflammatory signals.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
The research was funded by grants from Italian Cystic Fibrosis Research Foundation FFC#12/2010 and #19/2014, Italian Association for Cancer Research (AIRC, IG-23670), Telethon (GGP11139B), local funds from the University of Ferrara, and the Italian Ministry of Education, University and Research (PRIN grant 2017E5L5P3) to PP. SP is supported by Fondazione Umberto Veronesi. AR is supported by Italian Cystic Fibrosis Research Foundation grant FFC #20/2015, local funds from University of Ferrara, FIR-2017, the Italian Ministry of Health (GR-2016-02364602), and the Italian Ministry of Education, University and Research (PRIN grant 2017XA5J5N). PP is grateful to Camilla degli Scrovegni for continuous support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abdulrahman, B. A., Khweek, A. A., Akhter, A., Caution, K., Kotrange, S., Abdelaziz, D. H., et al. (2011). Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy 7, 1359–1370. doi: 10.4161/auto.7.11.17660
Antigny, F., Norez, C., Becq, F., Vandebrouck, C. (2011). CFTR and Ca Signaling in Cystic Fibrosis. Front. Pharmacol. 2, 67. doi: 10.3389/fphar.2011.00067
App, E. M., Baran, D., Dab, I., Malfroot, A., Coffiner, M., Vanderbist, F., et al. (2002). Dose-finding and 24-h monitoring for efficacy and safety of aerosolized Nacystelyn in cystic fibrosis. Eur. Respir. J. 19, 294–302. doi: 10.1183/09031936.02.00025802
Assani, K., Tazi, M. F., Amer, A. O., Kopp, B. T. (2014). IFN-gamma stimulates autophagy-mediated clearance of Burkholderia cenocepacia in human cystic fibrosis macrophages. PloS One 9, e96681. doi: 10.1371/journal.pone.0096681
Atlante, A., Favia, M., Bobba, A., Guerra, L., Casavola, V., Reshkin, SJ. (2016). Characterization of mitochondrial function in cells with impaired cystic fibrosis transmembrane conductance regulator (CFTR) function. J. Bioenerg. Biomembr. 48, 197–210. doi: 10.1007/s10863-016-9663-y
Banoth, B., Cassel, S. L. (2018). Mitochondria in innate immune signaling. Transl. Res. 202, 52–68. doi: 10.1016/j.trsl.2018.07.014
Barriere, H., Poujeol, C., Tauc, M., Blasi, J. M., Counillon, L., Poujeol, P. (2001). CFTR modulates programmed cell death by decreasing intracellular pH in Chinese hamster lung fibroblasts. Am. J. Physiol. Cell Physiol. 281, C810–C824. doi: 10.1152/ajpcell.2001.281.3.C810
Battino, M., Rugolo, M., Romeo, G., Lenaz, G. (1986). Kinetic alterations of cytochrome-c oxidase in cystic fibrosis. FEBS Lett. 199, 155–158. doi: 10.1016/0014-5793(86)80470-7
Bonora, M., Patergnani, S., Rimessi, A., De Marchi, E., Suski, J. M., Bononi, A., et al. (2012). ATP synthesis and storage. Purinergic Signal 8, 343–357. doi: 10.1007/s11302-012-9305-8
Brookes, P. S., Yoon, Y., Robotham, J. L., Anders, M. W., Sheu, S. S. (2004). Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 287, C817–C833. doi: 10.1152/ajpcell.00139.2004
Broz, P., Dixit, V. M. (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420. doi: 10.1038/nri.2016.58
Campello, S., Scorrano, L. (2010). Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Rep. 11, 678–684. doi: 10.1038/embor.2010.115
Cantin, A. M. (2004). Potential for antioxidant therapy of cystic fibrosis. Curr. Opin. Pulm. Med. 10, 531–536. doi: 10.1097/01.mcp.0000138997.29276.a1
Cho, D. Y., Zhang, S., Lazrak, A., Grayson, J. W., Pena Garcia, J. A., Skinner, D. F., et al. (2019). Resveratrol and ivacaftor are additive G551D CFTR-channel potentiators: therapeutic implications for cystic fibrosis sinus disease. Int. Forum Allergy Rhinol. 9, 100–105. doi: 10.1002/alr.22202
Coll, R. C., Robertson, A., Butler, M., Cooper, M., O’Neill, L. A. (2011). The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PloS One 6, e29539. doi: 10.1371/journal.pone.0029539
Congdon, P. J., Littlewood, J. M., Aggarwal, R. K., Shapiro, H. (1981). Glucose 6-phosphate dehydrogenase deficiency and cystic fibrosis. Postgrad. Med. J. 57, 453–454. doi: 10.1136/pgmj.57.669.453
Conrad, C., Lymp, J., Thompson, V., Dunn, C., Davies, Z., Chatfield, B., et al. (2015). Long-term treatment with oral N-acetylcysteine: affects lung function but not sputum inflammation in cystic fibrosis subjects. A phase II randomized placebo-controlled trial. J. Cyst. Fibros 14, 219–227. doi: 10.1016/j.jcf.2014.08.008
Csanady, L., Vergani, P., Gadsby, D. C. (2019). Structure, Gating, and Regulation of the Cftr Anion Channel. Physiol. Rev. 99, 707–738. doi: 10.1152/physrev.00007.2018
De Meer, K., Jeneson, J. A., Gulmans, V. A., van der Laag, J., Berger, R. (1995). Efficiency of oxidative work performance of skeletal muscle in patients with cystic fibrosis. Thorax 50, 980–983. doi: 10.1136/thx.50.9.980
Dhooghe, B., Bouckaert, C., Capron, A., Wallemacq, P., Leal, T., Noel, S., et al. (2015). Resveratrol increases F508del-CFTR dependent salivary secretion in cystic fibrosis mice. Biol. Open 4, 929–936. doi: 10.1242/bio.010967
Eisner, V., Picard, M., Hajnoczky, G. (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 20, 755–765. doi: 10.1038/s41556-018-0133-0
Escames, G., Lopez, L. C., Garcia, J. A., Garcia-Corzo, L., Ortiz, F., Acuna-Castroviejo, D. (2012). Mitochondrial DNA and inflammatory diseases. Hum. Genet. 131, 161–173. doi: 10.1007/s00439-011-1057-y
Fan, L. C., Lin, J. L., Yang, J. W., Mao, B., Lu, H. W., Ge, B. X., et al. (2017). Macrolides protect against Pseudomonas aeruginosa infection via inhibition of inflammasomes. Am. J. Physiol. Lung Cell Mol. Physiol. 313, L677–L686. doi: 10.1152/ajplung.00123.2017
Feigal, R. J., Shapiro, B. L. (1979). Mitochondrial calcium uptake and oxygen consumption in cystic fibrosis. Nature 278, 276–277. doi: 10.1038/278276a0
Fiorese, C. J., Schulz, A. M., Lin, Y. F., Rosin, N., Pellegrino, M. W., Haynes, C. M. (2016). The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 26, 2037–2043. doi: 10.1016/j.cub.2016.06.002
Galli, F., Battistoni, A., Gambari, R., Pompella, A., Bragonzi, A., Pilolli, F., et al. (2012). Oxidative stress and antioxidant therapy in cystic fibrosis. Biochim. Biophys. Acta 1822, 690–713. doi: 10.1016/j.bbadis.2011.12.012
Gao, L., Kim, K. J., Yankaskas, J. R., Forman, H. J. (1999). Abnormal glutathione transport in cystic fibrosis airway epithelia. Am. J. Physiol. 277, L113–L118. doi: 10.1152/ajplung.1999.277.1.L113
Gatica, D., Lahiri, V., Klionsky, D. J. (2018). Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242. doi: 10.1038/s41556-018-0037-z
Giorgi, C., Danese, A., Missiroli, S., Patergnani, S., Pinton, P. (2018a). Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 28, 258–273. doi: 10.1016/j.tcb.2018.01.002
Giorgi, C., Marchi, S., Pinton, P. (2018b). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730. doi: 10.1038/s41580-018-0052-8
Green, D. R., Kroemer, G. (2004). The pathophysiology of mitochondrial cell death. Science 305, 626–629. doi: 10.1126/science.1099320
Hamdaoui, N., Baudoin-Legros, M., Kelly, M., Aissat, A., Moriceau, S., Vieu, D. L., et al. (2011). Resveratrol rescues cAMP-dependent anionic transport in the cystic fibrosis pancreatic cell line CFPAC1. Br. J. Pharmacol. 163, 876–886. doi: 10.1111/j.1476-5381.2011.01289.x
Iannitti, R. G., Napolioni, V., Oikonomou, V., De Luca, A., Galosi, C., Pariano, M., et al. (2016). IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat. Commun. 7, 10791. doi: 10.1038/ncomms10791
Iyer, S. S., He, Q., Janczy, J. R., Elliott, E. I., Zhong, Z., Olivier, A. K., et al. (2013). Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39, 311–323. doi: 10.1016/j.immuni.2013.08.001
Jabir, M. S., Hopkins, L., Ritchie, N. D., Ullah, I., Bayes, H. K., Li, D., et al. (2015). Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11, 166–182. doi: 10.4161/15548627.2014.981915
Jai, Y., Shah, K., Bridges, R. J., Bradbury, N. A. (2015). Evidence against resveratrol as a viable therapy for the rescue of defective DeltaF508 CFTR. Biochim. Biophys. Acta 1850, 2377–2384. doi: 10.1016/j.bbagen.2015.08.020
Jendrossek, V., Grassme, H., Mueller, I., Lang, F., Gulbins, E. (2001). Pseudomonas aeruginosa-induced apoptosis involves mitochondria and stress-activated protein kinases. Infect. Immun. 69, 2675–2683. doi: 10.1128/IAI.69.4.2675-2683.2001
Kamdar, O., Le, W., Zhang, J., Ghio, A. J., Rosen, G. D., Upadhyay, D. (2008). Air pollution induces enhanced mitochondrial oxidative stress in cystic fibrosis airway epithelium. FEBS Lett. 582, 3601–3606. doi: 10.1016/j.febslet.2008.09.030
Kelly-Aubert, M., Trudel, S., Fritsch, J., Nguyen-Khoa, T., Baudouin-Legros, M., Moriceau, S., et al. (2011). GSH monoethyl ester rescues mitochondrial defects in cystic fibrosis models. Hum. Mol. Genet. 20, 2745–2759. doi: 10.1093/hmg/ddr173
Kleme, M. L., Sane, A., Garofalo, C., Seidman, E., Brochiero, E., Berthiaume, Y., et al. (2018). CFTR Deletion Confers Mitochondrial Dysfunction and Disrupts Lipid Homeostasis in Intestinal Epithelial Cells. Nutrients 10. doi: 10.3390/nu10070836
Kon, N., Murakoshi, M., Isobe, A., Kagechika, K., Miyoshi, N., Nagayama, T. (2017). DS16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discovery 3, 17045. doi: 10.1038/cddiscovery.2017.45
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kushwah, R., Gagnon, S., Sweezey, N. B. (2013). Intrinsic predisposition of naive cystic fibrosis T cells to differentiate towards a Th17 phenotype. Respir. Res. 14, 138. doi: 10.1186/1465-9921-14-138
Lamothe, J., Valvano, M. A. (2008). Burkholderia cenocepacia-induced delay of acidification and phagolysosomal fusion in cystic fibrosis transmembrane conductance regulator (CFTR)-defective macrophages. Microbiology 154, 3825–3834. doi: 10.1099/mic.0.2008/023200-0
Lavelle, E. C., Murphy, C., O’Neill, L. A., Creagh, E. M. (2010). The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol. 3, 17–28. doi: 10.1038/mi.2009.124
Lazarou, M., Sliter, D. A., Kane, L. A., Sarraf, S. A., Wang, C., Burman, J. L., et al. (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. doi: 10.1038/nature14893
Lee, S., Min, K. T. (2018). The Interface Between ER and Mitochondria: Molecular Compositions and Functions. Mol. Cells 41, 1000–1007. doi: 10.14348/molcells.2018.0438
Legendre, C., Mooij, M. J., Adams, C., O’gara, F. (2011). Impaired expression of hypoxia-inducible factor-1alpha in cystic fibrosis airway epithelial cells - a role for HIF-1 in the pathophysiology of CF? J. Cyst. Fibros 10, 286–290. doi: 10.1016/j.jcf.2011.02.005
Levy, H., Murphy, A., Zou, F., Gerard, C., Klanderman, B., Schuemann, B., et al. (2009). IL1B polymorphisms modulate cystic fibrosis lung disease. Pediatr. Pulmonol. 44, 580–593. doi: 10.1002/ppul.21026
Lopez-Armada, M. J., Carames, B., Martin, M. A., Cillero-Pastor, B., Lires-Dean, M., Fuentes- Boquete, I., et al. (2006). Mitochondrial activity is modulated by TNFalpha and IL-1beta in normal human chondrocyte cells. Osteoarthritis Cartilage 14, 1011–1022. doi: 10.1016/j.joca.2006.03.008
L’hoste, S., Chargui, A., Belfodil, R., Corcelle, E., Duranton, C., Rubera, I., et al. (2010). CFTR mediates apoptotic volume decrease and cell death by controlling glutathione efflux and ROS production in cultured mice proximal tubules. Am. J. Physiol. Renal Physiol. 298, F435–F453. doi: 10.1152/ajprenal.00286.2009
Lu, B., Corey, D. A., Kelley, T. J. (2020). Resveratrol restores intracellular transport in cystic fibrosis epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 318, L1145–L1157. doi: 10.1152/ajplung.00006.2020
Luciani, A., Villella, V. R., Esposito, S., Brunetti-Pierri, N., Medina, D., Settembre, C., et al. (2010). Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 12, 863–875. doi: 10.1038/ncb2090
Malhotra, S., Hayes, D., Jr., Wozniak, D. J. (2019). Cystic Fibrosis and Pseudomonas aeruginosa: the Host-Microbe Interface. Clin. Microbiol. Rev. 32, e00138–18. doi: 10.1128/CMR.00138-18
Manna, S. K., Mukhopadhyay, A., Aggarwal, B. B. (2000). Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-kappa B, activator protein-1, and apoptosis: potential role of reactive oxygen intermediates and lipid peroxidation. J. Immunol. 164, 6509–6519. doi: 10.4049/jimmunol.164.12.6509
Marson, F. A. L., Bertuzzo, C. S., Ribeiro, J. D. (2016). Classification of CFTR mutation classes. Lancet Respir. Med. 4, e37–e38. doi: 10.1016/S2213-2600(16)30188-6
McElvaney, O. J., Zaslona, Z., Becker-Flegler, K., Palsson-Mcdermott, E. M., Boland, F., Gunaratnam, C., et al. (2019). Specific Inhibition of the NLRP3 Inflammasome as an Antiinflammatory Strategy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 200, 1381–1391. doi: 10.1164/rccm.201905-1013OC
McNamara, N., Gallup, M., Sucher, A., Maltseva, I., Mckemy, D., Basbaum, C. (2006). AsialoGM1 and TLR5 cooperate in flagellin-induced nucleotide signaling to activate Erk1/2. Am. J. Respir. Cell Mol. Biol. 34, 653–660. doi: 10.1165/rcmb.2005-0441OC
Mejia, E. M., Hatch, G. M. (2016). Mitochondrial phospholipids: role in mitochondrial function. J. Bioenerg. Biomembr. 48, 99–112. doi: 10.1007/s10863-015-9601-4
Melber, A., Haynes, C. M. (2018). UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res. 28, 281–295. doi: 10.1038/cr.2018.16
Michalek, R. D., Gerriets, V. A., Jacobs, S. R., Macintyre, A. N., Maciver, N. J., Mason, E. F., et al. (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186, 3299–3303. doi: 10.4049/jimmunol.1003613
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. doi: 10.1126/science.1223560
Nichols, D. P., Chmiel, J. F. (2015). Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 50 Suppl 40, S39–S56. doi: 10.1002/ppul.23242
Nunnari, J., Suomalainen, A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145–1159. doi: 10.1016/j.cell.2012.02.035
Painter, R. G., Valentine, V. G., Lanson, N. A., Jr., Leidal, K., Zhang, Q., Lombard, G., et al. (2006). CFTR Expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 45, 10260–10269. doi: 10.1021/bi060490t
Park, S., Juliana, C., Hong, S., Datta, P., Hwang, I., Fernandes-Alnemri, T., et al. (2013). The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 191, 4358–4366. doi: 10.4049/jimmunol.1301170
Pedriali, G., Rimessi, A., Sbano, L., Giorgi, C., Wieckowski, M. R., Previati, M., et al. (2017). Regulation of Endoplasmic Reticulum-Mitochondria Ca(2+) Transfer and Its Importance for Anti-Cancer Therapies. Front. Oncol. 7, 180. doi: 10.3389/fonc.2017.00180
Pellegrino, M. W., Nargund, A. M., Kirienko, N. V., Gillis, R., Fiorese, C. J., Haynes, C. M. (2014). Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516, 414–417. doi: 10.1038/nature13818
Picci, L., Brentagni, L., Mastella, G., Scarso, E., Pizzochero, P., Mattiazzo, P., et al. (1991). 2D-electrophoresis of mitochondrial proteins from cystic fibrosis patients. Adv. Exp. Med. Biol. 290, 379–381. doi: 10.1007/978-1-4684-5934-0_44
Pinton, P., Leo, S., Wieckowski, M. R., Di Benedetto, G., Rizzuto, R. (2004). Long-term modulation of mitochondrial Ca2+ signals by protein kinase C isozymes. J. Cell Biol. 165, 223–232. doi: 10.1083/jcb.200311061
Rafeeq, M. M., Murad, H. A. S. (2017). Cystic fibrosis: current therapeutic targets and future approaches. J. Transl. Med. 15, 84. doi: 10.1186/s12967-017-1193-9
Ratner, D., Mueller, C. (2012). Immune responses in cystic fibrosis: are they intrinsically defective? Am. J. Respir. Cell Mol. Biol. 46, 715–722. doi: 10.1165/rcmb.2011-0399RT
Reilly, R., Mroz, M. S., Dempsey, E., Wynne, K., Keely, S. J., Mckone, E. F., et al. (2017). Targeting the PI3K/Akt/mTOR signalling pathway in Cystic Fibrosis. Sci. Rep. 7, 7642. doi: 10.1038/s41598-017-06588-z
Rimessi, A., Bezzerri, V., Patergnani, S., Marchi, S., Cabrini, G., Pinton, P. (2015). Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 6 (19), eaax9093. doi: 10.1038/ncomms7201
Rimessi, A., Previati, M., Nigro, F., Wieckowski, M. R., Pinton, P. (2016). Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 81, 281–293. doi: 10.1016/j.biocel.2016.06.015
Rimessi, A., Pedriali, G., Vezzani, B., Tarocco, A., Marchi, S., Wieckowski, M. R., et al. (2020a). Interorganellar calcium signaling in the regulation of cell metabolism: A cancer perspective. Semin. Cell Dev. Biol. 98, 167–180. doi: 10.1016/j.semcdb.2019.05.015
Rimessi, A., Pozzato, C., Carparelli, L., Rossi, A., Ranucci, S., De Fino, I., et al. (2020b). Pharmacological modulation of Mitochondrial Calcium Uniporter controls lung inflammation in cystic fibrosis. Sci. Adv. 6 (19), eaax9093. doi: 10.1126/sciadv.aax9093
Rottner, M., Kunzelmann, C., Mergey, M., Freyssinet, J. M., Martinez, M. C. (2007). Exaggerated apoptosis and NF-kappaB activation in pancreatic and tracheal cystic fibrosis cells. FASEB J. 21, 2939–2948. doi: 10.1096/fj.06-7614com
Rottner, M., Tual-Chalot, S., Mostefai, H. A., Andriantsitohaina, R., Freyssinet, J. M., Martinez, M. C. (2011). Increased oxidative stress induces apoptosis in human cystic fibrosis cells. PloS One 6, e24880. doi: 10.1371/journal.pone.0024880
Rowland, A. A., Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Saadane, A., Eastman, J., Berger, M., Bonfield, T. L. (2011). Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. J. Inflammation (Lond). 8, 26. doi: 10.1186/1476-9255-8-26
Samson, C., Tamalet, A., Thien, H. V., Taytard, J., Perisson, C., Nathan, N., et al. (2016). Long-term effects of azithromycin in patients with cystic fibrosis. Respir. Med. 117, 1–6. doi: 10.1016/j.rmed.2016.05.025
Santo-Domingo, J., Vay, L., Hernandez-Sanmiguel, E., Lobaton, C. D., Moreno, A., Montero, M., et al. (2007). The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br. J. Pharmacol. 151, 647–654. doi: 10.1038/sj.bjp.0707260
Scambler, T., Jarosz-Griffiths, H. H., Lara-Reyna, S., Pathak, S., Wong, C., Holbrook, J., et al. (2019). ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife 8. doi: 10.7554/eLife.49248
Shapiro, B. L., Feigal, R. J., Lam, L. F. (1979). Mitrochondrial NADH dehydrogenase in cystic fibrosis. Proc. Natl. Acad. Sci. U. S. A. 76, 2979–2983. doi: 10.1073/pnas.76.6.2979
Sheng, Z., Ma, L., Sun, J. E., Zhu, L. J., Green, M. R. (2011). BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood 118, 2840–2848. doi: 10.1182/blood-2010-12-322537
Smith, R. A., Porteous, C. M., Gane, A. M., Murphy, M. P. (2003). Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. U. S. A. 100, 5407–5412. doi: 10.1073/pnas.0931245100
Sokol, R. J., Reardon, M. C., Accurso, F. J., Stall, C., Narkewicz, M., Abman, S. H., et al. (1989). Fat-soluble-vitamin status during the first year of life in infants with cystic fibrosis identified by screening of newborns. Am. J. Clin. Nutr. 50, 1064–1071. doi: 10.1093/ajcn/50.5.1064
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G., Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532. doi: 10.1091/mbc.e09-03-0252
Subramanian, N., Natarajan, K., Clatworthy, M. R., Wang, Z., Germain, R. N. (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. doi: 10.1016/j.cell.2013.02.054
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245. doi: 10.1084/jem.20071239
Tabary, O., Boncoeur, E., De Martin, R., Pepperkok, R., Clement, A., Schultz, C., et al. (2006). Calcium-dependent regulation of NF-(kappa)B activation in cystic fibrosis airway epithelial cells. Cell Signal 18, 652–660. doi: 10.1016/j.cellsig.2005.06.004
Tarique, A. A., Logan, J., Thomas, E., Holt, P. G., Sly, P. D., Fantino, E. (2015). Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir. Cell Mol. Biol. 53, 676–688. doi: 10.1165/rcmb.2015-0012OC
Tarique, A. A., Sly, P. D., Holt, P. G., Bosco, A., Ware, R. S., Logan, J., et al. (2017). CFTR-dependent defect in alternatively-activated macrophages in cystic fibrosis. J. Cyst. Fibros 16, 475–482. doi: 10.1016/j.jcf.2017.03.011
Tiku, V., Tan, M. W., Dikic, I. (2020). Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 30, 263–275. doi: 10.1016/j.tcb.2020.01.006
Tolle, L., Yu, F. S., Kovach, M. A., Ballinger, M. N., Newstead, M. W., Zeng, X., et al. (2015). Redundant and cooperative interactions between TLR5 and NLRC4 in protective lung mucosal immunity against Pseudomonas aeruginosa. J. Innate Immun. 7, 177–186. doi: 10.1159/000367790
Turrens, J. F., Freeman, B. A., Levitt, J. G., Crapo, J. D. (1982). The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch. Biochem. Biophys. 217, 401–410. doi: 10.1016/0003-9861(82)90518-5
Turrens, J. F. (2003). Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344. doi: 10.1113/jphysiol.2003.049478
Valdivieso, A. G., Marcucci, F., Taminelli, G., Guerrico, A. G., Alvarez, S., Teiber, M. L., et al. (2007). The expression of the mitochondrial gene MT-ND4 is downregulated in cystic fibrosis. Biochem. Biophys. Res. Commun. 356, 805–809. doi: 10.1016/j.bbrc.2007.03.057
Velsor, L. W., Kariya, C., Kachadourian, R., Day, B. J. (2006). Mitochondrial oxidative stress in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Respir. Cell Mol. Biol. 35, 579–586. doi: 10.1165/rcmb.2005-0473OC
Villella, V. R., Esposito, S., Bruscia, E. M., Vicinanza, M., Cenci, S., Guido, S., et al. (2013). Disease-relevant proteostasis regulation of cystic fibrosis transmembrane conductance regulator. Cell Death Differ. 20, 1101–1115. doi: 10.1038/cdd.2013.46
Walle, T. (2011). Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 1215, 9–15. doi: 10.1111/j.1749-6632.2010.05842.x
West, A. P., Brodsky, I. E., Rahner, C., Woo, D. K., Erdjument-Bromage, H., Tempst, P., et al. (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480. doi: 10.1038/nature09973
Wiegman, C. H., Michaeloudes, C., Haji, G., Narang, P., Clarke, C. J., Russell, K. E., et al. (2015). Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 136, 769–780. doi: 10.1016/j.jaci.2015.01.046
Wood, S. J., Goldufsky, J. W., Bello, D., Masood, S., Shafikhani, S. H. (2015). Pseudomonas aeruginosa ExoT Induces Mitochondrial Apoptosis in Target Host Cells in a Manner That Depends on Its GTPase-activating Protein (GAP) Domain Activity. J. Biol. Chem. 290, 29063–29073. doi: 10.1074/jbc.M115.689950
Woods, J. J., Nemani, N., Shanmughapriya, S., Kumar, A., Zhang, M., Nathan, S. R., et al. (2019). A Selective and Cell-Permeable Mitochondrial Calcium Uniporter (MCU) Inhibitor Preserves Mitochondrial Bioenergetics after Hypoxia/Reoxygenation Injury. ACS Cent. Sci. 5, 153–166. doi: 10.1021/acscentsci.8b00773
Xu, Y., Nie, L., Yin, Y. G., Tang, J. L., Zhou, J. Y., Li, D. D., et al. (2012). Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating SIRT1 in rat mesangial cells. Toxicol. Appl. Pharmacol. 259, 395–401. doi: 10.1016/j.taap.2011.09.028
Zang, Q. S., Sadek, H., Maass, D. L., Martinez, B., Ma, L., Kilgore, J. A., et al. (2012). Specific inhibition of mitochondrial oxidative stress suppresses inflammation and improves cardiac function in a rat pneumonia-related sepsis model. Am. J. Physiol. Heart Circ. Physiol. 302, H1847–H1859. doi: 10.1152/ajpheart.00203.2011
Keywords: mitochondria, inflammation, cystic fibrosis, mitochondrial stress response, lung
Citation: Patergnani S, Vitto VAM, Pinton P and Rimessi A (2020) Mitochondrial Stress Responses and “Mito-Inflammation” in Cystic Fibrosis. Front. Pharmacol. 11:581114. doi: 10.3389/fphar.2020.581114
Received: 07 July 2020; Accepted: 11 September 2020;
Published: 30 September 2020.
Edited by:
Noel Gerard McElvaney, Royal College of Surgeons in Ireland, IrelandReviewed by:
Simone Guarnieri, University of Studies G. d’Annunzio Chieti and Pescara, ItalyCinzia Parolini, University of Milan, Italy
Copyright © 2020 Patergnani, Vitto, Pinton and Rimessi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Rimessi, YWxlc3NhbmRyby5yaW1lc3NpQHVuaWZlLml0