 Ziyuan Chen
Ziyuan Chen Laurent Geffroy2
Laurent Geffroy2 Julie S. Biteen
Julie S. Biteen- 1Department of Biophysics, University of Michigan, Ann Arbor, MI, United States
- 2Department of Chemistry, University of Michigan, Ann Arbor, MI, United States
Single particle tracking (SPT) enables the investigation of biomolecular dynamics at a high temporal and spatial resolution in living cells, and the analysis of these SPT datasets can reveal biochemical interactions and mechanisms. Still, how to make the best use of these tracking data for a broad set of experimental conditions remains an analysis challenge in the field. Here, we develop a new SPT analysis framework: NOBIAS (NOnparametric Bayesian Inference for Anomalous Diffusion in Single-Molecule Tracking), which applies nonparametric Bayesian statistics and deep learning approaches to thoroughly analyze SPT datasets. In particular, NOBIAS handles complicated live-cell SPT data for which: the number of diffusive states is unknown, mixtures of different diffusive populations may exist within single trajectories, symmetry cannot be assumed between the x and y directions, and anomalous diffusion is possible. NOBIAS provides the number of diffusive states without manual supervision, it quantifies the dynamics and relative populations of each diffusive state, it provides the transition probabilities between states, and it assesses the anomalous diffusion behavior for each state. We validate the performance of NOBIAS with simulated datasets and apply it to the diffusion of single outer-membrane proteins in Bacteroides thetaiotaomicron. Furthermore, we compare NOBIAS with other SPT analysis methods and find that, in addition to these advantages, NOBIAS is robust and has high computational efficiency and is particularly advantageous due to its ability to treat experimental trajectories with asymmetry and anomalous diffusion.
Introduction
The biophysical dynamics of biomolecules reflect the biochemical interactions in the system, and these dynamics can be quantified within a dataset of single-particle trajectories obtained by tracking individual molecules. The invention of the super-resolution microscope (Moerner and Kador, 1989; Hell and Wichmann, 1994; Betzig et al., 2006; Hess et al., 2006; Rust et al., 2006) and single-particle tracking (SPT) methods (Yildiz et al., 2003; Deich et al., 2004; Elmore et al., 2005; Manley et al., 2008) have made possible investigations of biomolecular dynamics at a high temporal and spatial resolution both in vitro and in vivo. Moreover, quantitative SPT algorithms can connect the real-time dynamics from biophysical trajectories to biochemical roles to uncover whether a molecule interacts with other cellular components (Izeddin et al., 2014), freely diffuses (Badrinarayanan et al., 2012), is actively transported (Park et al., 2014), or is constrained to a certain region (Bayas et al., 2018).
Conventionally, SPT trajectory datasets have been assumed to be Brownian, such that the mean squared displacement, MSD, of each track is linearly proportional to the time lag,
For some well-studied biological systems in which the biochemical states of molecules have been determined through other methods, a fixed-state number analytical tool can be suitable for quantifying the dynamics and weight for each state (Elf et al., 2007; Hansen et al., 2017). However, SPT can also be used as the beginning step to investigate biomolecule dynamics without prior knowledge of how many diffusive states there supposed to be (Monnier et al., 2015; Sungkaworn et al., 2017; Biswas et al., 2021). In these cases, how to objectively determine the number of diffusive states is a great challenge. Moreover, these models provide a D value for each subpopulation, but they do not assign the diffusive state to each individual single-molecule step, nor do they quantify the transition probability between distinct diffusive states within one trajectory. However, these transition probabilities can reveal important biological meaning such as the presence of critical biochemical intermediates (Biswas et al., 2021).
Bayesian statistics and Hidden Markov Models (HMMs) have been applied to analyze SPT datasets without assuming a predetermined number of diffusive states and to access the probabilities of transitioning between distinct states (Persson et al., 2013; Monnier et al., 2015; Karslake et al., 2020; Heckert et al., 2021). vbSPT, which was one of the first applications of HMM for SPT analysis (Persson et al., 2013), uses a maximum-evidence criterion to select between models with different numbers of diffusive states; within each model, a fixed-order HMM is used to infer the diffusion coefficient, weight fraction, and transition probabilities for each state. More recently, nonparametric Bayesian models based on Dirichlet processes were combined with HMM to recover the number of diffusive states from SPT trajectory datasets, such as in SMAUG (Karslake et al., 2020) and DSMM (Heckert et al., 2021). In these models, the motion of the molecule is approximated to be symmetric and Brownian, which is an oversimplification considering the crowded environment and various interaction partners for biomolecules in cells.
To move beyond Brownian motion, here we consider a more general random walk family: anomalous diffusion. In anomalous diffusion, MSD and
Here we introduce the NOnparametric Bayesian Inference for Anomalous diffusion in Single-molecule tracking (NOBIAS) framework to address the assumptions and simplifications discussed above and provide a more physiologically relevant analysis algorithm to quantify the dynamics encoded in SPT datasets (Figure 1). In particular, NOBIAS recovers the number of diffusive states and predict the diffusion type for each diffusive state, even in heterogeneous trajectories. The NOBIAS framework consists of two modules. The first module uses a Hierarchical Dirichlet Process Hidden Markov Model (HDP-HMM) with multivariate Gaussian emission to recover the number of diffusive states and infer their corresponding diffusion coefficients and weight fractions. This module also assigns each single-molecule step a diffusive state label to provide the state label sequence and the matrix of transition probabilities. In the second module, the original trajectories are segmented by diffusive state label and a pre-trained Recurrent Neural Network (RNN) is used to classify these segments and assign the diffusion type (Brownian motion, Fractional Brownian motion, Continuous Time Random Walk, or Lévy Walk) for each diffusive state. We simulated trajectory datasets with mixtures of heterogeneous dynamics and diffusion types to validate the NOBIAS framework, and we analyzed the SPT dataset from experimental measurements of the SusG outer-membrane protein in living Bacteroides thetaiotaomicron to access its dynamics and anomalous diffusion behaviors, which are consistent with its role in starch catabolism in gut microbiome. This framework uses nonparametric Bayesian statistics and Deep learning to thoroughly analyze a single-molecule tracking dataset. It provides an objective method to determine the number of diffusive states in an SPT dataset and accesses the multidirectional dynamics of each state. A further diffusion type classification for each diffusive state is also included in the framework. The NOBIAS framework overcomes some oversimplified assumptions in SPT analysis and provides a powerful tool to fully make use of single-molecule tracking data.
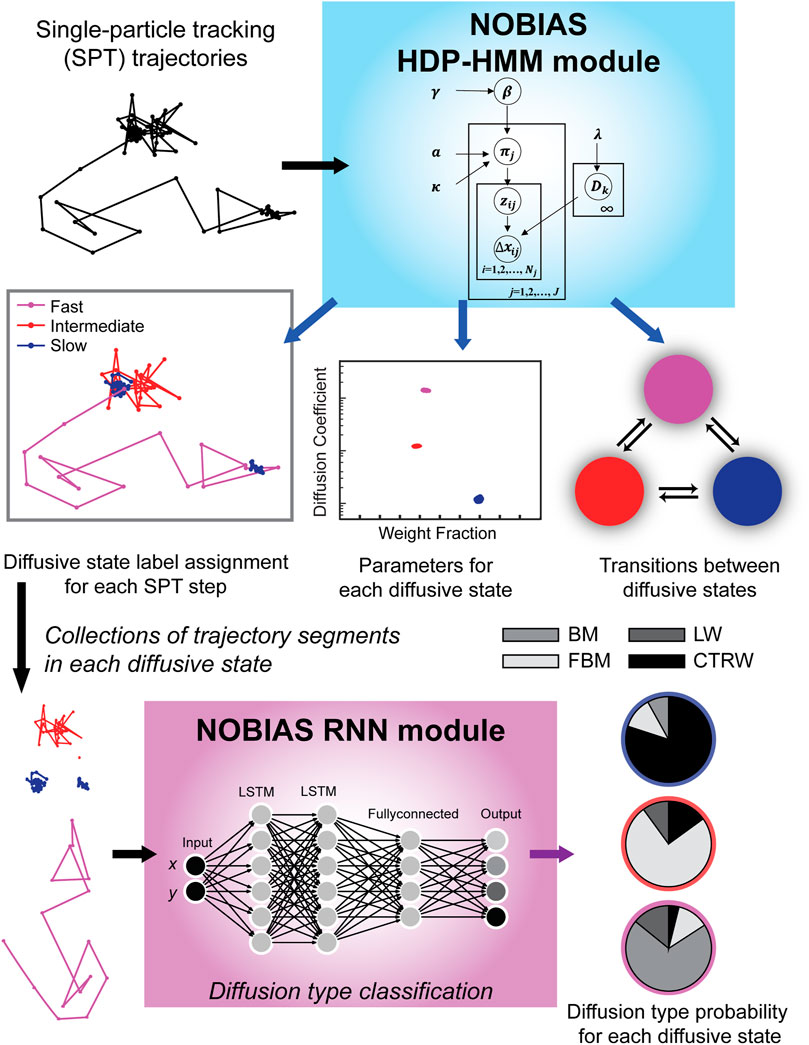
FIGURE 1. NOBIAS workflow. (1) Single-particle tracking (SPT) trajectory datasets are processed in the NOBIAS HDP-HMM module: the observed data (the displacements, Δx) are analyzed in the context of the emission parameters (the diffusion coefficients, D). The state sequence, z, indicates the diffusive state corresponding to each step, and the transition matrix, π, is estimated with a Hierarchical Dirichlet process prior using concentration hyperparameters a and γ and the sticky parameter, κ. The HDP-HMM module provides D and the weight fraction for each diffusive state, the π for transition probabilities between these states, and a state label assignment for each SPT step. (2) In the NOBIAS RNN module, trajectory segments of the same diffusive state are collected and put in a pre-trained Recurrent Neural Network (RNN) with two long short-term memory (LSTM) layers to classify the diffusion type for each diffusive state.
Methods
Hidden Markov Model
A HMM infers a system with a discrete-valued sequence of unobservable states that can be modeled as a Markovian process (Rabiner, 1989). The HMM assumes that the observed data have a hidden discrete-valued state sequence, and at each observed time, the observed data only depends on its hidden state. In our NOBIAS application of the HMM model, the observed data is the single-molecule displacements and the hidden state is the molecule’s distinct biophysical diffusive state.
Suppose
Here,
Dirichlet Process for Nonparametric Bayesian
In NOBIAS, the Dirichlet Process (DP) is used in the prior for the parameters of a mixture model with an unknown number of components. A random probability measure,
Here, Dir is a Dirichlet distribution, H is a base measurement,
From this definition follow two properties of Dirichlet processes. First, if
Here,
Second, based on the conjugacy of the finite Dirichlet distribution, given a set of observations
A stick-breaking process is used to construct the weight parameter
In this process, the weight
For each value of
Where K is the current unique number of values of z and
Hierarchical Dirichlet Process and Sticky Extension
In NOBIAS, the different single-molecule trajectories of multiple molecules under different biological condition and from different cells, so the groups of data are related but generated independently. Therefore, the DP is extended to a Hierarchical Dirichlet Process (HDP) (Teh et al., 2006). In the HDP, a first Dirichlet process, G0, is the base measure of a new Dirichlet process, Gj:
To apply a HDP as prior for a HMM model, a HDP-HMM model is generated according to:
In the NOBIAS parameter setting, the observed data
A common issue for the HDP-HMM model is that if the algorithm artificially divides a set of observations into an alternating pattern of rapid switching between several different states, then this alternating pattern will be reinforced by the DP (Fox et al., 2008). This assignment would result in an artificial over-splitting of one state into multiple substates characterized by a high probability of transitions between the substates. Because we would not expect such rapid transitions back and forth between two distinct but similar dynamical states in the single-molecule trajectory data studied here, a sticky parameter
Which add a self-transition bias to the jth components of the DP. The effects of
Different Markov Chain Monte Carlo (MCMC) sampling methods such as Direct Assignment Sampling, Beam Sampling, and Blocked Sampling have been developed for the HDP-HMM model (Teh et al., 2006; Fox et al., 2007; Van Gael et al., 2008). In NOBIAS, we apply the most computationally efficient Blocked Sampling method (Fox et al., 2007), which uses a fixed-order truncation with weak-limit approximation HDP-HMM. In this approach, the DP is L-degree approximated as:
with a truncation level, L, that is larger than the expected total number of mixture components. Increasing L does not affect the posterior results, but L does affect the running time (Supplementary Figure S1C). The Blocked Sampling method algorithm is detailed in the Supplementary Note, which describes how the state sequence is generated and how the parameters for each state are sampled.
Multivariate Normal Model
Bayes’ rule states that the posterior distribution is proportional to the product of the prior probability and the likelihood, i.e.,
In NOBIAS HDP-HMM module, we assume 2D Brownian motion trajectories. In this case, the displacements follow a zero-mean 2D Gaussian and the diffusion coefficients D determine the variance, Σ, of the 2D Gaussian. Without loss of generality, the mean, µ, is also included in the model,
In the 2D case, the observed data,
As derived in reference Gelman (2004), the general conjugate prior model for this multivariate normal model is the prior for the mean and the variance of the step displacement follow a Normal-inverse-Wishart distribution (NIW):
Specifically, the variance,
The posterior updates for this normal model with NIW prior follows (Gelman, 2004):
Where
To decrease the running time, we apply the conjugate prior for the Multivariate Normal Distribution, though a non-conjugate prior is permissible. For further discussion of choice of prior see (Gelman, 2004).
Trajectory Simulation
A state label sequence was firstly simulated with a given transition matrix through a Markov chain process. Then according the state label and the D of corresponding diffusive state, the 2D displacement step is generated, and cumulatively summed to get a single trajectory. Standard trajectory datasets are simulated by generate 2D Gaussian random variable where mean is 0 and variance is determined by the set diffusion coefficients with symmetry and no correlation in two directions.
Motion blur trajectory datasets are generated by simulating a state label sequence that is
Anomalous Diffusion
In the NOBIAS RNN module, trajectory segments of the same diffusive state (identified by the HDP-HMM module) are evaluated to classify the diffusion type for each diffusive state. In Brownian Motion, the mean squared displacement (MSD) is linearly proportional to the time lag, τ. In anomalous diffusion, MSD is related to τ according to a power law (Metzler et al., 2014):
Here,
FBM is a Gaussian process with correlated increments such that MSD is related to τ according to:
CTRW defines a random walk family in which the particle displacement, ∆x, follows a wait at its current position for a random waiting time t that is a stochastic variable (Scher and Montroll, 1975). NOBIAS considers the case where t follows a power-law distribution,
LW is a special case of CTRW in which the waiting time,
We simulated these three types of anomalous diffusion with the open-source Python package from the recent AnDi challenge (Muñoz-Gil et al., 2020).
Recurrent Neural Network for NOBIAS
All segments 40 steps or greater identified in the HDP-HMM module were further analyzed by the NOBIAS Recurrent Neural Network (RNN) consisting of two long short-term memory (LSTM) layers (Hochreiter and Schmidhuber, 1997). We trained this RNN to classify trajectory segments identified to have the same diffusive state from the HDP-HMM module. We implemented this architecture, which is based on the design of the RANDI package classification task (Bo et al., 2019; Argun et al., 2021) with the MATLAB Deep Learning Toolbox™. The two LSTM layers have 100 and 50 units, respectively, and these two LSTM layers are followed by a fullyconnected layer, and the output classification layer order is given in Figure 1.
The input to the network is the set of 2D coordinates from the track segments; these coordinates are normalized to have zero mean and unit variance. Despite a much higher classification performance when using tracks > 50 steps long to train and validate (Argun et al., 2021; Gentili and Volpe, 2021; Muñoz-Gil et al., 2021), we trained two networks with 20-step tracks and with 40-step tracks, respectively, after considering the typical segment lengths from real biological trajectories. The training data of 750,000 trajectories were simulated with the open-source Python package from the AnDi challenge (Muñoz-Gil et al., 2020). Regression networks with similar two LSTM layers architecture were also trained for FBM and CTRW to estimate the anomalous exponent
Single-Molecule Tracking in Living Bacteroides thetaiotaomicron Cells
B. thetaiotaomicron cells expressing SusG-HaloTag fusions at the native SusG promoter were grown as previously described (Karunatilaka et al., 2014). Briefly, cells were cultured overnight in 0.5% tryptone-yeast-extract-glucose medium and incubated at 37°C under anaerobic conditions (85% N2, 10% H2, 5% CO2) in a Coy chamber. Approximately 24 h before imaging, cells were diluted into B. theta minimal medium (MM) (Martens et al., 2008) containing 0.25% (wt/vol) amylopectin. On the day of the experiment, cells were diluted into fresh MM and carbohydrate and grown until reaching OD600 nm 0.55–0.60 (Tuson et al., 2018).
Before labeling, 900 μL of cells were washed twice by pelleting (6,000 G, 2 min) followed by resuspension in MM. Cells were then incubated in MM supplemented with 100 nM PAJF549 dye (Grimm et al., 2016) for 15 min in the dark. Cells were then washed five times in MM, transferring to a new tube on every step, to remove excess dye (Lepore et al., 2019). Finally, 100 μL cells were resuspended in MM containing 0.25% (wt/vol) amylopectin for 30 min in the dark. 1.5 μL labeled cells were pipetted onto a pad of 2% agarose in MM and placed between a large and a small coverslip. The two coverslips were sealed together with epoxy (Devcon 31345 2 Ton Clear Epoxy, 25 ml) to keep the media anaerobic (Karunatilaka et al., 2014).
Cells were imaged on an Olympus IX71 inverted epifluorescence microscope with a 1.45 numerical aperture, 100× oil immersion phase-contrast objective (Olympus UPLXAPO100XOPH) and a 3.3× beam expander. Frames were collected continuously on a 512 × 512 pixel electron-multiplying charge-coupled device camera (Photometrics Evolve 512) at 50 frames/s. In this microscopy geometry, one camera pixel corresponds to 48.5 nm PAJF549 dyes were photo-activated one at a time with a 200–400 ms exposure by a 406-nm laser (Coherent Cube 405-100; 0.1 μW/μm2) and imaged with a 561-nm laser (Coherent-Sapphire 561-50; 1 μW/μm2) using appropriate filters as previously described (Tuson et al., 2018).
In each movie, each cell was analyzed separately by using an appropriate mask. The collected frames were processed with SMALL-LABS (Isaacoff et al., 2019) to detect single molecules frame-by-frame and localize their position with typically ∼30 nm uncertainty. Single molecules were identified as non-overlapping punctuate spots of diameter larger than seven pixels and with pixel intensities larger than the 92nd percentile intensity of the fame. The punctate spots were fit to a 2D Gaussian and true single-molecule localizations satisfied the following conditions: 1) standard deviation > 1 pixel and 2) fit error
Results
The NOBIAS HDP-HMM Module Recovers the Number of Diffusive States and the Associated Diffusion Parameters
We first validated the NOBIAS HDP-HMM module with simulated single-molecule tracks, beginning from the most basic case: a mixture of Brownian motion trajectories. Figures 2A–D depicts the results for a mixture of two distinct diffusive states with D1 = 0.135 µm2/s and D2 = 1.8 µm2/s (Supplementary Table S1). A sequence of state labels (1 or 2) was first simulated with a given transition matrix (probability of transitioning from state 1 to 2 or from state 2 to 1) through a Markov chain process (Methods). Then, according the state label and the apparent diffusion coefficient, D, of the corresponding diffusive state, each 2D displacement step was generated, and cumulatively summed to get a single trajectory. Similar state label sequences were simulated to generate other trajectory datasets with four diffusive states (Figures 2E–G; Supplementary Table S2).
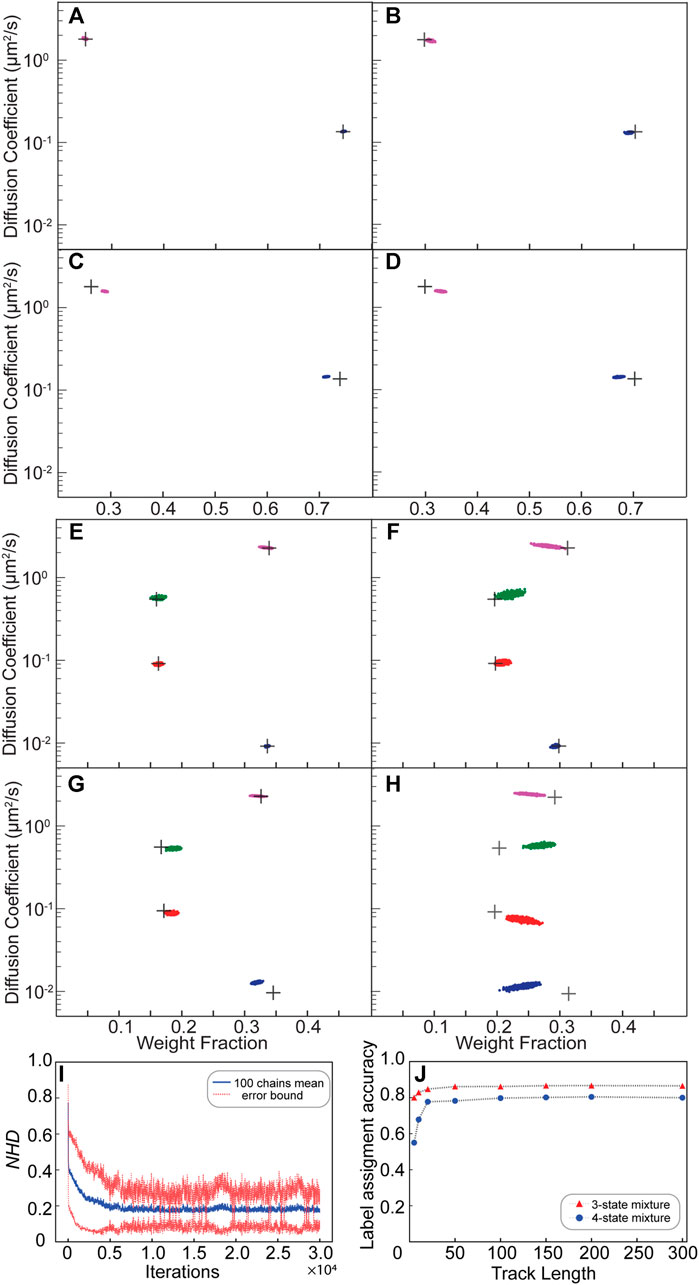
FIGURE 2. Validation of the NOBIAS HDP-HMM module with simulated trajectories. (A–H) The HDP-HMM module identifies distinct mobility states (colored clusters). All scatter plots include at least 500 uncorrelated samples. Each point represents the average apparent single-molecule diffusion coefficient vs. weight fraction in each distinct mobility state at each iteration of the Bayesian algorithm saved after convergence. The black crosses indicate the ground truth input for these simulated trajectories. (A–D) Results for two-state mixture simulated trajectories results: (A) Standard (no motion blur) and abundant (500 100-step trajectories) simulations, (B) Standard and sparse (2,000 10-step trajectories) simulations, (C) Motion blur and abundant simulations, and (D) Motion blur and sparse simulations. (E,H) Results for four-state mixture simulated trajectories results: (E) Standard (no motion blur) and abundant (500 100-step trajectories) simulations, (F) Standard and sparse (2,000 10-step trajectories) simulations, (G) Motion blur and abundant simulations, and (H) Motion blur and sparse simulations. (I) The normalized Hamming distance (NHD) decreases and converges with the number of iterations. All 100 chains use the same dataset under the settings in panel (E). (J) The final label assignment accuracy increases with the track length for three- and four-state mixture datasets. The number of trajectories decreases as the track lengths increase such that the total amount of steps is 30,000 for all track lengths.
The posterior results of the HDP-HMM module are shown in scatter plots of the inferred D and weight fraction from each iteration after the inferred number of states converges. Figure 2A shows the result for a dataset of 500 trajectories each with 100 steps. Here, the black crosses indicate the ground truth diffusion coefficient and weight fraction for each diffusive state; the posterior samples of the HDP-HMM model for the two states after convergence are distributed around the true values. Based on the posterior sample autocorrelation function (ACF) analysis (Supplementary Figure S3), the posterior samples are thinned by saving every 10 iterations; this setting is the same for all results in this paper and was chosen by considering the effective sample sizes and the ACF analysis for all the diffusive states. The number could be updated accordingly depending on the correlation of posterior samples from output. The mean values and standard deviations for the estimation of D and weight fractions for the two states are listed in Supplementary Table S1. The estimated number of unique states for this simulated dataset converges quickly over the course of iterations to the true number of states and remains mostly stable at that number (Supplementary Figure S4). Next, we considered the less ideal case that often occurs experimentally: much shorter trajectory lengths (10 steps) and many fewer total steps (2,000 10-step trajectories). We refer to the 2,000 10-step trajectories as a sparse dataset and the 500 100-step trajectories are an abundant dataset. Figure 2B shows that the HDP-HMM model still successfully converges to the true number of states (two) for this dataset, and the posterior samples of the diffusive parameters are still distributed near the true inputs (black crosses).
We further considered the true form of collected microscope experimental data by including the localization error due to finite photon counts and noise and motion blur due to the finite image acquisition time (Methods). We refer these datasets “Motion blur dataset” in contrast with the more ideal “Standard” dataset. In the case of motion blur, the sticky parameter is increased to avoid oversampling a single diffusive state into multiple state with similar dynamics. The hyperparameter settings for this sticky HDP-HMM model are listed in Supplementary Table S3. For both the abundant dataset (Figure 2C: 500 100-step trajectories) and the sparse dataset (Figure 2D: 2,000 10-step trajectories), the true number of states (two) is recovered with our sticky HDP-HMM model, and despite these added errors, the estimated parameters deviate only slightly from the true inputs (black crosses).
We extended our simulations of standard and motion blur Brownian motion track mixtures to a more complicated 4-state scenarios for abundant (500 100-step trajectories) and sparse (2,000 10-step trajectories) datasets (Figures 2E–H). Even with four diffusive states, the performance of the HDP-HMM module is still excellent for the standard mixture (Figures 2E,F). For the 4-state mixture simulation that includes localization error and motion blur, the HDP-HMM still successfully recovers the true number of states, and the parameters for the four distinct states are still estimated well, though the posterior samples have increased variance and deviation from the true value (Figures 2G,H). The statistics of the posterior samples for estimated parameters of the 4-state simulation result are listed in Supplementary Table S2, and the transition matrices for all the simulations in Figure 2 are shown in Supplementary Tables S1, S2.
The NOBIAS HDP-HMM module also assigns diffusive state labels to each single-molecule step within the trajectories dataset; we call this the state sequence for each track. We quantified the performance of the state sequence assignment relative to the ground truth simulated state sequence with the Hamming distance: the Hamming distance between two 1D sequences with equal length is the number of points where the components are different (Hamming, 1950). The resulting distances were normalized to the total length to demonstrate the Normalized Hamming Distance (NHD) convergence over iterations (Figure 2I). The NHD decreases with increasing iteration number and converges to approximately 0.18. This final converged NHD depends on the dataset size, the true transition matrix, and how separable the diffusive state are from one another.
The true number of diffusive states can be recovered for datasets of both abundant and sparse tracks, but the HDP-HMM module performance depends strongly on the length of the individual tracks. Using the overall state sequence assignment accuracy (1
The NOBIAS RNN Module Predicts the Diffusion Type for Each Diffusive State
To analyze anomalous diffusion in an SPT dataset, NOBIAS includes a second module: we built an RNN to classify the type of motion [Brownian motion (BM), Fractional Brownian motion (FBM), Continuous Time Random Walk (CTRW), or Lévy Walk (LW)] corresponding to the track segments within each diffusive state identified by HDP-HMM module. The RNN consists of two LSTM layers, a fullyconnected layer, and data input/output layer (Methods). Although the HDP-HMM module is based on BM, for some anomalous diffusion types, for example FBM, if the dynamics level for each state is distinct, the HDP-HMM module still performs well.
We simulated a mixture of BM and FBM with distinct apparent diffusion coefficients for the two states (
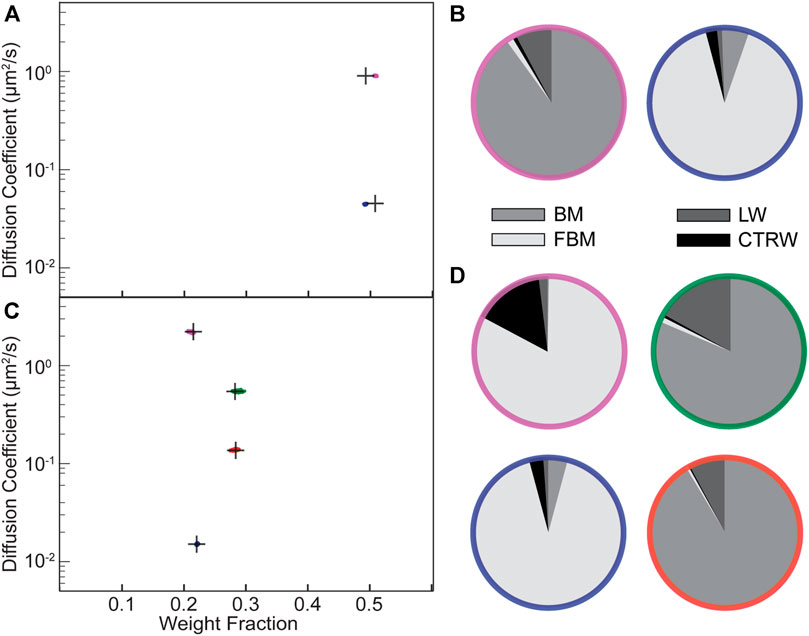
FIGURE 3. Validation of the NOBIAS-RNN module with simulated trajectories containing mixtures of different diffusion types. (A,C) The HDP-HMM module identifies distinct mobility states (colored clusters). Each point represents the average apparent single-molecule diffusion coefficient, D, vs. weight fraction in each distinct mobility state at each iteration of the Bayesian algorithm saved after convergence. The black crosses indicate the ground truth input for these simulated trajectories. (A) Two-state mixture comprising a subdiffusive Fractional Brownian Motion (FBM) state with lower D and a Brownian Motion (BM) state with higher D. (B) The NOBIAS-RNN determines the probability that the diffusion type for each diffusive state in (A) is classified as BM, FBM, Continuous Time Random Walk (CTRW), or Lévy Walk (LW). The final probability for each diffusive state is the average of the classification probability of its track segments weighted by the segment length. The color of each pie chart indicates the diffusive state corresponding to the color in (A). (C) Four-state mixture comprising a subdiffusive FBM state, two BM states, and a superdiffusive FBM state with D in ascending order. (D) Diffusion type classification probability pie chart for each diffusive state in (C). The final probability for each diffusive state is the average of the classification probability of its track segments weighted by the segment length and the color of each pie chart indicates the diffusive state corresponding to the corresponding color in (C).
We further simulated a 4-state mixture (500 100-step trajectories) corresponding to subdiffusive FBM, BM, BM, and superdiffusive FBM (in order of increasing D). The HDP-HMM module still successfully recovers the four states and make excellent estimations for D and weight fraction for each state (Figure 3C). The NOBIAS RNN module also predicts the true diffusion type for the segments from each of the four states (Figure 3D; Supplementary Table S4). Note that all track segments are normalized before being put into the RNN to avoid dynamics information bias in the diffusion type prediction (Methods). One limitation for this RNN classification analysis methodology is that only track segments with at least certain length (20 or 40 in our analysis depending on the trained network) could be classified with high accuracy; it is very challenging to use very short track segments to identify these modes of diffusion. Therefore, when the overall trajectory length is short (∼10 steps), the network classification module might not be usable. Another limitation of the HDP-HMM module is that the current implementation is based on BM displacement distributions, thus it would fail for anomalous diffusion types like LW, which does not have a similar Gaussian distribution of displacements.
Performance of NOBIAS on Experimental Data for the Diffusion of SusG-HaloTag in Bacteroides thetaiotaomicron Cells
After validating the performance of the two NOBIAS modules on simulated data, we applied this framework to experimental single-molecule trajectories. The SusG amylase recognizes and binds starch on the surface of B. thetaiotaomicron cells to enable starch catabolism (Koropatkin and Smith, 2010). We measured the motion of 7,897 trajectories (minimum length of 6 and average length of 64) of single SusG molecules in 226 movies of 149 B. thetaiotaomicron cells based on imaging photoactivatable fluorescently labeled SusG-HaloTag fusions (Methods).
We analyzed this data with NOBIAS to infer the number of diffusive states and to estimate the diffusion coefficient, weight fraction, and type of motion for each state as was done for the simulated data (Figures 2, 3). Additionally, NOBIAS analyzes 2D trajectories with a 2D Gaussian function and can therefore infer the diffusion coefficients for the x and y directions separately and estimate the potential correlation between the two directions. Though the simulations used symmetric tracks in an unbound domain, the experiments measure motion on the surface of cells with a long axis and a short axis, which may create an asymmetry in the diffusion. We rotated the cell orientations to orient the long axis in the x direction without rescaling (Figure 4A). We analyzed this rotated dataset with NOBIAS and found that it converged to a 3-state model, with a very small (1.8%) fast state fraction (Figure 4B). Interestingly, we found that the
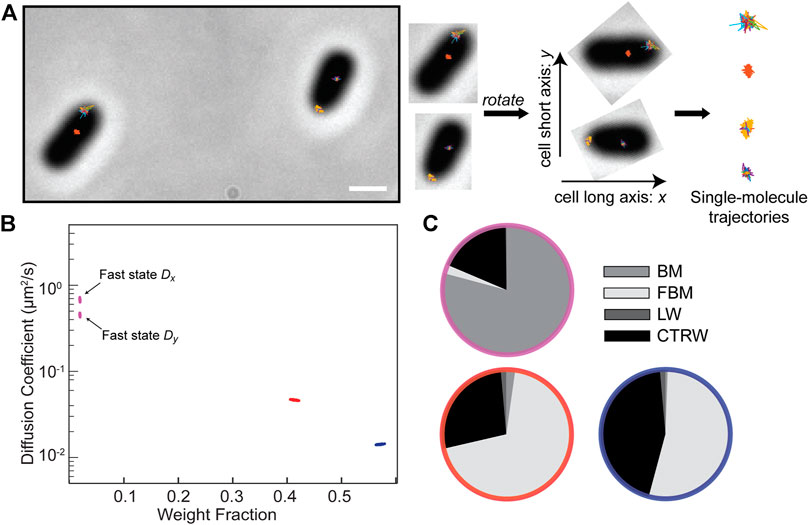
FIGURE 4. Application of NOBIAS to single-molecule trajectories of the SusG protein in living Bacteroides thetaiotaomicron cells. (A) Single-molecule trajectories of SusG-HaloTag overlaid on the phase-contrast image of the corresponding B. thetaiotaomicron cells, scale bar: 1 μm. The long axis of the phase mask for each cell was detected and a rotation transform was applied to all the trajectories in each cell such that the x-axis is the cell long axis for all cells. (B) The NOBIAS HDP-HMM module identifies three diffusive states for SusG (colored clusters). Each point represents the average apparent singlemolecule diffusion coefficient vs. weight fraction in each distinct mobility state at each iteration of the Bayesian algorithm saved after convergence. The blue and red points clusters average the x- and y-diffusion coefficients as they are symmetric (Supplementary Table S4); the asymmetric fast state (purple) shows a different DxandDy. (C) The NOBIAS-RNN determines the probability that the diffusion type for each diffusive state in (B) is classified as Brownian Motion (BM), Fractional Brownian Motion (FBM), Continuous Time Random Walk (CTRW), or Lévy Walk (LW). The color of each pie chart indicates the diffusive state corresponding to the color in (B). The fast state (purple) is predicted with high probability to be BM; the two slower states (red and blue) are predicted to be FBM or CTRW.
We separated the track segments by the state sequence label from the HDP-HMM module and placed each group into the RNN classification module. The fastest state was predicted with high probability (80%) to be Brownian motion (Figure 4C; Supplementary Table S4), consistent with the asymmetry between Dx and Dy that was attributed to free diffusion (Figure 4B). The two slower states were predicted to be either FBM or CTRW. We used a RNN regression network (Methods) to estimate the anomalous exponent
Discussion
Single-molecule tracking measures dynamics in biological systems at high spatial and temporal resolution, but how to make the best use of these tracking data for a broad set of experimental conditions remains an analysis challenge in the field (Shen et al., 2017; Elf and Barkefors, 2019). Here, we have introduced NOBIAS to quantify single-molecule dynamics and to associate these biophysical measurements with the underlying biochemical function and biological processes. NOBIAS handles complicated live-cell SPT datasets for which: 1) the number of diffusive states is unknown, 2) mixtures of different diffusive populations may exist, even within single trajectories, 3) symmetry cannot be assumed between the x and y directions, and 4) anomalous diffusion is possible. These features are enabled based on applying Nonparametric Bayesian statistics (Teh et al., 2006; Fox et al., 2008; Johnson and Willsky, 2013) to SPT datasets that have the same means but different variance with a HDP-HMM module that has a 2D Gaussian as the emission function and then by further investigating the anomalous diffusion types in the RNN module of NOBIAS.
Compared with previous applications of nonparametric Bayesian statistics in this field (Persson et al., 2013; Karslake et al., 2020; Heckert et al., 2021), the NOBIAS HDP-HMM module is more robust and has high computational efficiency (Supplementary Table S6). NOBIAS and SMAUG both consider motion blur effects and their estimation of D for each state is closer to the ground truth then other methods. As Bayesian method with similar principle NOBIAS is almost 10 times faster than SMAUG. This HDP-HMM module also provides a multivariate output to quantify and correlate dynamics in multiple directions instead of assuming symmetry (Supplementary Table S7). We observed that for asymmetric simulated trajectories, vbSPT overestimates the true number of states, and SMAUG can only provide the average D of for each diffusive state while NOBIAS provides the respective diffusion coefficients in two directions. The high accuracy of step state sequence prediction also enables the classification of anomalous diffusion type in the NOBIAS RNN module. We also applied SMAUG and vbSPT on the experimental dataset (Supplementary Table S8): SMAUG ran slow on large datasets and suggested four diffusive state, while vbSPT suggested the best model to be 10 diffusive state which is hard to explain their corresponding biological meanings.
A further advantage of NOBIAS lies in its ability to treat sets of relatively short trajectories (10-step trajectories in the simulated data of Figures 2, 3 and minimal 6-step trajectories in the experimental data of Figure 4). The recent AnDi (Anomalous Diffusion) Challenge (Muñoz-Gil et al., 2021) demonstrated that Deep Learning and Neural Network methods are currently the most powerful tools to study anomalous diffusion (Argun et al., 2021; Gentili and Volpe, 2021). However, in this challenge, the target dataset was an ideal collection of simulated anomalous diffusion trajectories with 100–1,000 steps, and only the simple case of one state transition in the middle part of a track was considered. There are also probability-based models that consider confinement and anomalous diffusion (Robson et al., 2013) and Bayesian methods that directly predict the diffusion type (Thapa et al., 2018; Cherstvy et al., 2019), but these analyses, like the neural network-based methods, are used for very long trajectories or assume a single diffusive state in each track. To apply a deep learning-based diffusion type classifier to realistic simulated trajectories and real experimental trajectories, NOBIAS segments the raw trajectories into collections of track segments that belong to the same diffusive state (as identified by the HDP-HMM module) and then predicts the diffusion type of the long segments in the RNN module. Since different biophysical diffusive states correspond to different biochemical functions which will exhibit different diffusion types due to interactions like confinement, binding, directional motion, NOBIAS enables a thorough investigation of these biochemical roles by revealing the diffusion coefficients, the transition probabilities between states, and the anomalous diffusion behaviors. Ultimately, NOBIAS will enable investigators to extract a complete information set from SPT data and to understand the role of each tracked molecule, even in the living cell.
Despite these strengths, NOBIAS has several limitations. Firstly, as an HMM-based method, NOBIAS is limited by the length of each track. Under the extreme case where only very short trajectories (∼2–5 steps) are available, the HDP-HMM module may suggest a number of states and posterior results with extremely high uncertainty; probability-based models (Rowland and Biteen, 2017; Hansen et al., 2018) or the histogram-based Bayesian method DPMM (Heckert et al., 2021) should be applied for these short trajectories. The track length also limits the RNN module, as the trained network need tracks with at least 20 steps for good classification performance because some anomalous diffusion types are characterized by memory of previous steps (Metzler et al., 2014). Therefore the application of the RNN module is limited for short experimental tracks. Secondly, NOBIAS performs the diffusive state estimation based on apparent diffusion coefficient in the HDP-HMM module and then carries out the anomalous diffusion classification in the RNN module. NOBIAS therefore assumes that each biochemical state has a unique average apparent diffusion coefficient. Although the RNN module can classify the diffusion types of two different diffusive states with the same diffusion coefficient, the HDP-HMM module would fail to separate these processes. Furthermore, for some diffusion types like LW, the trajectory displacements may exhibit different types of dynamics even though the trajectories are generated from one process. Finally, even for Brownian trajectories, a single biochemical state might not be represented by a single diffusion coefficient value. Thus, the actual number of biochemical states may not be equal to the number of diffusive states. Future development of NOBIAS could use spatial filtering to distinguish between these similar biochemical states.
NOBIAS provides a pioneering and compatible framework for the analysis of dynamical mixtures that also classifies the anomalous diffusion types. Future development of NOBIAS could include more types of diffusion and could integrate the anomalous distributions directly into the Bayesian framework for more accurate prediction of the stepwise state labels and the diffusion types. Furthermore, extra experimental corrections corresponding to the specific microscope setting (Berglund, 2010; Lindén et al., 2017; Hansen et al., 2018) could also help adapt NOBIAS more broadly to different types of SPT datasets. Overall, NOBIAS has provided a powerful framework to analyze of SPT dataset with unknown number of diffusive states and potential asymmetric diffusion, and to access the anomalous diffusion type for each diffusive state. The combination of nonparametric Bayesian statistics and Deep learning enables NOBIAS to fully extract the rich dynamics information from the SPT dataset.
Data Availability Statement
The original contributions presented in the study as well as simulated and experimental raw data, are included in the article/Supplementary Material. The Open-source Matlab code for implementing NOBIAS (GNU General Public License) and sometest datasets are provided at https://github.com/BiteenMatlab/NOBIAS; further development and expansion of the code post-publication will be hosted at that website as well. Further inquiries can be directed to the corresponding author.
Author Contributions
ZC and JB conceived of the idea. ZC developed the theory, implemented the algorithm, performed simulations, and analyzed simulated and experimental data. LG carried out the experiments. ZC and JB wrote the manuscript with input from all authors.
Funding
This work was supported by National Institutes of Health grant R21-GM128022 and National Science Foundation grant EF-1921677 to JB.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Thanks to Christopher Azaldegui and Guoming Gao for helpful discussions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fbinf.2021.742073/full#supplementary-material
References
Argun, A., Volpe, G., and Bo, S. (2021). Classification, inference and segmentation of anomalous diffusion with recurrent neural networksfication, inference and segmentation of anomalous diffusion with recurrent neural networks. J. Phys. A: Math. Theor. 54, 294003. doi:10.1088/1751-8121/ac070a
Badrinarayanan, A., Reyes-Lamothe, R., Uphoff, S., Leake, M. C., and Sherratt, D. J. (2012). In Vivo Architecture and Action of Bacterial Structural Maintenance of Chromosome Proteins. Science 338, 528–531. doi:10.1126/science.1227126
Bauer, M., and Metzler, R. (2012). Generalized Facilitated Diffusion Model for DNA-Binding Proteins with Search and Recognition States. Biophys. J. 102, 2321–2330. doi:10.1016/j.bpj.2012.04.008
Bayas, C. A., Wang, J., Lee, M. K., Schrader, J. M., Shapiro, L., and Moerner, W. E. (2018). Spatial organization and dynamics of RNase E and ribosomes in Caulobacter crescentus. Proc. Natl. Acad. Sci. U S A. 115, E3712–E3721. doi:10.1073/pnas.1721648115
Berglund, A. J. (2010). Statistics of camera-based single-particle tracking. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 82, 011917. doi:10.1103/PhysRevE.82.011917
Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., et al. (2006). Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 313, 1642–1645. doi:10.1126/science.1127344
Biswas, S., Karslake, J. D., Chen, Z., Farhat, A., Freddolino, P. L., Biteen, J. S., et al. (2021). HP1 oligomerization compensates for low-affinity H3K9me recognition and provides a tunable mechanism for heterochromatin-specific localization. bioRxiv 01 (26), 428151. doi:10.1101/2021.01.26.428151
Bo, S., Schmidt, F., Eichhorn, R., and Volpe, G. (2019). Measurement of anomalous diffusion using recurrent neural networks. Phys. Rev. E 100, 010102. doi:10.1103/PhysRevE.100.010102
Caspi, A., Granek, R., and Elbaum, M. (2002). Diffusion and directed motion in cellular transport. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 66, 011916. doi:10.1103/PhysRevE.66.011916
Cherstvy, A. G., Thapa, S., Wagner, C. E., and Metzler, R. (2019). Non-Gaussian, non-ergodic, and non-fickian diffusion of tracers in mucin hydrogels. Soft Matter 15, 2526–2551. doi:10.1039/C8SM02096E
Deich, J., Judd, E. M., McAdams, H. H., and Moerner, W. E. (2004). Visualization of the movement of single histidine kinase molecules in live Caulobacter cells. Proc. Natl. Acad. Sci. U S A. 101, 15921–15926. doi:10.1073/pnas.0404200101
Deschout, H., Neyts, K., and Braeckmans, K. (2012). The influence of movement on the localization precision of sub-resolution particles in fluorescence microscopy. J. Biophotonics 5, 97–109. doi:10.1002/jbio.201100078
Elf, J., and Barkefors, I. (2019). Single-Molecule Kinetics in Living Cells. Annu. Rev. Biochem. 88, 635–659. doi:10.1146/annurev-biochem-013118-110801
Elf, J., Li, G. W., and Xie, X. S. (2007). Probing Transcription Factor Dynamics at the Single-Molecule Level in a Living Cell. Science 316, 1191–1194. doi:10.1126/science.1141967
Elmore, S., Müller, M., Vischer, N., Odijk, T., and Woldringh, C. L. (2005). Single-particle tracking of oriC-GFP fluorescent spots during chromosome segregation in Escherichia coli. J. Struct. Biol. 151, 275–287. doi:10.1016/j.jsb.2005.06.004
Ferguson, T. S. (1973). A Bayesian Analysis of Some Nonparametric Problems. Ann. Stat. 1, 209–230. doi:10.1214/aos/1176342360
Fox, E. B., Sudderth, E. B., Jordan, M. I., and Willsky, A. S. (2008). “An HDP-HMM for systems with state persistence,” in Proceedings of the 25th international conference on Machine learning - ICML ’08 (Helsinki, Finland: ACM Press), 312–319. doi:10.1145/1390156.1390196
Fox, E. B., Sudderth, E. B., and Willsky, A. S. (2007). “Hierarchical Dirichlet processes for tracking maneuvering targets,” in 2007 10th International Conference on Information Fusion (Quebec City, QC: Canada: IEEE). 1–8. doi:10.1109/ICIF.2007.4408155
Gentili, A., and Volpe, G. (2021). Characterization of anomalous diffusion classical statistics powered by deep learning (CONDOR). J. Phys. A: Math. Theor. 54, 314003. doi:10.1088/1751-8121/ac0c5d
Granik, N., Weiss, L. E., Nehme, E., Levin, M., Chein, M., Perlson, E., et al. (2019). Single-Particle Diffusion Characterization by Deep Learning. Biophys. J. 117, 185–192. doi:10.1016/j.bpj.2019.06.015
Grimm, J. B., English, B. P., Choi, H., Muthusamy, A. K., Mehl, B. P., Dong, P., et al. (2016). Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 13, 985–988. doi:10.1038/nmeth.4034
Hamming, R. W. (1950). Error Detecting and Error Correcting Codes. Bell Syst. Tech. J. 29, 147–160. doi:10.1002/j.1538-7305.1950.tb00463.x
Hansen, A. S., Pustova, I., Cattoglio, C., Tjian, R., and Darzacq, X. (2017). CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 6, e25776. doi:10.7554/eLife.25776
Hansen, A. S., Woringer, M., Grimm, J. B., Lavis, L. D., Tjian, R., and Darzacq, X. (2018). Robust model-based analysis of single-particle tracking experiments with Spot-On. eLife 7, e33125. doi:10.7554/eLife.33125
Heckert, A., Dahal, L., Tjian, R., and Darzacq, X. (2021). Recovering mixtures of fast diffusing states from short single particle trajectories. bioRxiv. 05.03.442482. doi:10.1101/2021.05.03.4424822021
Hell, S. W., and Wichmann, J. (1994). Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782. doi:10.1364/OL.19.000780
Hess, S. T., Girirajan, T. P., and Mason, M. D. (2006). Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy. Biophys. J. 91, 4258–4272. doi:10.1529/biophysj.106.091116
Hochreiter, S., and Schmidhuber, J. (1997). Long Short-Term Memory. Neural Comput. 9, 1735–1780. doi:10.1162/neco.1997.9.8.1735
Isaacoff, B. P., Li, Y., Lee, S. A., and Biteen, J. S. (2019). SMALL-LABS: Measuring Single-Molecule Intensity and Position in Obscuring Backgrounds. Biophys. J. 116, 975–982. doi:10.1016/j.bpj.2019.02.006
Ishwaran, H., and James, L. F. (2001). Gibbs Sampling Methods for Stick-Breaking Priors. J. Am. Stat. Assoc. 96, 161–173. doi:10.1198/016214501750332758
Ishwaran, H., and Zarepour, M. (2002). Dirichlet Prior Sieves in Finite Normal Mixtures. Stat. Sinica 12, 941–963.
Izeddin, I., Récamier, V., Bosanac, L., Cissé, I. I., Boudarene, L., Dugast-Darzacq, C., et al. (2014). Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. eLife 3, e02230. doi:10.7554/eLife.02230
Jeon, J.-H., Javanainen, M., Martinez-Seara, H., Metzler, R., and Vattulainen, I. (2016). Protein Crowding in Lipid Bilayers Gives Rise to Non-gaussian Anomalous Lateral Diffusion of Phospholipids and Proteins. Phys. Rev. X 6, 021006. doi:10.1103/PhysRevX.6.021006
Jeon, J. H., and Metzler, R. (2010). Fractional Brownian motion and motion governed by the fractional Langevin equation in confined geometries. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 81, 021103. doi:10.1103/PhysRevE.81.021103
Johnson, M. J., and Willsky, A. S. (2013). Bayesian Nonparametric Hidden Semi-markov Models. J. Machine Learn. Res. 14, 673–701.
Karslake, J. D., Donarski, E. D., Shelby, S. A., Demey, L. M., DiRita, V. J., Veatch, S. L., et al. (2021). SMAUG: Analyzing single-molecule tracks with nonparametric Bayesian statistics. Methods 193, 16–26. doi:10.1016/j.ymeth.2020.03.008
Karunatilaka, K. S., Cameron, E. A., Martens, E. C., Koropatkin, N. M., and Biteen, J. S. (2014). Superresolution Imaging Captures Carbohydrate Utilization Dynamics in Human Gut Symbionts. mBio 5, e02172. doi:10.1128/mBio.02172-14
Klafter, J., and Zumofen, G. (1994). Lévy statistics in a Hamiltonian system. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 49, 4873–4877. doi:10.1103/PhysRevE.49.4873
Koropatkin, N. M., and Smith, T. J. (2010). SusG: a unique cell-membrane-associated alpha-amylase from a prominent human gut symbiont targets complex starch molecules. Structure 18, 200–215. doi:10.1016/j.str.2009.12.010
Kusumi, A., Sako, Y., and Yamamoto, M. (1993). Confined lateral diffusion of membrane receptors as studied by single particle tracking (nanovid microscopy). Effects of calcium-induced differentiation in cultured epithelial cells. Biophys. J. 65, 2021–2040. doi:10.1016/S0006-3495(93)81253-0
Lepore, A., Taylor, H., Landgraf, D., Okumus, B., Jaramillo-Riveri, S., McLaren, L., et al. (2019). Quantification of very low-abundant proteins in bacteria using the HaloTag and epi-fluorescence microscopy. Sci. Rep. 9, 7902. doi:10.1038/s41598-019-44278-0
Lindén, M., Ćurić, V., Amselem, E., and Elf, J. (2017). Pointwise error estimates in localization microscopy. Nat. Commun. 8, 15115. doi:10.1038/ncomms15115
Mandelbrot, B. B., and Van Ness, J. W. (1968). Fractional Brownian Motions, Fractional Noises and Applications. SIAM Rev. 10, 422–437. doi:10.1137/1010093
Manley, S., Gillette, J. M., Patterson, G. H., Shroff, H., Hess, H. F., Betzig, E., et al. (2008). High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 5, 155–157. doi:10.1038/nmeth.1176
Martens, E. C., Chiang, H. C., and Gordon, J. I. (2008). Mucosal Glycan Foraging Enhances Fitness and Transmission of a Saccharolytic Human Gut Bacterial Symbiont. Cell Host Microbe 4, 447–457. doi:10.1016/j.chom.2008.09.007
Mazza, D., Abernathy, A., Golob, N., Morisaki, T., and McNally, J. G. (2012). A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. 40, e119. doi:10.1093/nar/gks701
Metzler, R., Jeon, J. H., Cherstvy, A. G., and Barkai, E. (2014). Anomalous diffusion models and their properties: non-stationarity, non-ergodicity, and ageing at the centenary of single particle tracking. Phys. Chem. Chem. Phys. 16, 24128–24164. doi:10.1039/C4CP03465A
Michalet, X., and Berglund, A. J. (2012). Optimal diffusion coefficient estimation in single-particle tracking. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 85, 061916. doi:10.1103/PhysRevE.85.061916
Moerner, W. E., and Kador, L. (1989). Optical detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett. 62, 2535–2538. doi:10.1103/PhysRevLett.62.2535
Monnier, N., Barry, Z., Park, H. Y., Su, K. C., Katz, Z., English, B. P., et al. (2015). Inferring transient particle transport dynamics in live cells. Nat. Methods 12, 838–840. doi:10.1038/nmeth.3483
Muñoz-Gil, G., Volpe, G., Garcia-March, M. A., Aghion, E., Argun, A., Hong, C. B., et al. (2021). Objective comparison of methods to decode anomalous diffusion. arXiv:2105.06766 [cond-mat, physics:physics, q-bio]. Available at: http://arxiv.org/abs/2105.06766 (Accessed July 12, 2021).
Muñoz-Gil, G., Volpe, G., García-March, M. A., Metzler, R., Lewenstein, M., and Manzo, C. (2020). The anomalous diffusion challenge: single trajectory characterisation as a competition. Emerging Top. Artif. Intelligence 2020, 44. doi:10.1117/12.2567914
Park, H. Y., Lim, H., Yoon, Y. J., Follenzi, A., Nwokafor, C., Lopez-Jones, M., et al. (2014). Visualization of Dynamics of Single Endogenous mRNA Labeled in Live Mouse. Science 343, 422–424. doi:10.1126/science.1239200
Persson, F., Lindén, M., Unoson, C., and Elf, J. (2013). Extracting intracellular diffusive states and transition rates from single-molecule tracking data. Nat. Methods 10, 265–269. doi:10.1038/nmeth.2367
Pitman, J. (2006). “Sequential constructions of random partitions,” in Combinatorial Stochastic Processes: Ecole d’Eté de Probabilités de Saint-Flour XXXII – 2002. Editors J. Pitman, and J. Picard (Berlin, Heidelberg: Springer Berlin Heidelberg), 55–75. doi:10.1007/3-540-34266-4_4
Qian, H., Sheetz, M. P., and Elson, E. L. (1991). Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophys. J. 60, 910–921. doi:10.1016/S0006-3495(91)82125-7
Rabiner, L. R. (1989). A tutorial on hidden Markov models and selected applications in speech recognition. Proc. IEEE 77, 257–286. doi:10.1109/5.18626
Robson, A., Burrage, K., and Leake, M. C. (2013). Inferring diffusion in single live cells at the single-molecule level. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20120029. doi:10.1098/rstb.2012.0029
Rowland, D. J., and Biteen, J. S. (2017). Measuring molecular motions inside single cells with improved analysis of single-particle trajectories. Chem. Phys. Lett. 674, 173–178. doi:10.1016/j.cplett.2017.02.052
Rust, M. J., Bates, M., and Zhuang, X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–795. doi:10.1038/nmeth929
Saxton, M. J. (1997). Single-particle tracking: the distribution of diffusion coefficients. Biophys. J. 72, 1744–1753. doi:10.1016/S0006-3495(97)78820-9
Scher, H., and Montroll, E. W. (1975). Anomalous transit-time dispersion in amorphous solids. Phys. Rev. B 12, 2455–2477. doi:10.1103/PhysRevB.12.2455
Schütz, G. J., Schindler, H., and Schmidt, T. (1997). Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys. J. 73, 1073–1080. doi:10.1016/S0006-3495(97)78139-6
Shen, H., Tauzin, L. J., Baiyasi, R., Wang, W., Moringo, N., Shuang, B., et al. (2017). Single Particle Tracking: From Theory to Biophysical Applications. Chem. Rev. 117, 7331–7376. doi:10.1021/acs.chemrev.6b00815
Sungkaworn, T., Jobin, M. L., Burnecki, K., Weron, A., Lohse, M. J., and Calebiro, D. (2017). Single-molecule imaging reveals receptor-G protein interactions at cell surface hot spots. Nature 550, 543–547. doi:10.1038/nature24264
Teh, Y. W., Jordan, M. I., Beal, M. J., and Blei, D. M. (2006). Hierarchical Dirichlet Processes. J. Am. Stat. Assoc. 101, 1566–1581. doi:10.1198/016214506000000302
Thapa, S., Lomholt, M. A., Krog, J., Cherstvy, A. G., and Metzler, R. (2018). Bayesian analysis of single-particle tracking data using the nested-sampling algorithm: maximum-likelihood model selection applied to stochastic-diffusivity data. Phys. Chem. Chem. Phys. 20, 29018–29037. doi:10.1039/C8CP04043E
Tuson, H. H., Foley, M. H., Koropatkin, N. M., and Biteen, J. S. (2018). The Starch Utilization System Assembles around Stationary Starch-Binding Proteins. Biophys. J. 115, 242–250. doi:10.1016/j.bpj.2017.12.015
Van Gael, J., Saatci, Y., Teh, Y. W., and Ghahramani, Z. (2008). “Beam sampling for the infinite hidden Markov model,” in Proceedings of the 25th international conference on Machine learning - ICML ’08 (Helsinki, Finland: ACM Press), 1088–1095. doi:10.1145/1390156.1390293
Keywords: single-molecule tracking (SPT), nonparametric Bayesian statistics, hierarchical Dirichlet process (HDP), hidden Markov model (HMM), recurrent neural network (RNN), anomalous diffusion
Citation: Chen Z, Geffroy L and Biteen JS (2021) NOBIAS: Analyzing Anomalous Diffusion in Single-Molecule Tracks With Nonparametric Bayesian Inference. Front. Bioinform. 1:742073. doi: 10.3389/fbinf.2021.742073
Received: 15 July 2021; Accepted: 25 August 2021;
Published: 10 September 2021.
Edited by:
Thomas Pengo, University of Minnesota Twin Cities, United StatesReviewed by:
Colin Kinz-Thompson, Rutgers University, Newark, United StatesCarl-Magnus Svensson, Leibniz Institute for Natural Product Research and Infection Biology, Germany
Copyright © 2021 Chen, Geffroy and Biteen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie S. Biteen, anNiaXRlZW5AdW1pY2guZWR1