 Wei Zhou1†
Wei Zhou1† Wei Chi1†
Wei Chi1† Wanting Shen1Wanying Dou2
Wanting Shen1Wanying Dou2 Junyi Wang1
Junyi Wang1 Xuechen Tian1
Xuechen Tian1 Christoph Gehring3
Christoph Gehring3 Aloysius Wong1,4*
Aloysius Wong1,4*- 1Department of Biology, College of Science and Technology, Wenzhou-Kean University, Wenzhou, China
- 2Department of Computer Science, College of Science and Technology, Wenzhou-Kean University, Wenzhou, China
- 3Department of Chemistry, Biology and Biotechnology, University of Perugia, Perugia, Italy
- 4Zhejiang Bioinformatics International Science and Technology Cooperation Center of Wenzhou-Kean University, Wenzhou, China
In proteins, functional centers consist of the key amino acids required to perform molecular functions such as catalysis, ligand-binding, hormone- and gas-sensing. These centers are often embedded within complex multi-domain proteins and can perform important cellular signaling functions that enable fine-tuning of temporal and spatial regulation of signaling molecules and networks. To discover hidden functional centers, we have developed a protocol that consists of the following sequential steps. The first is the assembly of a search motif based on the key amino acids in the functional center followed by querying proteomes of interest with the assembled motif. The second consists of a structural assessment of proteins that harbor the motif. This approach, that relies on the application of computational tools for the analysis of data in public repositories and the biological interpretation of the search results, has to-date uncovered several novel functional centers in complex proteins. Here, we use recent examples to describe a step-by-step guide that details the workflow of this approach and supplement with notes, recommendations and cautions to make this protocol robust and widely applicable for the discovery of hidden functional centers.
Key Points
- Functional centers have key roles in catalysis, ligand-binding, hormone- and gas-sensing, and are difficult to identify through standard homology approaches.
- Functional centers are often hidden in complex multi-domain proteins where they perform important molecular and cellular functions thereby enabling temporal and spatial modulation of signaling molecules and networks.
- Here we present a method for the detection of functional centers and provide a step-by-step guide that systematically describes the workflow and the computational tools employed in this protocol.
- This protocol also provides informative notes, recommendations and cautions at the appropriate steps to allow for a broad and robust application.
Introduction
Functional centers are at the core of domains that contain the key amino acids required to perform a molecular function including, but not limited to, catalysis, ligand-binding, hormone- and gas-sensing (Wong et al., 2018). They are often embedded within complex multi-domain proteins and offer cryptic but important cellular signaling functions that afford intricate temporal and spatial regulation of signaling molecules and their signaling networks (Brady et al., 2007; Levskaya et al., 2009; Irving et al., 2012; Zhang and Ma, 2012; Freihat et al., 2014; Muleya et al., 2014). The lack of overall conservation of functional centers and the fact that they are often only a small part of complex proteins, have left them undetected by standard homology searches (Ludidi and Gehring, 2003; Guo and Fang, 2014; Wong et al., 2018). For instance, in higher plants, enzymes that generate and degrade cyclic mononucleotides or heme-based gas-sensing proteins were not identified until recently although their corresponding homologs exist in diverse groups of organisms ranging from bacteria to animals and humans (Martinez-Atienza et al., 2007; Domingos et al., 2015; Gehring and Turek, 2017; Swiezawska et al., 2018; Wong et al., 2020; Swiezawska-Boniecka et al., 2021; Turek and Irving, 2021). This is perplexing since the products and biological effects of the functional centers such as cyclic nucleotides-mediated cellular events (Bowler et al., 1994; Neuhaus et al., 1997; Durner et al., 1998; Maathuis and Sanders, 2001; Donaldson et al., 2004; Ederli et al., 2009; Isner and Maathuis, 2011; Pasqualini et al., 2011; Isner et al., 2012; Hartwig et al., 2014; Hussain et al., 2016; Marondedze et al., 2016) and nitric oxide mediated pollen tube chemotropic responses (Feijo et al., 2004; Prado et al., 2004, 2008; McInnis et al., 2006; Wang et al., 2009, 2012; Pasqualini et al., 2011, 2015; Domingos et al., 2015; Wong et al., 2020, 2021) have long been documented. Lately, a fundamentally different approach to the discovery of these molecular functions has been employed and has identified novel signaling components in complex proteins (Wong and Gehring, 2013b; Wong et al., 2015, 2018).
Based on the assumption that in complex multi-domain proteins only amino acids that directly perform a molecular function are conserved in the function centers, consensus sequence motifs that include only these key conserved amino acid residues, have been constructed and applied (Ludidi and Gehring, 2003; Gehring, 2010; Wong and Gehring, 2013a). These motifs are constructed by alignments of annotated functional centers from different and distantly related species and can then be used to query target proteomes—e.g., a model plant like Arabidopsis thaliana—to retrieve candidate proteins. The candidate proteins can subsequently be further assessed using homology modeling and molecular docking simulations prior to experimental testing. Functional annotation can also be done through the construction of sequence profiles on PROSITE which is a database of protein domains, families, and functional sites (Sigrist et al., 2013). These regions are better conserved throughout evolution especially in proteins from the same families and have largely similar three-dimensional structures which are crucial for a common molecular function (Wu et al., 2003; Lee et al., 2007; Marchler-Bauer et al., 2011; Mahlich et al., 2018). Functional centers deriving from annotated domains are either less obvious or have evolved beyond recognition in complex proteins because they often occupy a small part (<5%) of the entire protein and only harbor amino acids that are critical for functionality, which may explain why they were unaccounted for during de novo and homology sequence annotation. As they get incorporated into complex proteins of varying primary domains, their tertiary structures may also differ (Jeffery, 1999; Irving et al., 2018; Turek and Irving, 2021) (see Conclusion and Future Perspective for a specific example).
The sequential use of a motif search and structural assessment can overcome the challenges associated with identifying functional centers in complex proteins and indeed, several been identified in recent years as emerging evidence also suggests that many more await discovery (Turek and Gehring, 2016; Ooi et al., 2017; Wheeler et al., 2017; Al-Younis et al., 2018; Chatukuta et al., 2018; Bianchet et al., 2019; Freihat et al., 2019; Ruzvidzo et al., 2019; Wong et al., 2020). Given the highly varied nature of the functional centers, an automated pipeline is currently not feasible for all applications. Here, we use recent examples to document a step-by-step guide of the workflow and supplement it with detailed application notes, recommendations, and cautions. This will enable users to seamlessly apply this approach to the discovery of novel hidden functional centers in reference proteomes.
Materials and Methods
Required Software
In this protocol, the computer program required to generate the protein 3D models by homology modeling, is MODELLER (Sali and Blundell, 1993) which is available for download at https://salilab.org/modeller/download_installation.html. Users are required to register with a valid institutional email at https://salilab.org/modeller/registration.html prior to installation. To simulate docking of small molecules to the functional centers of the 3D models, AutoDock Vina (Trott and Olson, 2010) is used. This open-source program is available for download at http://vina.scripps.edu/download.html. AutoDockTools (ADT) (Morris et al., 2009) is a required graphical front-end for setting up and running Vina and can be downloaded as a package known as MGLTools at http://mgltools.scripps.edu/downloads. The package includes structure analysis and visualization programs such as Python Molecular Viewer (PMV) (Sanner, 1999). Alternatively, UCSF Chimera (Pettersen et al., 2004), which is available for download at https://www.cgl.ucsf.edu/chimera/download.html, can also be used to visualize protein structures and docking outcomes.
General Workflow
The computational approach to identify functional centers in complex proteins includes five general steps which are detailed in this section (Figure 1).
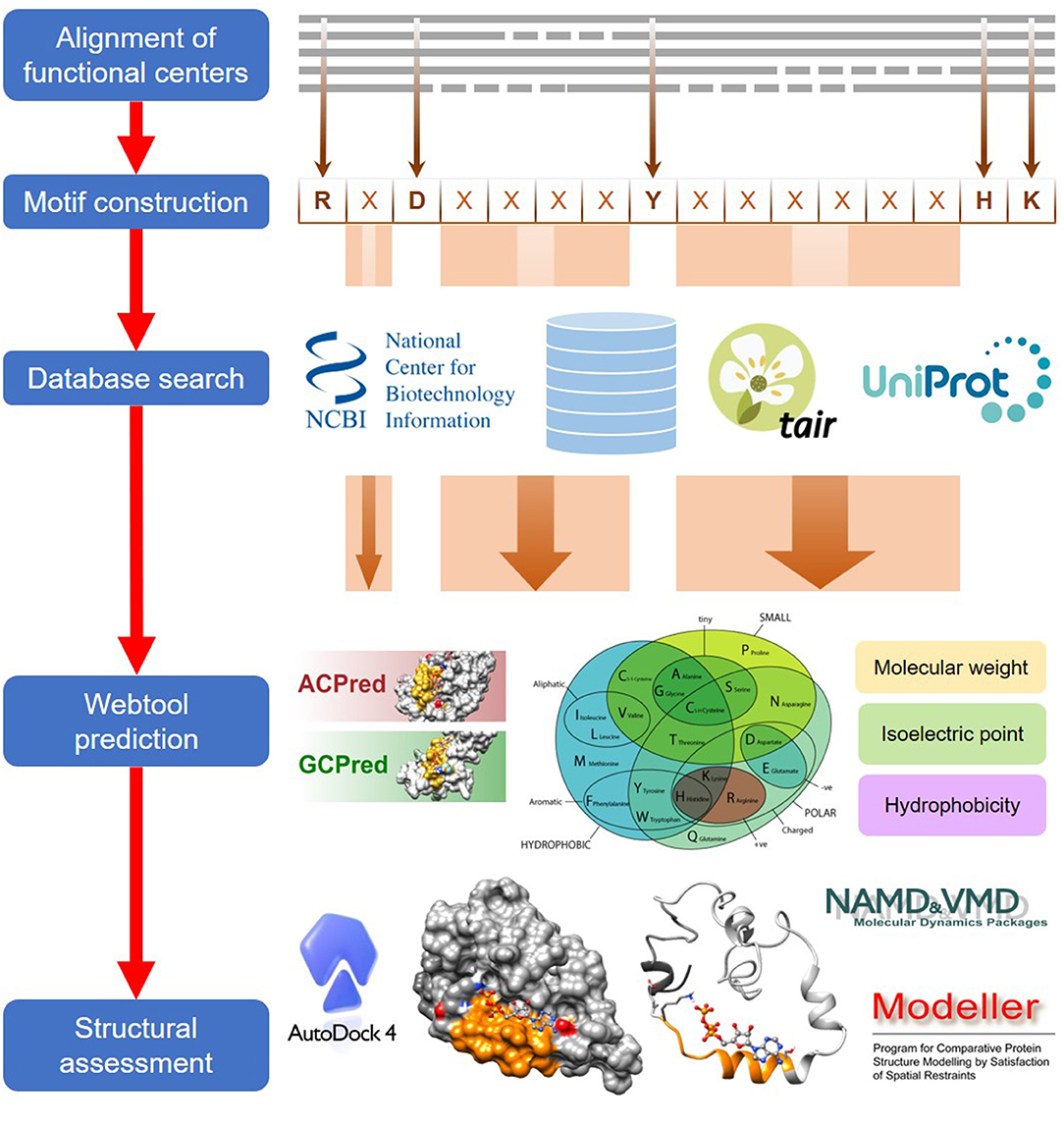
Figure 1. A general workflow for the computational identification of functional centers in complex proteins. The approach begins with an alignment of functional centers from proteins across species and followed by the construction of a consensus sequence that includes only conserved key amino acids which are separated by gaps as determined from the alignment. The consensus sequence now serves as a search motif to be queried on various databases to identify candidate functional centers and after which, they are screened on webtools if available, to assign confidence levels to the retrieved candidates. In the final step, top candidates are subjected to structural assessments that include model generations and docking simulations.
Step I: Alignment of Functional Centers
The first step of this approach involves the alignment of functional centers from a wide range of distantly related organisms. NOTE: The functional centers do not have to come from orthologs, and it is important to include sequences of experimentally validated proteins preferably from both prokaryotes and eukaryotes. This will increase the confidence of prediction by the amino acid motif constructed in the next phase. Functional centers may include catalytic sites, ligand- and hormone-binding sites or gas-sensing regions, as long as they are annotated as having a specific molecular function. NOTE: Only regions that directly participate in the molecular function should be considered as this protocol assumes a minimalistic strategy for functionality in highly diverse protein architectures. CAUTION: Functional centers typically range from 12 to 50 amino acids; an overly long sequence may reduce the chances of identifying promising candidates while a shorter sequence increases the chances of false positives.
Step II: Motif Construction
From the aligned sequences, highly conserved amino acids at each position in the alignment are included in a consensus sequence for the particular functional center. Amino acids that have been experimentally proven to perform key functions such as direct binding to ligands or essential for preserving certain charge and spatial configurations at the centers, are also included. These amino acids are indicated in square brackets []. NOTE: Key amino acids performing crucial molecular functions are also normally highly conserved at the functional centers of non-orthologous sequences and it is not uncommon for two or three amino acids with similar physicochemical properties to occupy a position. RECOMMENDATION: To be inclusive, we recommend including amino acids with similar chemical properties in the consensus sequence e.g., [RK] or [DE]. Detailed description of motif construction and its application have been described in Wong and Gehring (2013a,b), Wong et al. (2015, 2018).
Amino acids between the conserved residues and whose biochemical functions are unknown, may be excluded from the consensus sequence. They are indicated as “X” which stands for any of the 20 amino acids. From the aligned sequences, “X” may be several amino acids long and could assume differing ranges in centers from different species or non-orthologous sequences and the gap size is noted in brackets e.g., (N,M) or {N,M}, with “N” and “M” representing the minimum and maximum number of amino acids. CAUTION: Consult the pattern syntax of the respective tools to avoid errors.
After evaluating the conserved and non-conserved amino acids at each position of the alignment, the consensus sequence for the particular functional center is ready for application. CAUTION: Check the consensus sequence for errors and inconsistencies, e.g., odd or contrasting amino acid properties, at each position. RECOMMENDATION: A 12–50 amino acid long consensus sequence is typically a good starting point and motifs can be modified to increase or loosen stringency. NOTE: Shorter sequences can be considered if there are many specifically conserved residues and longer sequences can be considered if the conserved residues are too few and far apart. For a specific example of motif construction, please refer to the “Identification of heme-containing gas sensors in complex proteins” section in Results and Discussion and the sequence alignment of H-NOX domains in Supplementary Figure 1 or contact the corresponding author directly to obtain the most up to date datasets and output files.
Step III: Database Query
The consensus sequence now serves as a search motif to query any protein databases for candidate proteins harboring given functional centers. RECOMMENDATION: We recommend the use of the ScanProsite tool (Gattiker et al., 2002) available at: https://prosite.expasy.org/scanprosite that allows a search against protein database for known motifs (option 1) or with custom motifs (option 2). For proteins from Arabidopsis thaliana, the PatMatch tool (Yan et al., 2005) available at: https://www.arabidopsis.org/cgi-bin/patmatch/nph-patmatch.pl can be used. CAUTION: Consult the pattern syntax of the respective tools to avoid errors. NOTE: If the retrieved candidates are few, the consensus sequence (see Step II) can be revisited to relax the stringency of the motif by varying the gap sizes between conserved amino acids and/or by adding amino acids with similar chemical and physical properties to the conserved positions (e.g., instead of [IL] expand to [VIL]). CAUTION: A long list of candidates may indicate high false positive rates but at this point, this is not a concern because the retrieved hits can be conveniently screened and filtered with various webtools that provide statistics for confidence levels and candidate rankings in the next step. The motif only needs to be revised to increase stringency if no such webtools are currently available.
Step IV: Webtool Screening and Filtering
Candidates retrieved from the motif search can be screened with predictive tools for the particular molecular function if such tools are available. These webtools consider the physical and chemical properties of non-conserved amino acids in experimentally validated functional centers in addition to their conserved residues where their algorithms generate statistics that compare the queried sequence to the mean values of the experimentally validated pool of proteins (Xu et al., 2018a,b). Users can therefore use these statistics to rank retrieved hits from high to low confidence levels. RECOMMENDATION: We recommend the use of these webtools not only to increase confidence in the retrieved candidates but also to enable high-throughput processing of a long list of candidates. CAUTION: Although these webtools are designed to predict hits with high confidence, they remain predictive in nature and thus may not detect some positive hits especially in poorly characterized proteins. RECOMMENDATION: If the number of retrieved candidates is small and there are biochemical and cellular justifications supporting the functions of those hits, we recommend users to proceed with the next step involving structural assessment even if the webtool screening returns no hits. NOTE: At this point, it may be advantageous to inspect the candidate list and sort them into gene ontology (GO) categories to help selection of proteins with functions of particular interest before proceeding to the structural examination.
Step V: Structural Assessment
Candidates will now be assessed structurally and since this is the most computationally intense part and requires the most interpretation, the list of candidates should ideally be small. RECOMMENDATION: For a longer list of candidates, the top 5–10 proteins determined with the webtool (Step IV) can be selected for structural assessments. The assessment includes the generation of three-dimensional (3D) structures in case there are no known crystal structures for the candidates and conducting molecular docking simulations to predict functionality. NOTE: This phase requires installation of several software such as MODELLER, AutoDock Vina, MGLTools and UCSF Chimera, and a level of familiarity with these software. RECOMMENDATION: Users are recommended first to familiarize themselves with the operations of the required software packages before attempting this step, as this is a crucial phase for predicting functionality.
3D models should first be generated for candidates that have no known crystal structures by homology modeling using MODELLER (Sali and Blundell, 1993). This software generates protein 3D structures from amino acid sequences based on spatial restraints in template structures provided by the user. NOTE: See required software section for installation and registration guide. The five steps in homology modeling by MODELLER are: selection of crystal structures related to the candidate, template structure selection, alignment of candidate amino acid sequence to the template, model generation, and model evaluations. A detailed tutorial, the required program files, example input and output files in.zip format (for Windows) or.tar.gz format (for Unix/Linux), and the relevant scripts to run MODELLER can be found at: https://salilab.org/modeller/tutorial/basic.html. NOTE: These are the complete steps for homology modeling, but users can choose to perform alignments and template selection with other programs such as BLASTp (McGinnis and Madden, 2004) available at: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins. If using BLASTp, the “Protein Data Bank” database should be selected to only retrieve results with known crystal structures. CAUTION: Selection of template structure is a critical step in determining the quality of the model. If using BLASTp, a crystal structure with high identity with the queried candidate in the particular region where the functional center resides will be more suitable than another structure with higher overall coverage and/or max. score but lower identity at the corresponding functional center region. This is particularly relevant since moonlighting functional centers constitute only a small part of complex multi-domain proteins (Su et al., 2019).
Once a high-quality 3D model is obtained, molecular docking simulation can be performed using AutoDock Vina (Trott and Olson, 2010) which is an automated docking software for the prediction of the binding ligands to a receptor protein. NOTE: See required software for download and installation guides. CAUTION: Prior to running AutoDock Vina, necessary files must be prepared using AutoDockTools (ADT) (Morris et al., 2009) which is a graphical front-end software for setting up and running AutoDock. RECOMMENDATION: We recommend users to download the entire MGLTools package which include ADT and Python Molecular Viewer (PMV) (Sanner, 1999) for visualization of protein structures. Alternatively, users can also install UCSF Chimera (Pettersen et al., 2004) which is a tool for interactive visualization and analysis of molecular structures. All software downloads and installations are detailed in the software description. A detailed step-by-step tutorial on “.pdbqt” file preparation with ADT, example input and output files in.zip format, and running docking simulations using Vina and PMV is available at: http://vina.scripps.edu/tutorial.html. CAUTION: When setting up the grid box, the position and size determination is critical for the success of molecular docking. An overly small or off-position grid box will result in failure to identify suitable binding poses during docking simulation. The grid box volume should not only cover the entire area of the functional center but also be large enough to allow free rotation of the ligand at its most extended configuration.
After docking simulations with AudoDock Vina, binding poses can be evaluated on PMV or Chimera. By default, AutoDock Vina will rank binding poses based on free energies. NOTE: Binding poses of ligands at some functional centers are already known based on experimental data and, in such instances, these ligand orientations should be given priority over their free energy calculations. Further docking simulations can be conducted to evaluate the effect of mutations of key residues at the functional centers.
Results and Discussion
Using the general workflow as a guide, we describe specific applications using recent examples to detail the step-by-step process that led of the identification of catalytic centers and gas-sensing and hormone-interacting sites in complex proteins.
Identification of Catalytic Centers
Cyclic mononucleotide cyclases (guanylate cyclase and adenylate cyclase; GC and AC) are enzymes that catalyze the conversion of GTP and ATP to their cyclic forms, cGMP, and cAMP, respectively. In line with the concept of functional centers (Wong et al., 2018), only catalytic centers of canonical GCs and ACs are conserved non-orthologous cyclases. Currently available experimental data indicate that these enzymatic centers are often part of complex proteins with other primary domains and highly varied domain architectures (Figure 2).

Figure 2. Domain architecture of proteins containing nucleotide cyclase functional centers. The domain organizations of experimentally validated GCs and ACs identified using a motif-based approach from Arabidopsis thaliana (At), Brachypodium distachyon (Bd), Solanum lycopersicum (Sl), Hippeastrum (Hp), Pharbitis nil (Pn), and Homo sapiens (Hs), are illustrated as 2-dimensional bars and aligned at their corresponding GC/AC domains. Protein UniProt IDs are as follows: AtLRR (Q9LRR5), AtNCED (Q9LRR7), AtDGK4 (Q1PDI2), AtKUP5 (Q8LPL8), AtKUP7 (Q9FY75), AtClAP (Q9C9X5), AtPPR (Q9SXD8), HpAC1 (E1AQY1), BdTTM3 (I1I2P2), AtBRI1 (O22476) (see Supplementary Figure 2 for an illustration of AtBRI1 protein topology), AtPSKR1 (Q9ZVR7), AtPepR1 (Q9SSL9), AtWAKL10 (Q8VYA3), SlGC17 (A0A3Q7FS62), SlGC18 (A0A3Q7FY08), HsIRAK3 (Q9Y616), HpPepR1 (A0A1U9X9S6), HpGC1 (D9MWM6), PnGC1 (Q0PY32), AtGC1 (Q8L870), AtPNPR1 (F4HR92), and AtNOGC1 (Q9SXD9). *AC center undetermined.
A Step-by-Step Guide for the Identification of Cyclic Mononucleotide Cyclases
1. Alignment of catalytic centers of GCs or ACs from organisms across species including prokaryotes and eukaryotes. NOTE: For a specific example of motif construction, please refer to the “Identification of heme-containing gas sensors in complex proteins” section in Results and Discussion and the sequence alignment of H-NOX domains in Supplementary Figure 1 or contact the corresponding author directly to obtain the most up to date datasets and output files.
2. Determining the consensus sequence of GC or AC catalytic centers by including only the conserved key amino acids and separated by rational gaps made up of non-conserved amino acids. NOTE: The current consensus sequences for GC and AC functional centers are 14-amino acids long with negatively charged amino acids D or E (written as [DE] in the motif to indicate that both amino acids can function in this position) 0–3 positions downstream of the centers (Figure 3A).
3. Querying protein databases with GC or AC motifs to identify candidates GCs and ACs. NOTE: The currently tested motifs for GC and AC functional centers are [KS]x[CGS]x(10)[KR] and [RKS]x[DE]x(10)[KR], respectively (Figure 3). CAUTION: Consult the pattern syntax of the respective tools to avoid errors. Derivatives of these motifs justified by experimental evidence or rational modifications have also been described in detail (Kwezi et al., 2007; Mulaudzi et al., 2011; Bianchet et al., 2019; Ruzvidzo et al., 2019).
4. Application of GCPred and/or ACPred webtools [available at: http://gcpred.com (Xu et al., 2018a) and http://gcpred.com/acpred (Xu et al., 2018b)] for the assessment of confidence levels of retrieved candidates. These webtools contain algorithms that compare the physicochemical values of amino acids such as their molecular weights, isoelectric points and hydrophobicity to mean values of experimentally validated functional centers. RECOMMENDATION: We propose selecting the “cation-binding” option because current experimental data indicate that GCs and ACs of such nature have higher activities when these negatively charged amino acids [DE] are present at 1, 2, or 3 amino acids downstream of the functional centers. NOTE: According to the interpretation guide (Xu et al., 2018b), a score of 0-1 for each physicochemical parameter is generated, where 1 is closest to the mean of experimentally validated GC or AC functional center. Green scores represent “high” confidence and red ones represent “low” confidence. We also recommend that the users select candidates with at least two green physicochemical values in addition to the compulsory green overall mean score. Good candidates should contain no red scores. CAUTION: Although these webtools are designed to predict hits with high confidence, they remain predictive in nature and thus may not detect some positive hits especially in lesser studied proteins. RECOMMENDATION: If the retrieved list of candidates is small and there are justifications, e.g., from the literature, supporting the plausibility of the functions, we recommend that users proceed with the next phase involving structural assessment.
5. Model generation and docking simulation with MODELLER and AutoDock Vina to evaluate the structures of individual candidates and to predict the binding of substrate i.e., GTP or ATP, since this is a prerequisite for catalysis. NOTE: Based on experimentally validated GC and AC centers, they contain a characteristic alpha-helix barrel followed by a loop secondary fold (Wong and Gehring, 2013b; Wong et al., 2015, 2018; Al-Younis et al., 2018) (Figure 3A). Furthermore, experimentally validated GC and AC centers have the following substrate binding pose: purine bases adenine or guanine facing into the cavity toward amino acid at the first position of the motif in the hydrophobic interior while the phosphate end facing the positively charged amino acid [KR] at the opening of the cavity (Al-Younis et al., 2018; Ruzvidzo et al., 2019). If structural evaluations determine that such substrate orientations are feasible in new candidates, then the confidence level is further increased.
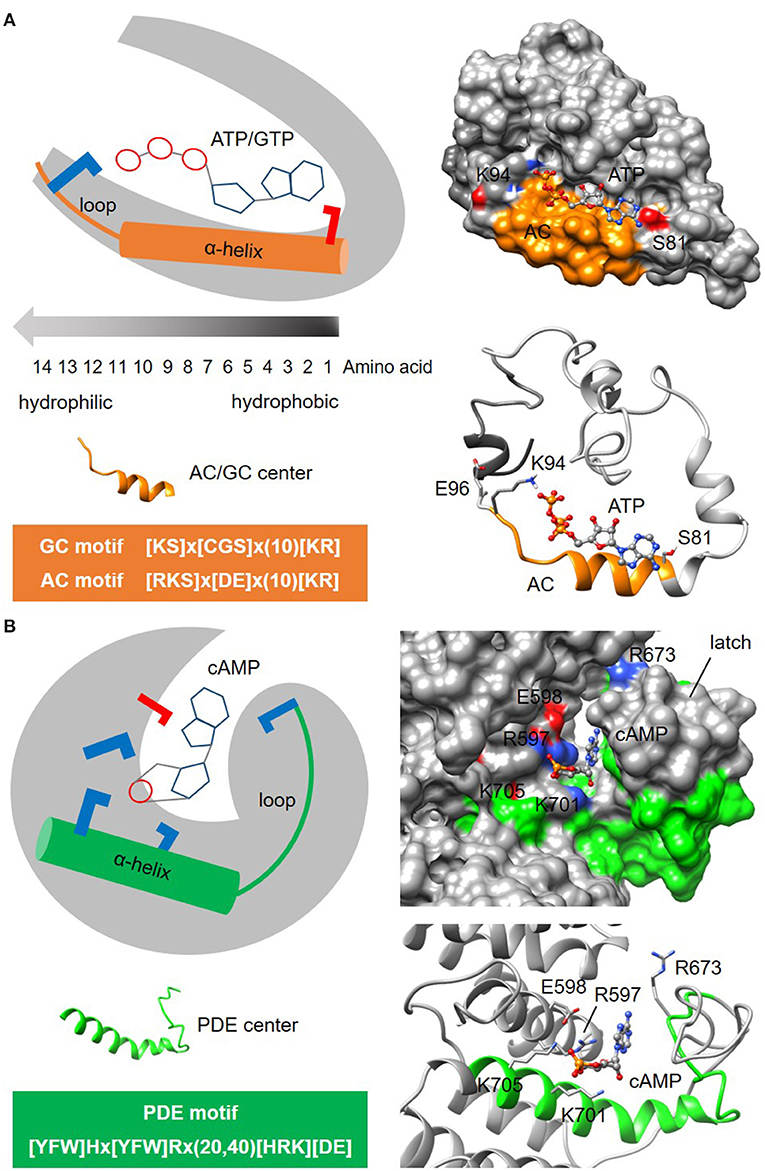
Figure 3. Representative structures of nucleotide cyclase and phosphodiesterase functional centers. (A) The typical nucleotide cyclase center identified through a 14-amino acid long search motif as exemplified by an adenylyl cyclase (AC) in an Arabidopsis potassium channel AtKUP5 (Al-Younis et al., 2018), assumes an alpha-helical secondary fold that is followed by a loop. At the tertiary level, the AC center typically forms a clear cavity that could dock with the substrate ATP in a binding pose where the adenine points into the cavity toward the amino acid at the first position of the motif, and the phosphate points outwards toward the positively charged [KR] amino acid at the solvent exposed region of the cavity. Negatively and positively charged amino acids that are crucial for the interactions with the substrate are colored red and blue, respectively. (B) The putative phosphodiesterase center in an Arabidopsis potassium channel AtKUP5 identified through a 27-47 amino acid long search motif, assumes an alpha-helical secondary fold that is followed by a loop which forms the latch region enclosing the docked cAMP substrate within a distinct cavity (Kwiatkowski et al., 2021). Negatively and positively charged amino acids crucial for the interactions with the substrate are colored red and blue, respectively.
In general, candidates retrieved from the motif search that rank highly in the webtool predictions and display the described structural features, will be considered suitable candidates for experimental verifications to ascertain their GC or AC activities both in vitro and in vivo.
Another example to illustrate the search for enzymatic functional centers are cyclic nucleotide phosphodiesterases (PDEs) which are enzymes that degrade the cyclic mononucleotides cGMP or cAMP. Much like nucleotide cyclase functional centers, PDEs may only retain the key conserved amino acids at the catalytic centers in complex multi-domain proteins and are therefore beyond the detection limit of BLAST searches. The steps involved in the identification of PDE centers follow the same guide as the cyclic mononucleotide cyclases. NOTE: Currently, the motif for PDE center is 27-47 amino acid long and is determined to be [YFW]Hx[YFW]Rx(20,40)[HRK][DE] (Figure 3B). CAUTION: The current PDE motif is stringent and identifies candidates with high confidence. As experimental evidence for PDE centers begin to emerge, the motif may eventually be relaxed to allow for the identification of novel candidates. In the absence of webtools for the prediction of PDE centers, retrieved candidates will be structurally evaluated in the next step. NOTE: Current model for PDE centers has a distinct cavity that accommodate cyclic mononucleotides within a latch. The PDE motif can form an alpha-helix secondary fold that is followed by a loop that makes up the latch (Kwiatkowski et al., 2021) (Figure 3B). Since PDE centers are less characterized than the centers of mononucleotide cyclases, the motif and structure presented here may yet undergo modifications that will improve prediction accuracy and coverage of hidden PDE centers.
Identification of Heme-Containing Gas Sensors in Complex Proteins
Heme-nitric oxide/oxygen (H-NOX) centers are heme-containing regions that can sense gases including the signaling molecule nitric oxide (NO). In line with the concept of functional centers (Wong et al., 2018), only amino acids at the H-NOX center occupied by the heme moiety, are conserved. The steps involved in the identification of heme-containing gas sensors follow the same guide as the cyclic mononucleotide cyclases. In this case, we show how an H-NOX consensus sequence can be constructed from the alignment of the heme-binding centers of H-NOX proteins from prokaryotes and eukaryotes. From the alignment, the “H” and “P” amino acids, as well as the “YxSxR” signature, are highly conserved across distantly related species (Figure 4A). Furthermore, these amino acids have been determined experimentally to have direct heme- and/or gas-binding functions at the H-NOX centers. For instance, the “H” residue in the motif serves as the distal ligand for NO (Olea et al., 2010) while the YxSxR signature stabilizes the porphyrin ring (Pellicena et al., 2004). Therefore, these amino acids are included in the consensus sequence with the non-conserved flanking amino acids that are not known to play functional roles at the H-NOX centers, assigned as “X” which stands for any of the 20 amino acids, and the gap size noted in brackets (N,M) or {N,M}, with “N” and “M” representing the minimum and maximum number of amino acids (Figure 4A). The constructed H-NOX consensus sequence was then used as a search term to identify heme-containing gas sensors in plants by querying the proteome of Arabidopsis thaliana and from which three candidates have since been experimentally confirmed to sense NO. NOTE: The current consensus sequence H-NOX center is 33–35 amino acids long and is determined to be Hx(12)Px(14,16)YxSxR (Figure 4). CAUTION: Consult the pattern syntax of the respective tools to avoid errors. Since there is currently no known webtools for the prediction of H-NOX centers, retrieved candidates will be directly subjected to structural evaluations in the next step. Current experimental data indicate that these H-NOX centers exist in complex proteins with other primary domains and varied architectures (Figure 4B). Mutations to either the “H” or “Y” residues impaired heme binding and concomitantly also the ability to sense NO (Figure 4C) (Zarban et al., 2019; Wong et al., 2020). In general, candidates retrieved from the motif search that display the described structural features, will be considered suitable candidates for experimental verifications to ascertain their gas-binding affinities both in vitro and in vivo. Currently, H-NOX motif is intentionally constructed to be rigid to identify candidates of high confidence especially in the absence of other predictive tools. Relaxation of the motif based on more data are likely to identify more heme-containing H-NOX proteins (Wong et al., 2021). RECOMMENDATION: We recommend omitting the “P” residue from the motif to increase coverage as this residue appears the least essential in terms of functionality.
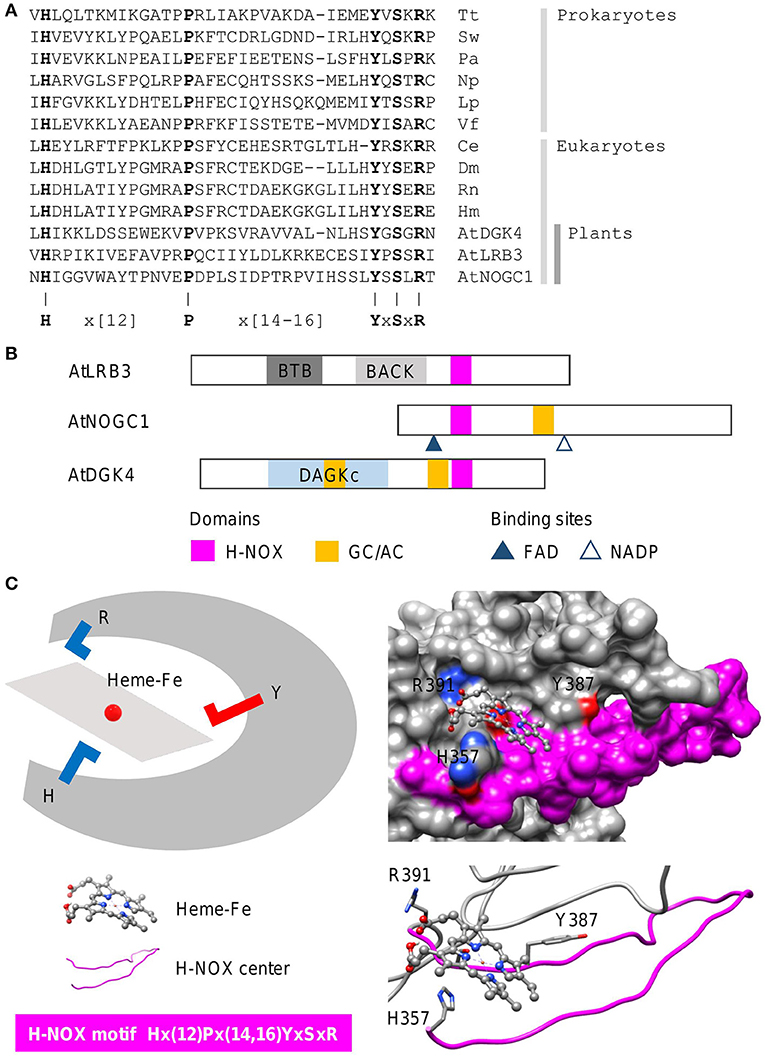
Figure 4. Sequence alignment and domain architecture of proteins containing H-NOX centers, and a representative structure of the H-NOX center. (A) Alignment of the heme-binding centers of H-NOX proteins from organisms across species. Tt, Thermoanaerobacter tengcongensis (UniProt ID: Q8RBX6); Sw, Shewanella woodyi (UniProt ID: B1KIH6); Pa, Pseudoalteromonas atlantica (UniProt ID: Q15VN4); Np, Nostoc punctiforme (UniProt ID: B2IZ76); Lp, Legionella pneumophila (UniProt ID: Q5WTZ5); Vf, Vibrio fischeri (UniProt ID: Q5E1F5); Ce, Caenorhabditis elegans (UniProt ID: Q86C56); Dm, Drosophila melanogaster (UniProt ID: Q24086); Rn, Rattus norvegicus (UniProt ID: P20595); Hs, Homo sapiens (UniProt ID: Q02153) and At, Arabidopsis thaliana. Bolded letters are conserved amino acids that are also experimentally shown to be crucial for heme-binding and stabilization. (B) The domain organizations of experimentally validated H-NOX centers identified using a motif-based approach from Arabidopsis thaliana (At), are illustrated as 2-dimensional bars, and aligned at their corresponding H-NOX centers (see Supplementary Figure 1 for full alignment of H-NOX domains). Protein UniProt IDs are as follows: AtLRB3 (O04615), AtDGK4 (Q1PDI2), and AtNOGC1 (Q9SXD9). (C) A representative structure of a protein containing the H-NOX center. The H-NOX center in an Arabidopsis BTB/POZ domain-containing protein AtLRB (Zarban et al., 2019), identified through a 33–35 amino acid long search motif, assumes a long loop that wraps the docked heme-Fe moiety within a clearly defined pocket. The “H” residue in the motif is the distal ligand that binds to the iron and the YxSxR signature stabilizes the heme through hydrogen bonding. Negatively and positively charged amino acids crucial for the interactions with the substrate are colored red and blue, respectively.
Identification of Hormone Binding Sites
In this case, we use the amino acids directly involved in binding to the plant hormone abscisic acid (ABA) from the well-characterized START protein family PYR/PYLs (Melcher et al., 2009; Park et al., 2009), to identify other plant proteins that can bind to or interact with ABA (Figure 5A). The steps involved in the identification of ABA-interacting centers follow the same guide as the cyclic mononucleotide cyclases. NOTE: The current consensus sequence for the ABA-interacting center is 26–28 amino acids long and is determined to be [DE]x(7,8)Rx(3,4)[DE]x(5)Yx(6)H (Figure 5). CAUTION: Consult the pattern syntax of the respective tools to avoid errors. Since there is currently no known webtools for the prediction of ABA-interacting centers, retrieved candidates will be directly subjected to structural evaluations in the next step. NOTE: Based on experimentally validated proteins containing the ABA-interacting centers, the “Y” residue in the motif together with the “K” located three amino acids upstream, are crucial for ABA binding (Figure 5B). Mutations to either the “Y” or “K” residues impair ABA binding and concomitantly other functions of the candidate protein e.g., K+ transport (Ooi et al., 2017). Currently, the ABA-interacting motif is intentionally constructed to be relatively rigid to identify candidates of high confidence especially in the absence of predictive tools, but it can conceivably be made more relaxed to identify more ABA-interacting proteins. RECOMMENDATION: We recommend increasing the gaps upstream of the “Y” and the “H” residues in the motif to allow for a more inclusive prediction.
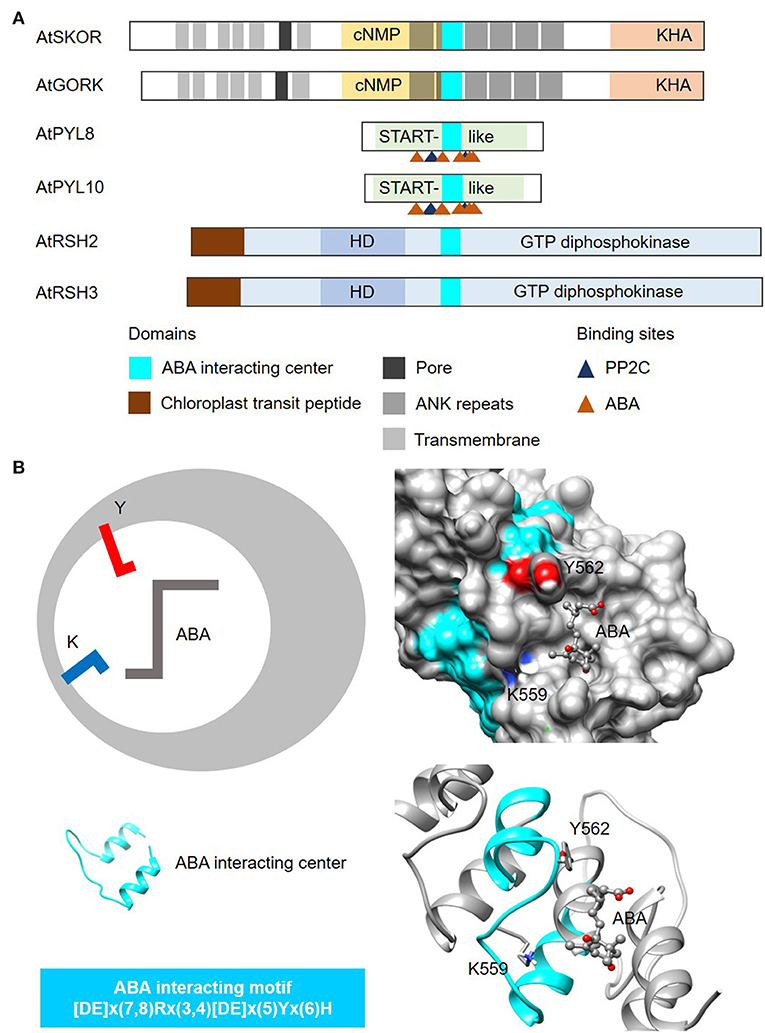
Figure 5. Domain architecture of proteins containing ABA-interacting centers and a representative structure of the ABA center. (A) The domain organizations of ABA-interacting centers identified using a motif-based approach from Arabidopsis thaliana (At), are illustrated as 2-dimensional bars and aligned at their corresponding H-NOX centers. Protein UniProt IDs are as follows: AtGORK (Q94A76), AtPYL8 (Q9FGM1), AtPYL10 (Q8H1R0), AtSKOR (Q9M8S6), AtRSH2 (Q9LVJ3) and AtRSH3 (Q9SYH1). AtGORK, AtPYL8 and AtPYL10 have been confirmed experimentally to be ABA receptors while AtSKOR, AtRSH2 and AtRSH3 harbor the ABA center motif and are known to response to ABA. (B) The ABA-interacting center in an Arabidopsis potassium transporter AtGORK (Ooi et al., 2017) identified through a 26-28 amino acid long search motif occupies a clear cavity that could dock with ABA with the “Y” and “K” residues being crucial for maintaining ABA affinity. Negatively and positively charged amino acids crucial for the interactions with the substrate are colored red and blue, respectively.
Conclusion and Future Perspective
We have developed a general workflow and presented a step-by-step guide for the applications of this computational approach to identify functional centers in complex proteins using recent examples as case studies. While this protocol leverages on existing software and follows standard methods in protein functional annotations, the focus is however on the identification of functional centers that unlike canonical domains, consist of only the key amino acids required to perform certain molecular functions in complex proteins. Instead of detecting broadly the common patterns or motifs in proteins, we derived our motifs from key residues of functional centers in existing domains that have been experimentally proven to be fully functional.
The initial motivation was to identify the corresponding signaling components of animals and bacteria (Loewenstein et al., 2009) in plant systems many of which, are long taught to be absent because there were no orthologs in plants. It was hypothesized that these domains have become markedly altered to retain only their functional centers as they get incorporated into complex multi-domain proteins such as that in the well-characterized Arabidopsis BRASSINOSTEROID INSENSITIVE 1 (AtBRI1; UniProt ID: O22476). The 1,196 amino acid long AtBRI1 consists of an extracellular brassinosteroid receptor region, a transmembrane, and an intracellular kinase domain with the GC center embedded within the primary kinase domain. Both the hormone receptor region and kinase domain take up ~62.7 and 23.0% of the protein while the GC center makes up only 0.12% of the entire protein (Supplementary Figure 2). Due to the diverse architecture of complex proteins, this protocol identifies functional centers that are more targeted and may not necessarily have similar 3D structures as it would be with canonical domains. Their incorporation into multi-domain complex proteins of varying primary functions, make these functional centers “hidden” or undetectable, hence not annotated in protein domains and families of existing databases.
Our application-specific CAUTIONS, NOTES and RECOMMENDATIONS will enable users to seamlessly apply the out-lined protocol and thereby increase the chances of success in the discovery of hidden functional centers or ligands in complex proteins as it informs users of potential pitfalls and gives suggestions for optimization. For instance, the recent identifications of functional nucleotide cyclase functional centers in crop plants such as tomato (Rahman et al., 2020) and rice (Malukani et al., 2020), and in other economically important plants such as ornamentals Hippeastrum sp. (Swiezawska et al., 2015, 2017) and Japanese morning glory (Szmidt-Jaworska et al., 2009) and in the monocot grass model Brachypodium (Swiezawska et al., 2020), as well as in a human immune-responsive protein IRAK3 (Freihat et al., 2019), have demonstrated the robustness of this approach and its significance in biological discoveries across different systems. Furthermore, the method enables candidate selections for follow-up in vivo and in planta studies that will eventually reveal the biological roles of these functional centers such as those reported in Joudoi et al. (2013), Shen et al. (2019), Vaz Dias et al. (2019), Angkawijaya et al. (2020), Lee et al. (2020), and Turek et al. (2020). We foresee that emerging experimental data will inform and strengthen motif refinement efforts, and enable the development of modern machine learning techniques that incorporate multiple features ranging from the classical physicochemical properties of protein domains and protein-protein interaction (PPI) networks to GO based function predictions, to not only automate annotations for uncharacterized proteins, but also to identify hidden functional centers in complex multi-functional proteins (Rifaioglu et al., 2019; Bonetta and Valentino, 2020; Cai et al., 2020; Littmann et al., 2021).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
AW conceived the project. WC, WZ, WS, WD, JW, and AW collected the data. AW, XT and CG analyzed the data. AW and CG wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
Work in the authors' laboratory was supported by the National Natural Science Foundation of China (31850410470) and the Zhejiang Provincial Natural Science Foundation of China (LQ19C130001) awarded to AW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Lee Staff for proofreading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2021.652286/full#supplementary-material
References
Al-Younis, I., Wong, A., Lemtiri-Chlieh, F., Schmöckel, S., Tester, M., Gehring, C., et al. (2018). The Arabidopsis thaliana K+-Uptake Permease 5 (AtKUP5) contains a functional cytosolic adenylate cyclase essential for K+ transport. Front. Plant Sci. 9:1645. doi: 10.3389/fpls.2018.01645
Angkawijaya, A. E., Nguyen, V. C., Gunawan, F., and Nakamura, Y. (2020). A pair of Arabidopsis diacylglycerol kinases essential for gametogenesis and endoplasmic reticulum phospholipid metabolism in leaves and flowers. Plant Cell 32, 2602–2620. doi: 10.1105/tpc.20.00251
Bianchet, C., Wong, A., Quaglia, M., Alqurashi, M., Gehring, C., Ntoukakis, V., et al. (2019). An Arabidopsis thaliana leucine-rich repeat protein harbors an adenylyl cyclase catalytic center and affects responses to pathogens. J. Plant Physiol. 232, 12–22. doi: 10.1016/j.jplph.2018.10.025
Bonetta, R., and Valentino, G. (2020). Machine learning techniques for protein function prediction. Proteins 88, 397–413. doi: 10.1002/prot.25832
Bowler, C., Neuhaus, G., Yamagata, H., and Chua, N. H. (1994). Cyclic GMP and calcium mediate phytochrome phototransduction. Cell 77, 73–81. doi: 10.1016/0092-8674(94)90236-4
Brady, S. M., Orlando, D. A., Lee, J.-Y., Wang, J. Y., Koch, J., Dinneny, J. R., et al. (2007). A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318, 801–806. doi: 10.1126/science.1146265
Cai, Y., Wang, J., and Deng, L. (2020). SDN2GO: an integrated deep learning model for protein function prediction. Front. Bioeng. Biotechnol. 8:391. doi: 10.3389/fbioe.2020.00391
Chatukuta, P., Dikobe, T. B., Kawadza, D. T., Sehlabane, K. S., Takundwa, M. M., Wong, A., et al. (2018). An Arabidopsis clathrin assembly protein with a predicted role in plant defense can function as an adenylate cyclase. Biomolecules 8:15. doi: 10.3390/biom8020015
Domingos, P., Prado, A. M., Wong, A., Gehring, C., and Feijo, J. A. (2015). Nitric oxide: a multitasked signaling gas in plants. Mol. Plant 8, 506–520. doi: 10.1016/j.molp.2014.12.010
Donaldson, L., Ludidi, N., Knight, M. R., Gehring, C., and Denby, K. (2004). Salt and osmotic stress cause rapid increases in Arabidopsis thaliana cGMP levels. FEBS. Lett. 569, 317–320. doi: 10.1016/j.febslet.2004.06.016
Durner, J., Wendehenne, D., and Klessig, D. F. (1998). Defense gene induction in tobacco by nitric oxide, cyclic GMP, and cyclic ADP-ribose. Proc. Natl. Acad. Sci. U.S.A. 95, 10328–10333. doi: 10.1073/pnas.95.17.10328
Ederli, L., Meier, S., Borgogni, A., Reale, L., Ferranti, F., Gehring, C., et al. (2009). Ozone and nitric oxide induce cGMP-dependent and -independent transcription of defence genes in tobacco. New. Phytol. 181, 860–870. doi: 10.1111/j.1469-8137.2008.02711.x
Feijo, J. A., Costa, S. S., Prado, A. M., Becker, J. D., and Certal, A. C. (2004). Signalling by tips. Curr. Opin. Plant.Biol. 7, 589–598. doi: 10.1016/j.pbi.2004.07.014
Freihat, L., Muleya, V., Manallack, D. T., Wheeler, J. I., and Irving, H. R. (2014). Comparison of moonlighting guanylate cyclases: roles in signal direction? Biochem. Soc. Trans. 42, 1773–1779. doi: 10.1042/BST20140223
Freihat, L. A., Wheeler, J. I., Wong, A., Turek, I., Manallack, D. T., and Irving, H. R. (2019). IRAK3 modulates downstream innate immune signalling through its guanylate cyclase activity. Sci. Rep. 9:15468. doi: 10.1038/s41598-019-51913-3
Gattiker, A., Gasteiger, E., and Bairoch, A. (2002). ScanProsite: a reference implementation of a PROSITE scanning tool. Appl. Bioinformatics 1, 107–108.
Gehring, C. (2010). Adenyl cyclases and cAMP in plant signaling - past and present. Cell Commun. Signal. 8:15. doi: 10.1186/1478-811X-8-15
Gehring, C., and Turek, I. S. (2017). Cyclic nucleotide monophosphates and their cyclases in plant signaling. Front. Plant Sci. 8:1704. doi: 10.3389/fpls.2017.01704
Guo, T., and Fang, Y. (2014). Functional organization and dynamics of the cell nucleus. Front. Plant Sci. 5:378. doi: 10.3389/fpls.2014.00378
Hartwig, C., Bahre, H., Wolter, S., Beckert, U., Kaever, V., and Seifert, R. (2014). cAMP, cGMP, cCMP and cUMP concentrations across the tree of life: High cCMP and cUMP levels in astrocytes. Neurosci. Lett. 579, 183–187. doi: 10.1016/j.neulet.2014.07.019
Hussain, J., Chen, J., Locato, V., Sabetta, W., Behera, S., Cimini, S., et al. (2016). Constitutive cyclic GMP accumulation in Arabidopsis thaliana compromises systemic acquired resistance induced by an avirulent pathogen by modulating local signals. Sci. Rep. 6:36423. doi: 10.1038/srep36423
Irving, H. R., Cahill, D. M., and Gehring, C. (2018). Moonlighting Proteins and Their Role in the Control of Signaling Microenvironments, as Exemplified by cGMP and Phytosulfokine Receptor 1 (PSKR1). Front. Plant Sci. 9:415. doi: 10.3389/fpls.2018.00415
Irving, H. R., Kwezi, L., Wheeler, J., and Gehring, C. (2012). Moonlighting kinases with guanylate cyclase activity can tune regulatory signal networks. Plant Signal. Behav. 7:201–204. doi: 10.4161/psb.18891
Isner, J. C., and Maathuis, F. J. (2011). Measurement of cellular cGMP in plant cells and tissues using the endogenous fluorescent reporter FlincG. Plant J. 65:329–334. doi: 10.1111/j.1365-313X.2010.04418.x
Isner, J. C., Nuhse, T., and Maathuis, F. J. (2012). The cyclic nucleotide cGMP is involved in plant hormone signalling and alters phosphorylation of Arabidopsis thaliana root proteins. J. Exp. Bot. 63, 3199–3205. doi: 10.1093/jxb/ers045
Jeffery, C. J. (1999). Moonlighting proteins. Trends Biochem. Sci. 24, 8–11. doi: 10.1016/S0968-0004(98)01335-8
Joudoi, T., Shichiri, Y., Kamizono, N., Akaike, T., Sawa, T., Yoshitake, J., et al. (2013). Nitrated cyclic GMP modulates guard cell signaling in Arabidopsis. Plant Cell 25, 558–571. doi: 10.1105/tpc.112.105049
Kwezi, L., Meier, S., Mungur, L., Ruzvidzo, O., Irving, H., and Gehring, C. (2007). The Arabidopsis thaliana brassinosteroid receptor (AtBRI1) contains a domain that functions as a guanylyl cyclase in vitro. PLoS ONE 2:e449. doi: 10.1371/journal.pone.0000449
Kwiatkowski, M., Wong, A., Kozakiewicz, A., Gehring, C., and Jaworski, K. (2021). A tandem motif-based and structural approach can identify hidden functional phosphodiesterases. Comput, Struct. Biotechnol. J. 19, 970–975. doi: 10.1016/j.csbj.2021.01.036
Lee, D., Redfern, O., and Orengo, C. (2007). Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 8, 995–1005. doi: 10.1038/nrm2281
Lee, K. P., Liu, K., Kim, E. Y., Medina-Puche, L., Dong, H., Duan, J., et al. (2020). Plant natriuretic peptide A and its putative receptor PNP-R2 antagonize salicylic acid-mediated signaling and cell death. Plant Cell 32, 2237–2250. doi: 10.1105/tpc.20.00018
Levskaya, A., Weiner, O. D., Lim, W. A., and Voigt, C. A. (2009). Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001. doi: 10.1038/nature08446
Littmann, M., Heinzinger, M., Dallago, C., Olenyi, T., and Rost, B. (2021). Embeddings from deep learning transfer GO annotations beyond homology. Sci. Rep. 11:1160. doi: 10.1038/s41598-020-80786-0
Loewenstein, Y., Raimondo, D., Redfern, O. C., Watson, J., Frishman, D., Linial, M., et al. (2009). Protein function annotation by homology-based inference. Genome Biol. 10, 207. doi: 10.1186/gb-2009-10-2-207
Ludidi, N., and Gehring, C. (2003). Identification of a novel protein with guanylyl cyclase activity in Arabidopsis thaliana. J. Biol. Chem. 278, 6490–6494. doi: 10.1074/jbc.M210983200
Maathuis, F. J., and Sanders, D. (2001). Sodium uptake in Arabidopsis roots is regulated by cyclic nucleotides. Plant Physiol. 127, 1617–1625. doi: 10.1104/pp.010502
Mahlich, Y., Steinegger, M., Rost, B., and Bromberg, Y. (2018). HFSP: high speed homology-driven function annotation of proteins. Bioinformatics 34, i304–i312. doi: 10.1093/bioinformatics/bty262
Malukani, K. K., Ranjan, A., Hota, S. J., Patel, H. K., and Sonti, R. V. (2020). Dual activities of receptor-like kinase OsWAKL21.2 induce immune responses. Plant Physiol. 183, 1345–1363. doi: 10.1104/pp.19.01579
Marchler-Bauer, A., Lu, S., Anderson, J. B., Chitsaz, F., Derbyshire, M. K., DeWeese-Scott, C., et al. (2011). CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39(Database issue), D225–D229. doi: 10.1093/nar/gkq1189
Marondedze, C., Groen, A. J., Thomas, L., Lilley, K. S., and Gehring, C. (2016). A quantitative phosphoproteome analysis of cGMP-dependent cellular responses in Arabidopsis thaliana. Mol. Plant 9:621–623. doi: 10.1016/j.molp.2015.11.007
Martinez-Atienza, J., Van Ingelgem, C., Roef, L., and Maathuis, F. J. (2007). Plant cyclic nucleotide signalling: facts and fiction. Plant Signal. Behav. 2, 540–543. doi: 10.4161/psb.2.6.4789
McGinnis, S., and Madden, T. L. (2004). BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–25. doi: 10.1093/nar/gkh435
McInnis, S. M., Desikan, R., Hancock, J. T., and Hiscock, S. J. (2006). Production of reactive oxygen species and reactive nitrogen species by angiosperm stigmas and pollen: Potential signalling crosstalk? New Phytol. 172, 221–228. doi: 10.1111/j.1469-8137.2006.01875.x
Melcher, K., Ng, L. M., Zhou, X. E., Soon, F. F., Xu, Y., Suino-Powell, K. M., et al. (2009). A gate-latch-lock mechanism for hormone signalling by abscisic acid receptors. Nature 462, 602–608. doi: 10.1038/nature08613
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). Autodock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 16, 2785–2791. doi: 10.1002/jcc.21256
Mulaudzi, T., Ludidi, N., Ruzvidzo, O., Morse, M., Hendricks, N., Iwuoha, E., et al. (2011). Identification of a novel Arabidopsis thaliana nitric oxide-binding molecule with guanylate cyclase activity in vitro. FEBS Lett. 585, 2693–2697. doi: 10.1016/j.febslet.2011.07.023
Muleya, V., Wheeler, J. I., Ruzvidzo, O., Freihat, L., Manallack, D. T., Gehring, C., et al. (2014). Calcium is the switch in the moonlighting dual function of the ligand-activated receptor kinase phytosulfokine receptor 1. Cell Commun. Signal. 12:60. doi: 10.1186/s12964-014-0060-z
Neuhaus, G., Bowler, C., Hiratsuka, K., Yamagata, H., and Chua, N. H. (1997). Phytochrome-regulated repression of gene expression requires calcium and cGMP. EMBO J. 16, 2554–2564. doi: 10.1093/emboj/16.10.2554
Olea, C., Herzik, M. A., Kuriyan, J., and Marletta, M. A. (2010). Structural insights into the molecular mechanism of H-NOX activation. Protein Sci. 19, 881–887. doi: 10.1002/pro.357
Ooi, A., Lemtiri-Chlieh, F., Wong, A., and Gehring, C. (2017). Direct modulation of the guard cell outward-rectifying potassium channel (GORK) by abscisic acid. Mol. Plant 10, 1469–1472. doi: 10.1016/j.molp.2017.08.010
Park, S. Y., Fung, P., Nishimura, N., Jensen, D. R., Fujii, H., Zhao, Y., et al. (2009). Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 324, 1068–1071. doi: 10.1126/science.1173041
Pasqualini, S., Cresti, M., Del Casino, C., Faleri, C., Frenguelli, G., Tedeschini, E., et al. (2015). Roles for NO and ROS signalling in pollen germination and pollen-tube elongation in Cupressus arizonica. Biol. Plant. 59, 735–744. doi: 10.1007/s10535-015-0538-6
Pasqualini, S., Tedeschini, E., Frenguelli, G., Wopfner, N., Ferreira, F., D'Amato, G., et al. (2011). Ozone affects pollen viability and NAD(P)H oxidase release from Ambrosia artemisiifolia pollen. Environ Pollut. 159:2823–30. doi: 10.1016/j.envpol.2011.05.003
Pellicena, P., Karow, D. S., Boon, E. M., Marletta, M. A., and Kuriyan, J. (2004). Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc. Natl. Acad. Sci. U.S.A. 101, 12854–12859. doi: 10.1073/pnas.0405188101
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Prado, A. M., Colaço, R., Moreno, N., Silva, A. C., and Feijó, J. A. (2008). Targeting of pollen tubes to ovules is dependent on nitric oxide (NO) signaling. Mol. Plant 1, 703–714. doi: 10.1093/mp/ssn034
Prado, A. M., Porterfield, D. M., and Feijó, J. A. (2004). Nitric oxide is involved in growth regulation and re-orientation of pollen tubes. Development 131, 2707–2714. doi: 10.1242/dev.01153
Rahman, H., Wang, X. Y., Xu, Y. P., He, Y.-H., and Cai, X.-Z. (2020). Characterization of tomato protein kinases embedding guanylate cyclase catalytic center motif. Sci. Rep. 10:4078. doi: 10.1038/s41598-020-61000-7
Rifaioglu, A. S., Dogan, T., Martin, M. J., Cetin-Atalay, R., and Atalay, V. (2019). DEEPred: automated protein function prediction with multi-task feed-forward deep neural networks. Sci. Rep. 9:7344. doi: 10.1038/s41598-019-43708-3
Ruzvidzo, O., Gehring, C., and Wong, A. (2019). New perspectives on plant adenylyl cyclases. Front. Mol. Biosci. 6:136. doi: 10.3389/fmolb.2019.00136
Sali, A., and Blundell, T. L. (1993). Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815. doi: 10.1006/jmbi.1993.1626
Sanner, M. F. (1999). Python: a programming language for software integration and development. J. Mol. Graphics Mod. 17, 57–61.
Shen, Q., Zhan, X., Yang, P., Li, J., Chen, J., Tang, B., et al. (2019). Dual activities of plant cGMP-dependent protein kinase and its roles in gibberellin signaling and salt stress. Plant Cell 31, 3073–3091. doi: 10.1105/tpc.19.00510
Sigrist, C. J., de Castro, E., Cerutti, L., Cuche, B. A., Hulo, N., Bridge, A., et al. (2013). New and continuing developments at PROSITE. Nucleic Acids Res. 41(Database issue), D344–D347. doi: 10.1093/nar/gks1067
Su, B., Qian, Z., Li, T., Zhou, Y., and Wong, A. (2019). PlantMP: a database for moonlighting plant proteins. Database 2019:baz050. doi: 10.1093/database/baz050
Swiezawska, B., Duszyn, M., Jaworski, K., and Szmidt-Jaworska, A. (2018). Downstream targets of cyclic nucleotides in plants. Front. Plant Sci. 9:1428. doi: 10.3389/fpls.2018.01428
Swiezawska, B., Duszyn, M., Kwiatkowski, M., Jaworski, K., Pawełek, A., and Szmidt-Jaworska, A. (2020). Brachypodium distachyon triphosphate tunnel metalloenzyme 3 is both a triphosphatase and an adenylyl cyclase upregulated by mechanical wounding. FEBS Lett. 594, 1101–1111. doi: 10.1002/1873-3468.13701
Swiezawska, B., Jaworski, K., Duszyn, M., Pawełek, A., and Szmidt-Jaworska, A. (2017). The Hippeastrum hybridum PepR1 gene (HpPepR1) encodes a functional guanylyl cyclase and is involved in early response to fungal infection. J. Plant Physiol. 216, 100–107. doi: 10.1016/j.jplph.2017.05.024
Swiezawska, B., Jaworski, K., Szewczuk, P., Pawełek, A., and Szmidt-Jaworska, A. (2015). Identification of a Hippeastrum hybridum guanylyl cyclase responsive to wounding and pathogen infection. J. Plant Physiol. 189, 77–86. doi: 10.1016/j.jplph.2015.09.014
Swiezawska-Boniecka, B., Duszyn, M., Kwiatkowski, M., Szmidt-Jaworska, A., and Jaworski, K. (2021). Cross talk between cyclic nucleotides and calcium signaling pathways in plants-achievements and prospects. Front. Plant Sci. 12:643560. doi: 10.3389/fpls.2021.643560
Szmidt-Jaworska, A., Jaworski, K., Pawelek, A., and Kopcewicz, J. (2009). Molecular cloning and characterization of a guanylyl cyclase, PNGC-1, involved in light signaling in Pharbitis nil. J. Plant Growth Regul. 28, 367–380. doi: 10.1007/s00344-009-9105-8
Trott, O., and Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. doi: 10.1002/jcc.21334
Turek, I., and Gehring, C. (2016). The plant natriuretic peptide receptor is a guanylyl cyclase and enables cGMP-dependent signaling. Plant Mol. Biol. 91, 275–286. doi: 10.1007/s11103-016-0465-8
Turek, I., and Irving, H. (2021). Moonlighting proteins shine new light on molecular signaling niches. Int. J. Mol. Sci. 22:1367. doi: 10.3390/ijms22031367
Turek, I., Wheeler, J., Bartels, S., Szczurek, J., Wang, Y. H., Taylor, P., et al. (2020). A natriuretic peptide from Arabidopsis thaliana (AtPNP-A) can modulate catalase 2 activity. Sci. Rep. 10:19632. doi: 10.1038/s41598-020-76676-0
Vaz Dias, F., Serrazina, S., Vitorino, M., Marchese, D., Heilmann, I., Godinho, M., et al. (2019). A role for diacylglycerol kinase 4 in signalling crosstalk during Arabidopsis pollen tube growth. New Phytol. 222, 1434–1446. doi: 10.1111/nph.15674
Wang, Y., Chen, T., Zhang, C., Hao, H., Liu, P., Zheng, M., et al. (2009). Nitric oxide modulates the influx of extracellular Ca2+ and actin filament organization during cell wall construction in Pinus bungeana pollen tubes. New Phytol. 182, 851–862. doi: 10.1111/j.1469-8137.2009.02820.x
Wang, Y.-H., Li, X.-C., Zhu-Ge, Q., Jiang, X., Wang, W. D., Fang, W. P., et al. (2012). Nitric oxide participates in cold-inhibited Camellia sinensis pollen germination and tube growth partly via cGMP in vitro. PLoS ONE 7:e52436. doi: 10.1371/journal.pone.0052436
Wheeler, J. I., Wong, A., Marondedze, C., Groen, A. J., Kwezi, L., Freihat, L., et al. (2017). The brassinosteroid receptor BRI1 can generate cGMP enabling cGMP-dependent downstream signaling. Plant J. 91, 590–600. doi: 10.1111/tpj.13589
Wong, A., Donaldson, L., Portes, M. T., Eppinger, J., Feijó, J. A., and Gehring, C. (2020). Arabidopsis Diacylglycerol Kinase4 is involved in nitric oxide-dependent pollen tube guidance and fertilization. Development 147:dev183715. doi: 10.1242/dev.183715
Wong, A., and Gehring, C. (2013a). Computational identification of candidate nucleotide cyclases in higher plants. Methods Mol. Biol. 1016, 195–205. doi: 10.1007/978-1-62703-441-8_13
Wong, A., and Gehring, C. (2013b). The Arabidopsis thaliana proteome harbors undiscovered multi-domain molecules with functional guanylyl cyclase catalytic centers. Cell Commun. Signal. 11:48. doi: 10.1186/1478-811X-11-48
Wong, A., Gehring, C., and Irving, H. R. (2015). Conserved functional motifs and homology modelling to predict hidden moonlighting functional sites. Front. Bioeng. Biotechnol. 3:82. doi: 10.3389/fbioe.2015.00082
Wong, A., Tian, X., Gehring, C., and Marondedze, C. (2018). Discovery of novel functional centers with rationally designed amino acid motifs. Comput. Struct. Biotechnol. J. 16, 70–76. doi: 10.1016/j.csbj.2018.02.007
Wong, A., Tian, X., Yang, Y., and Gehring, C. (2021). Identification of potential nitric oxide sensing proteins using the H-NOX motif. Mol. Plant 14, 195–197. doi: 10.1016/j.molp.2020.11.015
Wu, C. H., Huang, H., Yeh, L. S., and Barker, W. C. (2003). Protein family classification and functional annotation. Comput. Biol. Chem. 27, 37–47. doi: 10.1016/S1476-9271(02)00098-1
Xu, N., Fu, D., Li, S., Wang, Y., and Wong, A. (2018a). GCPred: a web tool for guanylyl cyclase functional centre prediction from amino acid sequence. Bioinformatics 34, 2134–2135. doi: 10.1093/bioinformatics/bty067
Xu, N., Zhang, C., Lim, L. L., and Wong, A. (2018b). “Bioinformatic analysis of nucleotide cyclase functional centers and development of ACPred webserver,” in Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics (BCB '18). (New York, NY: Association for Computing Machinery), 122–129. doi: 10.1145/3233547.3233549
Yan, T., Yoo, D., Berardini, T. Z., Mueller, L. A., Weems, D. C., Weng, S., et al. (2005). PatMatch: a program for finding patterns in peptide and nucleotide sequences. Nucleic Acids Res. 33, W262–W266. doi: 10.1093/nar/gki368
Zarban, R., Vogler, M., Wong, A., Eppinger, J., Al-Babili, S., and Gehring, C. (2019). Discovery of a nitric oxide-responsive protein in Arabidopsis thaliana. Molecules 24, 2691. doi: 10.3390/molecules24152691
Keywords: functional centers, moonlighting proteins, hidden domains, guanylate cyclase, adenylyl cyclase, nitric oxide sensors, abscisic acid receptor, H-NOX
Citation: Zhou W, Chi W, Shen W, Dou W, Wang J, Tian X, Gehring C and Wong A (2021) Computational Identification of Functional Centers in Complex Proteins: A Step-by-Step Guide With Examples. Front. Bioinform. 1:652286. doi: 10.3389/fbinf.2021.652286
Received: 12 January 2021; Accepted: 02 March 2021;
Published: 25 March 2021.
Edited by:
Jiangning Song, Monash University, AustraliaReviewed by:
Selvaraj Samuel, Bharathidasan University, IndiaFrank Eisenhaber, Bioinformatics Institute (A*STAR), Singapore
Copyright © 2021 Zhou, Chi, Shen, Dou, Wang, Tian, Gehring and Wong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aloysius Wong, YWx3b25nQGtlYW4uZWR1
†These authors have contributed equally to this work