 Poonam Sikka1*
Poonam Sikka1* Kunwar Pal Singh1Inderjeet Singh1
Kunwar Pal Singh1Inderjeet Singh1 Dwijesh Chandra Mishra2Shyam Sundar Paul1
Dwijesh Chandra Mishra2Shyam Sundar Paul1 Ashok K. Balhara1
Ashok K. Balhara1 Jerome Andonissamy1
Jerome Andonissamy1 Krishna Kumar Chaturvedi2
Krishna Kumar Chaturvedi2 Atmakuri Ramakrishna Rao2
Atmakuri Ramakrishna Rao2 Anil Rai2
Anil Rai2- 1Central Institute for Research on Buffaloes, Indian Council of Agricultural Research (ICAR), Hisar, Haryana, India
- 2Indian Agricultural Statistics Research Institute, Indian Council of Agricultural Research (ICAR), New Delhi, India
Functional genome profiling of Murrah buffaloes (Bubalus bubalis) was performed for milk-production trait by whole blood transcriptome analysis comparing RNA-seq data assembled from high and low milk producing multiparous (5 -6 parity) animals. These buffaloes reflected the genetic merit inherited as daughters born to extremely high- and low-end bulls evaluated under a progeny testing scheme and ranked by the estimated breeding value. The average standard milk yield (SMY) over the 305 d during the parity was recorded as 2909.50L ± 492.63 and 1869.57 ± 189.36L in high- and low-performance buffaloes, respectively. The “reference” assembly data was assembled from transcriptome libraries of a group of buffaloes (n=16), comprising of animals in different physiological states. Replicates selected within each category of the high and low genetic merit animals showed a correlation coefficient of high order (R2=0.98) while comparing with the `reference' assembly. The sequence data of selected buffaloes, mapped over the Mediterranean water buffalo genome, revealed differentially expressed genes (DEGs) distinctly depicted via heat maps and volcano plots obtained for two categories of animals, determining more than 25,000 genes via the Cufflink analysis. DEGs included 83 down-regulating and 142 up-regulating genes (p<0.05, FDR<0.05). Functional classification of the DEGs revealed a fine networking of biological processes, primarily cell signaling, cell proliferation, cell differentiation, RNA splicing, fat metabolism, and inflammasome generation. These processes are regulated by transcription factors and binding proteins covered under the network of TNF alpha signaling, NF-kappa B signaling and MAPK PI3K-AKT signaling pathways/ cascade emerged as main biological pathways. Emerged pathways revealed remarkably intricate tuning of metabolic and cell development processes converging into milk production in buffaloes. Segregated patterns of gene expression obtained for high and low milk producing buffaloes using the non-invasive method of whole blood transcriptome analysis has emerged as a promising resource comprising gene network and protein -protein interactions, primarily involved in lactation. Synergism of transcription factors and binding proteins promoting epigenetic regulation at all development stages of mammary tissue induce mammogenic and lactogenic responses for subsequent milk secretion under optimum feeding management. These findings may help improve breeding strategies to achieve the desired milk yield in Murrah buffaloes.
1 Introduction
Milk yield efficiency reflects the integrated functional ability of multi-organ systems to biosynthesize fat, protein, and lactose to increase the milk volume by utilizing blood constituents (Bauman et al., 2006). The synthesis and secretion of 1L of milk is estimated to require a flow of 400–500L blood through the mammary gland (Xue et al., 2016). Milk fat and protein yield can be improved by selective breeding of mates with high estimated breeding values (EBVs), favoring higher milk yield; however, the contribution of paternal inheritance towards female progeny performance has been determined to be greater than that of maternal inheritance (Schaeffer, 2006) in Murrah buffaloes (Bubalus bubalis). This warrants further understanding of genomic differences underlying the metabolic changes translating into high or low milk yield to identify genomic determinants for efficient milk production that are less known in buffaloes (B. bubalis).Energy flow to the mammary tissue is of paramount importance as milk protein synthesis utilizes approximately 35% of the total energy (Osorio et al., 2016), supported by circulating insulin (Gan et al., 2013) and glucose in mammary, liver, adipose, muscle, and immunity cells. Compared with non-lactating animals, lactating animals exhibit five-fold higher energy utilization (Schingoethe et al., 1988) and four to seven-fold greater mRNA translation (Kühn et al., 1999) in the mammary tissue alone during milk protein synthesis. Nevertheless, milk fat is secreted by mammary epithelial cells (MECs) as the result of an energy-driven metabolic shift within the mammary layer through a synergistic control of cell homeostasis and integrated functionality of multiple tissues mediated through blood. Therefore, the blood may exhibit differential expression of genes associated with milk synthesis, and whole-blood transcriptomics may be used as a non-invasive method to identify gene variants, unraveling the complex physiological interactions between various tissues during lactation.
2 Materials and methods
2.1 Animals and study design
We evaluated two groups of divergent animals, with high and low genetic value based on the respective sire’s EBV estimated over 150 bulls and phenotype records on the standard milk yield (SMY) of multi-parous (6 to 7 parities) animals at the government farm of ICAR-Central Institute for Research on Buffaloes, Hisar, India. The EBV for the SMY for each bull comprised the first lactation records of 100 daughters of the respective bull, thus comprising 4.575 million milk records compared with available records for contemporary daughters at the farm. Parental inheritance contribution from bulls is more than 35% toward their daughter buffaloes as per progeny testing, thus expressing high and/or low genetic merits of production as per the inherited potential from their respective sires. All the animals were in mid-lactation physiologically and were kept on a uniform feeding system. Conclusive inference is based on the functional expression of milk yield trait with respect to high and low genetic merit (sire’s EBV and phenotypic records for milk production) in buffaloes. The average SMY over parities was 1869.57 ± 189.36 and 1893.00 ± 275.45 in low milk yielding animals, i.e. MY1 and MY2, respectively. The SMY for high milk yielding individuals was 2909.50 ± 492.63 and 2419.83 ± 361.49 in MY3 and MY4, respectively, as replicates in the two study groups were compared for their RNA-seq data to identify differentially expressed genes(DEGs). RNA-seq data were obtained selectively, considering high and low performance extremes with respect to four economically important traits: residual feed intake, age at first calving, inter-calving period between the first two calvings, and milk volume for growth to develop the “reference” assembly.
2.2 RNA isolation and complementary DNA library preparation for sequencing
RNA extraction was performed using whole blood from B. bubalis. RNA quality was assessed using an Agilent 2100 Bioanalyzer. mRNA was purified using oligo-dT beads (TruSeq RNA Kit, Illumina) with 1 μg of RNA (RIN = 8.0). The pure RNA was fragmented at 90°C and reverse transcribed using random hexamers and Superscript II Reverse Transcriptase (Life Technologies). Complementary DNA (cDNA) was synthesized using RNaseH and DNA polymerase I and was cleaned for SPRI cleanup using Beckman Coulter Agencourt Ampure XP SPRI beads. The cDNA molecules were ligated with Illumina adapters after end repair and were subsequently polyadenylated. The cDNA library was further amplified using PCR to enrich the adapter-ligated fragments. Nucleic acid quantification was performed for individual libraries using a NanoDrop spectrophotometer, and the quality was validated using a Bioanalyzer (Agilent Technologies). Sequencing was performed on an IlluminaHiSeq 2500 platform. Paired-end FASTQ files with Phred scores >20 were used to obtain high-quality (HQ)-filtered reads.
2.3 Reference-based transcriptome assembly, annotation, and global expression profiling
Considering the vast natural diversity with respect to major physiological criteria of genetic merit indicating the economic significance of buffaloes, the “reference” assembly was generated for animals exhibiting extremely low and high performances with respect to early maturity, those achieving first calving at a younger age with the ability to further produce a calf per year, and those taking optimum time for establishing subsequent pregnancy during the reproductive life span as a result of efficient feed utilization selectively to achieve better growth and milk production status.
Therefore, the reference assembly was prepared using paired-end RNA-seq library, as per the protocol recommended by Illumina, (GenBank NCBI databases under SRA ID SUB5692555 and the Bio Project ID: PRJNA546485) including 16 test animals of B.bubalis selected from both low- and high-performance extremes with respect to the following criteria of physiological merit to achieve true assembly covering vast diversity: milk production, age at first calving, inter-calving period, and feed efficiency. Replicates in each of the high- and low-performing groups in each physiological category were sequenced using 100 bp paired end module sequencing and re-assembled into 40–60 million reads for each library. The reads were subjected to quality control using the NGSQC tool kit (Patel and Jain, 2012). Paired end sequence reads with Phred score >Q30 were selected for further analysis.
The HQ-filtered reads obtained from each group were provided as input to a Trinity assembly with an auto-kmer setting with default parameters. The output of the Trinity assembly was subjected to an improved amelioration pipeline to filter putative false positive transcripts using coverage and depth criteria in each library. This resulted in true positive transcripts in each library, which were subjected to CD-HIT EST clustering pipeline for eliminating redundant transcripts, thereby creating a reference unigene transcriptome for each library of B.bubalis. This reference unigene transcriptome was further clustered into a single non-redundant transcriptome assembly used as a “reference” assembly. HQ reads obtained from individual libraries generated from low milk yield performers (MY1 & MY2) and high performers (MY3 & MY4) were aligned with the reference buffalo genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_003121395.1/UOA_WB_1) using the Bowtie tool for quantification of expressed transcripts. Transcripts with RPKM ≥1 were considered as expressed in each of the sequenced library.
The resultant “reference” transcriptome assembly was used as reference, and reads from each individual libraries were aligned to the reference using the Bowtie tool for quantification of expressed transcripts to validate sequencing. Replicate reproducibility was assessed using a correlation matrix (R2). Distribution of inter and intra stages of physiology specifically expressing transcripts (in replicates) were checked via the Venn diagram approach. Read count for each transcript was normalized to obtain RPKM≥1, which qualified it as an expressed transcript in each sequenced library. The final transcriptome was subjected to SNP calling using SAM tools and VarScan and filtered (Depth≥30). SNPs were divided based on the four physiological-stage-specific groups. Milk production specific transcripts and SNPs in the genes were annotated with Gene Ontology (GO) and Pathways to perform biological enrichment analysis.
2.4 RNA-seq data analysis
The reference genome of the Mediterranean water buffalo (B. bubalis) (https://www.ncbi.nlm.nih.gov/assembly/GCF_003121395.1/) was used for read alignment of transcripts, identification of coding regions, and annotation of transcripts using Kallisto (Nicolas et al., 2016). Replicate transcripts obtained from the low- and high-merit subgroups were used for differential expression analysis using DESEQ2 (Love et al., 2014). Transcript log2ratio ≥2.0 was the limit to select differentially expressed transcripts. Volcano plots were generated using the R package “Enhanced Volcano” to identify the differential gene clusters. Gene ontology (GO) and pathways involved in high or low levels of milk production were elucidated using the DAVID Functional Annotation Tool (DAVID Bioinformatics Resources 6.8, NIAID/NIH) (Da et al., 2007). Differentially expressed transcripts obtained from individuals of low and high merit for milk production were identified.
2.5 Gene Ontology and KEGG pathways
GO and KEGG pathways were identified using the DAVID Functional Annotation Tool. Integrated networks were deduced using the Cytoscape software (Shannon, 2003).
3 Results and discussion
3.1 Next-generation sequencing
Deep sequencing of RNA obtained from HiSeq platform resulted in an average of approximately 55 million raw reads of 210bp average insert size per sample library (Table 1). The raw FASTQ sequences were filtered using NGSQC tool kit to obtain HQ reads, an average of approximately 50 million HQ reads per sample (~90%). Each library datapoint had an SD of <30% within and across indicating uniformity in data generation, which is critical for estimating transcript coverage and normalization, while the minimum transcript length cutoff of the assembled transcriptome was 500 bases and the maximum transcript length observed was 27 kbp. N50 of the assembled transcriptome was approximately 1.97 kbp, indicating good integrity of the transcriptome available for expression profiling, resulting in identification of an average of approximately 57,000 transcripts expressed in one sample.
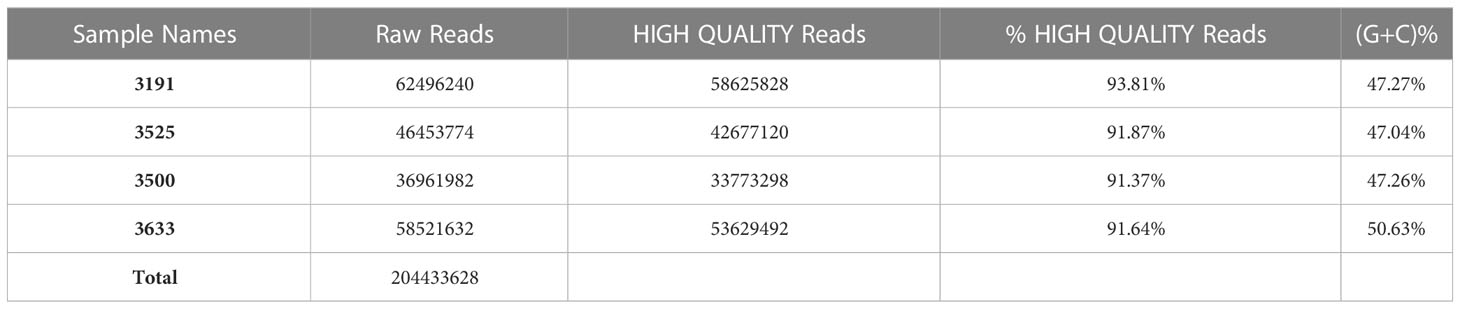
Table 1 QC Statistics of RNA Seq data obtained for high and low milk yield merit.
3.2 Global expression profiling
Transcript distribution provided the expression dynamics specific to high and low milk yield. Furthermore, the transcripts obtained with respect to differentials between and within high and low milk yield subgroups were annotated using GO and KEGG pathways for biological enrichment analysis. Transcripts obtained from RNA-seq were subjected to differential expression analysis. Data retrieval from the HiSeq platform with respect to the replicates between the high and low milk production subgroups yielded 0.2044 billion reads. An average of 51.1 million raw paired-end reads (Table 1) with an average insert size of 210bp was used to determine DEGs.
3.3 Reference-based assembly, validation, and annotation
The transcriptome assembly comprised 83,783 sequences with an average sequence length of 1518.46 and G + C% of 48.93, based on which replicates were compared. HQ reads obtained from the individual library were mapped over the B. bubalis reference genome, and around 88.7% of the HQ reads were mapped to the human reference genome using KALLISTO pipeline, suggesting an acceptable alignment (Figure 1). Approximately 20% difference in alignment of reads with reference genome indicated underlying diversity in the genetic merit of individual animals under study, as the ratio of HQ reads to total reads obtained in each compared sample library was uniform (0.91–0.93). A high degree of correlation between biological replicates within the performance subgroup, with an average R2 value of 0.98 among all the profiled replicates, indicated acceptable phenotype selection of replicates and reliable quality of experimentation and sequencing.
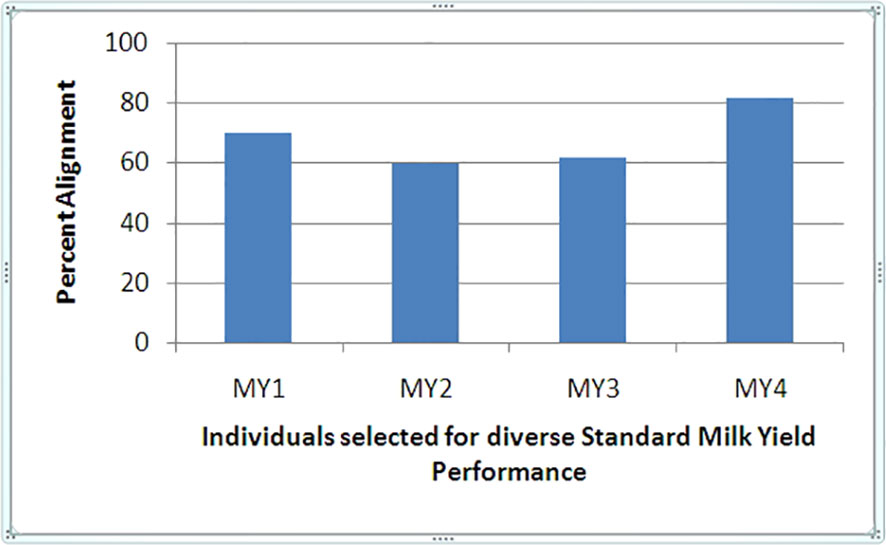
Figure 1 Alignment percentage of Reference Bubalus bubalis divergent to high and low SMY (MY) performance.
3.4 Differential gene expression
Notable differences were observed among the transcripts of animals of diverse genetic merit referred through the de-regulated genes (Figure 2). Distinct gene patterns obtained from the RNAseq data of low and high milk yield subgroups’ animals depicted through heat maps (Figure 3) covered 30,551 transcripts, representing over 25,000 genes, among which 887 were differentially expressed transcripts (fold-change value <-2 and padj value <0.05). Significantly differentiated transcripts were ascribed to the functional expression of low and high merit in lactating buffaloes.
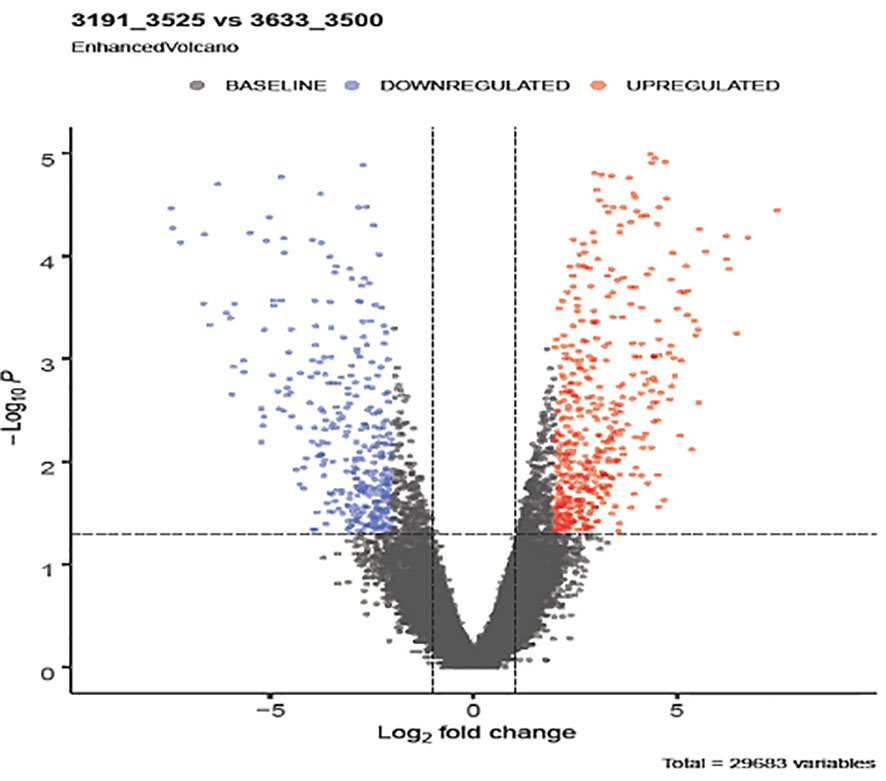
Figure 2 Differentially expressed genes (DEGs) emerged from comparing low and high yielding buffaloes. The y-axis corresponds to the mean expression value of log10 (q-value), and the x-axis displays the log2 fold change value. Differential expression of transcripts (padj value < 0.05) is shown in red dots circled in the frame. Blue dots represent the non-significant differential expression of transcripts between the two study groups.
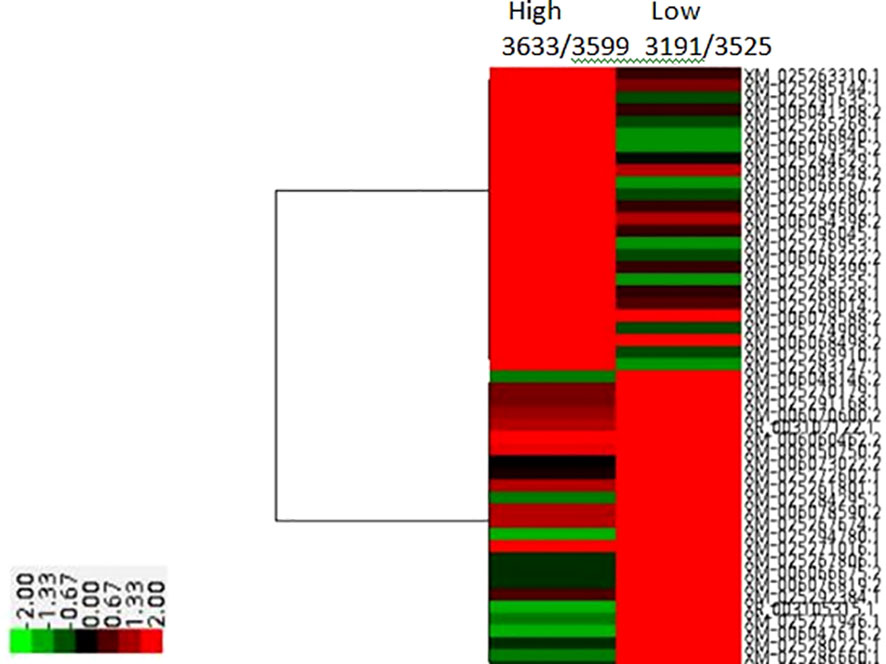
Figure 3 HeatMap of RNA Seq reads comparing High and Low genetic merit buffalo Blood transcriptomes for Milk Volume.
The downregulated expression of genes was apparently less, including for 373 genes, than the upregulated expression of 514 genes (Figure 4); 83 and 147 genes were significantly(p<0.05) downregulated (Table 2) and upregulated (Table 3), respectively.
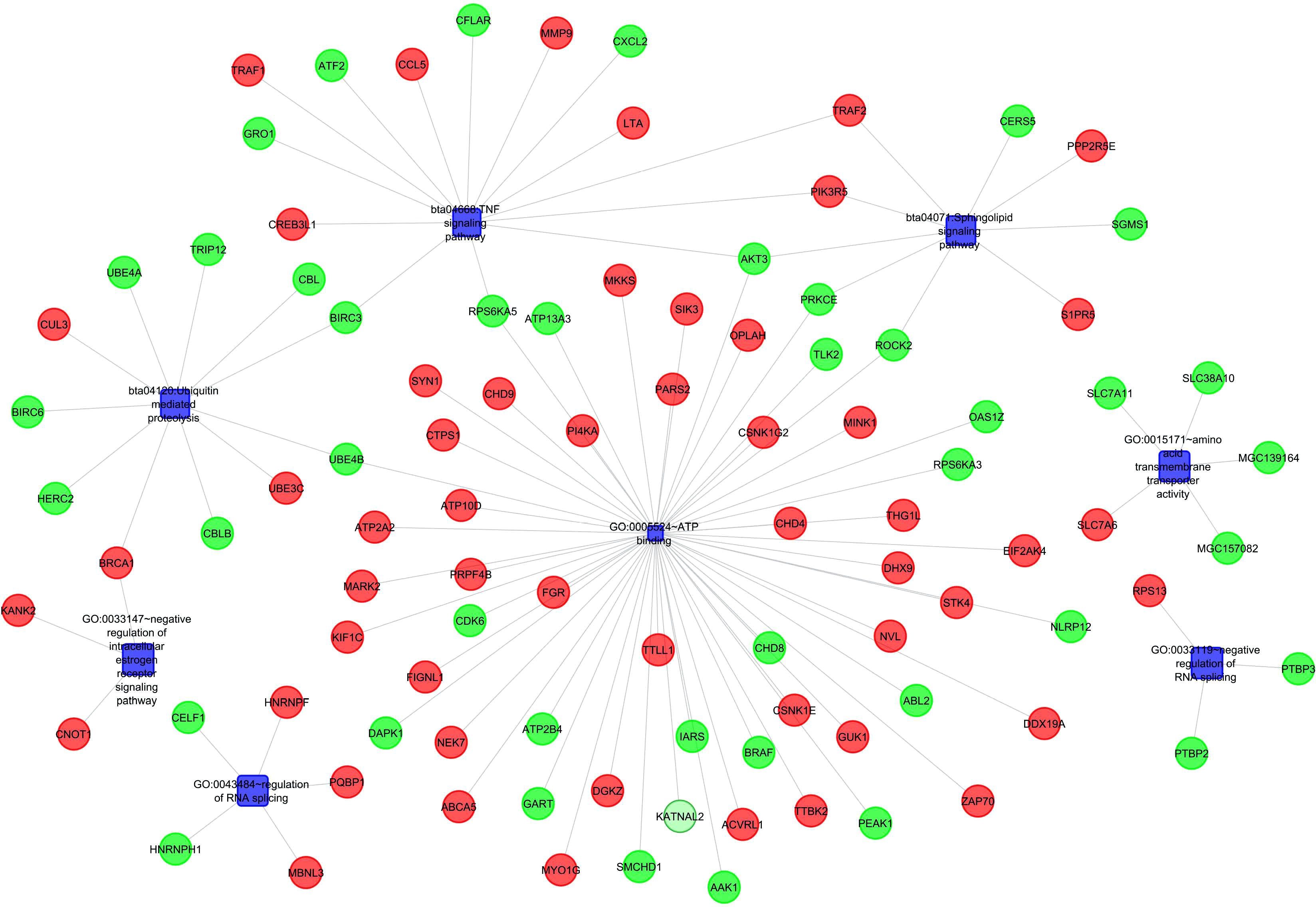
Figure 4 Gene Network and protein interactions in response to Milk volume production in Murrah Buffaloes.
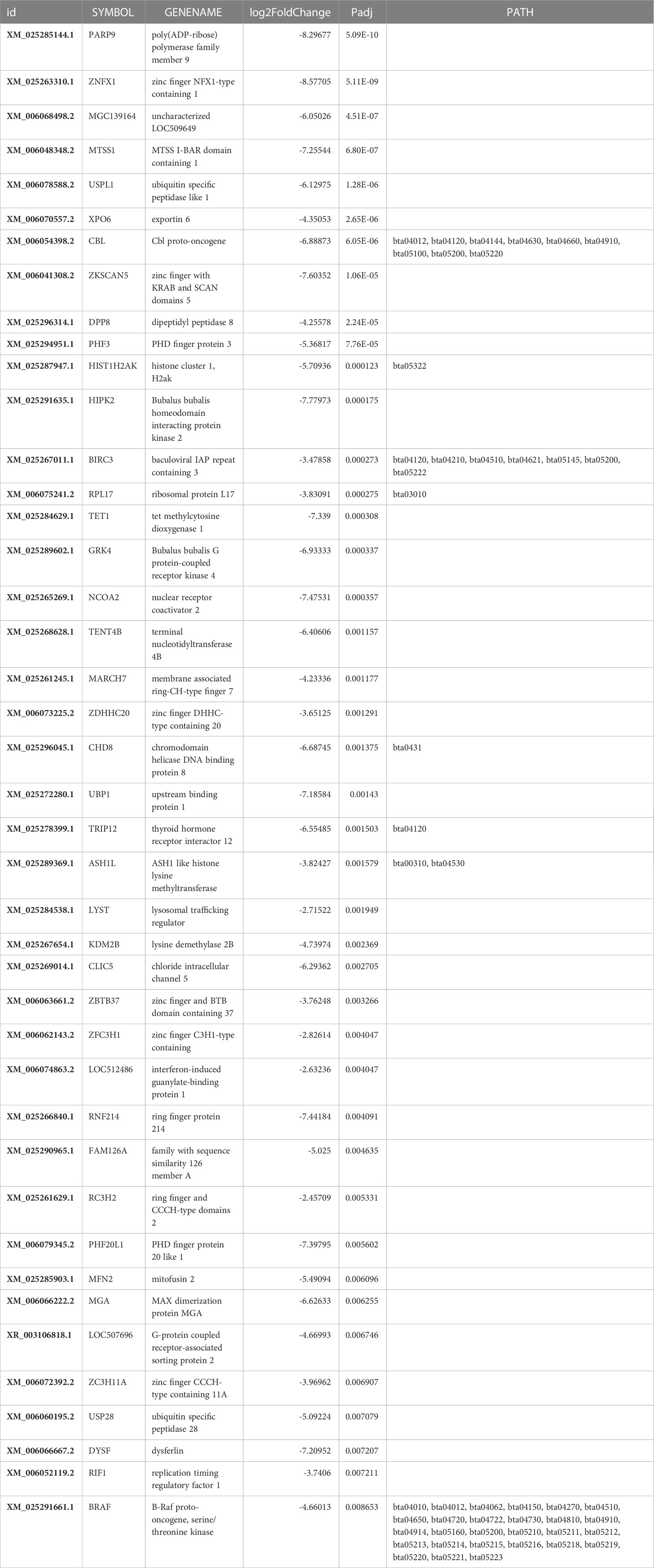
Table 2 Top downregulated (padj<0.05) genes.
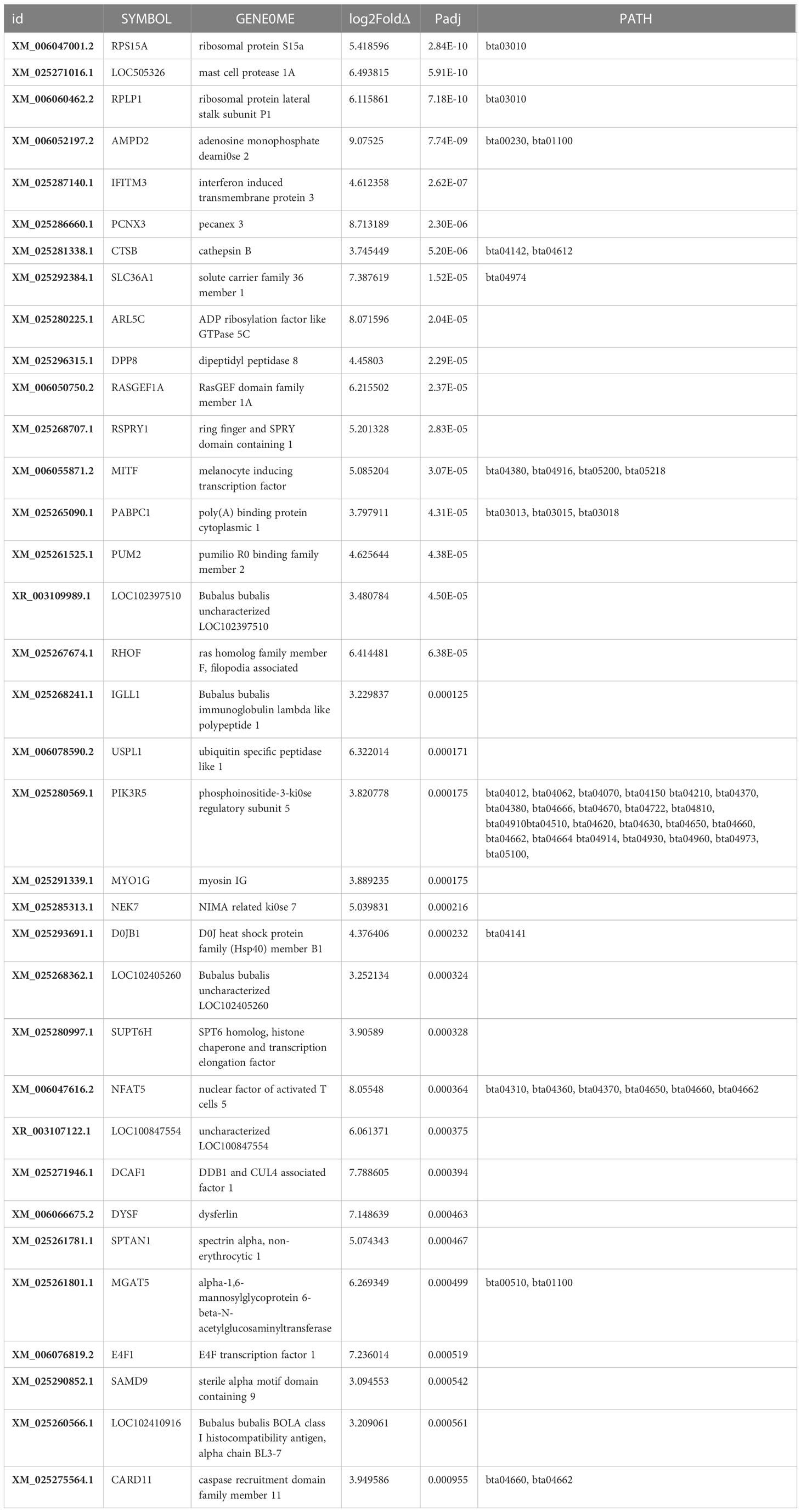
Table 3 Top Upregulated (padj<0.05) genes.
3.4.1 Downregulated genes and pathways
Poly (ADP-ribose) polymerase member 9 (PARP9), also known as BAL/BAL1/ARTD9/MGC, was the most significantly downregulated (padj=5.09486035306163E-10), and phosphodiesterase was the least significantly (padj=0.776373155513801) downregulated among the 373 differentially transcribed genes in this study. The top 35 significantly (padj.<0.05) downregulated genes (Table 2) identified in this study were generally associated with various functional processes, such as DNA repair during cell cycle, mammary cell development, lactogenesis, apoptosis, and oxidative stress, as discussed in following sub-sections.
3.4.1.1 Downregulation of genes for DNA repair and mammary cell development
Downregulated activity of poly(ADP-ribose) polymerase (PARP) II saves energy in mammary cells, sustaining its survival against different metabolic or chemical agents to propagate milk synthesis (Desvergne et al., 2005). Downregulated PARP9 propagates synthesis of casein and alpha lactalbumin by employing the ubiquitination of transcription repressor protein/downregulation of ribosomal proteins L17, which inhibits RNA polymerase binding (Askarian-Amiri et al., 2011), involving downregulation of the zinc finger and BTB domain containing 37 genes. Downregulation of the transcription repressor chromodomain helicase D0 binding protein 8 may reduce chromatin re-modeling activity, favoring milk protein synthesis. Downregulation of E4F transcription factor 1 (p120E4F, p50E4F) (Moison et al., 2021), concomitant with PARP1 gene, is involved in DNA repair for cell cycling during lactation (Bauman et al., 2006),mediating through energy (ATP) saving activity. DNA repair maintains genomic integrity and cell survival. Downregulation of NCOA6 in this study may limit milk fat synthesis, consequently allowing high milk production (Desvergne et al., 2005) by regulating the binding of transcription factors to peroxisome proliferator-activator receptor. The tetra methyl cytosine dioxygenase1 gene, zinc finger NFX1-type long chain (lnc) RNA, and exportin may support mammary cell proliferation and lactogenesis when downregulated via DNA de-methylation through acetylation, i.e., binding of histone H2B GlcN to CpG-rich sequences at promoters of either transcriptionally active (Wu et al., 2017) or Polycomb-repressed genes. Thus, they play a role in post-puberty mammary gland development (Liu et al., 2017), indicating that tetra methyl cytosine dioxygenase1 serves as a potent epigenetic enhancer for milk gene expression. Furthermore, exportin protein regulates mitosis, mediating the nuclear export of actin and profilin-actin complexes to somatic cells (Stüven et al., 2003) and inhibiting several mitotic assembly events (Nord et al., 2020). Thus, its downregulation enhances cell proliferation and differentiation in mammary tissues and facilitates milk production in high-yielding buffaloes. Downregulated zinc finger with KRAB protein gene maintains the nucleolus, cell differentiation, and proliferation by reducing the expression of a repressor protein to facilitate lactation. Downregulation of the growth inversing (Smolock et al., 2012) RpL17 may aid in cell proliferation and milk synthesis. Additionally, it stabilizes the cellular proteins as a subunit of the poly(A) tail complex (Wu et al., 2020).
The expression of some genes associated with cell proliferation in the mammary gland of high-yielding buffaloes may depend on the lactation stage (Sorensen et al., 2006). They may be downregulated at the onset of lactation and their activity may plateau during the pre-lactation stage, as a consequence of their higher expression during late pregnancy to support higher milk production during lactation.
3.4.1.2 Genes involved in lactogenesis
Down-regulated MTSS I-BAR domain-containing 1 (MTSS1) (Table 2) plays a role in the proliferation and development of MECs and promotion of milk production through glucose uptake from blood, in association with catenin, promoting lactose synthesis and increasing milk volume (Wu et al., 2022). Downregulation of ASH1, a histone lysine methyltransferase gene and negative regulator of chromatin activation, enhances expression by reducing H3K36 methylation of chromatin in the nucleoplasm, including homeobox genes (Okamoto et al., 2016).
High rates of demethylation of H3K4me2 at the promoter region of milk-synthesis-related genes support lactation, and this effect is inversed in late lactation (Bionaz et al., 2012).This highlights the important role of epigenetic regulation of lactogenesis in cell transcription sites, which regulates the functionality of the mammary gland (Rijnkels et al., 2013). Downregulated zinc finger C3H1-type protein, ring finger protein 214, and ring finger CCCH-type domain 2 protein lower the rate of protein ubiquitination, thus channelizing energy (ATP) into processes of cell division, differentiation, and apoptosis, favoring milk production. Chromatin compression is likely to thus reduce and promote milk protein expression through downregulation of PHD finger protein (transcription regulator) and histone H2A type 1 protein genes carrying epigenetic heredity (Singh et al., 2012).
Phosphorylation pattern of proteins that favor high milk production is altered by down-regulation of B.bubalis G protein-coupled receptor kinase 4. Downregulated homeodomain-interacting protein kinase 2 (HIPK2) regulates snRNA transcription in response to DNA damage in the nucleus, which induces apoptosis, activating various intracellular signal pathways (Kuwano et al., 2016), including p53, transforming growth factor-β, Notch, Wnt, JNK, Hedgehog, and Hippo. Additionally, HIPK2governs phosphorylation of downstream transcription factors, epigenetic regulators, and ubiquitin ligases to regulate cellular development and differentiation of the neural, muscular, and circulatory system in addition to the mammary tissue.
Downregulation of B.bubalis HIPK2 indicates low DNA damage in the nucleus and apoptosis, which is expected to improve cell development and differentiation of a healthy udder. Downregulation of membrane-associated protein genes, such as thyroid hormone receptor interactor 12 and E3 ubiquitin-protein ligase, delays ubiquitination and reduces milk production, matching the physiological status of late lactation.
3.4.1.3 Genes regulating apoptosis
Downregulated dipeptidyl peptidase 8 (DPP8) and ubiquitin specific peptidase 1-like genes inhibit pyroptosis, promoting cell proliferation and lactogenesis. BIRC3 inhibits apoptosis induced by serum binding to tumor necrosis factor receptor-associated factors, interfering with the activity of proteases known as caspase(s), thus promoting lactation by preventing apoptosis.
3.4.1.4 Oxidative stress
Downregulation of GTPase-mitofusin 2 (Mfn2) indicates a state of low oxidative stress in cells, which attenuates mitochondrial fragmentation (Gao et al., 2018), thus increasing the energy available for lactogenesis in MECs. The present study revealed that downregulation of zinc finger DHHC-type containing 20 proteins lowered the transport of energy-carrying moieties, such as carnitine acyl coA, from the cytosol to inter-membrane space of the mitochondria for oxidation, indicating the onset of an alternate energy-generation pathway other than the oxidative pathway to facilitate the energy conservation for building milk volume. Gluconeogenesis associated with a decrease in oxygen consumption induced by altered insulin signaling (Gan et al., 2013) has been reported (Bach et al., 2003) as an alternative pathway to maintain glucose homeostasis (Yang et al., 2016) in high-yield animals. PHD finger protein 20 like 1 and MFN2 have been reported to regulate lipid and glucose metabolism for mitochondrial function propagation (Pich et al., 2005) in hepatic tissue, which indicates synergistic functioning of mammary and hepatic tissues toward cell energy optimization (Wathes et al., 2021), favoring milk synthesis.
3.4.2 Genes emerged as upregulated and related pathways
The top 35 significantly (p<0.05) upregulated transcripts (Table 3) of the total 514 upregulated genes related to milk production are discussed in following subsections. Ribosomal protein S15a emerged as the most significant (padj=2.84032091633935E-10) gene and C-C motif chemokine ligand 5 as the least significant (padj = 0.0494934823949582) gene in this study. The top upregulated genes discussed below are mostly related to the biological processes of epigenetic regulation of protein synthesis, cell growth, and pathogenicity in order to optimize mammary functions for increased milk synthesis (Figure 4).
3.4.2.1 Protein-coding genes
Upregulation of ribosomal protein genes, such as ribosomal protein S15a (DBA20), a component of the 40S subunit located in the cytoplasm, enhances cell proliferation. Concomitantly, ribosomal phosphoprotein lateral stalk subunit P1 gene, a component of the 60S subunit of the ribosome, promotes milk protein synthesis in lactating buffaloes. Upregulated adenosine monophosphate deaminase 2 in this study may enhance ADP ribosylation via guanine nucleotide biosynthesis (Naiara et al., 2013), cell signaling, and intracellular protein transport. Up-regulated poly A binding protein cytoplasmic 1 gene promotes cellular protein synthesis by binding to the 3’ poly(A) tail of eukaryotic messenger RNAs. Up-regulation of melanocyte-inducing transcription factor genes may induce expression and release of melanocyte hormone (Battagello et al., 2020), thus increasing milk secretion by the mammary gland.
3.4.2.2 Mammary cell growth
In the present study, upregulation of the RNA-associated protein DDB1 and CUL4, an associated factor protein, indicates enhanced cell proliferation, cell growth (Shan et al., 2022), and energy metabolism (Pryce et al., 2012) through nuclear transcription co-activator, leading to high milk production in buffaloes. Upregulation of NIMA-related kinase 7 gene plays a critical role in mitotic cell cycle progression via mitotic spindle formation (Yissachar et al., 2006) and cytokinesis (Adib et al., 2019)by dissociating RPS6KB1 and EML4 proteins from microtubules for optimum chromosome compression. Upregulated interferon-induced trans-membrane protein 3 gene is likely to elevate mammary cell growth and proliferation; PECANEX 3 promotes self-renewal of mammary stem cells (Notch signaling) and optimizes ER assembly; and spectrin alpha non-erythrocytic 1, a filamentous cytoskeletal protein, stabilizes the plasma membrane and intracellular organelles, promoting DNA repair and cell cycle regulation for smooth lactation.
3.4.2.3 Milk synthesis
Upregulated ADP ribosylation factors, i.e., GTP-binding proteins (Ras superfamily) “might bind to cell-surface GTP receptors, activating these receptors and modulating downstream effecter pathways, which in turn promotes membrane transport, favoring milk production. The upregulation of Pumilio R0 binding family member 2 gene in this study might affect mitochondrial infrastructure and mitochondrial homeostasis as a negative regulator, favoring increased milk production (Davide et al., 2019). Upregulated amino acid transporter gene, the solute carrier family 36 member 1, may potentiate milk synthesis by developing the secretary cells and optimizing the energy homeostasis of MECs for milk secretion.
3.4.2.4 Pathogenic control
Upregulated mast cell protease 1 might increase the activity of typtase, a pro-inflammatory protease, and impede pathogenesis in the MEC layer. Up-regulation of the DPP8 protease gene in this study might have played a role in improving energy metabolism, homeostasis, and immune function through T-cell activation in support of milk production. The caspase recruitment domain 15 (CARD15) is a cytosolic protein that develops inflammasomes in response to microbial infection (Martinon and Tschopp, 2005), involving NF-κB activation (Ogura et al., 2001). Accordingly, up-regulation of the CARD15 might be associated with increased somatic cell count (SCC), which reflects the somatic cell score phenotype expressed in high-lactating animals (Pant et al., 2007). Upregulation of sterile alpha motif domain-containing protein 9 gene indicates a high level of cell proliferation in actively lactating mammary tissue, showing high inflammation under the influence of interferon (TNF-α signaling) in high milk-producing animals. Upregulation of caspase 1 recruitment domain family member 11L gene indicates positive regulation of apoptosis and NF-κB activation, improving immune expression as generation of an inflammasome in association with NLRP3 upregulated gene, thus protecting mammary cells in high-yielding buffaloes. Upregulated B. bubalis BOLA class I histocompatibility antigen gene confirms that immunity and mammary development play a synergistic role during milk synthesis and its secretion. Upregulated myosin IG gene corroborates with T cell migration at the expense of ATP to detect and eliminate pathogens and regulate phagocytosis in B cells as an immune function during lactation.
Elevated Ras Homolog Family Member F(in filopodia), a Rho guanosine triphosphatase family protein, indicates an alternate pathway to glycolysis for ATP generation under glucose deprivation. Metabolic shift, which occurs because of the elevated function of glycolytic enzymes (Warburg effect) in cells, might affect the cell morphology, resulting in epithelial–mesenchymal transition, cell migration, and invasion, similar to low-potential lactating animals.
3.4.2.5 Ubiquitylation
Upregulated ring finger and SPRY domain 1-containing gene is likely to enhance post-translational regulation by scavenging underutilized mRNAs and methylated DNA regions, regulating overall protein synthesis in mammary cells. Nuclear factor of activated T cells 5 is a transcription factor that regulates gene transcription in response to T cell receptor-mediated signals in lymphocytes.
3.4.2.6 Other genes involved in cellular events
Upregulated DNAJB1 in this study acts as a molecular chaperone induced by environmental and lactation stress in mammary tissues by activating HSP70 (dnaK) heat shock protein via G protein-coupled receptor signaling and protein degradation in cells (Kastenhuber et al., 2017). Upregulated phosphoinositide-3-kinase regulatory subunit 5 of the PI3K complex (Figure 4) is critical for promoting G protein-mediated functions of cell growth, proliferation, differentiation, motility, survival, and intracellular trafficking through insulin signaling, glucose homeostasis, and neuroendocrine function involved in lactogenesis.
3.4.2.7 Histone modification
Upregulation of the transcription elongation factor SPT6 homolog gene may promote transcription of proteins essential for lactation, coupled with histone-modifying enzymes for mRNA splicing in the nucleoplasm and its subsequent translocation in cytoplasm. Upregulation of dysferlin and spectrin alpha genes promotes membrane repair (Leigh et al., 2011), which is required in proportion to the severity of cell membrane damage during milk secretion.
Other upregulated (p<0.05) genes that emerged as stress-combating and immunity-developing entities in this study include activin A, receptor-like type 1 stress-induced transcription factor, IKAROS family zinc finger 3, which participates in T and B cell differentiation, and transforming acidic coiled-coil containing protein 1 gene,which influences mitosis. Proteins that emerged as epigenetic regulators were translated from 5-azacytidine-induced DNA methyltransferase inhibitors, Menin1, a component of the SET1 histone methyltransferase complex, methylating ‘Lys-4’ of histone H3 (H3K4), ubiquitin-associated protein 1, and a regulator of vesicular trafficking processes. Methylase inhibitors do inhibit the histone methylation, thus blocking the associated silencing and permitting the onset of vascular trafficking and related processes. The energy-associated determinant genes for GTPase, IMAP family member 7, regulators of lymphocyte survival and homeostasis, zinc finger protein 335, transcriptional regulators in neurogenesis, and zinc finger protein 665 were identified as enhancers of energy, immunity, and epigenetic control in milk synthesis. LRP1 is a transmembrane receptor modulating cell signaling molecules, including integrins and receptor tyrosine kinases.
3.5 Gene enrichment analysis and functional classification of DEGs
Gene enrichment analysis was performed using standard procedures (https://david.ncifcrf.gov/). Significant (p ≤ 0.05) biological processes, cellular locations, and molecular functions that are related to the production of milk volume (Table 4) were selected to plot the networks using System Biology of Milk Yield in Buffaloes. The gene clusters (p<0.05) identified in this study represented 57 categories of annotated GO terms and KEGG pathways govern cell functions, including the cytoplasm, nucleus, nucleolus, and membrane, to propagate milk production (Figure 5). GO:0005654~nucleoplasm (FDR=0.000257993148089497), bta04668: TNF α signaling pathway (FDR=0.00685236886627249), and GO:0016020~membrane (FDR= 0.0814138896939487) were among the most significant (p <0.05) functional categories. The number of genes annotated under each functional category along with the enlisted significant genes are listed in Table 4. The main biological processes and pathways were zinc ion binding (GO:0008270), negative regulation of extrinsic apoptotic signaling pathway(GO:2001237), negative regulation of intracellular estrogen receptor signaling pathway(GO:0033147), ATP binding (GO:0005524), ubiquitin-mediated proteolysis, UB4B(bta04120), TNF αsignaling (bta:04668), sphingolipid signaling (bta04071), amino acid transmembrane transporter activity (GO:0015171), and RNA splicing regulation (GO:0043484), which are involved in regulating the milk volume (Figure 5). The gene network and protein–protein interaction determined via Cytoscape analysis are depicted in Figure 4; Table 4.
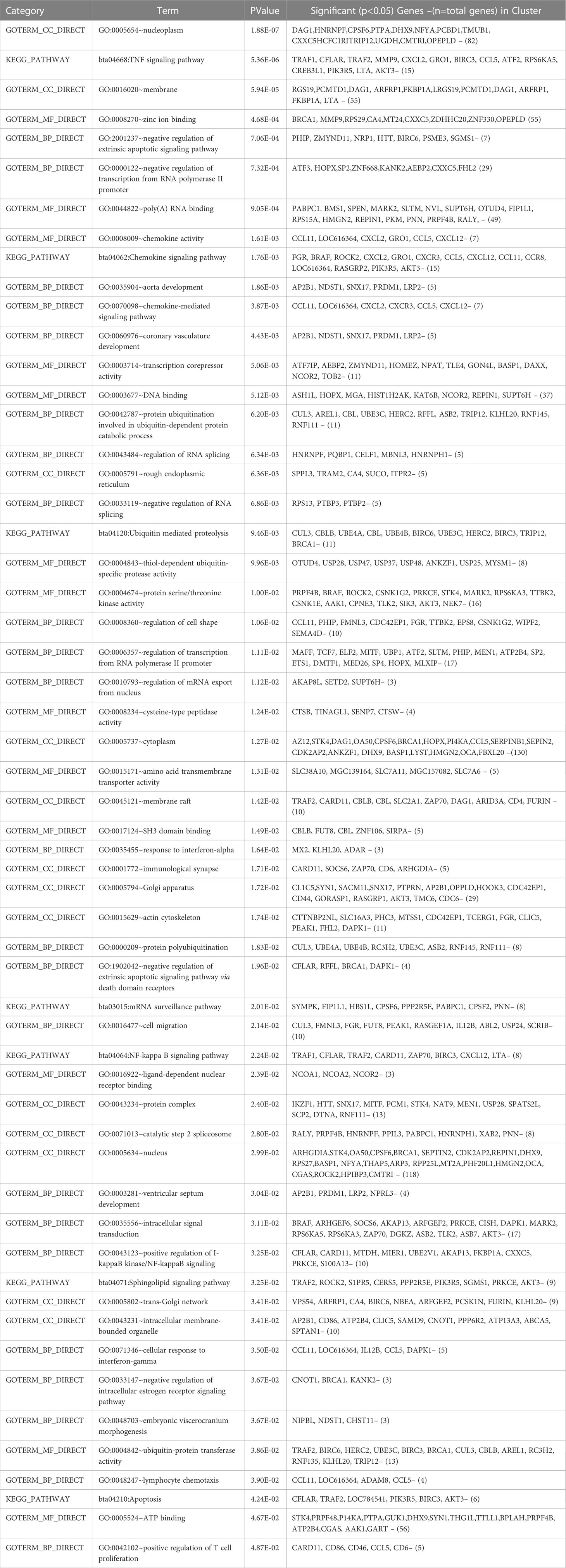
Table 4 Gene ontology terms and biological pathways identified from differential genes in high and low milk yield buffaloes.
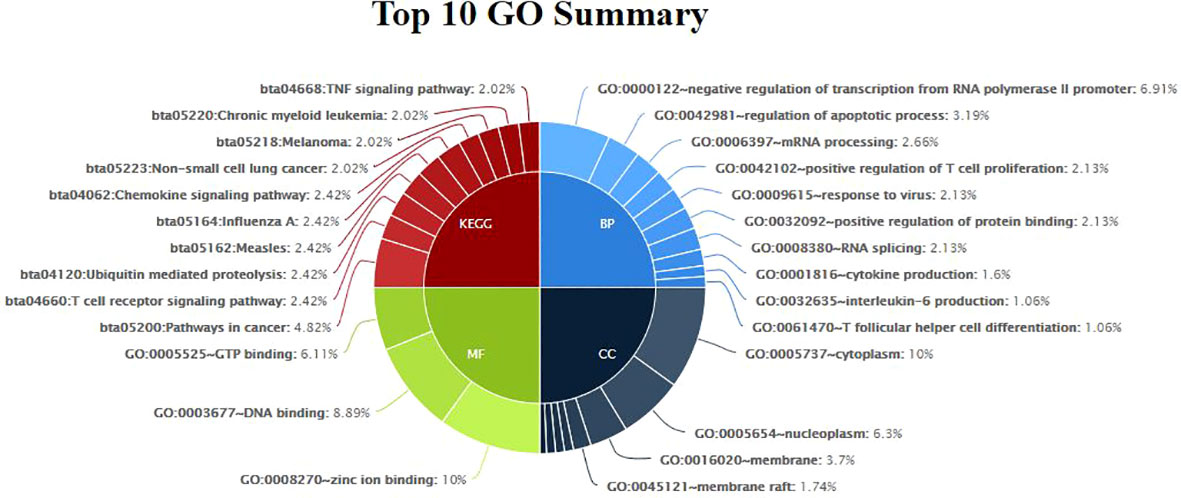
Figure 5 Functional Classification Showing Significant Gene Ontology Terms (p<0.05) emerged from Murrah Buffaloes diversified in milk production.
3.6 Emerging pathways
GO:0005654~nucleoplasm emerged as a significant function location (Table 4) while inferring the differences related to high and low milk yield in buffaloes. Other significant (padj<0.05) processes included dystroglycan 1, heterogeneous nuclear ribonucleoprotein F, cleavage and polyadenylation specific factor 6, protein phosphatase 2 phosphatase activator, DExH-box helicase 9, nuclear transcription factor Y subunit alpha, pterin-4 alpha-carbinolamine dehydratase 1, transmembrane and ubiquitin-like domain containing 1, CXXC finger protein 5, and host cell factor C1 regulator 1.TNF signaling pathway (bta04668) emerged as a significant (p< 0.05, FDR<0.05) pathway; the up-regulated genes involved were TRAF1, TRAF2 (TNF ligand), MMP9 (SCC in milk), CCL5 (recruiting leukocytes,T-cells, and macrophages to inflammatory sites), CREB3L1 (homeostasis of the secretary pathway), PIK3R5 (cell growth, proliferation, differentiation, and motility), and LTA (innate immunity). Similarly, down-regulated (p< 0.05) genes were CXCL2 (chemokine antimicrobial protein), BIRC3, ATF2 (inhibits cellular proliferation and induces cell death), RPS6KA5, AKT3, CFLAR (apoptosis regulation), and GRO (Table 4).
TNF α holds significance because it regulates apoptosis, cell survival, inflammation, and immunity as a cytokine, thus propagating the onset of lactation and enhancing the prolactin promoter in the pituitary. The NF-κB signaling pathway (bta04064) corroborates with TNF α signaling (Soünke et al., 2006) in these animals. Estrogen interacts with TNF-α (GO:0033147), regulating PRL expression. Active TNF emerged as a marker protein for milk production in this study. Binding of TRAF2 to TNFR protein molecules may trigger signal transduction to activate genes of the NF-kappa B pathway and the MAPK PI3K-AKT signaling cascade, determining MEC survival and its capacity for milk production. PIK3R5was differentially up-regulated (p<0.05), supporting PI3K-AKT signaling cascades regulating the cytoskeleton and cell organization (GO:0015629)(Figure 4). ActivatedPI3K phosphorylates AKT and regulates apoptosis, protein synthesis, metabolism, and the cell cycle. PI3K executes actin reorganization and cellular polarization through G-protein-coupled receptors and the secretion of chemokines and signaling proteins (GO:0008009 and bta04062).The PI3K-Akt-mTOR-signaling pathway thus creates a structural link between membrane receptors and the actin cytoskeleton, harboring signaling molecules (kinases and phosphatases) to regulate gene functions and cell survival. Associated pathways initiate the innate immune response against pathogens via Toll-like receptors (TLRs), producing pro-inflammatory cytokines using activated NF-κB and MAPK pathways.TLR ligand binding triggers responses, such as protein modifiers, helper cofactors, and post-transcriptional/epigenetic regulators.
Baculoviral IAP repeat-containing gene (BIRC3) emerged as a significantly downregulated gene in this study. BIRC3 regulates caspases, apoptosis, inflammatory signaling, immunity, mitogenic kinase signaling, cell proliferation, and cell invasion. BIRC3 acts as an E3 ubiquitin-protein ligase, regulating the NF-kappa-B signaling. The present study confirmed the collaborative role of metabolic and immune regulatory pathways to establish the genetic merit of milk production, thus corroborating with a previous hypothesis (Bu et al., 2017) regarding mutual crosstalk between the liver and mammary tissue for optimum lactation. Nevertheless, the whole blood transcriptome, a non-invasive resource for study, enabled projections of lactation physiology corroborative with the genes and functions that emerged as lactation enhancers with potential as markers for selecting high-yielding (high EBV) buffaloes.
In the present study, whole blood transcriptome reflected differential gene expression with respect to buffaloes with high and low genetic merit for milk production. Identification of DEGs using non-invasive methods is useful in large dairy animals. Most transcripts were associated with transcriptional regulation, primarily RNA synthesis, mRNA translation, cell signaling, and proliferative capacity of mammary cells either regulated by hormones and/or growth factors, whereas cell metabolic activity is controlled by the liver and is thus regulated by diet (Bu et al., 2017). These findings indicate the major role of transcription factors in the nucleus as regulators for the repression or activation of biological processes of cell/ribosomes, and epigenetic regulation by amino acids and amino acid transporters as inducers and availability of energy (NADH) across the membranes which are the key factors to achieve adequate milk production, consistent with previous reports (Dai et al., 2018). Other than the structural variation depicted as genomic variants reported earlier (Mishra et al., 2021). Pre-mRNA modifications introduce expression variants at the level of individual animals. Differential gene expression reflects the physiological differences in mammary tissue functioning between high and low milk producing buffaloes. A decrease in methyl-deoxycytidine levels in buffaloes is associated with increased milk production due to de-condensation of chromatin, consistent with previous reports (Choi et al., 1998). Casein production is enhanced by reduced cytidine methylation, indicating differential epigenetic regulation in high and low milk producing animals.
The present study revealed the impact of cell growth and differentiation factors, cytokines/chemokines, and transcription repressors as signaling molecules on epithelial cell differentiation and proliferation, migration, vasculogenesis, apoptosis, innate immune cells, inflammation (i.e., the NFκB pathway), and translocation of signal carriers from external membranes to nuclear regulatory factors through receptors/signal sensors in the mammary tissue. Enrichment of these functions indicate that lactation results from the combined reciprocal control of signals induced as metabolites/molecules originating from the pituitary, liver, and mammary tissue in synergy with immune function regulation in buffaloes.
4 Conclusion
Over 25,000 DEGs were determined with respect to physiological differences between low and high milk production in buffaloes using non-invasive whole blood transcriptomic analysis. Transcriptomic data revealed that post-transcriptional and post-translational modifications play an important regulatory role in lactogenesis via energy balancing and stress combating in favor of high milk production, emulating high genetic merit in buffaloes. Reduced ribosomal activity and uncontrolled protein degradation regulate milk synthesis in addition to the insufficiency of energy or its utilization. This study revealed that lactogenesis is vulnerable to extrinsic inducers of pathogenicity and oxidative stress. The genetic markers identified in this study may be informative for identifying quantitative trait loci associated with milk production traits and developing markers for controlled breeding programs of Murrah buffaloes. Genomic sequences encompassing the DEGs with respect to milk production performance is a potential resource for generating genomic tools for identifying high-merit germplasm.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The animal study was reviewed and approved by Institute Animal Ethics Committee (IAEC) of CIRB Hisar. India (Reg. No. 406/GO/RBI/L/01/CPCSEA).
Author contributions
PS: Project formulation, data analysis, and manuscript writing, KS: Genetic merit determination and phenotype recording, IS: Project formulation and facilitation, AB: Farm data analysis, KC, DM, ARR, and AR: RNAseq data curation and SNP analysis, JA and SP: Project formulation, AR: Funding. All authors contributed to the article and approved the submitted version.
Funding
This research work was supported by the Network Project on Agricultural Bioinformatics and Computation Biology under Centre for Agricultural Bioinformatics Scheme (CABin Scheme), at ICAR-CIRB-IASRI Collaboration/Project Code AGENIASRICIP201500900046.
Acknowledgments
Assistance given in the development of Gene networks by Bionivid Technology private Limited, Bengaluru, India is acknowledged. Services obtained from professional editors ‘Editage’ is acknowledged.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adib R., Montgomery J. M., Atherton J., O’Regan L., Richards M. W., Straatman K. R., et al. (2019). Mitotic phosphorylation by NEK6 and NEK7 reduces the microtubule affinity of EML4 to promote chromosome congression. Sci. Signal 12 (594), eaaw2939. doi: 10.1126/scisignal.aaw2939
Askarian-Amiri M. E., Crawford J., French J. D., Smart C. E., Smith M. A., Clark M. B., et al. (2011). Snord-host rna zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA 17, 878–891. doi: 10.1261/rna.2528811
Bach D., Pich S., Soriano F. X., Vega N., Baumgartner B., Oriola J., et al. (2003). Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. a novel regulatory mechanism altered in obesity. J. Biol. Chem. 278 (19), 17190–17197. doi: 10.1074/jbc.M212754200
Battagello D. S., Lorenzon A. R., Diniz G. B., Motta-Teixeira L. C., Klein M. O., Ferreira J. G. P., et al. (2020). The rat mammary gland as a novel site of expression of melanin-concentrating hormone receptor 1 mRNA and its protein immunoreactivity. Front. Endocrinol. 11. doi: 10.3389/fendo.2020.00463
Bauman D. E., Mather I. H., Wall R. J., Lock A. L. (2006). Major advances associated with the biosynthesis of milk. J. Dairy. Sci. 89 (4), 1235–1243. doi: 10.3168/jds.s0022-0302(06)72192-0
Bionaz M., Periasamy K., Rodriguez-Zas S. L., Everts R. E., Lewin H. A., Hurley W. L., et al. (2012). Old and new stories: revelations from functional analysis of the bovine mammary transcriptome during the lactation cycle. PloS One 7, e33268. doi: 10.1371/journal.pone.0033268
Bu D., Bionaz M., Wang M., Nan X., Ma L., Wang J. (2017). Transcriptome difference and potential crosstalk between liver and mammary tissue in mid-lactation primiparous dairy cows. PloS One 12 (3), e0173082. doi: 10.1371/journal.pone.0173082
Choi Y. J., Jang K., Yim D. S., Baik M. G., Myung K. H., Kim Y. S., et al. (1998). Effect of compensatory growth on the expression of milk protein geneand biochemical changes of the mammary gland in Holstein cows. J. Nutr. Biochem. 9, 380–387. doi: 10.1016/S0955-2863(98)00027-8
Da W., Huang B. T., Sherman Q. T., Jack R., Collins W., Alvord G., et al. (2007). The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8 (9), R183. doi: 10.1186/gb-2007-8-9-r183
Dai W., Wang Q., Zhao, Fengqi Z., Jianxin L., Hongyun L. (2018). Understanding the regulatory mechanisms of milk production using integrative transcriptomic and proteomic analyses: improving inefficient utilization of crop by-products as forage in dairy industry. BMC Genomics 19, 403. doi: 10.1186/s12864-018-4808-5)
Davide D, Adrienne M., Francesca P., Vincenzo S., Hao L, Romani M., et al. (2019). The RNA-binding protein PUM2 impairs mitochondrial dynamics and mitophagy during aging Molecular cell 73, 775–787. doi: 10.3389/fendo.2020.00463
Desvergne B., Michalik L., Wahli W. (2005). Transcriptional regulation of metabolism. Physiol. Rev. 86, 465–514. doi: 10.1152/physrev.00025
Gan K. X., Wang C., Chen J. H., Zhu C. J., Song G. Y. (2013). Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J. Gastroenterol. 19 (23538485), 1572–1581. doi: 10.3748/wjg.v19.i10.1572
Gao W., Du X., Lei L., Wang H., Zhang M., Wang Z., et al. (2018). NEFA-induced ROS impaired insulin signalling through the JNK and p38MAPK pathways in non-alcoholic steatohepatitis. J. Cell. Mol. Med. 22 (29602237), 3408–3422. doi: 10.1111/jcmm.13617
Kastenhuber E. R., Lalazar G., Shauna L., Houlihan D., Tschaharganeh F., Baslan T., et al. (2017). DNAJB1-PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. 114, (50) 13076–13084. doi: 10.1073/pnas.1716483114
Kühn C., Freyer G., Weikard R., Goldammer T., Schwerin M. (1999). Detection of QTL for milk production traits in cattle by application of a specifically developed marker map of BTA6. Anim. Genet. 30 (5), 333–340. doi: 10.1046/j.1365-2052.1999.00487.x
Kuwano Y., Nishida K., Akaike Y., Kurokawa K., Nishikawa T., Masuda K., et al. (2016). Homeodomain-interacting protein kinase-2: a critical regulator of the DNA damage response and the epigenome. Int. J. Mol. Sci. 17 (10):1638–1650. doi: 10.3390/ijms17101638
Leigh B., Waddell F. A., Xi L., Zheng F., Tran J., Frances J., et al. (2011). Dysferlin, annexin A1, and mitsugumin 53 are up-regulated in muscular dystrophy and localize to longitudinal tubules of the T-system with stretch. J. Neuropathol. Exp. Neurol. 70 (4), 302–313. doi: 10.1097/NEN.0b013e31821350b0
Liu S., Wang Z., Chen D., Zhang B., Tian R. R., Zhang Y., et al. (2017). Annotation and cluster analysis of spatiotemporal- and sex-related lncRNA expression in rhesus macaque brain. Genome Res. 27, 1608–1620. doi: 10.1101/gr.217463.116
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15 (12), 550. doi: 10.1186/s13059-014-0550-8
Martinon F., Tschopp J. (2005). NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 26, 447–454. doi: 10.1016/j.it.2005.06.004
Mishra D. C., Yadav S., Sikka P., Jerome A., Paul S. S., Rao A. R., et al. (2021). SNPRBb: economically important trait specific SNP resources of buffalo (Bubalus bubalis) conservation genetics resources 13 (3), 283–289. doi: 10.1007/s12686-021-01210-
Moison C., Chagraoui J., Caron M. C., Gagné J. P., Coulombe Y., Poirier G. G., et al. (2021). Zinc finger protein E4F1 cooperates with PARP-1 and BRG1 to promote DNA double-strand break repair. Proc. Natl. Acad. Sci. U. S. A. 118 (11), e2019408118. doi: 10.1073/pnas.2019408118
Naiara A., Cantagre V., Schroth J., Cai N., Vaux K., McCloskey D., et al. (2013>). AMPD2 regulates GTP synthesis and is mutated in a PotentiallyTreatable neurodegenerative brainstem disorder cell 154 (3), 505–17. doi: 10.1016/j.cell.2013.07.005
Nicolas L. ,. B., Pimentel H., Melsted P., LiorPachter (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Nord M. S., Bernis C., Carmona S., Garland D. C., Travesa A., Forbes D. J. (2020). Exportins can inhibit major mitotic assembly events in vitro: membrane fusion, nuclear pore formation, and spindle assembly. NUCLEUS 11 (1), 178–193). doi: 10.1080/19491034.2020.1798093
Ogura Y., Inohara N., Benito A., Chen F. F., Yamaoka S., Nunez G. (2001). Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 276, 4812–4818. doi: 10.1074/jbc.M008072200
Okamoto M., Miyata T., Konno D., Ueda H. R., Kasukawa T., Hashimoto M., et al. (2016). Cell-cycle-independent transitions in temporal identity of mammalian neural progenitor cells. Nat. Commun. 7, 11349. doi: 10.1038/ncomms11349
Pant S. D., Schnkel F. S., Leyva-Baca I., Sharma B. S., Karrow N. A. (2007). Identification of single nucleotide polymorphisms in bovine CARD15 and their associations with health and production traits in Canadian holsteins. BMC Genomics 8, 421. doi: 10.1186/1471-2164-8-421
Patel R. K., Jain M. (2012). NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PloS One 7 (2), e30619. doi: 10.1371/journal.pone.0030619
Pich S., Bach D., Briones P., Liesa M., Camps M., Testar X., et al. (2005). The charcot–Marie–Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 14 (11), 1405–1415. doi: 10.1093/hmg/ddi149
Pryce J. E., Arias J., Bowman P. J., Davis S. R., Macdonald K. A., Waghorn G. C., et al. (2012). Accuracy of genomic predictions of residual feed intake and 250-day body weight in growing heifers using 625,000 single nucleotide polymorphism markers. J. Dairy. Sci. 95, 2108–2119. doi: 10.3168/jds.2011-4628
Rijnkels M., Freeman-Zadrowsk i C., Hernandez J., Potluri V., Wang L., Li W., et al. (2013). Epigenetic modifications unlock the milk protein gene loci during mouse mammary gland development and differentiation. PloS One 8, e53270. doi: 10.1371/journal.pone.0053270
Schaeffer L. R. (2006). Strategy for applying genome-wide selection in dairy cattle. J. Anim. Breed. Genet. 123 (4), 218–223. doi: 10.1111/j.1439-0388.2006.00595.x
Schingoethe D. J., Byers F. M., Schelling G. T. (1988). “Nutrient needs during critical periods of the life cycle,” in The ruminant animal: digestive, physiology, and nutrition. Ed. Church D. (Illinois, USA: Waveland Press Inc), 421–447.
Shan B. Q., Wang X. M., Zheng L., Han Y., Gao J., Lv M. D., et al. (2022). DCAF13 promotes breast cancer cell proliferation by ubiquitin inhibiting PERP expression. Cancer Sci. 113 (5), 1587–1600. doi: 10.1111/cas.15300
Shannon P. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Singh K., Molenaar A. J., Swanson K. M., Gudex B., Arias J. A., Erdman R. A., et al. (2012). Epigenetics: a possible role in acute and transgenerational regulation of dairy cow milk production. Animal 6, 375–381. doi: 10.1017/S1751731111002564
Smolock E. M., Korshunov V. A., Glazko G., Qiu X., Gerloff J., Berk B. C. (2012). Ribosomal protein L17, RpL17, is an inhibitor of vascular smooth muscle growth and carotid intima formation. Circulation 126 (20), 2418–2427. doi: 10.1161/CIRCULATIONAHA.112.125971
Soünke F., Harper C. V., Semprini S., Wilding M., Adamson A. D., Spiller D. G., et al. (2006). Tumor necrosis factor-α activates the human prolactin gene promoter via nuclear factor-κB signaling. Endocrinology 147 (2), 773–781. doi: 10.1210/en.2005-0967
Sorensen M. T., Norgaard J. V., Theil P. K., Vestergaard M., Sejrsen K. (2006). Cell turnover and activity in mammary tissue during lactation and the dry period in dairy cows. J. Dairy. Sci. 89, 4632–4639. doi: 10.3168/jds.S0022-0302(06)72513-9
Stüven T., Hartmann E., Görlich D. (2003). Exportin 6: a novel nuclear export receptor that is specific for profilin·actin complexes. EMBO J. 22, 5928–5940. doi: 10.1093/emboj/cdg565
Wathes D. C., Cheng Z., Salavati M., Buggiotti L., Takeda H., Tang L., et al. (2021). Consortium. relationships between metabolic profiles and gene expression in liver and leukocytes of dairy cows in early lactation. J. Dairy. Sci. 104 (3), 3596–3616. doi: 10.3168/jds.2020-19165
Wu D., Cai Y., Jin J. (2017). Potential coordination role between O-GlcNAcylation and epigenetics. Protein Cell 8, 713–723. doi: 10.1007/s13238-017-0416-4
Wu M., Qiu Q., Zhou Q., Li J., Yang J., Zheng C., et al. (2022). circFBXO7/miR-96-5p/MTSS1 axis is an important regulator in the wnt signaling pathway in ovarian cancer. Mol. Cancer 21, 137. doi: 10.1186/s12943-022-01611-y
Wu G., Schmid M., Rib L., Polak P., Meola N., Sandelin A., et al. (2020). A two-layered targeting mechanism underlies nuclear RNA sorting by the human exosome. Cell Rep. 30 (7), 2387–2401.e5. doi: 10.1016/j.celrep.2020.01.068
Xue B., Zheng Z., Liu B., Ji X., Bai Y., Zhang W. (2016). Whole blood transcriptional profiling comparison between different milk yield of Chinese Holstein cows using RNA-seq data. BMC Genomics 17 (Suppl 7), 512. doi: 10.1186/s12864-016-2901-1
Yang J., Jiang J., Liu X., Wang H., Guo G., Zhang Q., et al. (2016). Differential expression of genes in milk of dairy cattle during lactation. Anim. Genet. 47 (2), 174–180. doi: 10.1111/age.12394
Keywords: gene expression, Murrah buffaloes, RNA-Seq, milk volume, genetic variation
Citation: Sikka P, Singh KP, Singh I, Mishra DC, Paul SS, Balhara AK, Andonissamy J, Chaturvedi KK, Rao AR and Rai A (2023) Whole blood transcriptome analysis of lactating Murrah buffaloes divergent to contrasting genetic merits for milk yield. Front. Anim. Sci. 4:1135429. doi: 10.3389/fanim.2023.1135429
Received: 31 December 2022; Accepted: 23 May 2023;
Published: 22 June 2023.
Edited by:
Rajendran Ramanujam, Tamil Nadu Veterinary and Animal Sciences University, IndiaReviewed by:
Seyed Abbas Rafat, University of Tabriz, IranVikas Vohra, National Dairy Research Institute (ICAR), India
Jiangjiang Zhu, Southwest Minzu University, China
Copyright © 2023 Sikka, Singh, Singh, Mishra, Paul, Balhara, Andonissamy, Chaturvedi, Rao and Rai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Poonam Sikka, ZHJzaWthcHVuYW1AZ21haWwuY29t