 Annina Lyly
Annina Lyly Tanya M. Laidlaw3
Tanya M. Laidlaw3 Marie Lundberg
Marie Lundberg- 1Department of Otorhinolaryngology – Head and Neck Surgery, Helsinki University Hospital, University of Helsinki, Helsinki, Finland
- 2Inflammation Center, Skin and Allergy Hospital, Helsinki University Hospital, University of Helsinki, Helsinki, Finland
- 3Department of Medicine, Division of Allergy and Clinical Immunology, Brigham and Women's Hospital and Harvard Medical School, Boston, MA, United States
The pathomechanisms behind NSAID-exacerbated respiratory disease are complex and still largely unknown. They are presumed to involve genetic predisposition and environmental triggers that lead to dysregulation of fatty acid and lipid metabolism, altered cellular interactions involving transmetabolism, and continuous and chronic inflammation in the respiratory track. Here, we go through the recent advances on the topic and sum up the current understanding of the background of this illness that broadly effects the patients' lives.
Introduction
AERD or aspirin/non-steroidal anti-inflammatory drug (NSAID)-exacerbated respiratory disease is an adult-onset triad characterized by asthma, eosinophilic chronic rhinosinusitis with nasal polyposis (CRSwNP), and respiratory reactions upon ingestion of COX-1 inhibitors such as NSAIDs or aspirin (ASA). Both the upper and lower respiratory symptoms are typically difficult-to-treat, and many patients suffer from frequent asthma exacerbations and require multiple endoscopic sinus surgeries despite good medical treatment including local and oral corticosteroids.
The acute inflammatory symptoms that occur within 30–180 min after ingestion of COX-1 inhibitors is a non-immunoglobulin (Ig)E-mediated hypersensitivity reaction that causes a release of multiple mast cell mediators, such as tryptase, cysteinyl leukotrienes (CysLTs), and prostaglandin D2 (PDG2) (1–3). The typical symptoms of reaction can include nasal congestion, rhinorrhea, sneezing, coughing, wheezing, and drop in lung function, though pruritic rash and gastrointestinal discomfort have also been noted.
Arachidonic Acid Metabolism
Arachidonic acid (AA) is a C20 polyunsaturated fatty acid derived from phospholipid hydrolysis at the inner surface of the cell membrane by phospholipase PLA2. AA regulates phospholipase and protein kinase signaling as a second messenger, and acts as a key inflammatory intermediate, being a precursor for eicosanoids. Eicosanoids, which encompass prostaglandins (PGs), prostacyclins, thromboxanes (TXs) and leukotrienes (LTs), are hormone-like compounds signaling via G-coupled receptors and are involved in variety of biological processes, such as inflammation, platelet aggregation, electrolyte balance, and smooth-muscle contraction (4). Leukotrienes are synthesized via lipoxygenase (LOX)-dependent route, while PGs, prostacyclins and TXs are derived from the cyclooxygenase (COX)-dependent pathway (Figure 1). AA can also be metabolized by cytochrome P450 (CYP) enzyme, resulting in hydroxyeicosatetraneoic acids (HETEs) and epoxyeicosatrienoic acids (EETs), important in cardiovascular biology (4). AA metabolism is a complex entity, resulting in variety of lipid mediators that can be anti-inflammatory, proinflammatory, or both, depending on the target receptors.
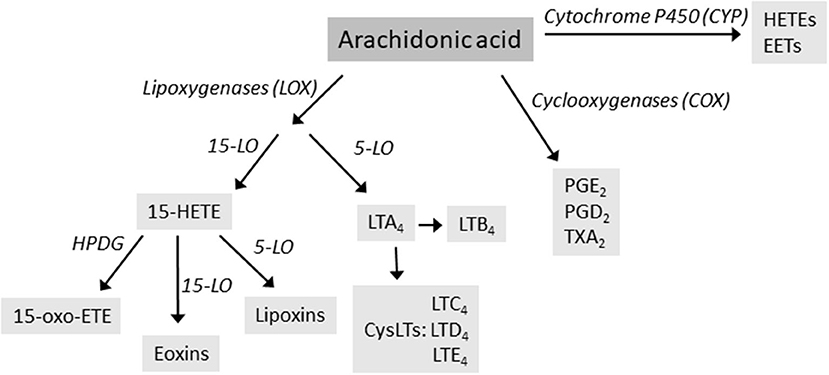
Figure 1. The main routes of arachidonic acid metabolism, focusing on lipoxygenase (LOX) pathway. Enzymes are in italics. HETE, hydroxyeicosatetraneoic acid; LT, leukotrienes; PG, prostaglandins; TX, thromboxanes.
Pathophysiologic Findings of Arachidonic Acid Metabolism in AERD Patients
In AERD, AA metabolism is chronically imbalanced and both the COX- and the LOX-pathways are dysregulated. There is overproduction of CysLTs, increase in the expression of CysLT receptors in respiratory mucosa and in the LTC4 synthase enzyme, decrease in the lipoxin levels, decrease in prostaglandin PGE2 and increase in PGD2 [reviewed in (5)]. 5-lipoxygenase (5-LO) has gained a lot of attention because of the 30-year-old finding that AERD patients have increased levels of the 5-LO pathway end-product CysLTs, as measured by high levels of the stable end-metabolite LTE4 in urine and nasal fluids (6). Inhibiting the COX1 enzyme with NSAIDs further increases these levels. LT antagonists, such as montelukast, inhibit the actions of CysLTs by blocking their receptor CysLT1R, and have been shown to improve AERD-patients' asthma (7) and in some patients also inhibit the lower airway symptoms induced by NSAID ingestion (8).
ATAD—Changes in Clinical Features
Another treatment option for non-responsive CRSwNP disease is aspirin treatment after desensitization (ATAD) (9). Successful desensitization has been shown to suppress the levels of PGD2, while the levels of CysLTs remain unaltered or even increase, and the levels of blood eosinophils and basophils increase (2). PGD2 induces chemotaxis in Th2 cells, eosinophils, and basophils (10), and the increased levels of circulating eosinophils might be due to their decreased tissue recruitment (2). A prospective, placebo-controlled, double-blind study comparing the effects of ATAD between ASA-intolerant and ASA-tolerant patients with asthma and CRSwNP (34 patients altogether) has shown improvement in the nasal and bronchial symptoms in the ASA-intolerant group (11). Clinical improvements were shown in SNOT-20 and Asthma Control Questionnaire scores, peak nasal inspiratory flows and as reduced doses of inhaled corticosteroids. In this study, the levels of LTE4 or PGD2 metabolites showed no changes after ATAD (11). It has been estimated, that over 80% of patients would benefit from ATAD (12) as it can reduce the need for surgeries, oral corticosteroids, and emergency room visits (13).
The downside of ATAD are the side effects: gastric pain or bleeding, bruising, tinnitus, urticaria or worsening of the respiratory symptoms, leading to discontinuation in 15% of cases (13, 14). Side effects may even prevent the desensitization, and acutely intolerable side effects during the aspirin desensitization procedure have been associated with elevated levels of PGD2 (2). A recent real-life study in the Finnish population reported ATAD-discontinuation rates of nearly 30%, in spite of lower ASA dose than the average described in literature (15). Another study from Finland reported similar results with a discontinuation rate of 22% (16), suggesting that there might be ethnic or genetic variability in ASA tolerance.
15-LO
Another enzyme that metabolizes AA is 15-lipoxygenase (15-LO), encoded by ALOX-15 and expressed in airway epithelial cells, eosinophils and mast cells (17). It converts AA to 15-HETE, lipoxins and eoxins. 15-HETE can act as an independent anti-inflammatory mediator, or it can be converted to lipoxins by 5-LO, or to 15-oxo-eicosatetraeonic (15-oxo-ETE) by hydroxyprostaglandin dehydrogenase (HPGD) (18). However, epithelial cells lack HPGD and instead the enzyme is found in tissue mast cells in close proximity to the epithelial cells. The cells work transmetabolically, passing on the 15-HETE from the epithelial cell to the mast cell, which in turn produces 15-oxo-ETE (19). The role of 15-oxo-ETE in AERD progression is not known but it may contribute to the dysregulation of AA metabolism.
Increased levels of 15-HETE have been associated with pulmonary eosinophilia in asthmatics (20). Eoxins are proinflammatory metabolites capable of causing severe asthma and allergic reactions. They are produced by eosinophils and mast cells within the nasal polyp tissue (21). 15-LO activity was found to be increased in eosinophils isolated from asthmatics with either severe disease or AERD, and the levels of eoxins were specifically increased in asthmatics with AERD (22). ALOX-15 is also upregulated in the epithelium in other type 2 inflammatory mucosal diseases such as eosinophilic esophagitis (23). Microarray experiments in human peripheral monocytes showed that the expression of 15-LO is strongly induced by interleukins (IL)-4 and IL-13, and real-time PCR indicated that IL-4 induced more than 100-fold upregulation of 15-LO expression (24).
Recently, a genome-wide association study (GWAS) of patients with CRSwNP across several cohorts showed that a missense variant of ALOX-15, causing a p.Thr560Met alteration and leading to near total loss of 15-LO enzymatic activity was associated with a reduction in the risk of CRSwNP (25). This was the first GWAS, and so far, the only one, to report a significant association with nasal polyps. Results from mouse models support this finding, as a mouse model deficient in 12/15-LO, an ortholog for human 15-LO, has been shown to have reduced airway inflammation and remodeling in allergen provocation tests (26).
Furthermore, a single-cell RNA sequencing analysis from nasal polyp tissue revealed dysregulated arachidonic metabolism in the 15-LO pathway in patients with AERD (19). When the polyp tissue isolated from CRSwNP patients was compared to that of AERD patients, ALOX-15 expression was significantly elevated in patients with AERD, particularly within apical epithelial cells (19). Patients with CRSwNP and asthma had higher enhancement of whole-tissue ALOX-15 expression compared to CRSwNP non-asthmatics. The same correlation was not observed in patients with non-polyp CRS. Patients with higher ALOX-15 expression suffered from a worse CRSwNP disease with higher number of sinus operations and worse inflammation in the sinus CT scans (19). The expression level of ALOX-15 mRNA was significantly higher in eosinophilic polyps and could distinguish between eosinophilic and non-eosinophilic nasal polyps (27). The expression of 15-LO was seen in both the epithelial cells and eosinophils in nasal polyp tissue detected by immunohistochemical staining (27). Dupilumab, a biological drug for treating asthma, atopic dermatitis and CRSwNP, suppresses IL-4 and IL-13 signaling, on which the expression of ALOX-15 is strictly dependent (28). Dupilumab has been shown to be particularly therapeutically effective in AERD patients (29).
Local Immunoglobulin Levels in AERD Nasal Polyps
Activation of B cells and local antibody production may play a key role in nasal polyp severity and AERD pathogenesis (30). The airway mucosa can function as a tertiary lymphoid organ where antibody production and class switching are facilitated as high levels of active B cells, plasma cells and plasmablasts have been identified locally (31, 32). IgE is a link between antibodies and inflammatory disease as it activates and can prime mast cells, basophils and other Fc-receptor bearing effector cells in nasal tissue (30). Other local antibodies may also promote inflammation. IgA enhances eosinophil survival (33) and IgG can activate local complement cascades leading to destruction of the epithelial barrier (34). One theory is that the presence of autoantibodies contribute to the destruction of this barrier, but no single antigen has consistently been linked to AERD.
Recently Buchheit et al. showed that the local antibody profile of patients with AERD differs from that of other CRSwNP patients, non-polyp CRS, and controls (35). In AERD, higher amounts of all subclasses of immunoglobulins were found, but especially of IgG4 and IgE. Local IgE levels did not correlate with serum IgE levels, indicating the presence of local antibody production within the tissues (35). Also, IGHG4 encoding the constant region of IgG4 is overexpressed locally in AERD patients, strengthening the theory of local antibody production (35). Further, local IgE levels are associated with a worse disease and fast regrowth of polyposis whereas IgG4 associates with lifetime disease duration of AERD (35). It is speculated that IgG4 might prevent polyp regrowth, possibly causing a fibrotic disease in the sinuses (36, 37) but IgG4 might also be a step in the class switching into local IgE as IL10 encoding IL10 cytokine that drives immunoglobulin production toward IgG4 has been shown in RNA-sequencing analysis of nasal polyp cells of AERD patients (35).
IgE antibody production has also been proposed to be driven by superantigens derived from bacteria such as Staphylococcus aureus, as enterotoxin-specific antibodies that may play a role in the inflammation cascade have been identified (38). A role for IL-5 has also been suspected in this eosinophilic disease, as IL-5 is an eosinophil activator that stimulates IgA production, produced by Th2 cells and mast cells. In AERD patients, antibody-expressing cells expressed IL5AR encoding for the IL-5 alfa receptor (35). This finding supports a possible additional pathway toward the presence of increased antibody levels in nasal polyps.
The positive effect of the IgE-binding monoclonal antibody omalizumab on CRSwNP showed in randomized, placebo-controlled studies POLYP1 and POLYP2 further strengthens the evidence of IgE playing part in the pathomechanism of AERD (39). In these identical studies that included 265 patients, 27% had AERD. After 24 weeks of treatment with omalizumab, polyp score decreased by 2 points or more in 31% of the patients in the active treatment group. Significant improvement compared to placebo was shown in all primary and secondary end points, i.e., nasal congestion score, SNOT-22, olfactory test UPSIT, and total nasal symptom score (39).
Cells Involved in AERD and Their Interactions
AERD is characterized by an imbalance in eicosanoid levels, especially CysLTs, PGD2 and PGE2. Interactions between the cells that produce eicosanoids and the cells that respond to them play a key role in the AERD pathomechanism. Mast cells, eosinophils, epithelial cells, platelets, and innate lymphoid cell type 2 (ILC2) cells are thought to be the major players involved in these actions (Figure 2).
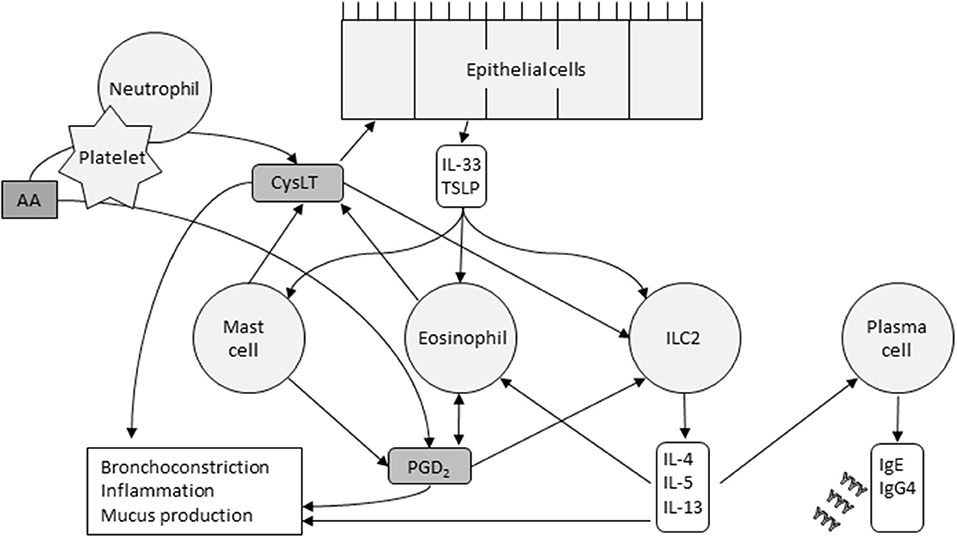
Figure 2. Cells and their major interactions in AERD. CysLTs released by mast cells, eosinophils, and platelets cause epithelial release of IL-33 and TSLP. These innate cytokines in turn activate mast cells, eosinophils, and ILC2s. The cytokines released by ILC2s, the pathways of which are now targeted by several monoclonal antibodies, stimulate plasma cells to produce IgE and IgG4, and promote, recruit and activate eosinophils. Eosinophils and ICL2s are also activated by PGD2. Platelets metabolize arachidonic acid (AA) into PGD2, and, in collaboration with neutrophils, CysLTs. The inflammatory response is driven by multiple factors.
Eosinophils, abundant in nasal polyps, bronchial mucosa, and peripheral blood, produce PGD2 and are attracted to PGD2 by chemotaxis (40). Choi et al. found that eosinophils interact with epithelial cells through surfactant protein D, and may mediate smooth muscle remodeling, a clinical feature of AERD (41). Eosinophils are activated by many effectors including PGD2, IL-5, IL-3, IL-33 and thymic stromal lymphopoietin (TSLP). Upon activation, they release CysLTs and other mediators that promote type 2 inflammatory reactions and tissue damage (42).
Mast cells and their activation through CysLT is central in AERD-related inflammation and aspirin-induced reactions. Mast cells can be activated through IgE cross-linking or via the innate alarmin IL-33. Recently, Pan et al. demonstrated that in mouse models, COX-1 activity is required for IL-33-dependent mast cell release of AA (43), a mechanism that may explain aspirin desensitization. IL-33 and the innate alarmin TSLP induce mast cells' production of PGD2 (3) and potentiate each other's actions. PGD2 promotes bronchospasm and inflammatory cell recruitment, which is typical in AERD but mast cells also release inflammatory mediators such as histamine and tryptase, and CysLTs.
CysLTs released by mast cells and eosinophils regulate the release of IL-33 and TSLP from the basal epithelial cell layer of the airways. In addition to activating mast cells, TSLP and IL-33 also activate eosinophils and ILC2 cells. These cells are a rare subset of lymphocytes and upon stimulation, release cytokines such as IL-4, IL-5, IL-13, and IL-9. The three first mentioned may stimulate plasma cells to produce IgE and IgG4. IL-5 also promotes recruitment, survival and activation of eosinophils and IL-9 may increase the amount of mast cells recruited to the tissue (40). Several prostaglandins also regulate ILC2 function. PGD2 activates ILC2s and induces chemotaxis and cytokine production, whereas PGE2 and PGI2 have inhibitory actions (40). During aspirin desensitization, ILC2s in nasal fluid increased in correlation with symptom scores and with increases in PGD2 metabolites in urine, indicating a potential connection between PGD2 release, ILC2 recruitment, and symptom severity during aspirin-induced reactions in AERD (44).
Platelets and neutrophils also work transmetabolically in AERD (45). Activated platelets in AERD express high surface levels of P-selectin (46) which acts as an adhesion molecule (47). Platelets adhere to neutrophils, cells that generate LTA4. Neutrophils, however, lack LTC4 synthase, which is needed to metabolize LTA4 further into LTC4 (45). The adherent platelets express LTC4 synthase, and the neutrophil-platelet aggregates can then function together to allow for CysLT overproduction.
Lipid Dysregulation in AERD
Because the fatty acid metabolism is dysregulated in AERD, dietary modifications that could balance the distortion and decrease the symptoms have been investigated. The dietary intake of AA and its precursor, linoleic acid, correlates to the amount of AA in inflammatory cells, while supplementation with omega-3-rich fatty acids has been shown to decrease the bodies' production of inflammatory leukotrienes (48). A 2-week diet high in omega-3 and low in omega-6 fatty acids significantly decreased the urinary end-metabolites of LTE4 and PGD2, as well as the clinical end-points of upper respiratory symptom control (SNOT-22 score) and asthma control (ACQ score) (49). Low-salicylate diet has been evaluated in a trial of 30 patients, showing benefits for both subjective and objective clinical end-points (50). However, the effects of non-acetylated salicylates on AERD end-metabolites have not been investigated and the mechanism by which dietary salicylates would affect the respiratory symptoms is not clear.
A totally new finding of macrophage memory and their epigenetic reprogramming in AERD was recently reported by Haimerl et al. (51), assessed by RNA sequencing, metabolomics flux assays and LC-MS/MS. Together with reported dysregulation in sphingolipid metabolism (52), they report increased release of acylcarnitine metabolites, indicating a broader dysregulation in fatty acid metabolism in AERD. Haimerl et al. also reported that although the expression profile of macrophages showed upregulation of proinflammatory genes, it also showed reduction on host-protective molecules. They speculated that a dysfunctional macrophage activation state may contribute to defect in pathogen clearance and higher risk of exacerbations in AERD patients (51).
Discussion
AERD is characterized by chronic inflammation with an imbalance in eicosanoids, high levels of inflammatory mediators such as CysLTs and PGD2, and mast cell, platelet, and ILC2 activation. Upon activation, ILC2s release interleukins that can be blocked with new monoclonal antibodies that are used in the treatment of severe asthma and CRSwNP (39, 53, 54). The inflammation type in asthma and CRSwNP is mostly type 2, but significant heterogeneity in the inflammatory background in AERD patients has been reported (55). The more we learn about the pathomechanisms of these inflammatory diseases, and especially about the subset AERD, the more we can personalize treatment.
In the future, systems biology methods could give us more insight on the cellular level changes in this disease. Already now, the pathways behind CRS and asthma have been investigated with different omics approaches (genomics, transcriptomics, proteomics and metabolomics) [reviewed in (56, 57)]. The findings have made biomarker analysis and point-of-care technology development realistic, thus allowing more precise and quicker diagnoses and personalized treatment to be available in the future clinical practice.
Author Contributions
AL and ML contributed to conception and design of the study and wrote the first draft of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bochenek G, Nagraba K, Nizankowska E, Szczeklik A. A controlled study of 9α,11β-PGF2 (a prostaglandin D2 metabolite) in plasma and urine of patients with bronchial asthma and healthy controls after aspirin challenge. J Allergy Clin Immunol. (2003) 111:743–9. doi: 10.1067/mai.2003.1387
2. Cahill KN, Bensko JC, Boyce JA, Laidlaw TM. Prostaglandin D2: a dominant mediator of aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2015) 135:245–52. doi: 10.1016/j.jaci.2014.07.031
3. Buchheit KM, Cahill KN, Katz HR, Murphy KC, Feng C, Lee-Sarwar K, et al. Thymic stromal lymphopoietin controls prostaglandin D2 generation in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2016) 137:1566–76.e5. doi: 10.1016/j.jaci.2015.10.020
4. Wang B, Wu L, Chen J, Dong L, Chen C, Wen Z, et al. Metabolism pathways of arachidonic acids: mechanisms and potential therapeutic targets. Signal Transduct Target Ther. (2021) 6:1–30. doi: 10.1038/s41392-020-00443-w
5. Parker AR, Ayars AG, Altman MC, Henderson WR. Lipid mediators in aspirin-exacerbated respiratory disease. Immuno Allergy Clin North Am. (2016) 36:749–63. doi: 10.1016/j.iac.2016.06.009
6. Laidlaw TM. Pathogenesis of NSAID-induced reactions in aspirin-exacerbated respiratory disease. World J Otorhinolaryngol Head Neck Surg. (2018) 4:162–8. doi: 10.1016/j.wjorl.2018.08.001
7. Dahlén SE, Malmström K, Nizankowska EWA, Dahlén B, Kuna P, Kowalski M, et al. Improvement of aspirin-intolerant asthma by montelukast, a leukotriene antagonist: a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med. (2002) 165:9–14. doi: 10.1164/ajrccm.165.1.2010080
8. Stevenson DD, Simon RA, Mathison DA, Christiansen SC. Montelukast is only partially effective in inhibiting aspirin responses in aspirin-sensitive asthmatics. Ann Allergy Asthma Immunol. (2000) 85:477–82. doi: 10.1016/S1081-1206(10)62575-6
9. Stevenson DD, Szczeklik A. Clinical and pathologic perspectives on aspirin sensitivity and asthma. J. Allergy Clin Immunol. (2006) 118:773–86. doi: 10.1016/j.jaci.2006.07.024
10. Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. (2001) 193:255–61. doi: 10.1084/jem.193.2.255
11. Swierczyńska-Krepa M, Sanak M, Bochenek G, Strek P, Cmiel A, Gielicz A, et al. Aspirin desensitization in patients with aspirin-induced and aspirin-tolerant asthma: A double-blind study. J Allergy Clin Immunol. (2014) 134:883–90. doi: 10.1016/j.jaci.2014.02.041
12. White AA, Woessner K, Simon R. Aspirin-exacerbated respiratory disease: update on medical management. World J Otorhinolaryngol Head Neck Surg. (2020) 6:241–7. doi: 10.1016/j.wjorl.2020.07.009
13. Berges-Gimeno MP, Simon RA, Stevenson DD. Long-term treatment with aspirin desensitization in asthmatic patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2003) 111:180–6. doi: 10.1067/mai.2003.7
14. Walters KM, Waldram JD, Woessner KM, White AA. Long-term clinical outcomes of aspirin desensitization with continuous daily aspirin therapy in aspirin-exacerbated respiratory disease. Am J Rhinol Allergy. (2018) 32:280–6. doi: 10.1177/1945892418770260
15. Laulajainen-Hongisto A, Turpeinen H, Vento SI, Numminen J, Sahlman J, Kauppi P, et al. High discontinuation rates of peroral ASA treatment for CRSwNP: a real-world multicenter study of 171 N-ERD patients. J Allergy Clin Immunol Pract. (2020) 8:3565–74. doi: 10.1016/j.jaip.2020.06.063
16. Turpeinen H, Laulajainen-Hongisto A, Lyly A, Numminen J, Penttilä E, Sahlman J, et al. Lung function and side effects of Aspirin desensitization: a real world study. Eur Clin Respir J. (2021) 8:1869408. doi: 10.1080/20018525.2020.1869408
17. Claesson HE. On the biosynthesis and biological role of eoxins and 15-lipoxygenase-1 in airway inflammation and hodgkin lymphoma. Prostagl Other Lipid Mediat. (2009) 89:120–5. doi: 10.1016/j.prostaglandins.2008.12.003
18. Singh NK, Rao GN. Emerging role of 12/15-Lipoxygenase (ALOX15) in human pathologies. Prog Lipid Res. (2019) 73:28–45. doi: 10.1016/j.plipres.2018.11.001
19. Stevens WW, Staudacher AG, Hulse KE, Carter RG, Winter DR, Abdala-Valencia H, et al. Activation of the 15-lipoxygenase pathway in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2021) 147:600–12. doi: 10.1016/j.jaci.2020.04.031
20. Chu HW, Balzar S, Westcott JY, Trudeau JB, Sun Y, Conrad DJ, et al. Expression and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition. Clin Exp Allergy. (2002) 32:1558–65. doi: 10.1046/j.1365-2222.2002.01477.x
21. Feltenmark S, Gautam N, Brunnström Å, Griffiths W, Backman L, Edenius C, et al. Eoxins are proinflammatory arachidonic acid metabolites produced via the 15-lipoxygenase-1 pathway in human eosinophils and mast cells. Proc Natl Acad Sci USA. (2008) 105:680–5. doi: 10.1073/pnas.0710127105
22. James A, Daham K, Backman L, Brunnström Å, Tingvall T, Kumlin M, et al. The influence of aspirin on release of eoxin C4, leukotriene C4 and 15-HETE, in eosinophilic granulocytes isolated from patients with asthma. Int Arch Allergy Immunol. (2013) 162:135–42. doi: 10.1159/000351422
23. Sherrill JD, Kiran KC, Blanchard C, Stucke EM, Kemme KA, Collins MH, et al. Analysis and expansion of the eosinophilic esophagitis transcriptome by RNA sequencing. Genes Immun. (2014) 15:361–9. doi: 10.1038/gene.2014.27
24. Chaitidis P, O'Donnell V, Kuban RJ, Bermudez-Fajardo A, Ungethuem U, Kühn H. Gene expression alterations of human peripheral blood monocytes induced by medium-term treatment with the TH2-cytokines interleukin-4 and−13. Cytokine. (2005) 30:366–77. doi: 10.1016/j.cyto.2005.02.004
25. Kristjansson RP, Benonisdottir S, Davidsson OB, Oddsson A, Tragante V, Sigurdsson JK, et al. A loss-of-function variant in ALOX15 protects against nasal polyps and chronic rhinosinusitis. Nat Genet. (2019) 51:267–76. doi: 10.1038/s41588-018-0314-6
26. Andersson CK, Claesson HE, Rydell-Törmänen K, Swedmark S, Hällgren A, Erjefält JS. Mice lacking 12/15-lipoxygenase have attenuated airway allergic inflammation and remodeling. Am J Respir Cell Mol Biol. (2008) 39:648–56. doi: 10.1165/rcmb.2007-0443OC
27. Imoto Y, Takabayashi T, Sakashita M, Kato Y, Yoshida K, Kidoguchi M, et al. Enhanced 15-lipoxygenase 1 production is related to periostin expression and eosinophil recruitment in eosinophilic chronic rhinosinusitis. Biomolecules. (2020) 10:1–13. doi: 10.3390/biom10111568
28. Harb H, Chatila TA. Mechanisms of dupilumab. Clin Exp Allergy. (2020) 50:5–14. doi: 10.1111/cea.13491
29. Laidlaw TM, Mullol J, Fan C, Zhang D, Amin N, Khan A, et al. Clinical communications dupilumab improves nasal polyp burden and asthma control in patients with CRSwNP and AERD. J Allergy Clin Immunol Pract. (2019) 7:2462–5.e1. doi: 10.1016/j.jaip.2019.03.044
30. Buchheit KM, Hulse KE. Local immunoglobulin production in nasal tissues: a key to pathogenesis in chronic rhinosinusitis with nasal polyps and aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol. (2021) 126:127–34. doi: 10.1016/j.anai.2020.09.016
31. Feldman S, Kasjanski R, Poposki J, Hernandez D, Chen JN, Norton JE, et al. Chronic airway inflammation provides a unique environment for B cell activation and antibody production. Clin Exp Allergy. (2017) 47:457–66. doi: 10.1111/cea.12878
32. Hulse KE, Norton JE, Suh L, Zhong Q, Mahdavinia M, Simon P, et al. Chronic rhinosinusitis with nasal polyps is characterized by B-cell inflammation and EBV-induced protein 2 expression. J Allergy Clin Immunol. (2013) 131:1075–83. doi: 10.1016/j.jaci.2013.01.043
33. Bartemes KR, Cooper KM, Drain KL, Kita H. Secretory IgA induces antigen-independent eosinophil survival and cytokine production without inducing effector functions. J Allergy Clin Immunol. (2005) 116:827–35. doi: 10.1016/j.jaci.2005.07.014
34. Van Roey GA, Vanison CC, Wu J, Huang JH, Suh LA, Carter RG, et al. Classical complement pathway activation in the nasal tissue of patients with chronic rhinosinusitis. J. Allergy Clin Immunol. (2017) 140:89–100.e2. doi: 10.1016/j.jaci.2016.11.015
35. Buchheit KM, Dwyer DF, Ordovas-Montanes J, Katz HR, Lewis E, Vukovic M, et al. IL-5Rα marks nasal polyp IgG4- and IgE-expressing cells in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2020) 145:1574–84. doi: 10.1016/j.jaci.2020.02.035
36. Moteki H, Yasuo M, Hamano H, Uehara T, Usami SI. IgG4-related chronic rhinosinusitis: a new clinical entity of nasal disease. Acta Otolaryngol. (2011) 131:518–26. doi: 10.3109/00016489.2010.533699
37. Vandjelovic ND, Humphreys IM. Immunoglobulin G4-related sclerosing disease of the paranasal sinuses: a case report and literature review. Allergy Rhinol. (2016) 7:85–9. doi: 10.2500/ar.2016.7.0154
38. Chen JB, James LK, Davies AM, Wu YCB, Rimmer J, Lund VJ, et al. Antibodies and superantibodies in patients with chronic rhinosinusitis with nasal polyps. J. Allergy Clin Immunol. (2017) 139:1195–204.e11. doi: 10.1016/j.jaci.2016.06.066
39. Gevaert P, Omachi TA, Corren J, Mullol J, Han J, Lee SE, et al. Efficacy and safety of omalizumab in nasal polyposis: 2 randomized phase 3 trials. J Allergy Clin Immunol. (2020) 146:595–605. doi: 10.1016/j.jaci.2020.05.032
40. Badrani JH, Doherty TA. Cellular interactions in aspirin-exacerbated respiratory disease. Curr Opin Allergy Clin Immunol. (2021) 21:65–70. doi: 10.1097/ACI.0000000000000712
41. Choi Y, Lee DH, Trinh HKT, Ban GY, Park HK, Shin YS, et al. Surfactant protein D alleviates eosinophil-mediated airway inflammation and remodeling in patients with aspirin-exacerbated respiratory disease. Allergy Eur J Allergy Clin Immunol. (2019) 74:78–88. doi: 10.1111/all.13458
42. Choi Y, Lee Y, Park HS. Which factors associated with activated eosinophils contribute to the pathogenesis of aspirin-exacerbated respiratory disease? Allergy Asthma Immunol Res. (2019) 11:320–9. doi: 10.4168/aair.2019.11.3.320
43. Pan D, Buchheit KM, Samuchiwal SK, Liu T, Cirka H, Raff H, et al. COX-1 mediates IL-33–induced extracellular signal-regulated kinase activation in mast cells: Implications for aspirin sensitivity. J Allergy Clin Immunol. (2019) 143:1047–57.e8. doi: 10.1016/j.jaci.2018.06.033
44. Eastman JJ, Cavagnero KJ, Deconde AS, Kim AS, Karta MR, Broide DH, et al. Group 2 innate lymphoid cells are recruited to the nasal mucosa in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2017) 140:101–8.e3. doi: 10.1016/j.jaci.2016.11.023
45. Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood. (2012) 119:3790–8. doi: 10.1182/blood-2011-10-384826
46. Mitsui C, Kajiwara K, Hayashi H, Ito J, Mita H, Ono E, et al. Platelet activation markers overexpressed specifically in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin Immunol. (2016) 137:400–11. doi: 10.1016/j.jaci.2015.05.041
47. Laidlaw TM, Boyce JA. Platelets in patients with aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2015) 135:1407–14. doi: 10.1016/j.jaci.2015.02.005
48. von Schacky C, Kiefl R, Jendraschak E, Kaminski WE. n-3 fatty acids and cysteinyl-leukotriene formation in humans in vitro, ex vivo, in vivo. J Lab Clin Med. (1993) 121:302–9.
49. Schneider TR, Johns CB, Palumbo ML, Murphy KC, Cahill KN, Laidlaw TM. Dietary fatty acid modification for the treatment of aspirin-exacerbated respiratory disease: a prospective pilot trial. J. Allergy Clin Immunol Pract. (2018) 6:825–31. doi: 10.1016/j.jaip.2017.10.011
50. Sommer DD, Rotenberg BW, Sowerby LJ, Lee JM, Janjua A, Witterick IJ, et al. A novel treatment adjunct for aspirin exacerbated respiratory disease: the low-salicylate diet: a multicenter randomized control crossover trial. Int Forum Allergy Rhinol. (2016) 6:385–91. doi: 10.1002/alr.21678
51. Haimerl P, Bernhardt U, Schindela S, Henkel FDR, Lechner A, Zissler UM, et al. Inflammatory macrophage memory in nonsteroidal anti-inflammatory drug–exacerbated respiratory disease. J Allergy Clin Immunol. (2021) 147:587–99. doi: 10.1016/j.jaci.2020.04.064
52. Ban G.-Y., Youn D.-Y., Ye Y.-M.-S, et al. Increased expression of serine palmitoyl transferase and ORMDL3 polymorphism are associated with eosinophilic inflammation and airflow limitation in aspirin-exacerbated respiratory disease. PLoS ONE. (2020) 15:e0240334. doi: 10.1371/journal.pone.0240334
53. Bachert C, Han JK, Desrosiers M, Hellings PW, Amin N, Lee SE, et al. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet. (2019) 394:1638–50. doi: 10.1016/S0140-6736(19)31881-1
54. Han JK, Bachert C, Fokkens W, Desrosiers M, Wagenmann M, Lee SE, et al. Mepolizumab for chronic rhinosinusitis with nasal polyps (SYNAPSE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. (2021). doi: 10.1016/S2213-2600(21)00097-7
55. Scott WC, Cahill KN, Milne GL, Li P, Sheng Q, Huang LC, et al. Inflammatory heterogeneity in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol. (2021) 147:1318–28.e5. doi: 10.1016/j.jaci.2020.11.001
56. Michalik M, Samet A, Dmowska-Koroblewska A, Podbielska-Kubera A, Waszczuk-Jankowska M, Struck-Lewicka W, et al. An overview of the application of systems biology in an understanding of chronic rhinosinusitis (CRS) development. J Pers Med. (2020) 10:1–22. doi: 10.3390/jpm10040245
Keywords: asthma, CRSwNP, samter's triad, N-ERD, aspirin-exacerbated respiratory disease, NSAID-exacerbated respiratory disease, nasal polyp, chronic rhinosinusitis
Citation: Lyly A, Laidlaw TM and Lundberg M (2021) Pathomechanisms of AERD—Recent Advances. Front. Allergy 2:734733. doi: 10.3389/falgy.2021.734733
Received: 02 July 2021; Accepted: 05 August 2021;
Published: 10 September 2021.
Edited by:
Peter Valentin Tomazic, Medical University of Graz, AustriaReviewed by:
Mongkhon Sompornrattanaphan, Mahidol University, ThailandAleksandar Peric, Military Medical Academy, Serbia
Copyright © 2021 Lyly, Laidlaw and Lundberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annina Lyly, YW5uaW5hLmx5bHlAaGVsc2lua2kuZmk=