 Yuji Mikata1,2,3*†
Yuji Mikata1,2,3*† Miyu Akedo2Yutaka Ohsedo1,2†Sunao Shoji1,2†Takashi Matsuo4†Attawit Jehdaramarn5Preeyanuch Sangtrirutnugul5*†Yasuhiro Funahashi6†
Miyu Akedo2Yutaka Ohsedo1,2†Sunao Shoji1,2†Takashi Matsuo4†Attawit Jehdaramarn5Preeyanuch Sangtrirutnugul5*†Yasuhiro Funahashi6†- 1Laboratory for Molecular and Functional Design, Department of Engineering, Nara Women’s University, Nara, Japan
- 2Cooperative Major in Human Centered Engineering, Nara Women’s University, Nara, Japan
- 3KYOUSEI Science Center, Nara Women’s University, Nara, Japan
- 4Division of Materials Science, Graduate School of Science and Technology, Nara Institute of Science and Technology (NAIST), Nara, Japan
- 5Center of Excellence for Innovation in Chemistry (PERCH-CIC), Department of Chemistry, Faculty of Science, Mahidol University, Bangkok, Thailand
- 6Department of Chemistry, Graduate School of Science, Osaka University, Osaka, Japan
A set of o-phenylenediamine-based pentadentate ligands having pyridine and quinoline binding sites was prepared in this work. The weak metal-binding abilities of anilinic nitrogen atoms and rigid chelate structure of the o-phenylenediamine skeleton serve as unique metal coordination properties of the ligand library presented herein. In addition to variations in the pyridine and quinoline binding sites, the non-coordinating alkyl groups (CH3 or CH2Ph) in the ligand structure cause significant differences in the coordination structures of copper(II) complexes. In the aerobic alcohol oxidation reaction in the presence of CuBr and 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), the ligand donating fewer electrons, namely, N-benzyl-N,N’,N’-tris(2-quinolylmethyl)-1,2-phenylenediamine (Bn-TQPHEN, L4), exhibited greater activity than N-methyl-N,N’,N’-tris(2-pyridylmethyl)-1,2-phenylenediamine (Me-TPPHEN, L1). Thus, the present study proposes future directions for the utilization of pentadentate ligands and their relevance to redox-active copper metalloenzymes.

GRAPHICAL ABSTRACT |
1 Introduction
The catalytic activities and selectivities of transition-metal complexes covering the active sites of metalloenzymes and artificial systems are strictly governed by the electronic and steric properties of the supporting ligands. Carefully designed ligands can be used to manipulate the structure, stability, as well as redox and electronic properties of the resulting metal complexes. For mimicking biological enzymatic systems, several researchers have extensively studied pyridine-based pentadentate nitrogen ligands, such as N-benzyl-N,N′,N′-tris(2-pyridylmethyl)ethylenediamine (Bn-TPEN) (Tamura et al., 2000; Hazell et al., 2002; Ortega-Villar et al., 2005; Zhang et al., 2014), N,N-bis(2-pyridylmethyl)-N-bis(2-pyridyl)methylamine (N4Py) (Lubben et al., 1995; Roelfes et al., 1999; Jackson et al., 2011; Ohzu et al., 2012; Chen et al., 2013; Mukherjee et al., 2019), and N-(2-(N,N-bis(2-pyridylmethyl)amino)ethyl)pyridine-2-carboxamide (PaPy3H) (Rowland et al., 2001; Patra et al., 2002; Patra and Mascharak, 2003; Ghosh et al., 2004; Gonzalez et al., 2011; Young et al., 2013), whose structures are shown in Figure 1. Based on the promising characteristics of these ligands, rational modifications have been implemented to enhance the reactivities and selectivities of the metal complexes utilized as catalysts for diverse applications.
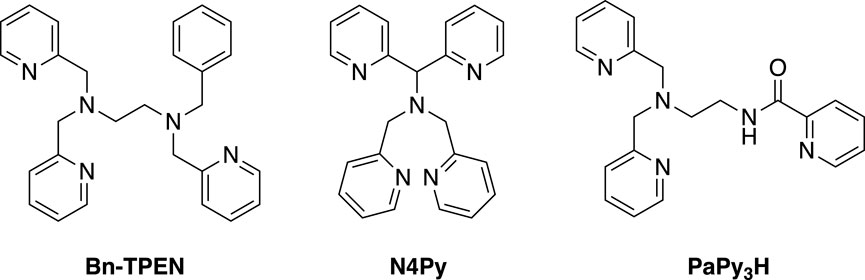
Figure 1. Structures of some pyridine-based pentanitrogen ligands.
One of the most important strategies used to modify existing ligand structures like those shown in Figure 1 is replacement of the widely used pyridines with quinolines. Compared to pyridine, quinoline has weaker metal-binding affinity and electron-donating ability owing to its weak basicity (pKa = 4.93 for quinoline vs. 5.23 for pyridine in H2O) (Lõkov et al., 2017; Hosmane and Liebman, 2009) as well as steric hindrance of the peri (H-8) hydrogen atom. Therefore, metal complexes with quinoline-based ligands usually exhibit electron-deficient natures, resulting in higher substrate oxidation reaction activities. Recently, our group systematically investigated the replacement of pyridines and the benzyl group of Bn-TPEN with other nitrogen-containing heteroaromatics and non-coordinating alkyl substituents, respectively, to comprehensively examine the effects of the ligands on the structural and electrochemical properties of copper(II) complexes (Mikata et al., 2024). Copper proteins and copper-containing metalloenzymes are ubiquitous in biological systems. The diverse coordination geometries and electronic properties of the copper centers enable specific functions, including electron transfer, dioxygen binding/activation, and oxygen atom transfer reactions (Rubino and Franz, 2012; Liu et al., 2014; Solomon et al., 2014). Extensive studies on synthetic model complexes based on structural and functional mimicry of the copper proteins via careful ligand design have revealed many unprecedented features of this biologically important metal ion (Itoh, 2015; Hong et al., 2017; Adam et al., 2018).
In the present study, in addition to the alteration of pyridine with quinoline, the ethylenediamine backbone of Bn-TPEN was replaced with o-phenylenediamine to construct a new ligand library with weak electron-donating abilities using the diminished basicity of anilinic nitrogen atoms compared to the alkyl amine nitrogen atoms (Figure 2). The ligand library presented herein has better synthetic accessibility than our previously reported work on a similar effort, where ethylenediamine was replaced with the 2-aminoethanol skeleton (Mikata et al., 2023). As shown in Figure 2, a total of four ligands were examined in this study, including the known ligand N-methyl-N,N′,N′-tris(2-pyridylmethyl)-1,2-phenylenediamine (L1) (Basu et al., 2015; Ekanayake et al., 2017), to investigate the ligand effects on structure, redox potential, absorption spectrum, and catalytic activity for alcohol oxidation reactions of copper complexes. These pentadentate ligands are appealing for Cu-catalyzed aerobic alcohol oxidation as they provide well-defined N5 environments that resist disproportionation and allow fine-tuning of the copper redox properties. Notably, Cu catalysts supported by pentadentate ligands have never been reported for this transformation, in contrast to the well-established (bpy)Cu(I)/TEMPO catalyst system (where bpy = 2,2′-bipyridine and TEMPO = 2,2,6,6-tetramethylpiperidine 1-oxyl) (Hoover and Stahl, 2011) that exhibits high activity for alcohol oxidation at room temperature under air as well as wide substrate scope.

Figure 2. Structures of the four ligands investigated in this study.
2 Materials and methods
2.1 General
All reagents and solvents used to prepare the ligands and complexes were procured from commercial sources and used without further purification. The 1H/13C nuclear magnetic resonance (NMR) spectra were recorded using a JEOL AL-400 spectrometer at 400/100 MHz and referenced to internal Si(CH3)4 or solvent signals. Furthermore, elemental analyses were performed on the J-Science JM10 micro corder, and the UV–vis spectra were recorded using a Jasco V-770 spectrophotometer equipped with a temperature control unit (Jasco ETCS-761). The perchlorate salts of metal complexes with organic ligands are known to be potentially explosive, so all due precautions were observed during the study.
2.2 Synthesis of ligands
2.2.1 N-methyl-N,N′,N′-tris(2-pyridylmethyl)-1,2-phenylenediamine (Me-TPPHEN, L1)
The preparation of this compound was reported previously by Basu et al. (2015). Briefly, a mixture of N-methyl-1,2-phenylenediamine (0.169 mL, 1.50 mmol), potassium carbonate (1.04 g, 7.53 mmol), potassium iodide (0.249 g, 1.50 mmol), and 2-chloromethylpyridine hydrochloride (0.740 g, 4.51 mmol) in acetonitrile (20 mL) was refluxed for 48 h under a nitrogen atmosphere in the dark. After the reaction solution was cooled to room temperature, the solvent was removed under reduced pressure, and the organic materials were extracted with chloroform/water. The chloroform was dried over Na2SO4 and evaporated, and the residue was purified by silica gel column chromatography (eluent: ethyl acetate and methanol in the ratio of 10:1) to yield Me-TPPHEN as an amorphous brown substance (0.245 g, 0.619 mmol, 41%). Further recrystallization from acetonitrile-ether afforded single crystals suitable for X-ray crystallography.
1H NMR (CDCl3, 400 MHz): δ 8.53 (dt, J = 4.9, 1.0 Hz, 1H), 8.49–8.51 (m, 2H), 7.42–7.49 (m, 3H), 7.17 (d, J = 7.8 Hz, 2H), 7.05–7.15 (m, 4H), 6.97–7.00 (m, 1H), 6.90–6.94 (m, 2H), 6.80–6.85 (m, 1H), 4.67 (s, 6H), 2.85 (s, 3H).
13C NMR (CDCl3, 100 MHz): δ 159.0, 158.8, 149.0, 148.8, 144.6, 142.1, 136.09, 136.05, 122.79, 122.75, 122.6, 122.4, 121.7, 121.6, 120.4, 60.0, 57.5, 40.0.
2.2.2 N-methyl-N,N′,N′-tris(2-quinolylmethyl)-1,2-phenylenediamine (Me-TQPHEN, L2)
A mixture of N-methyl-1,2-phenylenediamine (0.169 mL, 1.50 mmol), potassium carbonate (1.04 g, 7.53 mmol), potassium iodide (0.275 g, 1.66 mmol), and 2-chloromethylquinoline hydrochloride (0.945 g, 4.41 mmol) in acetonitrile (15 mL) was refluxed for 48 h under a nitrogen atmosphere in the dark. After the reaction solution was cooled to room temperature, the solvent was removed under reduced pressure, and the organic materials were extracted with chloroform/water. The chloroform was dried over Na2SO4 and evaporated, and the residue was purified by silica gel column chromatography (eluent: hexane and ethyl acetate in the ratio of 3:1) to yield Me-TQPHEN as an orange solid (0.778 g, 1.43 mmol, 97%).
1H NMR (CDCl3, 400 MHz): δ 8.08 (d, J = 8.3 Hz, 1H), 8.00 (d, J = 8.8 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.77 (d, J = 8.3 Hz, 1H), 7.60–7.70 (m, 6H), 7.43–7.52 (m, 3H), 7.37 (d, J = 8.3 Hz, 2H), 7.21 (t, J = 8.8 Hz, 1H), 7.03–7.12 (m, 2H), 6.92 (dt, J = 7.6, 1.5 Hz, 1H), 6.84 (dt, J = 7.6, 1.5 Hz, 1H), 4.94 (s, 4H), 4.92 (s, 2H), 2.94 (s, 3H).
13C NMR (CDCl3, 100 MHz): δ 159.8, 159.5, 147.64, 147.56, 144.7, 142.2, 136.0, 129.3, 129.2, 129.0, 128.9, 127.44, 127.38, 127.2, 127.1, 126.0, 122.7, 122.6, 121.7, 120.9, 120.5, 60.8, 58.5, 40.2.
High-resolution mass spectrometry (HRMS) via electrospray ionization (ESI) m/z: calculated value for C37H32N5 (L2 + H+) = 546.2658 and measured value = 546.2630; calculated value for C37H31N5Na (L2 + Na+) = 568.2477 and measured value = 568.2421.
Analytically calculated value for C37H33.4N5O1.2 (L2·1.2H2O): C, 78.33; H, 5.93; N, 12.34; measured value: C, 77.97; H, 5.61; N, 12.16.
2.2.3 N-benzyl-N,N′,N′-tris(2-pyridylmethyl)-1,2-phenylenediamine (Bn-TPPHEN, L3)
A mixture of N-benzyl-1,2-phenylenediamine (0.198 g, 1.00 mmol), potassium carbonate (0.691 g, 5.00 mmol), potassium iodide (0.166 g, 1.00 mmol), and 2-chloromethylpyridine hydrochloride (0.493 g, 3.01 mmol) in acetonitrile (15 mL) was refluxed for 48 h under a nitrogen atmosphere in the dark. After the reaction solution was cooled to room temperature, the solvent was removed under reduced pressure, and the organic materials were extracted with chloroform/water. The chloroform was dried over Na2SO4 and evaporated, and the residue was purified by silica gel column chromatography (eluent: ethyl acetate and methanol in the ratio of 30:1) to yield Bn-TPPHEN as a brown oil (0.178 g, 0.377 mmol, 38%).
1H NMR (CDCl3, 400 MHz): δ 8.51 (d, J = 4.4 Hz, 3H), 7.36–7.45 (m, 3H), 7.14–7.19 (m, 5H), 7.03–7.10 (m, 6H), 6.90–6.95 (m, 1H), 6.72–6.85 (m, 3H), 4.75 (s, 4H), 4.54 (s, 2H), 4.53 (s, 2H).
13C NMR (CDCl3, 100 MHz): δ 158.83, 158.76, 149.0, 148.9, 142.5, 142.3, 137.5, 136.05, 135.97, 129.5, 128.0, 126.8, 123.1, 122.9, 122.5, 122.2, 121.81, 121.76, 57.7, 56.4, 55.0.
HRMS (ESI) m/z: calculated value for C31H30N5 (L3 + H+) = 472.2501 and measured value = 472.2477; calculated value for C31H29N5Na (L3 + Na+) = 494.2321 and measured value = 494.2287.
Analytically calculated value for C31H31N5O (L3·H2O): C, 76.05; H, 6.38; N, 14.30; measured value: C, 75.93; H, 6.18; N, 14.09.
2.2.4 N-benzyl-N,N′,N′-tris(2-quinolylmethyl)-1,2-phenylenediamine (Bn-TQPHEN, L4)
A mixture of N-benzyl-1,2-phenylenediamine (0.297 g, 1.50 mmol), potassium carbonate (1.04 g, 7.53 mmol), potassium iodide (0.255 g, 1.54 mmol), and 2-chloromethylquinoline hydrochloride (0.966 g, 4.51 mmol) in acetonitrile (15 mL) was refluxed for 24 h under a nitrogen atmosphere in the dark. After the reaction solution was cooled to room temperature, the solvent was removed under reduced pressure, and the organic materials were extracted with chloroform/water. The chloroform was dried over Na2SO4 and evaporated, and the residue was purified by silica gel column chromatography (eluent: hexane and ethyl acetate in the ratio of 3:1) to yield Bn-TQPHEN as an orange solid (0.519 g, 0.835 mmol, 56%).
1H NMR (CDCl3, 400 MHz): δ 7.98–8.03 (m, 3H), 8.87 (d, J = 8.3 Hz, 2H), 7.76 (d, J = 8.8 Hz, 1H), 7.61–7.71 (m, 6H), 7.43–7.48 (m, 3H), 7.32 (d, J = 8.4 Hz, 2H), 7.13–7.25 (m, 6H), 7.07 (dd, J = 7.1, 2.2 Hz, 1H), 6.88 (dd, J = 7.3, 2.4 Hz, 1H), 6.76–6.87 (m, 2H), 5.04 (s, 4H), 4.79 (s, 2H), 4.66 (s, 2H).
13C NMR (CDCl3, 100 MHz): δ 159.4, 147.7, 147.6, 142.4, 142.3, 137.4, 136.0, 135.9, 129.7, 129.2, 129.0, 128.0, 127.4, 127.2, 126.9, 126.0, 122.7, 122.6, 122.3, 121.7, 121.2, 121.0, 58.6, 57.2, 55.1.
HRMS (ESI) m/z: calculated value for C43H36N5 (L4 + H+) = 622.2971 and measured value = 622.2902; calculated value for C43H35N5Na (L4 + Na+) = 644.2790 and measured value = 644.2705.
Analytically calculated value for C43H37N5O (L4·H2O): C, 80.72; H, 5.83; N, 10.95; measured value: C, 80.65; H, 5.67; N, 10.83.
2.3 Synthesis of copper(II) complexes
2.3.1 [Cu(L1)](ClO4)2 as L1-CuII
The hexafluorophosphate complex of L1-CuII has been synthesized and characterized previously (Ekanayake et al., 2017). Briefly, Cu(ClO4)2·6H2O (13 mg, 35 µmol) in ethanol (0.5 mL) was added to the solution of Me-TPPHEN (11.9 mg, 30 µmol) in ethanol (0.5 mL). The precipitated materials were then collected by filtration to yield [Cu(L1)](ClO4)2 as a blue powder (18.5 mg, 28 μmol, 93%).
2.3.2 [Cu(L2)(CH3OH)](ClO4)2 as L2-CuII
Cu(ClO4)2·6H2O (10 mg, 27 µmol) in methanol (0.5 mL) was added to the solution of Me-TQPHEN (13.6 mg, 25 µmol) in methanol (0.5 mL). The resulting mixture was maintained at 4 °C under ether diffusion condition to yield [Cu(L2)(CH3OH)](ClO4)2 as green crystals (12.3 mg, 16.9 μmol, 68%) suitable for X-ray crystallography.
HRMS (ESI) m/z: calculated value for C37H31CuN5 (L2 + CuII) = 608.1875 and measured value = 608.1836; calculated value for C37H31ClCuN5 (L2 + CuII + Cl−) = 643.1564 and measured value = 643.1527.
Analytically calculated value for C38.5H37Cl2CuN5O9.5 ([Cu(L2)(CH3OH)](ClO4)2·0.5CH3OH): C, 54.01; H, 4.36; N, 8.18; measured value: C, 53.91; H, 3.96; N, 8.32.
2.3.3 [Cu(L3)](PF6)2 as L3-CuII
Cu(ClO4)2·6H2O (11.1 mg, 30 µmol) in ethanol (0.5 mL) was added to the solution of Bn-TPPHEN (14.1 mg, 30 µmol) in ethanol (0.5 mL). The precipitated materials were collected by filtration and recrystallized from acetonitrile (0.8 mL) in the presence of NH4PF6 (14.5 mg, 89 µmol) under ether diffusion condition at 4 °C to yield [Cu(L3)](PF6)2 as blue crystals (9.77 mg, 11.8 μmol, 39%) suitable for X-ray crystallography.
HRMS (ESI) m/z: calculated value for C31H29CuN5 (L3 + CuII) = 534.1719 and measured value = 534.1706; calculated value for C31H29ClCuN5 (L3 + CuII + Cl−) = 569.1408 and measured value = 569.1389; calculated value for C31H29ClCuN5O4 (L3 + CuII + ClO4−) = 633.1204 and measured value = 633.1178.
Analytically calculated value for C34H33.5CuF12N6.5P2 ([Cu(L3)](PF6)2·1.5CH3CN): C, 46.06; H, 3.81; N, 10.27; measured value: C, 46.39; H, 3.92; N, 9.93.
2.3.4 [Cu(L4)](ClO4)2 as L4-CuII
Cu(ClO4)2·6H2O (10 mg, 27 µmol) in methanol (1.0 mL) was added to the solution of Bn-TQPHEN (15.5 mg, 25 µmol) in methanol (1.0 mL). The resulting mixture was maintained at 4 °C under ether diffusion condition to yield [Cu(L4)](ClO4)2 as green crystals (16.4 mg, 18.5 μmol, 74%) suitable for X-ray crystallography.
HRMS (ESI) m/z: calculated value for C43H35CuN5 (L4 + CuII) = 684.2188 and measured value = 684.2171; calculated value for C43H35ClCuN5 (L4 + CuII + Cl−) = 719.1877 and measured value = 719.1860.
Analytically calculated value for C43H35Cl2CuN5O8 ([Cu(L4)](ClO4)2): C, 58.41; H, 3.99; N, 7.92; measured value: C, 58.48; H, 3.76; N, 7.78.
2.4 X-ray crystallography
Single crystals of L1, L2-CuII·0.5CH3OH, L3-CuII, and L4-CuII·3CH3OH were placed on a mounted CryoLoop or MiTeGen MicroLoop with Paratone-N oil. Then, all data were collected at 150 or 153 K on a Rigaku Mercury charge-coupled device (CCD), Saturn CCD, or Synergy-S detector with monochromatic MoKα radiation at 50 kV/40 mA (Mercury), 50 kV/24 mA (Saturn), or 50 kV/1 mA (Synergy-S), respectively. The data were processed on a personal computer using CrystalClear Software or CrysAllisPro (Rigaku). The structures were solved by direct methods (SIR-92 (Altomare et al., 1994), SIR 2008 (Burla et al., 2007), or SHELXT 2018/2 (Sheldrick, 2015b)) and refined using the full-matrix least-squares method on F2 (SHELXL 2016/6 or 2019/3) (Sheldrick, 2015a). The hydrogen atoms were located at the ideal positions and treated as riding models. The disordered solvent molecules for L3-CuII were removed by PLATON SQUEEZE (Spek, 2015). The crystal data are summarized in Supplementary Tables S1, S2. The CCDC 2479086-2479089 repository contains the supplementary crystallographic data for this study; these data may be obtained free of charge from the Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk/datarequest/cif).
2.5 Cyclic voltammetry (CV)
CV measurements of the copper(II) complexes were performed using the ALS Electrochemical Analyzer (Model 6271E, Bioanalytical Systems Inc.) equipped with a glassy carbon working electrode, a platinum wire auxiliary electrode, and an Ag/Ag+ reference electrode (in acetonitrile containing 0.01 M of AgNO3 and 0.1 M of Bu4NClO4). The complex concentration was 1 mM in acetonitrile solution containing 0.1 M of Bu4NPF6 as a supporting electrolyte. The redox potentials were corrected against the FcH/FcH+ couple measured under each experimental condition.
2.6 Oxidation of benzyl alcohol
A stock solution of the CuBr/L catalyst (L = Me-TPPHEN or Bn-TQPHEN) was prepared by stirring CuBr (7.2 mg, 0.050 mmol) and L (0.050 mmol) in 5 mL of CH3CN at room temperature for 15 min. The resulting solution was filtered and then diluted with CH3CN to a total volume of 10.0 mL. For the typical catalytic reaction shown in Scheme 1, benzyl alcohol (0.11 g, 1.0 mmol) was dissolved in 5.0 mL of the 1:1 CuBr/L stock solution, followed by addition of TEMPO (7.9 mg, 0.050 mmol), N-methylimidazole (NMI; 8.0 mL, 0.10 mmol), and anisole (10.9 mL, 0.010 mmol). The reaction solution was stirred at room temperature with or without ultraviolet irradiation (PHILIPS© model 41-6782 H15.A, 300–600 V; wavelength: 315–380 nm). After a specified amount of time, the reaction solution was filtered through a short silica gel column using ethyl acetate as the eluent. The conversion percentages were determined by gas chromatography mass spectrometry (GC-MS; column ZB-5MS: 30 m × 0.25 mm × 0.25 mm) using anisole as an internal standard.

Scheme 1. Cu-catalyzed aerobic oxidation of benzyl alcohol to benzaldehyde.
3 Results and discussion
3.1 Synthesis and characterization of ligands
Three new ligands (L2–L4) and the known compound Me-TPPHEN (L1) (Basu et al., 2015) shown in Figure 2 were synthesized as shown in Scheme 2. Refluxing N-methyl- or N-benzyl-1,2-phenylenediamine with thrice the equivalent of 2-chloromethylpyridine or 2-chloromethylquinoline in the presence of potassium carbonate and potassium iodide in acetonitrile under a nitrogen atmosphere in the dark yielded the desired compounds in moderate to good amounts (38%–97%). The structures of the ligands were characterized by 1H/13C NMR spectroscopy and elemental analyses. The solid-state structure for L1 was determined by X-ray crystallography (Supplementary Table S1; Supplementary Figure S1).
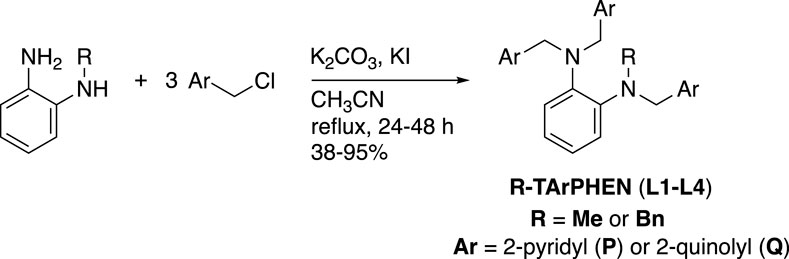
Scheme 2. Synthesis of the ligands.
3.2 Synthesis and X-ray crystal structures of the copper(II) complexes
The mononuclear copper(II) complexes ligated with L1–L4 were prepared from equimolar amounts of Cu(ClO4)2·6H2O in ethanol or methanol at room temperature (Scheme 3). The hexafluorophosphate salt of the L1-CuII complex has been prepared previously and characterized by X-ray crystallography (Ekanayake et al., 2017). For L2 and L4, the complexation with copper(II) perchlorate in methanol directly yielded single crystals suitable for X-ray crystallography. For the pyridine-based ligand L3, crystallization of the copper(II) complex was achieved as a hexafluorophosphate salt from acetonitrile solution. The new copper(II) complexes were then characterized by elemental analyses and X-ray crystallography. Supplementary Tables S1, S2 list the crystallographic parameters and diffraction conditions of these complexes. The interatomic distances and angles around the copper centers are listed in Supplementary Tables S3–S6. Figure 3 shows the solid-state structures of the copper(II) complexes with L1–L4, including the previously reported L1-CuII (Ekanayake et al., 2017) as the reference. Figure 3 also displays the τ5 values (Addison et al., 1984; Blackman et al., 2020) for convenient geometric assessments of the five-coordinate metal centers for pentacoordinate complexes. Briefly, the τ5 value is calculated as follows: {(largest L-M-L angle, β) – (second largest L-M-L angle, α)}/60. The ideal square pyramid exhibits τ5 = 0 (α = β = 180°), whereas the ideal trigonal bipyramid exhibits τ5 = 1 (α = 120° and β = 180°). The coordination distances are summarized in Table 1.

Scheme 3. Synthesis of the copper(II) complexes.

Figure 3. Perspective views of (a) L1-CuII (refcode: VAYDIL (Ekanayake et al., 2017)); (b) L2-CuII; (c) L3-CuII; (d) L4-CuII at 50% probability. The non-coordinating solvents, counter anions, and hydrogen atoms are omitted for clarity.
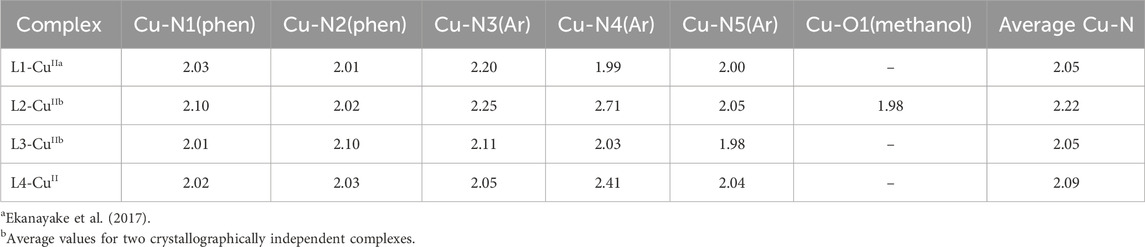
Table 1. Interatomic distances (Å) of the copper(II) complexes.
The L2-CuII complex exhibits octahedral geometry with five nitrogen atoms from the ligand and one oxygen atom from methanol. Our previous studies on copper(II) complexes with related pentacoordinate ligands revealed that the quinoline nitrogen atom at the N5 position binds strongly with the metal center because the steric hindrance of the peri hydrogen (H-8) of the quinoline ring prevents coordination of the counter anion (mostly perchlorate), yielding a pentacoordinate copper center (Mikata et al., 2023; Mikata et al., 2024). For L2-CuII, the electron deficiency of the copper center due to the weak electron-donating abilities of phenylenediamine and the quinoline nitrogen atoms allows coordination from a sixth binding site even for the N5-quinoline complex. The less-bulky structure of methanol compared to perchlorate results in the formation of the [Cu(L2)(CH3OH)]2+ complex. The Cu-N5 distance (2.05 Å) is short enough for quinoline coordination even in the hexacoordinate structure. The copper(II) center of L2-CuII exhibits typical Jahn–Teller distorted geometry with elongated Cu-N3 (2.25 Å) and Cu-N4 (2.71 Å) distances.
The L3-CuII complex exhibits trigonal-bipyramid-like geometry (τ5 = 0.63) with short Cu-N1 (2.01 Å) and Cu-N5 (1.98 Å) distances, which contrast with the square-pyramid-like geometry of L1-CuII (τ5 = 0.20). The steric hindrance of the benzyl substituent in L3 increases the N2-Cu distance (2.10 Å), resulting in shortening of the Cu-N3 distance (2.11 Å) compared to the apical bond of L1-CuII (Cu-N3 = 2.20 Å) and widening of the N3-Cu-N4 angle (123° for L3-CuII vs. 93° for L1-CuII). Such differences between the methyl and benzyl substituents were not observed in the previous investigation (Mikata et al., 2024), indicating that there is flexibility in the coordination structures of phenylenediamine complexes for efficient accommodation of cationic metal centers by weak electron-donating sites. This difference in geometry was also confirmed in the solution state (vide infra).
As discussed above, the quinoline nitrogen atom at the N5 position coordinates strongly with the metal center. Concomitantly, this movement shortens the N2-Cu distance, which in turn increases the steric clash between the benzyl substituent and quinoline ring at the N3 position in L4-CuII. As a result, there is significant widening of the N2-Cu-N3 angle (152°) and elongation of the Cu-N4 distance (2.41 Å) in the typical square-pyramid-like structure of the L4-CuII complex (τ5 = 0.19). The average Cu-N coordination distance of L4-CuII (2.09 Å) is larger than those of pyridine complexes (2.05 Å) and comparable to those of related copper(II) complexes with N-alkylethylenediamine ligands having three quinoline binding sites (2.09 Å) (Mikata et al., 2024). Although the phenylenediamine effect was not observed for the overall coordination distances in the copper(II) complexes herein, the redox property may be affected.
3.3 Redox potentials of copper(II) complexes
The redox potentials of all four copper(II) complexes were measured by CV in acetonitrile solution, where reversible or quasi-reversible one-electron redox couples were observed in all complexes (Figure 4). The redox potentials of the Cu(II)/Cu(I) couple (E1/2 (V) vs. FcH/FcH+) are summarized in Table 2, and the corresponding values of the ethylenediamine complexes are shown for comparison.

Figure 4. Cyclic voltammograms of (a) Me-TPPHEN-CuII (L1-CuII); (b) Me-TQPHEN-CuII (L2-CuII); (c) Bn-TPPHEN-CuII (L3-CuII); (d) Bn-TQPHEN-CuII (L4-CuII) in acetonitrile (1 mM, scan rate: 100 mV/s).

Table 2. Cu(II)/Cu(I) redox potentials (E1/2 (V) vs. FcH/FcH+) for the four copper complexesa,b.
In general, the quinoline complexes exhibit higher redox potentials than the pyridine complexes, and the benzyl substituent tends to increase the redox potentials from the methyl derivatives. Both effects were smaller for the phenylenediamine complexes than previously reported ethylenediamine complexes. Because the phenylenediamine skeleton already increases the potentials from the ethylenediamine derivatives (0.10–0.15 V for pyridine and 0.04–0.07 V for quinoline complexes), there is gradual leveling of all shifts toward positive potentials. Another important observation on the electrochemical measurements of copper complexes is that the peak-to-peak separation (ΔE, shown in parentheses in Table 2) increases as the redox potential shifts in the positive direction. This is also valid for the entire family of ethylenediamine/phenylenediamine complexes (Mikata et al., 2024); however, the L2-CuII complex exhibits an extremely large value (ΔE = 0.45 V) compared to the other quinoline complexes (ΔE = 0.29 V for L4-CuII, 0.12 V for Me-TQEN-CuII, and 0.14 V for Bn-TQEN-CuII), while the quinoxaline (Qx) complexes with extremely high redox potentials exhibit comparable ΔE values of 0.12 V for Me-TQxEN-CuII (E1/2 = −0.05 V) and 0.49 V for Bn-TQxEN-CuII (E1/2 = +0.05 V).
The possible reason for the large ΔE value of L2-CuII, which indicates a lack of full thermodynamic reversibility, could be the rigid skeletons of the phenylenediamine-quinoline complexes that require significant rearrangement of the coordination geometry around the copper center for one-electron redox processes. As shown in Table 3, the reduction peak potential (Ered) values for Me-TQPHEN-CuII (−0.35 V) and Bn-TQPHEN-CuII (−0.24 V) are more negative than those of the corresponding ethylenediamine complexes (−0.26 V for Me-TQEN-CuII and −0.19 V for Bn-TQEN-CuII), breaking the general trend of the EN/PHEN skeleton effect. Moreover, the oxidation peak potential (Eox) of Me-TQPHEN-CuII (0.10 V) is more positive than that of its benzyl counterpart Bn-TQPHEN-CuII (0.05 V), deviating from the general trend of the Me/Bn substituent effect. Such deviations were clearly observed in the peak potential differences between the pyridine and quinoline complexes (E(Q) – E(P), last column in Table 3) as well as between the phenylenediamine and ethylenediamine complexes (E (PHEN) – E (EN), bottom two rows in Table 3). The former values are at approximately 0.4 V for all ethylenediamine complexes, including pyridine and quinoline, but significant deviations were observed for Me-TArPHEN-CuII for both Ered (0.16 V) and Eox (0.51 V). The latter values are negative for only Ered for the quinoline complexes (−0.09 for Me and −0.05 for Bn). Thus, the redox potentials (E1/2) of the present phenylenediamine complexes follow the expected coordination abilities of the ligand structures, but the Ered value for the quinoline complex with the phenylenediamine skeleton exhibits a negative shift than the ethylenediamine complexes, resulting in large ΔE values, especially for Me-TQPHEN-CuII.
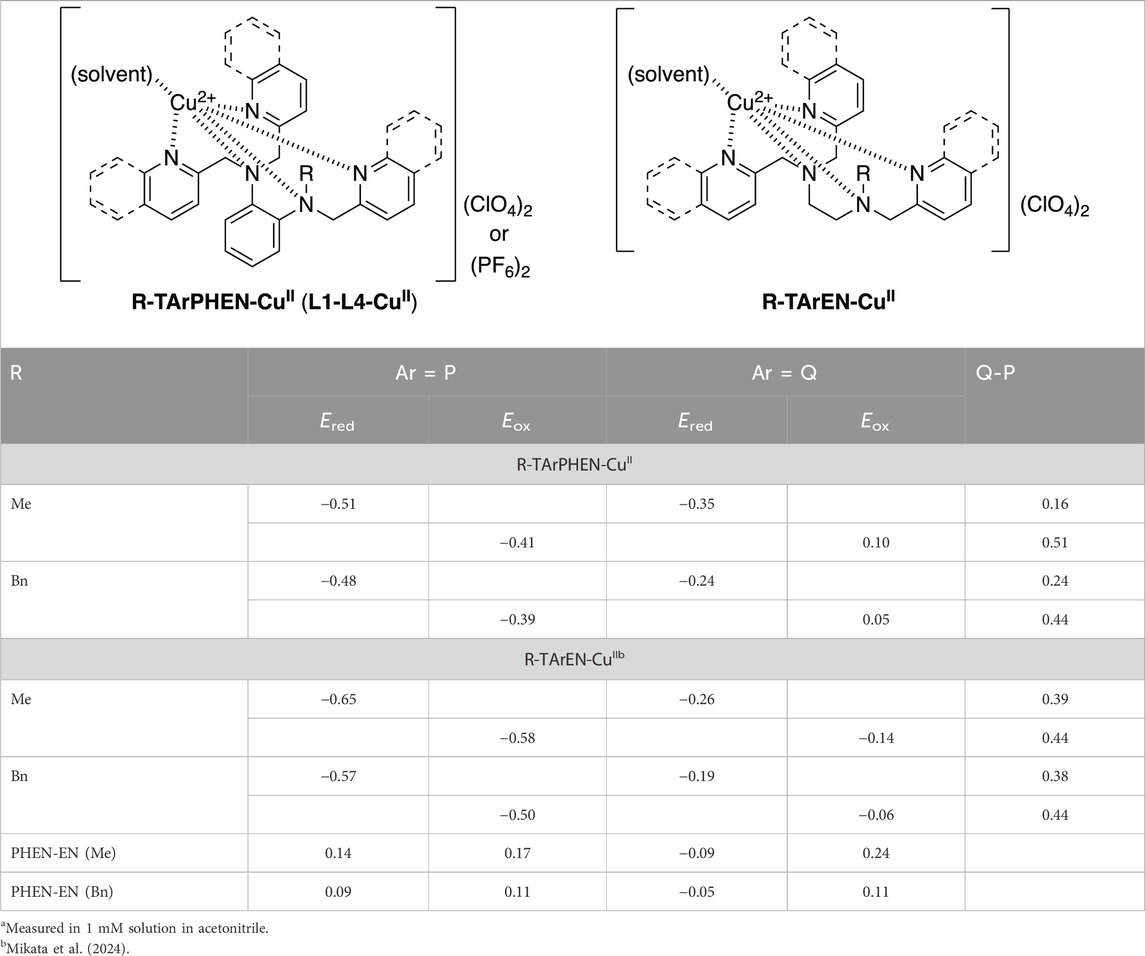
Table 3. Cu(II)/Cu(I) reduction and oxidation peak potentials (Ered and Eox (V) vs. FcH/FcH+) for the copper complexesa.
3.4 UV–vis absorption properties of the copper(II) complexes
The UV-vis absorption spectra of all the copper(II) complexes were measured in acetonitrile (Figure 5) and methanol (Supplementary Figure S2). All copper(II) complexes exhibited characteristic d-d transition bands at approximately 600–1000 nm, reflecting the coordination environment of the metal center in the solution state. The maximum absorption wavelengths in both solvents are summarized in Table 4, and the data indicate that negligible solvent effects were observed in the present copper complexes.

Figure 5. Absorption spectrum for (a) Me-TPPHEN-CuII (L1-CuII); (b) Me-TQPHEN-CuII (L2-CuII); (c) Bn-TPPHEN-CuII (L3-CuII); (d) Bn-TQPHEN-CuII (L4-CuII) in acetonitrile (1 mM).

Table 4. Maximum absorption wavelengths (nm) for Copper (II) complexesa.
As discussed previously (Mikata et al., 2023; Mikata et al., 2024), the maximum absorption wavelength in the d-d transition region is a potential indicator of the coordination geometry of the copper(II) center. The absorption spectrum of the L1-CuII complex shows three distinct maximum wavelengths at approximately 640, 770, and 930 nm (Figure 5A). The highest energy absorption corresponding to the square pyramidal and/or octahedral structure is slightly dominant for L1-CuII in acetonitrile, as emphasized in the methanol solution given the increased polarity and possible coordination of the solvent molecule or counter anion to the metal center (Supplementary Figure S2a). In contrast, the L3-CuII complex shows an absorption maximum at approximately 800 nm while maintaining the other two absorption components (640 and 930 nm) with weak intensities. The >800 nm absorption band and increased molar extinction coefficient at this wavelength (ε = 240 in acetonitrile) is indicative of the trigonal bipyramidal copper(II) center for this complex. These assessments of the solution structures of the complexes from the absorption spectra of L1-CuII and L3-CuII are in good agreement with the solid-state structures elucidated by X-ray crystallography (Figure 3; Table 4). The three component absorption spectra and rather half-way τ values for the phenylenediamine–pyridine complexes clearly indicate that their structures are highly flexible as well as in-between those of the trigonal bipyramid and square pyramid/octahedron.
The quinoline complexes L2-CuII and L4-CuII exhibit short absorption wavelength maxima corresponding to the square pyramidal or octahedral geometry, with no other absorption bands being observed in the 600–1100 nm range. The slightly red-shifted absorption maxima compared to the pyridine complex L1-CuII are attributable to the weak coordination of quinolines at the equatorial position, as reported previously (Mikata et al., 2023). From these analyses of the absorption spectra, the rigid structures of quinoline complexes were elucidated and shown to support the large ΔE values observed in the electrochemical measurements (Table 2).
3.5 Catalytic activity in CuI/TEMPO-mediated aerobic oxidation of benzyl alcohol in the presence of L1 and L4
To demonstrate the practical applications of the present ligand/complex library, we investigated the aerobic alcohol oxidation reaction. The oxidation of alcohols to carbonyl compounds is one of the most important molecular transformations in biological systems, and TEMPO/CuI-mediated aerobic catalytic oxidation of alcohol has recently shown some potential (Hoover and Stahl, 2011; Varala and Seema, 2023). This system is relevant to the galactose oxidase active site that contains copper and the phenolate radical from tyrosine residue (Whittaker, 2003; Oshita and Shimazaki, 2020). Mimicking the enzymatic systems that allow conversion of broadly available alcohols into useful carbonyl compounds has great importance. We chose L1 and L4 for preliminary assessments in the alcohol oxidation reaction in the presence of CuBr, TEMPO, and NMI in acetonitrile solution (Scheme 2; Table 5; Supplementary Figures S3–S8). The CuBr/L4 catalyst exhibited superior activity for aerobic oxidation of benzyl alcohol to benzaldehyde, achieving 97% conversion in CH3CN at room temperature after 24 h compared to the 25% conversion obtained for CuBr/L1 under identical conditions (entries 2 and 5 in Table 5). Ultraviolet irradiation provided moderate performance improvements for both CuBr/L catalysts, resulting in 51% and 30% conversions after 8 h (entries 3 and 6, respectively). The oxidation activity profiles of both catalysts with and without ultraviolet irradiation are depicted in Figure 6.

Table 5. Catalytic activities toward aerobic benzyl alcohol oxidation using Me-TPPHEN (L1) and Bn-TQPHEN (L4)a.

Figure 6. Oxidation profiles of the in situ generated CuBr/L (L = Me-TPPHEN and Bn-TQPHEN).
The higher catalytic performance of CuBr/L4 is ascribed to its more positive half-cell potential (E1/2 (CuI/CuII) = −0.09 V) relative to that of CuBr/L1 (−0.46 V) in acetonitrile (Table 2). This more-positive potential indicates greater reducibility (i.e., stronger oxidization capability) of the CuII species in the presence of L4, which facilitates the substrate oxidation step in the catalytic cycle as the possible rate-controlling step (Hoover et al., 2013). Although the relative catalytic activity of CuBr/L4 does not match those of leading catalyst systems (Hoover and Stahl, 2011; Al-Hunaiti et al., 2022), our catalytic study demonstrates the effect of ligand tuning on the CuI/CuII redox potential, which enhances the catalytic oxidation activity of CuBr/L4 by promoting efficient oxygen activation and subsequent alcohol dehydrogenation. Accordingly, the phenylenediamine ligand, especially Bn-TQPHEN, could be considered a competent pentadentate ligand platform for Cu-catalyzed aerobic alcohol oxidation. In addition to the redox potential, the relationship between the rigidity of the coordination geometry of the complex and its catalytic reactivity will be studied further to reproduce the observed flexibility of the coordination environment at the metalloenzyme active sites.
4 Conclusion
The o-phenylenediamine skeleton of the ligand library presented herein effectively enhances the electron abstracting ability of copper(II) complexes compared to the ethylenediamine ligands. In addition to the quinoline binding sites, alteration of the non-coordinating alkyl substituents largely perturbed the structures of the copper(II) complexes. The diverse feature in the absorption spectra of these copper(II) complexes in the d-d transition region (600–1100 nm) reflect the metal coordination environments and plausible structural flexibilities of the complexes in their solution states. The redox potential is independent of the structural diversity in the square pyramidal or trigonal bipyramidal geometry because the Cu(II)/Cu(I) redox potentials of the copper complexes depend solely on the ligand structures; this was shown through the positive shifts of 0.33–0.34 V and 0.03–0.04 V obtained by replacing the pyridine and methyl groups with quinoline and benzyl substituents, respectively. The poorly electron-donating ligand Bn-TQPHEN (L4) was found to be an efficient supporting ligand in the aerobic oxidation reaction of alcohol to aldehyde when catalyzed by the CuBr/TEMPO/NMI system as it facilitated electron abstraction of the copper(II) complexes from the metal-bound alkoxide intermediates. The present investigation provides an important case study in controlling the structural and redox properties of copper(II) complexes through rational design of the ligand structures. The extension of the present pentacoordinate ligand library is expected to enable the preparation of a wide variety of artificial metal complexes with diverse structural, electrochemical, and catalytic properties that could exceed those of natural copper-containing metalloenzymes in the formation of reactive aldehydes.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
YM: Conceptualization, Project administration, Supervision, Writing – original draft, Writing – review and editing. MA: Investigation, Writing – original draft. YO: Investigation, Writing – review and editing. SS: Writing – review and editing. TM: Investigation, Writing – review and editing. AJ: Investigation, Writing – original draft. PS: Supervision, Writing – review and editing. YF: Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the grant-in-aid for scientific research provided by JSPS KAKENHI (no. JP23K04808) and Nara Women’s University Intramural Grant for Project Research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchbi.2025.1688400/full#supplementary-material
References
Adam, S. M., Wijeratne, G. B., Rogler, P. J., Diaz, D. E., Quist, D. A., Liu, J. J., et al. (2018). Synthetic Fe/Cu complexes: toward understanding heme-copper oxidase structure and function. Chem. Rev. 118, 10840–11022. doi:10.1021/acs.chemrev.8b00074
Addison, A. W., Rao, T. N., Reedijk, J., van Rijn, J., and Verschoor, G. C. (1984). Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2'-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans., 1349–1356. doi:10.1039/DT9840001349
Al-Hunaiti, A., Abu-Radaha, B., Wraith, D., and Repo, T. (2022). Catalytic behaviour of the Cu(I)/L/TEMPO system for aerobic oxidation of alcohols – a kinetic and predictive model. RSC Adv. 12, 7864–7871. doi:10.1039/d1ra09359b
Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G., et al. (1994). SIR92 - a program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 27, 435. doi:10.1107/S002188989400021X
Basu, D., Mazumder, S., Shi, X., Baydoun, H., Niklas, J., Poluektov, O., et al. (2015). Ligand transformations and efficient proton/water reduction with cobalt catalysts based on pentadentate pyridine-rich environments. Angew. Chem. Int. Ed. 54, 2105–2110. doi:10.1002/anie.201409813
Blackman, A. G., Schenk, E. B., Jelley, R. E., Krenske, E. H., and Gahan, L. R. (2020). Five-coordinate transition metal complexes and the value of τ5: observations and caveats. Dalton Trans. 49, 14798–14806. doi:10.1039/D0DT02985H
Burla, M. C., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, G. L., De Caro, L., et al. (2007). IL MILIONE: a suite of computer programs for crystal structure solution of proteins. J. Appl. Crystallogr. 40, 609–613. doi:10.1107/S0021889807010941
Chen, J., Lee, Y.-M., Davis, K. M., Wu, X., Seo, M. S., Cho, K.-B., et al. (2013). A mononuclear non-heme manganese(IV)−oxo complex binding redox-inactive metal ions. J. Am. Chem. Soc. 135, 6388–6391. doi:10.1021/ja312113p
Ekanayake, D. M., Kulesa, K. M., Singh, J., Kpogo, K. K., Mazumder, S., Schlegel, H. B., et al. (2017). A pentadentate nitrogen-rich copper electrocatalyst for water reduction with pH-dependent molecular mechanisms. Dalton Trans. 46, 16812–16820. doi:10.1039/c7dt02711g
Ghosh, K., Eroy-Reveles, A. A., Avila, B., Holman, T. R., Olmstead, M. M., and Mascharak, P. K. (2004). Reactions of NO with Mn(II) and Mn(III) centers coordinated to carboxamido nitrogen: synthesis of a manganese nitrosyl with photolabile NO. Inorg. Chem. 43, 2988–2997. doi:10.1021/ic030331n
Gonzalez, M. A., Fry, N. L., Burt, R., Davda, R., Hobbs, A., and Mascharak, P. K. (2011). Designed iron carbonyls as carbon monoxide (CO) releasing molecules: rapid CO release and delivery to myoglobin in aqueous buffer, and vasorelaxation of mouse aorta. Inorg. Chem. 50, 3127–3134. doi:10.1021/ic2000848
Hazell, A., McKenzie, C. J., Nielsen, L. P., Schindler, S., and Weitzer, M. (2002). Mononuclear non-heme iron(III) peroxide complexes: syntheses, characterisation, mass spectrometric and kinetic studies. J. Chem. Soc. Dalton Trans., 310–317. doi:10.1039/B103844N
Hong, S., Lee, Y.-M., Ray, K., and Nam, W. (2017). Dioxygen activation chemistry by synthetic mononuclear nonheme iron, copper and chromium complexes. Coord. Chem. Rev. 334, 25–42. doi:10.1016/j.ccr.2016.07.006
Hoover, J. M., and Stahl, S. S. (2011). Highly practical copper(I)/TEMPO catalyst system for chemoselective aerobic oxidation of primary alcohols. J. Am. Chem. Soc. 133, 16901–16910. doi:10.1021/ja206230h
Hoover, J. M., Ryland, B. L., and Stahl, S. S. (2013). Copper/TEMPO-catalyzed aerobic alcohol oxidation: mechanistic assessment of different catalyst systems. ACS Catal. 3, 2599–2605. doi:10.1021/cs400689a
Hosmane, R. S., and Liebman, J. F. (2009). Paradoxes and paradigms: why is quinoline less basic than pyridine or isoquinoline? A classical organic chemical perspective. Struct. Chem. 20, 693–697. doi:10.1007/s11224-009-9464-6
Itoh, S. (2015). Developing mononuclear copper−active-oxygen complexes relevant to reactive intermediates of biological oxidation reactions. Acc. Chem. Res. 48, 2066–2074. doi:10.1021/acs.accounts.5b00140
Jackson, C. S., Schmitt, S., Dou, Q. P., and Kodanko, J. J. (2011). Synthesis, characterization, and reactivity of the stable iron carbonyl complex [Fe(CO)(N4Py)](ClO4)2: photoactivated carbon monoxide release, growth inhibitory activity, and peptide ligation. Inorg. Chem. 50, 5336–5338. doi:10.1021/ic200676s
Liu, J., Chakraborty, S., Hosseinzadeh, P., Yu, Y., Tian, S., Petrik, I., et al. (2014). Metalloproteins containing cytochrome, iron−sulfur, or copper redox centers. Chem. Rev. 114, 4366–4469. doi:10.1021/cr400479b
Lõkov, M., Tshepelevitsh, S., Heering, A., Plieger, P. G., Vianello, R., and Leito, I. (2017). On the basicity of conjugated nitrogen heterocycles in different media. Eur. J. Org. Chem. 2017, 4475–4489. doi:10.1002/ejoc.201700749
Lubben, M., Meetsma, A., Wilkinson, E. C., Feringa, B., and Que, L. (1995). Nonheme iron centers in oxygen activation: characterization of an iron(III) hydroperoxide intermediate. Angew. Chem. Int. Ed. 34, 1512–1514. doi:10.1002/anie.199515121
Mikata, Y., Uchida, M., Koike, H., Shoji, S., Ohsedo, Y., Kawai, Y., et al. (2023). Evaluation of oxygen-containing pentadentate ligands with pyridine/quinoline/isoquinoline binding sites via the structural and electrochemical properties of mononuclear copper(II) complexes. Dalton Trans. 52, 17375–17388. doi:10.1039/d3dt02814c
Mikata, Y., Akedo, M., Hamamoto, E., Yoshida, S., Shoji, S., Ohsedo, Y., et al. (2024). Structural and electrochemical properties of mononuclear copper(II) complexes with pentadentate ethylenediamine-based ligands with pyridine/quinoline/isoquinoline/quinoxaline binding sites. Dalton Trans. 53, 16716–16732. doi:10.1039/d4dt02363c
Mukherjee, G., Alili, A., Barman, P., Kumar, D., Sastri, C. V., and de Visser, S. P. (2019). Interplay between steric and electronic effects: a joint spectroscopy and computational study of nonheme iron(IV)-oxo complexes. Chem. A Eur. J. 25, 5086–5098. doi:10.1002/chem.201806430
Ohzu, S., Ishizuka, T., Hirai, Y., Jiang, H., Sakaguchi, M., Ogura, T., et al. (2012). Mechanistic insight into catalytic oxidations of organic compounds by ruthenium(IV)-oxo complexes with pyridylamine ligands. Chem. Sci. 3, 3421–3431. doi:10.1039/C2SC21195E
Ortega-Villar, N., Thompson, A. L., Muñoz, M. C., Ugalde-Saldívar, V. M., Goeta, A. E., Moreno-Esparza, R., et al. (2005). Solid- and solution-state studies of the novel µ-dicyanamide-bridged dinuclear spin-crossover system {[Fe(bztpen)]2[µ-N(CN)2]}(PF6)3·nH2O. Chem. A Eur. J. 11, 5721–5734. doi:10.1002/chem.200500171
Oshita, H., and Shimazaki, Y. (2020). Recent advances in one-electron-oxidized CuII diphenoxide complexes as models of galactose oxidase: importance of the structural flexibility in the active site. Chem. A Eur. J. 26, 8324–8340. doi:10.1002/chem.201905877
Patra, A. K., and Mascharak, P. K. (2003). A ruthenium nitrosyl that rapidly delivers NO to proteins in aqueous solution upon short exposure to UV light. Inorg. Chem. 42, 7363–7365. doi:10.1021/ic030110h
Patra, A. K., Afshar, R., Olmstead, M. M., and Mascharak, P. K. (2002). The first non-heme iron(III) complex with a ligated carboxamido group that exhibits photolability of a bound NO ligand. Angew. Chem. Int. Ed. 41, 2512–2515. doi:10.1002/1521-3773(20020715)41:14<2512::AID-ANIE2512>3.0.CO;2-7
Roelfes, G., Lubben, M., Chen, K., Ho, R. Y. N., Meetsma, A., Genseberger, S., et al. (1999). Iron chemistry of a pentadentate ligand that generates a metastable FeIII-OOH intermediate. Inorg. Chem. 38, 1929–1936. doi:10.1021/ic980983p
Rowland, J. M., Olmstead, M., and Mascharak, P. K. (2001). Syntheses, structures, and reactivity of low spin iron(III) complexes containing a single carboxamido nitrogen in a [FeN5L] chromophore. Inorg. Chem. 40, 2810–2817. doi:10.1021/ic001127s
Rubino, J. T., and Franz, K. J. (2012). Coordination chemistry of copper proteins: how nature handles a toxic cargo for essential function. J. Inorg. Biochem. 107, 129–143. doi:10.1016/j.jinorgbio.2011.11.024
Sheldrick, G. M. (2015a). Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 71, 3–8. doi:10.1107/S2053229614024218
Sheldrick, G. M. (2015b). SHELXT – integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 71, 3–8. doi:10.1107/S2053273314026370
Solomon, E. I., Heppner, D. E., Johnston, E. M., Ginsbach, J. W., Cirera, J., Qayyum, M., et al. (2014). Copper active sites in biology. Chem. Rev. 114, 3659–3853. doi:10.1021/cr400327t
Spek, A. L. (2015). PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. C Struct. Chem. 71, 9–18. doi:10.1107/S2053229614024929
Tamura, M., Urano, Y., Kikuchi, K., Higuchi, T., Hirobe, M., and Nagano, T. (2000). Synthesis and superoxide dismutase activity of novel iron complexes. J. Organomet. Chem. 611, 586–592. doi:10.1016/S0022-328X(00)00394-6
Varala, R., and Seema, V. (2023). Recent applications of TEMPO in organic synthesis and catalysis. SynOpen 07, 408–413. doi:10.1055/a-2155-2950
Whittaker, J. W. (2003). Free radical catalysis by galactose oxidase. Chem. Rev. 103, 2347–2364. doi:10.1021/cr020425z
Young, K. J., Takase, M. K., and Brudvig, G. W. (2013). An anionic N-donor ligand promotes manganese-catalyzed water oxidation. Inorg. Chem. 52, 7615–7622. doi:10.1021/ic400691e
Keywords: copper, o-phenylenediamine, pentadentate ligand, quinoline, pyridine, alcohol oxidation, 2,2,6,6-tetramethylpiperidine 1-oxyl
Citation: Mikata Y, Akedo M, Ohsedo Y, Shoji S, Matsuo T, Jehdaramarn A, Sangtrirutnugul P and Funahashi Y (2025) Copper complexes containing o-phenylenediamine-based pentadentate ligands catalyze TEMPO-mediated alcohol oxidation. Front. Chem. Biol. 4:1688400. doi: 10.3389/fchbi.2025.1688400
Received: 19 August 2025; Accepted: 13 October 2025;
Published: 20 November 2025.
Edited by:
Sylvia Mary Draper, Trinity College Dublin, IrelandReviewed by:
Aviva Levina, The University of Sydney, AustraliaKonrad Kowalski, University of Lodz, Poland
Copyright © 2025 Mikata, Akedo, Ohsedo, Shoji, Matsuo, Jehdaramarn, Sangtrirutnugul and Funahashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuji Mikata, bWlrYXRhQGNjLm5hcmEtd3UuYWMuanA=; Preeyanuch Sangtrirutnugul, cHNhbmd0cmlydXRAZ21haWwuY29t
†ORCID: Yuji Mikata, orcid.org/0000-0002-9450-0908; Yutaka Ohsedo, orcid.org/0000-0003-2762-0894; Sunao Shoji, orcid.org/0000-0002-0329-1136; Takashi Matsuo, orcid.org/0000-0002-5646-6251; Preeyanuch Sangtrirutnugul, orcid.org/0000-0001-6201-2814; Yasuhiro Funahashi, orcid.org/0000-0002-2237-579X