 Lianzhi Yang1,2†
Lianzhi Yang1,2† Yaping Wang1,2,3†Pan Yu1,2†Shunlin Ren3
Yaping Wang1,2,3†Pan Yu1,2†Shunlin Ren3 Zhuoying Zhu1,2Yinzhe Jin1,2
Zhuoying Zhu1,2Yinzhe Jin1,2 Jizhou Yan4
Jizhou Yan4 Xu Peng5
Xu Peng5 Lanming Chen1,2*
Lanming Chen1,2*- 1Key Laboratory of Quality and Safety Risk Assessment for Aquatic Products on Storage and Preservation (Shanghai), China Ministry of Agriculture, Shanghai, China
- 2College of Food Science and Technology, Shanghai Ocean University, Shanghai, China
- 3Department of Internal Medicine, Virginia Commonwealth University/McGuire VA Medical Centre, Richmond, VA, United States
- 4College of Fishers and Life Science, Shanghai Ocean University, Shanghai, China
- 5Archaea Centre, Department of Biology, University of Copenhagen, Copenhagen, Denmark
Vibrio parahaemolyticus is a leading seafood-borne pathogen that can cause acute gastroenteritis and even death in humans. In aquatic ecosystems, phages constantly transform bacterial communities by horizontal gene transfer. Nevertheless, biological functions of prophage-related genes in V. parahaemolyticus remain to be fully unveiled. Herein, for the first time, we studied one such gene VpaChn25_0724 encoding an unknown hypothetical protein in V. parahaemolyticus CHN25. This gene deletion mutant ΔVpaChn25_0724 was constructed by homologous recombination, and its complementary mutant ΔVpaChn25_0724-com was also obtained. The ΔVpaChn25_0724 mutant exhibited a sever defect in growth and swimming motility particularly at lower temperatures. Biofilm formation and cytotoxicity capacity of V. parahaemolyticus CHN25 was significantly lowered in the absence of VpaChn25_0724. Comparative secretomic analysis revealed an increase in extracellular proteins of ΔVpaChn25_0724, which likely resulted from its damaged cell membrane. Comparison of transcriptome data showed twelve significantly altered metabolic pathways in ΔVpaChn25_0724, suggesting inactive transport and utilization of carbon sources, repressed energy production and membrane biogenesis in ΔVpaChn25_0724. Comparative transcriptomic analysis also revealed several remarkably down-regulated key regulators in bacterial gene regulatory networks linked to the observed phenotypic variations. Overall, the results here facilitate better understanding of biological significance of prophage-related genes remaining in V. parahaemolyticus.
Introduction
Vibrio parahaemolyticus is a gram-negative bacterium and thrives in marine, riverine, and aquaculture environments worldwide (Ghenem et al., 2017). Consumption of raw, undercooked or mishandled seafood contaminated by pathogenic V. parahaemolyticus can cause acute gastroenteritis in humans and even death (Kim et al., 2017). The bacterium was originally identified in 1950 in Osaka, Japan, where an outbreak of acute gastroenteritis, caused by contaminated semidried juvenile sardines, sickened 272 and killed 20 people (Fujino et al., 1953). Since then, the infectious disease caused by V. parahaemolyticus has been reported in many Asian countries, and subsequently in Africa, America, and Europe, arguing a pandemic of V. parahaemolyticus worldwide (Ghenem et al., 2017). In China, V. parahaemolyticus is a leading cause of foodborne bacterial disease, especially among adults in coastal regions in recent years (Jiang et al., 2019). Most pathogenic V. parahaemolyticus strains of clinical origin have two major virulence factors, a thermostable direct hemolysin (TDH) and a TDH-related hemolysin (TRH). Both toxins have hemolytic activity, enterotoxin activity, cardiotoxicity and cytotoxicity to the host (Li et al., 2019). Nevertheless, some V. parahaemolyticus isolates of environmental origins lacking the tdh and/or trh genes also show cytotoxicity to human intestinal cells, suggesting additional virulence-associated factors exist in the bacterium (Raghunath, 2014). Thus, identification of risk factors in V. parahaemolyticus is imperative for assuming food safety.
In aquatic ecosystems, phages are the most abundant biological entity involved in numerous biological cycles and constantly transform bacterial communities by horizontal gene transfer (HGT) (Penades et al., 2015; Castillo et al., 2018). Phage genomic DNA can integrate into and replicate as part of bacterial chromosomes, which is a typical feature of mild phages for a lysogen cycle (Feiner et al., 2015; Howard-Varona et al., 2017). HGT can result in pandemic or pathogenic clones with expanded ecological persistence and dispersibility, e.g., the filamentous phage VfO3:K6 in V. parahaemolyticus O3:K6 (Loyola et al., 2015). It constitutes important driving forces in host evolution, and bestows a wide range of phenotypes upon hosts with transmitted gene cassettes, such as phage-coded virulence, cell adhesion, antibiotic resistance, and metabolizing enzyme determinants (Harrison and Brockhurst, 2017; Castillo et al., 2018). Previous studies have revealed phages or prophage gene clusters present in V. parahaemolyticus (e.g., Jensen et al., 2013; Gomez-Gil et al., 2014; Kalburge et al., 2014). For instance, the phage Vp58.5 enhances ultraviolet sensitivity of V. parahaemolyticus O3:K6 (Zabala et al., 2009), and phage Vp882 transmits DNA adenine methylase and quorum sensing (QS) transcription factors to V. parahaemolyticus O3:K6 (Lan et al., 2009). A recently reported lytic Vibrio phage VP06 can infect a broad range of hosts, including Vibrio alginolyticus, Vibrio azureus, Vibrio harveyi, and V. parahaemolyticus. This Vibrio phage is resistant to environmental stresses, displaying potential as a candidate biocontrol agent (Wong et al., 2019).
In our prior studies, V. parahaemolyticus CHN25 strain (serotype: O5:KUT) was isolated, identified, and characterized (Song et al., 2013; Sun et al., 2014; He et al., 2015; Zhu et al., 2017). The complete genome sequence of V. parahaemolyticus CHN25 contains 5,443,401 bp with 45.2% G+C content (Zhu et al., 2017). Comparative genomic analysis revealed five prophage gene clusters in V. parahaemolyticus CHN25. The largest one has sequence similarity with a 33,277-bp Vibrio phage Martha 12B12 (GenBank accession no. HQ316581). Nevertheless, this prophage sequence exists in a truncated version in the bacterial genome, where approximately 37.5% of the remaining genes encode predicted proteins of unknown function. Identification of these hypothetical proteins is yet to be determined. We, therefore, asked whether V. parahaemolyticus CHN25 would be affected by the absence of these prophage-related genes. In this study, we aimed to address possible function of one such gene VpaChn25_0724 in V. parahaemolyticus CHN25.
Materials and Methods
Bacterial Strains, Plasmids, and Culture Conditions
The V. parahaemolyticu CHN25 strain was used in this study. Escherichia coli DH5α λpir [BEINUO Biotech (Shanghai) Co. Ltd., China] was used as a host strain for DNA cloning. The pDS132 plasmid and E. coli β2155 λpir were used as a suicide vector and a donor strain in conjugation experiments, respectively (Zhu et al., 2017). The pMMB207 plasmid (Biovector Science Lab, Inc., China) was used as an expression vector to construct the reverse mutant. The E. coli strains were routinely incubated in Luria-Bertani (LB) medium (1% NaCl, pH 7.2) at 37°C, and the V. parahaemolyticus strains were grown in LB (3% NaCl, pH 8.5) or Tryptic Soy Broth (TSB) (3% NaCl, pH8.5) media. The growth medium was supplemented as needed with chloramphenicol to a final concentration of 30 μg/ml for E. coli and 5 μg/ml for V. parahaemolyticus (Zhu et al., 2017).
Construction of Deletion Mutant and Reverse Mutant of the VpaChn25_0724 Gene
Genomic DNA was prepared using TaKaRa MiniBEST Bacterial Genomic DNA Extraction Kit (Japan TaKaRa BIO, Dalian Company, China). Plasmid DNA was isolated using TIANpure Midi Plasmid Kit (Tiangen Biotech Beijing Co. Ltd., China). Oligonucleotide primers (Table 1) were designed using Primier 5.0 software (https://www.premierbiosoft.com), and synthesized by the Sangon Biotech (Shanghai) Co. Ltd., China. A markerless in-frame gene deletion mutant of the VpaChn25_0724 gene was constructed using the homologous recombination method (Zhu et al., 2017). Briefly, based on the VpaChn25_0724 gene sequence (294 bp) in V. parahaemolyticus CHN25 genome, the primer pairs (VpaChn25_0724-up-F/R and VpaChn25_0724-down-F/R) (Table 1) were designed to target the upstream (464 bp) and downstream (444 bp) sequences of the VpaChn25_0724 gene, respectively. The amplified products by polymerase chain reaction (PCR) were individually digested with corresponding restriction endonucleases (TaKaRa, Japan), purified, and ligated into XbaI and SacI cloning sites on the pDS132. The ligated DNA was transformed into E. coli DH5α λpir competent cells and positive transformants were screened (Zhu et al., 2017). The recombinant plasmid pDS132+VpaChn25_0724 was subsequently prepared and transformed into diaminopimelic acid (DAP) auxotroph E. coli β2155 competent cells grown in LB medium supplemented with 0.3 mM DAP (Sigma-Aldrich, USA). Plate mating assay was performed (Zhu et al., 2017). Exconjugants with successful double crossover deletions of the VpaChn25_0724 gene were screened by colony PCR assay using the VpaChn25_0724-up-ex-F and VpaChn25_0724-down-ex-R primer pair (Table 1). The obtained ΔVpaChn25_0724 mutant was confirmed by DNA sequencing, quantitative reverse transcription-PCR (RT-qPCR), and transcriptome analysis (see below). DNA sequences were determined by the Sangon (China).
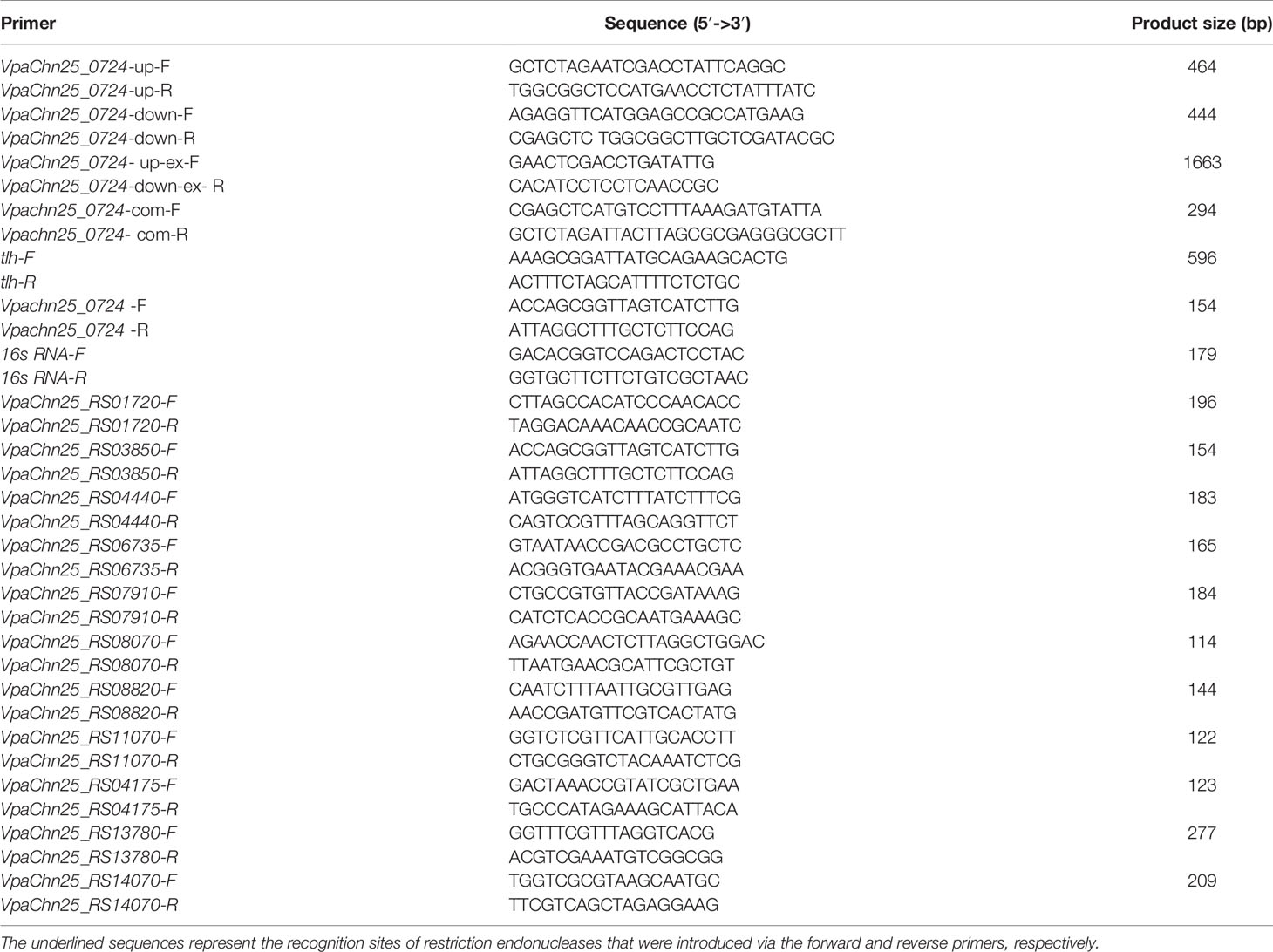
Table 1 Oligonucleotide primers designed and used in this study.
The 294-bp VpaChn25_0724 gene was amplified from the genomic DNA of V. parahaemolyticus CHN25 by PCR with the VpaChn25_0724-com-F/-R primers (Table 1). The PCR product was ligated into the expression vector pMMB207. The ligated DNA was transformed into E. coli DH5α and positive transformants were screened as described above. The recombinant plasmid pMMB207+VpaChn25_0724 was then prepared and transformed into the ΔVpaChn25_0724 mutant by electrotransformation as described previously (Zhu et al., 2017). The positive electrotransformants (ΔVpaChn25_0724-com mutant) were screened by colony PCR with primers VpaChn25_0724-com-F/R and tlh-F/R (Table 1), and confirmed using the aforementioned methods.
Swimming Mobility and Biofilm Formation Assays
Swimming motility was examined according to the method described previously (Huang et al., 2017; Yang et al., 2017). Briefly, V. parahaemolyticus strains were individually incubated at 37°C in the TSB medium (pH 8.5, 3% NaCl) to the middle-logarithmic growth phase (mid-LGP) with OD600nm values of about 0.8 to 1.0. Growth curves were measured using Bioscreen Automatic Growth Curve Analyzer (BioTek, USA). The differential growth phases were calculated on the basis of OD600nm values between the wild type and mutant strains (Zhu et al., 2017). A 0.5 μl of each cell culture were inoculated into semi-solid TSB agar plates containing 0.25% agar, and incubated at 15°C, 25°C, and 37°C for 48, 24, and 12 h, respectively. The bacterial clones formed on the plates were measured and recorded.
Biofilm formation was quantified using the crystal violet staining method (Tan et al., 2018). Briefly, V. parahaemolyticus strains from overnight culture were individually diluted in the TSB medium to an absorption value at OD600nm of about 0.4, and then 1 ml of the dilutions was individually inoculated into 24-well polystyrene microtiter plates (Sangon, China). After incubation at 37°C for 12 h, 24 h, 36 h, 48 h, and 60 h, the planktonic bacteria were removed, and biofilms were gently washed with 1 ml of 0.1M phosphate-buffered saline (PBS, pH 7.2 to 7.4, Sangon, China) for three times. The biofilms were then fixed using 0.1% (w/v) crystal violet (Sangon, China), washed, dissolved, and measured for absorbance values at OD600nm using BioTek Synergy 2 (BioTek, USA) (Tan et al., 2018).
Bacterial Cell Membrane Damage, Hydrophobicity, and Fluidity Assays
Cell membrane damage was analyzed using the method described by Collado et al. (Collado et al., 2017). The bacterial cell suspension was double dyed using propidium iodide (PI) (final concentration 10 mM) (Sangon, China) and 5(6)-carboxydiacetate fluorescein succinimidyl ester (CFDA) (final concentration 10 μM) (Beijing Solarbio Science & Technology Co. Ltd., China). Bacterial cell forward scatter, lateral astigmatism, and fluorescent channels FL1 (green) and FL2 (red) were determined using a flow cytometer BD FACSVerse™ (Becton, Dickinson and Company, USA). 1,000 cells were detected in each sample.
The cell membrane hydrophobicity and fluidity assays were performed as described by Pelletier et al. (Pelletier et al., 1997), and Voss and Montville (Voss and Montville, 2014), respectively.
Secretome Analysis
Extracellular proteins of V. parahaemolyticus strains were extracted according to the method described previously (He et al., 2015). The 2-dimensional gel electrophoresis (2-DE) was performed according to the method by Zhu et al. (Zhu et al., 2020). Briefly, approximately 20 μg of extracellular proteins was diluted with the rehydration buffer [8 M urea, 4% (w/v) CHAPS, 65 mM dithiothreitol, 0.2% (vol/vol) Bio-Lyte 3/10 ampholyte and 0.0001% (wt/vol) bromophenol blue (Sangon, China)] to a final volume of 200 μl per sample. The mixture of each sample was applied to the pH gradient gel (IPG) strips (pH 4–7, 7 cm, Bio-Rad, USA) and passive rehydrated for 16 h at 17°C. After rehydration, IEF (isoelectric focusing) was run with a six-step program (Zhu et al., 2020). Following the electrophoresis in the first dimension, the strips were first equilibrated for 15 min in equilibration buffer I, and then washed for a further 15 min with equilibration buffer II (Zhu et al., 2020). The second-dimension separation was performed using SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The strips were individually transferred onto 12.5% separation gel using a Mini-PROTEANW electrophoresis cell (Bio-Rad, USA) with a 2-step program (Zhu et al., 2020). The gels were stained, imaged, and analyzed as described previously (He et al., 2015). Additionally, amino acid sequences of protein spots were determined using liquid chromatography tandem mass spectrometry (LC-MS/MS) technique at Shanghai Houji Biology Co Ltd., China as described previously (Zhu et al., 2020).
Human Intestinal Epithelial Cell Viability and Apoptosis Assay
Human intestinal epithelial cell viability infected by V. parahaemolyticus strains was determined as described previously (Tsai et al., 2018) with minor modifications. Human rectal cancer epithelial cell line Caco-2 (ATCC number : HTB-37™) was purchased from Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). Briefly, Caco-2 cells were seeded into 96-well cell culture plates at a concentration of 5 × 104 cells/ml per well, and cultured in Dulbecco’s modified eagle medium (DMEM, Gibco, USA) at 37°C, 5% CO2 for 24 h using a CO2 Cell Incubator (Thermo, USA). The cell culture fluid was aspirated off, and cells were washed twice with 0.1 M PBS (pH 7.2–7.4, Sangon, China). Meanwhile, V. parahaemolyticus strains grown to the mid-LGP at 37°C were individually harvested, and cell pellet was washed twice with 0.1 M PBS, and resuspended with the phenol red-free DMEM medium to adjust OD490nm values of about 0.2 ± 0.02. A 100 μl of the bacterial suspension was added into each well containing the Caco-2 cells, and 10 μl of [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfonate)-2h-tetrazole monosodium salt (CCK-8, Sigma-Aldrich, USA)] was also added and incubated for 4 h at 5% CO2, 37°C. Caco-2 cell viability was calculated according to the following formula: cell viability (%) = [A (bacteria) − A (blank)]/[A (0 bacteria) − A (blank)] × 100, in which A (bacteria) represents an absorbance at OD450nm of the cell culture wells with Caco-2 cells, CCK-8 solution and bacterial suspension; A (blank) represents a OD450nm value without Caco-2 cells, DME medium and CCK-8 solution; A (0 bacteria) represents a OD450nm value with Caco-2 cells and CCK-8 solution but without bacterial suspension.
A 2 ml of Caco-2 cells (1 × 105 cells/ml) was inoculated into 6-well cell culture plates, and individually infected by V. parahaemolyticus strains for 4 h at 5% CO2, 37°C. Then a 200-μl trypsin (Gibco, USA) was added into each cell culture well to digest for 3 min. Then the supernatant was discarded, and cells were collected by centrifugation at 800 rpm for 4 min. Apoptosis of the Caco-2 cells was assayed using Annexin-V-FITC/PI Apoptosis Detection Kit (Solarbio, China), according to the manufacturer’s protocol. The treated samples were detected by BD FACSVerse™ flow cytometry (Becton, Dickinson and Company, USA), and data analysis was performed using FlowJo software (https://www.flowjo.com).
Illumina RNA Sequencing
Total RNA was prepared using RNeasy Protect Bacteria Mini Kit (QIAGEN Biotech Co. Ltd., Germany) and QIAGEN RNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocols. The DNA was removed from the samples using RNase-Free DNase Set (QIAGEN). Three independently prepared RNA samples were used in each Illumina RNA-sequencing experiment. The sequencing library construction and Illumina sequencing were conducted at Shanghai Majorbio Bio-pharm Technology Co. Ltd., China using Illumina HiSeq 2500 platform as described previously (Zhu et al., 2017). High quality reads that passed the Illumina quality filters were used for sequence analyses.
Real-Time Reverse Transcription-PCR Assay
The RT-qPCR was performed as described previously (Wang et al., 2013). V. parahaemolyticus cultures grown to the mid-LGP were harvested for RNA extraction. The RNeasy Mini Kit (Qiagen, Germany) was used to extract total RNA. Reverse transcription reactions were performed using PrimeScript™ RT reagent Kit with gDNA Eraser (Perfect Real Time) (TaKaRa, Japan) kit. Relative quantitative PCR reactions were performed with TB Green® Premix Ex Taq™ II (Tli RNaseH Plus) (TaKaRa, Japan) kit using 7500 Fast Real-Time PCR Instrument (Applied Biosystems, USA). The 16S rRNA gene was used as the internal reference gene, and 2−ΔΔCt method was used to calculate the relative expression between the target and the internal reference genes. The representative eighteen differentially expressed genes (DEGs) in the transcriptome of ΔVpaChn25_0724 mutant were confirmed by the RT-qPCR assay, and listed in Table S1.
Detection of VpaChn25_0724 Gene by PCR
Total 138 V. parahaemolyticus strains isolated from commonly consumed aquatic products collected in Shanghai, China (Su et al. unpublished) were used in this study. The VpaChn25_0724 gene was amplified with the primers VpaChn 25_0724 –F/R (Table 1) by PCR (Song et al., 2013). A 6 μl of each PCR product was analyzed by agarose gel (1%) electrophoresis, imaged and recorded, and validated by DNA sequencing as described above.
Transmission Electron Microscope Assay
The TEM observation of V. parahaemolyticus strains was conducted using ultrathin edge-cutting method (Morgelin, 2017; Xu et al., 2017) via a transmission electron (JEM2100, JEOL, Japan, ×80,000) at the Instrumental Analysis Center at Shanghai Jiao Tong University, Shanghai, China.
Enzyme Activity Assays
The wild type and mutant strains grown to the mid-LGP were individually harvested by centrifugation at 8,000 g for 10 min at 4°C. The supernant was removed, and the bacterial cell pellets were used for the malate dehydrogenase and citrate synthase activity assays using the corresponding kits (Product Nos. BC1040, and BC1060, Beijing Solarbio Science & Technology Co. Ltd., China), according to the manufecturer’s instructions. Approximately 0.05 ± 0.002 g and 0.1 ± 0.02 g of each sample was used in the malate dehydrogenase and citrate synthase reactions, and their OD340nm and OD412nm values were recorded using BioTek Synergy 2 (BioTek, USA), respectively.
Data Analysis
Quality filtration of raw RNA-seq data were performed using theSeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle version 1.33 software (https://github.com/najoshi/sickle) as described previously (Zhu et al., 2017). The resulting clean reads were aligned to the V. parahaemolyticus CHN25 genome using the Bowtie2 version 2.0.5 software (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). Expression of each gene was calculated using RNA-Seq by Expectation-Maximization (RSEM, http://deweylab.github.io/RSEM/). Genes with the criteria, fold-changes ≥ 2.0 or ≤ 0.5, and p-values by BH (fdr correction with Benjamini/Hochberg) < 0.05 relative to the control, were defined as DEGs. These DEGs were used for gene set enrichment analysis (GSEA) against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) and gene set annotation analysis (GSAA) against the Gene Ontology (GO) database (http://www.geneontology.org) as described previously (Zhu et al., 2017). Significantly changed GSEA were identified when the enrichment test p-value fell below 0.05.
Prophage gene clusters were searched and analyzed using Prophage Finder (http://phast.wishartlab.com/) and Basic Local Alignment Search Tool (BLAST) (http://www.ncbi.nlm.nih.gov/BLAST) software. All tests in this study were conducted in at least triplicate. The data were analyzed using SPSS statistical analysis software version 17.0 (SPSS Inc., USA).
Results
Gene Organization of the Largest Prophage Gene Cluster in Vibrio parahaemolyticus CHN25 Genome
Comparative genomic analysis revealed that the largest prophage gene cluster in V. parahaemolyticus CHN25 genome has sequence similarity to the Vibrio phage Martha 12B12 that contains 50 predicted genes. Approximately 24 genes thereof were present in chromosome 1 (3,416,467 bp) of V. parahaemolyticus CHN25 genome, where they located in the locus from 816,554 bp to 846,961 bp (Figure 1). Among the 24 genes, 7 coded for potential phage proteins (i.g., phage head, tail, and baseplate), 8 for predicted regulators, and 9 for hypothetical structural proteins of unknown function. Within the gene cluster, genes encoding DNA endonuclease, DNA transport protein, conjugal transfer protein TraR, and additional proteins were also identified, suggesting that the bacterium underwent extensive genetic recombination via HGT during its evolution (Zhu et al., 2017). Among the genes encoding unknown hypothetical proteins, the 294-bp VpaChn25_0724 gene was further investigated and reported in this study.
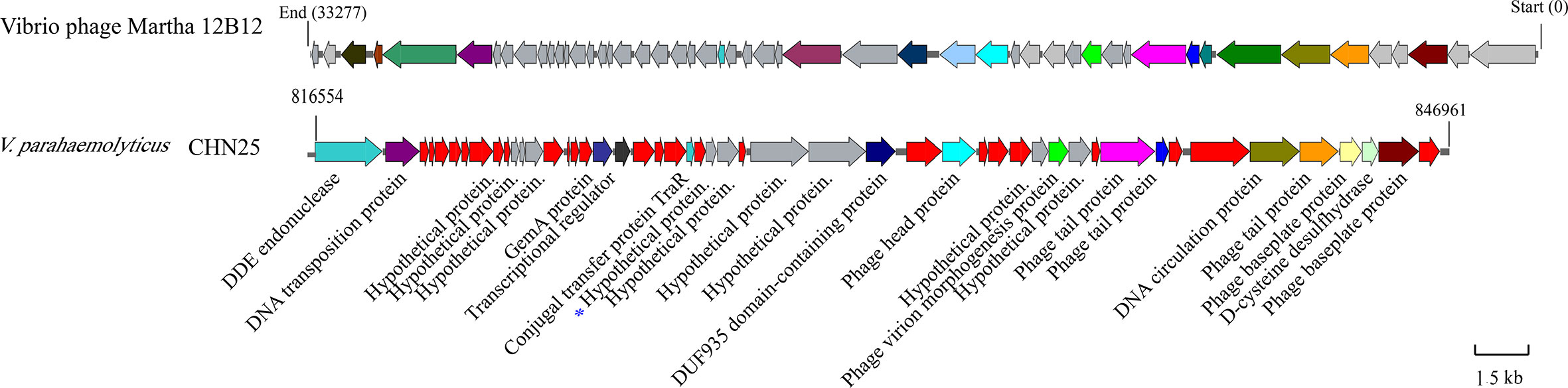
Figure 1 Gene organization of the Vibrio phage Martha 12B12-like sequence in chromosome 1 of V. parahaemolyticus CHN25 genome. Genes in gray color represent predicted hypothetical proteins, and those in red represent additional proteins absent from the Vibrio phage Martha 12B12. The VpaChn25_0724 gene was marked with a star in blue.
Deletion and Reverse Complementation of the VpaChn25_0724 Gene
To study biological function of the VpaChn25_0724 gene, we constructed an unmarked in-frame gene deletion mutant ΔVpaChn25_0724 using the homologous recombination method (Figure S1). The upstream and downstream sequences (approximately 0.5 kb) that flank the VpaChn25_0724 gene were obtained by PCR (Table 1), and then cloned into a suicide vector pDS132 to yield a recombinant vector pDS132+VpaChn25_0724. The 908-bp inserted sequence was validated by DNA sequencing (data not shown). The recombinant vector was transformed into E. coli β2155, and the chloramphenicol-resistant transformant was obtained and conjugated with V. parahaemolyticus CHN25. Positive exconjugants were obtained using the two-step allelic exchange method. Deletion of the 294-bp VpaChn25_0724 gene from V. parahaemolyticus CHN25 genome was validated by PCR and DNA sequencing assays (data not shown), as well as by RT-qPCR and transcriptomic analysis (see below).
To facilitate a complementation assay, a reverse mutant ΔVpaChn25_0724-com was also successfully constructed. The 294-bp VpaChn25_0724 gene was amplified from genomic DNA of V. parahaemolyticus CHN25 by PCR, and then cloned into an expression vector pMMB207, which yielded a recombinant vector pMMB207-VpaChn25_0724. The inserted 294-bp sequence was confirmed by DNA sequencing (data not shown). This recombinant vector was then electrotransformed into the ΔVpaChn25_0724 mutant, and generated the reverse mutant ΔVpaChn25_0724-com that was further confirmed using the aforementioned methods.
Growth of the ΔVpaChn25_0724 Mutant at Different Temperatures
To gain insights into possible impact of the VpaChn25_0724 gene deletion on bacterial growth, we determined growth curves of V. parahaemolyticus CHN25, ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains at 37°C, 25°C, and 15°C, which are within the temperature range experienced by V. parahaemolyticus during its life cycle (Desai and Kenney, 2019). As illustrated in Figure 2, the ΔVpaChn25_0724 mutant exhibited a significantly extended lag phase, which was 4-fold, 6-fold, and 2.5-fold of that of the wild type strain at 37°C, 25°C, and 15°C, respectively. Complementation with a plasmid-borne VpaChn25_0724 (ΔVpaChn25_0724-com) restored the growth almost completely at 37°C and partially at 25°C, but not at all at 15°C. These results highlighted the importance of VpaChn25_0724 gene for the growth of V. parahaemolyticus CHN25, particularly at the lower temperatures.
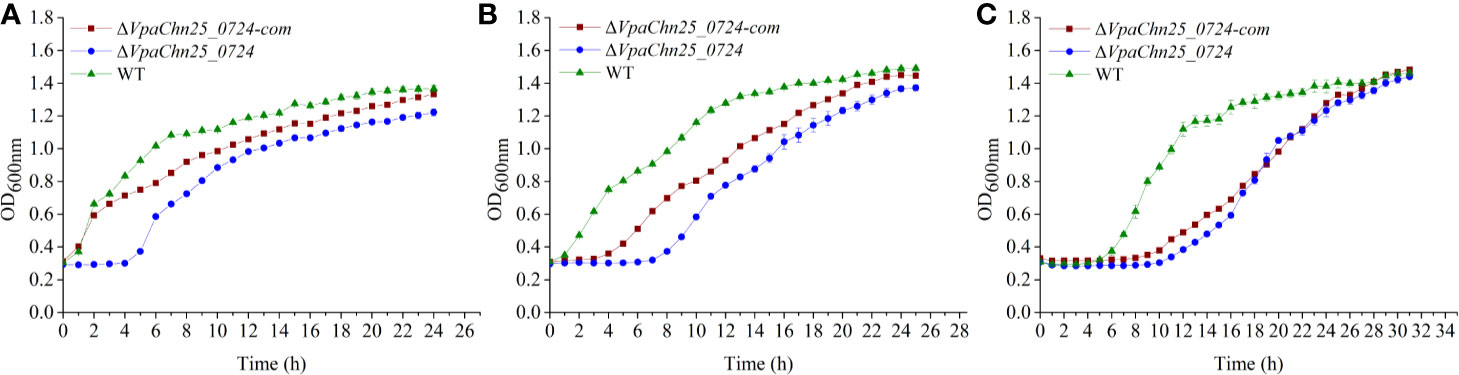
Figure 2 Survival of V. parahaemolyticus CHN25 (WT), ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains at different temperatures. The strains were incubated in the TSB medium at 37°C (A), 25°C (B), 15°C (C), respectively. WT, wild type.
Swimming Motility and Biofilm Formation of the ΔVpaChn25_0724 Mutant
As shown in Figure 3, on the semi-solid swimming plates, the lower temperatures notably shrunk swimming cycles of wild type, ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains. However, swimming capacity of the wild type was about 2-fold and 3-fold higher than that of the ΔVpaChn25_0724 mutant at 25°C and 15°C, respectively (p<0.05). No significant difference in swimming cycles was observed among the three strains at 37°C (p > 0.05). Additionally, the lower temperatures appeared to inhibit restoring the different phenotypes by the ΔVpaChn25_0724-com strain. Therefore, we focused on the optimal growth temperature 37°C in the following analysis in this study. These results indicated that the severe defect in growth elicited by the VpaChn25_0724 gene deletion likely led to the variant swimming motility of V. parahaemolyticus CHN25 at the lower temperatures.
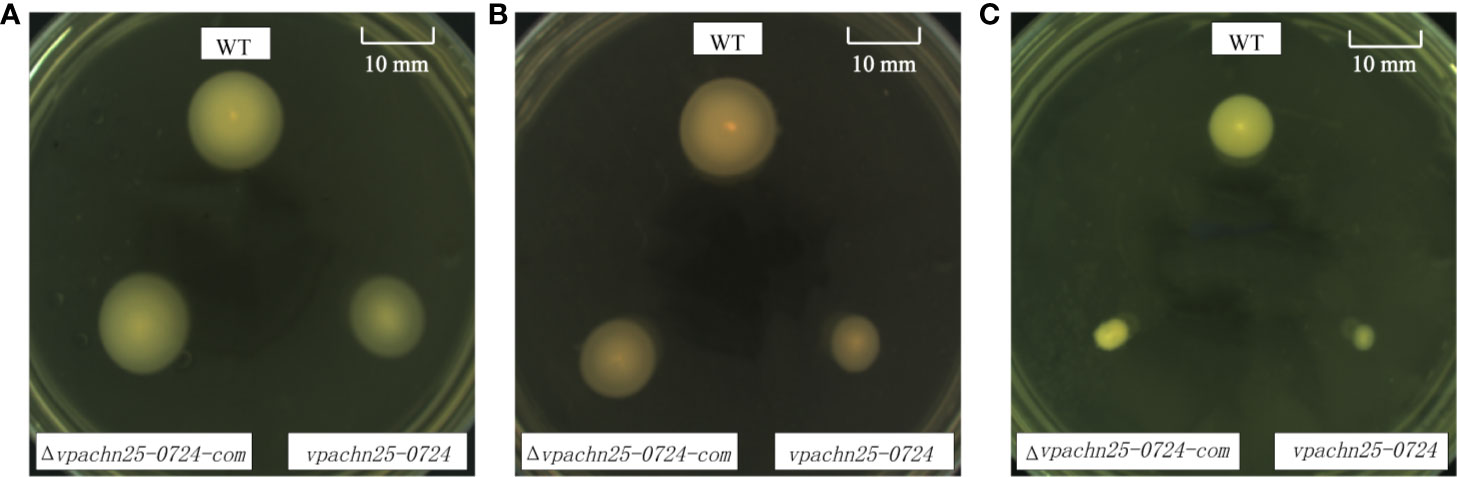
Figure 3 Swimming motility of V. parahaemolyticus CHN25 (WT), ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains at different temperature. A: 37°C; B: 25°C; C: 15°C; WT: wild type. The strains were incubated on the semi-solid TSB agar plates containing 0.25% agar at 37°C (A), 25°C (B), 15°C (C), respectively. WT, wild type.
In addition, we analyzed and quantified the bacterial biofilms formed under static incubation conditions using the crystal violet staining assay. The dynamic process of biofilm formation was followed for the wild type, ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains grown at 37°C for 60 h (Figure 4). We observed that biofilms were built at three different stages (development, maturation and diffusion) by all the three strains, consistent with previous studies. At the early formation stage (24 h), the ΔVpaChn25_0724 mutant exhibited a 1.5-fold decrease in biomass compared with the wild type (p<0.01). No significant difference was observed at the other stages among the three strains (p>0.05), except a notably decrease in biomass of ΔVpaChn25_0724-com at the latter stage. These results showed a defect in early biofilm formation of V. parahaemolyticus CHN25 in the absence of the VpaChn25_0724 gene.
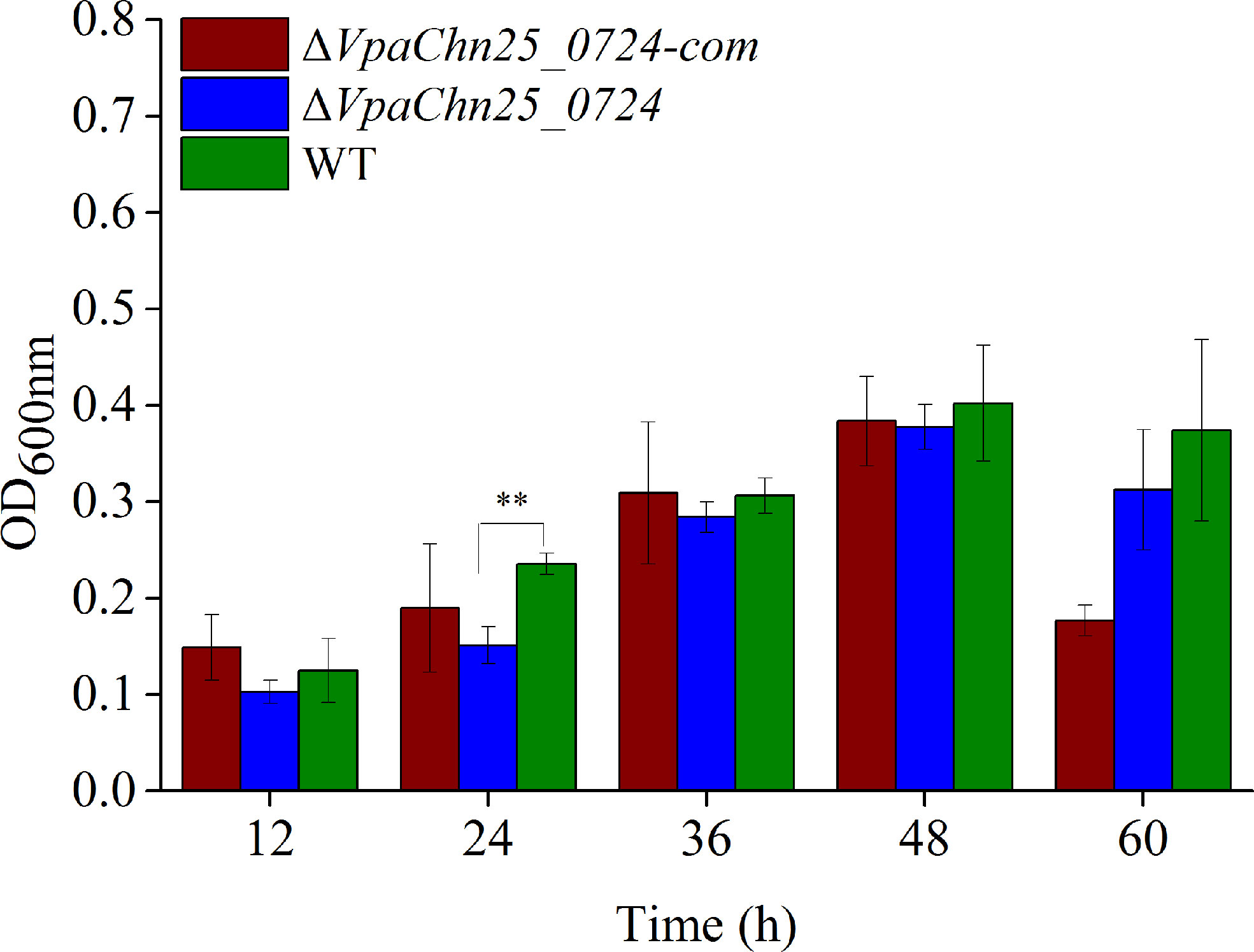
Figure 4 Biofilm formation of V. parahaemolyticus CHN25 (WT), ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains. The strains were incubated in the TSB medium at 37°C under static conditions. WT: wild type. **p<0.01 compared with the WT.
Cell Membrane Damage, Hydrophobicity, and Fluidity of the ΔVpaChn25_0724 Mutant
On the basis of the above results, we, therefore, asked whether bacterial cell membrane structure would be affected by the VpaChn25_0724 gene deletion. We evaluated cell membrane damage, hydrophobicity and fluidity of the three strains grown in the TSB medium to mid-LGP at 37°C. As shown in Figure 5, no significant difference in cell membrane fluidity was observed among the three strains (p>0.05). However, the proportion of ΔVpaChn25_0724 cells with damaged membrane was about twofold of that of the wild type (p < 0.01). The ΔVpaChn25_0724 mutant also showed a twofold cell surface hydrophobicity (p<0.01) in comparison to the wild type. In correlation to the almost full restoration of cellular growth by the plasmid-borne VpaChn25_0724 at 37°C (Figure 1), the complementation also restored the membrane integrity and hydrophobicity phenotypes. These results indicated that the VpaChn25_0724 gene is important for cell membrane integrity of V. parahaemolyticus CHN25.
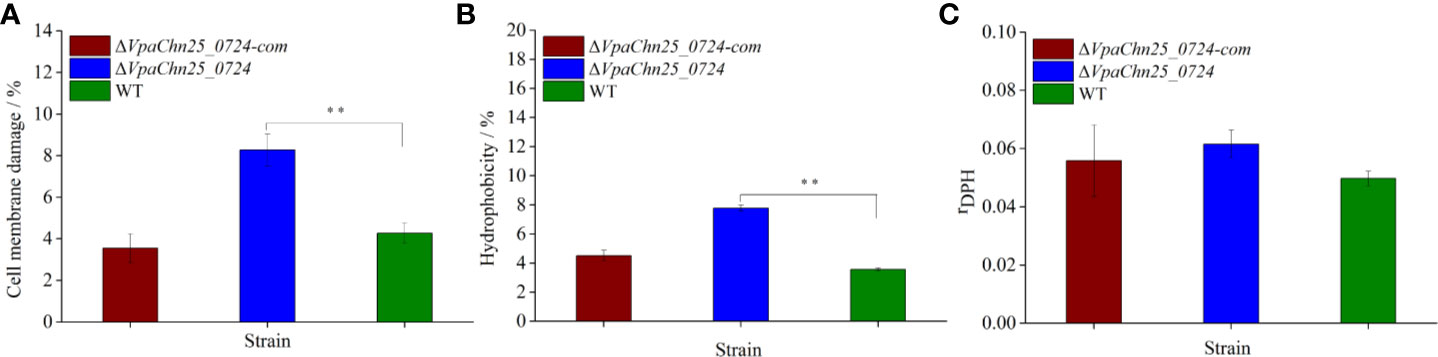
Figure 5 Cell membrane damage (A), hydrophobicity (B) and fluidity (C) of V. parahaemolyticus CHN25 (WT), ΔVpaChn25_0724, and ΔVpaChn25_0724-com. The strains were incubated in the TSB medium at 37°C to the mid-LGP. WT: wild type. **p<0.01 compared with the WT.
Differential Secretomes Mediated by the VpaChn25_0724 Gene Deletion
Given the altered cell membrane trait mediated by the VpaChn25_0724 gene deletion, we next conducted comparative secretomic analysis of the three strains. When incubated in the TSB medium at 37°C without shaking, the ΔVpaChn25_0724 mutant also grew more slowly than wild type (data not shown). The supernatant of bacterial cultures at the mid-LGP were collected, and extracellular proteins were isolated, and analyzed by 2-DE assay. This analysis revealed different secretome profiles among the wild type, ΔVpaChn25_0724 and ΔVpaChn25_0724-com strains, showing various numbers of visible protein spots (Figure 6). The patterns yielded from three independent 2-DE gels per biological sample were consistent (data not shown). These varied protein spots were excised from the 2-DE gels and digested with the trypsin. The resulting peptides were further identified by LC-MS/MS analysis.

Figure 6 The 2-DE analysis of extracellular proteins of V. parahaemolyticus CHN25, ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains. (A) Wild type; (B) ΔVpaChn25_0724; (C) ΔVpaChn25_0724-com.
The results yielded from the LC-MS/MS analysis were summarized in Table 2. Sequences of seven differentially expressed extracellular proteins among the three strains were obtained. Six proteins thereof were secreted by the ΔVpaChn25_0724 mutant. For instance, the protein Spot 24-b-1 was identified as an 8-stranded β-barrel protein (OmpW). The protein Spots 24-b-2 and 24-b-3 were identified as FlaB/D and FlaA flagellins, respectiely, while the 24-b-6 and 24-C-1 were identified as an aldehyde-alcohol dehydrogenase (AdhE), and a 2-hydroxyacid dehydrogenase, respectively. These results indicated that the VpaChn25_0724 gene deletion resulted in increased numbers of extracellular proteins, which was likely associated with damaged cell membrane structure of V. parahaemolyticus CHN25.
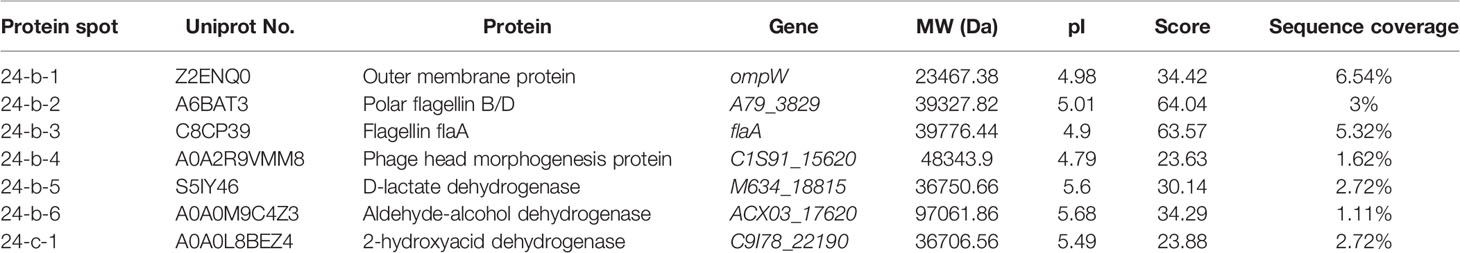
Table 2 Identification of the protein spots on the secretome profiles by LC-MS/MS analysis.
Effects of the VpaChn25_0724 Gene Deletion on V. parahaemolyticus CHN25-Host Intestinal Epithelial Cell Interaction
Consequently, we reasoned that the changed secretome may affect V. parahaemolyticus CHN25-host intestinal epithelial cell interaction, whereby the bacterium elicits gastroenteritis disease (O’Boyle and Boyd, 2013). The human rectal cancer epithelial cell line Caco-2 was used as an in vitro model for the cell interaction analysis in this study. As shown in Figure 7, after infected with the ΔVpaChn25_0724 mutant at 37°C for 4 h, the viability of Caco-2 cells was significantly higher (97.05% ± 0.84) than those infected with the wild type (66.97% ± 1.04) and ΔVpaChn25_0724-com (62.38% ± 1.34) strains (p<0.01). Moreover, apoptosis of Caco-2 cells was examined using Annexin V-FITC and propidium iodide (PI) double stainings by flow cytometry assay. Unexpectedly, the results showed that at 4 h post infection, ΔVpaChn25_0724 induced an early apoptosis in Caco-2 cells at a much higher rate (32.67% ± 2.12%) than the wild type (11.27% ± 0.94) and the ΔVpaChn25_0724-com (10.94% ± 2.12%) strains (p<0.01). Interestingly, an opposite pattern was observed where late apoptosis elicited by ΔVpaChn25_0724 was significantly less (48.50% ± 0.30%) than by wild type (77.53% ± 5.59%) and ΔVpaChn25_0724-com (74.57% ± 4.56%) strains (p<0.01).
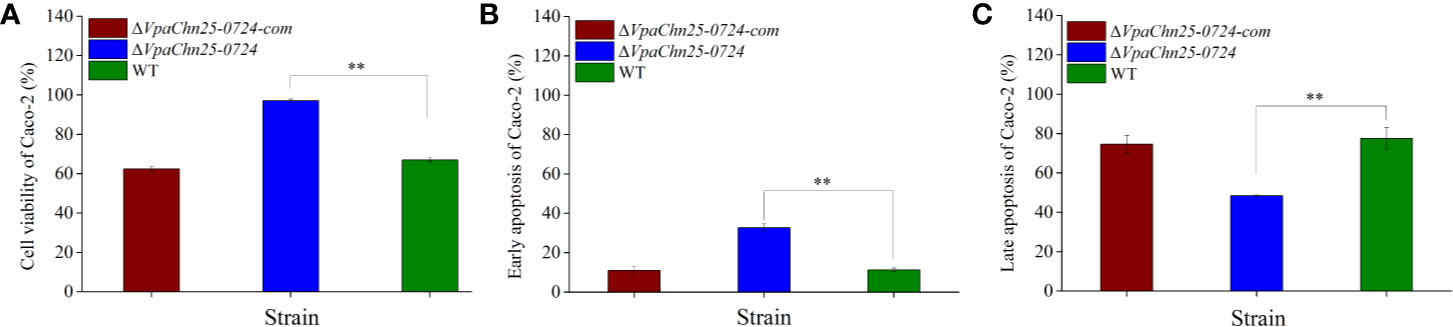
Figure 7 The viability and apoptosis of Caco-2 cells infected by V. parahaemolyticus CHN25 (WT), ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains. The infection was performed at 37°C for 4 h. (A) Cell viability; (B) Early stage of apoptosis; (C) Late stage of apoptosis. **p<0.01 compared with the WT.
Differential Transcriptomes Mediated by the VpaChn25_0724 Gene Deletion
To get insights into global-level gene expression change mediated by the VpaChn25_0724 gene deletion, we next determined transcriptomes of V. parahaemolyticus CHN25, ΔVpaChn25_0724 and ΔVpaChn25_0724-com strains using Illumina RNA sequencing technique. This analysis revealed that approximately 13.7% of the bacterial genes were differentially expressed in the ΔVpaChn25_0724 mutant, when compared with the wild type and ΔVpaChn25_0724-com strains grown at 37°C to the mid-LGP. Of these genes, 190 showed higher transcriptional levels (fold change ≥ 2.0), while 569 genes were down-regulated (fold change ≤ 0.5). These DEGs in ΔVpaChn25_0724 were grouped into one hundred and twenty-six gene functional catalogues in the KEGG database (data not shown). A complete list of the DEGs in the three strains is available in the NCBI SRA database (http://www.ncbi.nlm.nih.gov/sra/) under the accession number SRP258529. To validate the transcriptome data, we examined 18 representative genes (Table S1) in the ΔVpaChn25_0724 mutant by RT-qPCR analysis. The resulting data were correlated with those yielded from the transcriptome analysis (Table S1).
The Major Altered Metabolic Pathways in the ΔVpaChn25_0724 Mutant
Based on the GSEA of the transcriptome data against the KEGG database, approximately twelve significantly altered metabolic pathways were identified in the VpaChn25_0724 mutant, including the galactose, glyoxylate and dicarboxylate, fructose and mannose, butanoate and thiamine metabolisms; citrate cycle (TCA); pentose and glucuronate interconversions; valine, leucine, and isoleucine degradation and glycerolipid metabolism; QS; ATP-binding cassette (ABC) transporters; and phosphotransferase system (PTS) (Table 3).
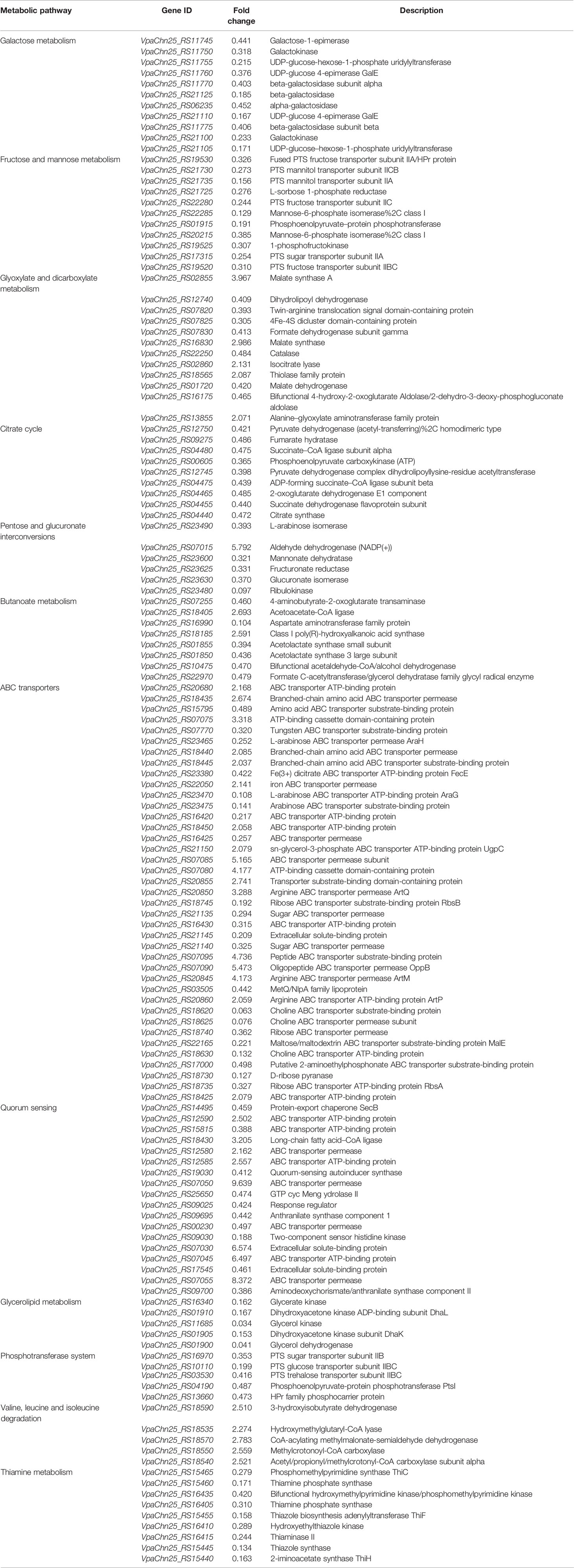
Table 3 Major altered metabolic pathways in the ΔVpaChn25_0724 mutant.
Remarkably, approximately sixty DEGs involved in the galactose metabolism, fructose and mannose metabolism, TCA, glycerolipid metabolism, PTS, and thiamine metabolism were all significantly down-regulated in the ΔVpaChn25_0724 mutant (0.034- to 0.487-fold) (p<0.05), when compared with the wild type and ΔVpaChn25_0724-com strains (Table 3). Also, all the DEGs in the pentose and glucuronate interconversions were greatly down-regulated (0.097- to 0.393-fold), except one encoding aldehyde dehydrogenase. These changes were directly related to the observed phenotypic variations of ΔVpaChn25_0724. For example, in the galactose metabolism, eleven DEGs encoding key metabolizing enzymes were significantly repressed at the transcriptional level (0.171–0.441 fold) (p<0.05), including the galactokinase (VpaChn25_RS11750 and VpaChn25_RS21100), galactose-1-epimerase (VpaChn25_RS11745), alpha and beta-galactosidase (VpaChn25_RS21125, VpaChn25_RS06235, VpaChn25_RS11770 and VpaChn25_RS11775), UDP-glucose 4-epimerase GalE (VpaChn25_RS21110 and VpaChn25_RS11760), and UDP-glucose-hexose-1-phosphate uridylyltransferase (VpaChn25_RS11755 and VpaChn25_RS21105), which may have resulted in reduced important metabolites (e.g., galactose-1-phosphate, and UDP-glucose/galactose/fructose 1,6-diphosphate) in the metabolic pathway. Similarly, in the fructose and mannose metabolism, the DEGs encoding key metabolizing enzymes, e.g., 1-phosphofructokinase (VpaChn25_RS19525) and mannose-6-phosphate isomerase (VpaChn25_RS22285, VpaChn25_RS20215), were also significantly down-regulated (0.156- to 0.326-fold) (p<0.05). Moreover, approximately all the DEGs linked to PTS showed a significant decrease in transcription in ΔVpaChn25_0724 (0.191- to 0.473-fold) (p<0.05). Interestingly, expression of the gene encoding glucose transporter subunit IIBC (VpaChn25_RS10110) was notably down-regulated (0.199-fold). Meanwhile, expression of about nine DEGs involved in TCA was slightly down-regulated (0.365- to 0.486-fold) (p<0.05). In addition, in the thiamine metabolism, expression of thiamine phosphorylase (VpaChn25_RS16405) was also down-regulated (0.310-fold) (p<0.05). These data suggested inactive transport and utilization of the carbon sources as well as repressed energy production in the ΔVpaChn25_0724 mutant.
Additionally, five DEGs involved in the glycerolipid metabolism were strikingly down-regulated (0.034- to 0.041-fold) (p<0.05), including the glycerol kinase (VpaChn25_RS16340 and VpaChn25_RS11685), glycerol dehydrogenase (VpaChn25_RS01900), dihydroxyacetone kinase ADP-binding subunit DhaL (VpaChn25_RS01910), and dihydroxyacetone kinase subunit DhaK genes (VpaChn25_RS01905). The reaction product (glycerol 3-phosphate, G3P) catalyzed by glycerol kinase is also an important metabolite in phospholipid biosynthesis under all growth conditions (Holtman et al., 2001), which plays a vital role in the regulation of membrane biogenesis.
On the other aspect, the deletion of VpaChn25_0724 also triggered significant changes in the other five metabolic pathways in the ΔVpaChn25_0724 mutant. Many DEGs thereof showed higher transcriptional levels. For instance, most DEGs linked to the valine, leucine and isoleucine degradation were slightly up-regulated in ΔVpaChn25_0724 (2.179- to 2.783-fold) (p<0.05), which may have resulted in an increases in acetyl-CoA and subsequent entry into TCA. Remarkably, approximately 39 DEGs associated with ABC transporters were significantly altered at the transcriptional level. Consistent with the down-regulated carbon metabolism as well as repressed energy production in ΔVpaChn25_0724, some ABC transporters for sugar uptake were also greatly down-regulated (0.192- to 0.362-fold) (p<0.05), e.g., sugar ABC transporter permease (VpaChn25_RS21140 and VpaChn25_RS21135), maltose/maltodextrin ABC transporter substrate-binding protein MalE (VpaChn25_RS22165), ribose ABC transporter permease (VpaChn25_RS18740), ribose ABC transporter ATP-binding protein RbsAB (VpaChn25_RS18735 and VpaChn25_RS187450), and extracellular solute-binding protein (VpaChn25_RS21145). In marked contrast, the DEGs encoding arginine ABC transporter permease ArtM (VpaChn25_RS20845), peptide ABC transporter substrate-binding protein (VpaChn25_RS07095), and oligopeptide ABC transporter permease (VpaChn25_RS07090) were notably up-regulated (4.173- to 5.473-fold). Likewise, some DEGs encoding ABC transporter ATP-binding protein (VpaChn25_RS07045), ABC transporter permease (VpaChn25_RS07085, VpaChn25_RS07050 and VpaChn25_RS07055), and extracellular solute-binding protein (VpaChn25_RS07030) were also greatly up-regulated in the QS (5.165- to 9.639-fold) (p<0.05), suggesting that the VpaChn25_0724 gene may act as a suppressor of these ABC transporters in V. parahaemolyticus CHN25.
Major Altered DGEs Related with Phenotypic Variations of the ΔVpaChn25_0724 Mutant
On the basis of the transcriptome data, the GSAA against the GO database also revealed major altered DGEs related with phenotypic variations of the ΔVpaChn25_0724 mutant compared with the wild type and ΔVpaChn25_0724-com strain.
Expression of the DEGs encoding flagellar basal body protein FliL (VpaChn25_RS22910) (0395-fold), rod protein FlgD (VpaChn25_RS17140) (0.389-fold), and export and assembly protein FliR (VpaChn25_RS22850) (0.094-fold) were all significantly down-regulated in the ΔVpaChn25_0724 mutant (p<0.05). The remarkably repressed FliR (0.094-fold) belongs to a membrane-embedded part of flagellar export apparatus. These data suggested a defective flagellar basal body in the ΔVpaChn25_0724 mutant that may have contributed to its affected swimming and biofilm formation.
Some biofilm formation-associated genes were also repressed at the transcriptional level in the ΔVpaChn25_0724 mutant, e.g., QS autoinducer synthase (VpaChn25_RS19030), anthranilate synthase component 1 (VpaChn25_RS09695), aminodeoxychorismate/anthranilate synthase component II (VpaChn25_RS09700), and PTS glucose transporter subunit IIBC (VpaChn25_RS10110) (0.199- to 0.442-fold) (p<0.05). QS impacts bacterial motility, biofilm formation, and construction (Whiteley et al., 2017). The conserved phage shock protein (Psp) system functions in cell envelope stress response, and links to antibiotic resistance, biofilm formation and virulence in a diverse group of bacteria (Flores-Kim and Darwin, 2016). In this study, the genes encoding core components of the Psp system PspB (VpaChn25_RS06290) (0.385-fold) and PspC (VpaChn25_RS06295) (0.342-fold) were significantly down-regulated in the ΔVpaChn25_0724 mutant (p<0.05).
The genes encoding T3SS chaperones SycN (VpaChn25_RS08695) (0.197-fold) and YopN-like gatekeeper (VpaChn25_RS08705) (0.399-fold), YopR-like regulator (VpaChn25_RS08820) (0.461-fold), and export apparatus protein (VpaChn25_RS08740) (0.244-fold) were all greatly down-regulated in ΔVpaChn25_0724 (p<0.05). Additionally, expression of the genes encoding OmpA (VpaChn25_RS22825) (0.164-fold) and OmpW (VpaChn25_RS16240) (0.331-fold) were significantly decreased (p<0.05). In contrast, the gene encoding T3SS chaperone CesT (VpaChn25_RS08785) was notably up-regulated in the ΔVpaChn25_0724 mutant (6.221-fold) (p<0.05).
Remarkably, several differentially expressed response and transcriptional regulators were greatly repressed in the ΔVpaChn25_0724 mutant, which are key components in bacterial gene regulatory networks, and can sense fluctuations under internal and external conditions (Brinkrolf et al., 2007). For example, two genes (VpaChn25_RS08860 and VpaChn25_RS22275) encoded DNA-binding transcriptional regulator AraC, one of the most common positive regulators in bacteria, were notably down-regulated (0.270-fold and 0.204-fold) (p<0.05). Regulators belonging to this family have three major regulatory functions in common: carbon metabolism, stress response, and pathogenesis (Gallegos et al., 1997). Moreover, the gene (VpaChn25_RS04915) encoding a TetR/AcrR family transcriptional regulator was strikingly down-regulated (0.021-fold) (p<0.05). Another interesting observation was that expression of a regulator BetI (VpaChn25_RS18605) was also strikingly down-regulated in the ΔVpaChn25_0724 mutant (0.022-fold) (p<0.05). Additionally, TEM images provided additional evidence for the global-level gene expression change in ΔVpaChn25_0724, as cell morphological characteristics of this mutant appeared different from those of the wild type and ΔVpaChn25_0724-com strains, such as the changed intracellular structure with more cytocysts in ΔVpaChn25_0724 (Figure 8).
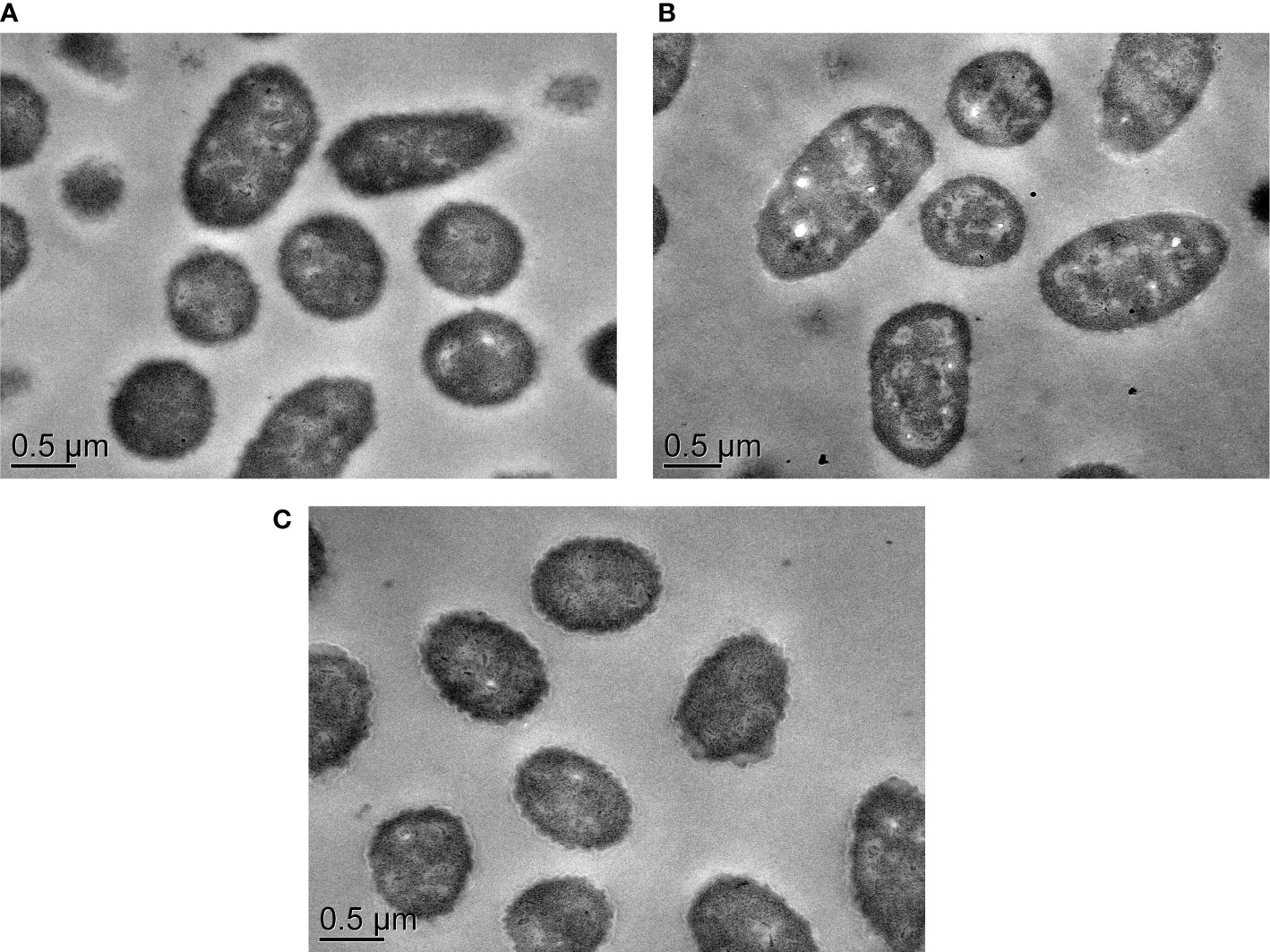
Figure 8 The TEM observation of cell structure of V. parahaemolyticus CHN25, ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains. (A) Wild type; (B) ΔVpaChn25_0724; (C) ΔVpaChn25_0724-com.
On the other hand, to further verify the differential transcriptomic data, two representative DEGs Vpachn25_RS01720 and Vpachn25_RS04440 (Table S1) encoding key enzymes were chosen for enzyme activity analyses, given their detection methods are available in literature to date. The results showed significantly reduced malate dehydrogenase and citrate synthase activities in ΔVpaChn25_0724 compared with the wild type strain (p<0.05) (Figure S2), which were encoded by the Vpachn25_RS01720 and Vpachn25_RS04440, respectively. These results were correlated with those yielded from the transcriptome analysis and confirmed by the RT-qPCR analyses (Table S1).
On the basis of the findings, we tried to locate the protein encoded by VpaChn25_0724 in V. parahaemolyticus CHN25 using routine immunochemistry method, but failed to find its exact cellular position. It will be interesting to investigate its protein property and regulation in the bacterium in the future research.
Transmission of the VpaChn25_0724 Gene in Bacteria
The VpaChn25_0724 gene was examined in 138 V. parahaemolyticus strains isolated from aquatic products collected in Shanghai, China (Su et al. under review) by PCR assay. The resulting data showed that approximately 5.8% (n=8) of the V. parahaemolyticus isolates carried the VpaChn25_0724 gene. Moreover, BLAST analysis against the GenBank database revealed that the VpaChn25_0724 gene is present in three Vibrio phages, four Vibrio species including Vibrio cholerae, Vibrio campbellii, Vibrio mimicus, and V. parahaemolyticus, as well as the other two bacterial genera such as Marinomonas primoryensis, and Shewanella oneidensis. These data indicated that transmission of the VpaChn25_0724 gene occurred among V. parahaemolyticus population in aquatic products, within Vibrio genus, and even across bacterial genera during the evolution history.
Discussion
Vibrio parahaemolyticus is a leading seafood-borne pathogen worldwide. In aquatic ecosystems, phages constantly transform bacterial communities by HGT (Penades et al., 2015; Castillo et al., 2018). Nevertheless, biological functions of prophage-related genes remaining in V. parahaemolyticus are not yet fully understood. In this study, for the first time, we studied one such gene VpaChn25_0724 encoding an unknown hypothetical protein in the largest prophage gene cluster identified in V. parahaemolyticus CHN25 genome (Zhu et al., 2017). An unmarked in-frame gene deletion mutant ΔVpaChn25_0724 was successfully constructed, and its complementary mutant ΔVpaChn25_0724-com was also obtained in this study. Our data unveiled that the VpaChn25_0724 gene deletion resulted in a sever defect in growth of V. parahaemolyticus CHN25, particularly at the lower temperatures.
Motility is closely associated with bacterial virulence and affects their attachment, colonization and invasion toward host cells (Guo et al., 2019). V. parahaemolyticus is motile by means of a single, sheathed polar flagellum that propels the swimmer cell in liquid environments (Kim and McCarter, 2000). The bacterial flagellum is essential in forming biofilm (i.e., matrix enclosed and surface-associated communities), which is critical for V. parahaemolyticus persistence in aquatic environments and pathogenicity in the host (Yildiz and Visick, 2009). In this study, we observed significantly lowered swimming capacity of the ΔVpaChn25_0724 mutant at 25°C and 15°C, when compared with the wild type (p < 0.05). Moreover, our data showed a decrease in the development of biofilm by V. parahaemolyticus CHN25 in the absence of the VpaChn25_0724 gene. The severe defect in growth elicited by the VpaChn25_0724 gene deletion likely led to the variant swimming motility and biofilm formation of V. parahaemolyticus CHN25.
Bacterial secretion systems play a vital role in virulence, symbiosis, interbacterial interactions, and environmental stress (De Nisco et al., 2017). In this study, we found differential secretomes mediated by the VpaChn25_0724 gene deletion. Among the differentially expressed extracellular proteins, the protein Spot 24-b-1 is an important outer membrane protein and protects bacteria against host phagocytosis (Li et al., 2016). The protein Spots 24-b-2 and 24-b-3, identified as FlaB/D and FlaA flagellins, respectiely, are involved in polar flagellar biosynthesis in V. parahaemolyticus (Kim and McCarter, 2000). The latter is involved in P-ring assembly of flagellar structure and swimming motility (Kim and McCarter, 2000). Recently, Echazarreta et al. reported that FlaA also facilitated filament formation of V. cholerae flagellum (Echazarreta et al., 2018). The protein Spot 24-b-6 was identified as an AdhE that forms a high-order spirosome architecture for its activity (Kim et al., 2019). This multi-functional enzyme is essential for the fermentation of glucose to sustain the glycolytic pathway, and the deletion of adhE gene in pathogenic E. coli O157:H7 strongly suppressed type III secretion systems (T3SS) and induced over-expression of non-functional flagella (Kim et al., 2019). The Spot 24-C-1, identified as a 2-hydroxyacid dehydrogenase, plays an important role in cell stability at high saline concentrations (Bonete et al., 2000). These results indicated that the VpaChn25_0724 gene deletion resulted in increased numbers of extracellular proteins, which was likely associated with damaged cell membrane of the ΔVpaChn25_0724 mutant.
The damaged cell membrane elicited by the VpaChn25_0724 gene deletion also significantly affected V. parahaemolyticus CHN25-host intestinal epithelial cell interaction. Based on the in vitro Caco-2 cell model, our data showed that the ΔVpaChn25_0724 mutant induced much higher viability and early apoptosis rate of Caco-2 cells than the wild type and the ΔVpaChn25_0724-com strains (p < 0.01). Nevertheless, an opposite pattern was observed in the late apoptosis elicited by ΔVpaChn25_0724. The cell membrane of Caco-2 cells at the early apoptosis phase was still intact, whereas at the later stage was damaged. The most possible explanation for the above observation was that the increased number of extracellular proteins secreted by the ΔVpaChn25_0724 mutant may have contributed to the higher apoptosis occurrence at the early stage, however, the significantly changed membrane surface structure and cellular process (see below) of the mutant likely lowered its cytotoxicity to the host cells at the later stage.
Transcriptomes of V. parahaemolyticus CHN25, ΔVpaChn25_0724 and ΔVpaChn25_0724-com strains were determined to get insights into global-level gene expression change mediated by the VpaChn25_0724 gene deletion. Based on the GSEA of the transcriptome data against the KEGG database, approximately twelve significantly altered metabolic pathways were identified in the VpaChn25_0724 mutant (Table 3). For instance, PTS is known as a major sugar transport multicomponent system in bacteria, by which many sugars are transported into bacteria, concomitantly phosphorylated, and then fed into glycolysis (Postma et al., 1993). In this study, approximately all the DEGs linked to PTS showed a significant decrease in transcription in ΔVpaChn25_0724 (0.191- to 0.473-fold) (p<0.05), which encode fructose, glucose. mannitol, trehalose, and sugar transporter subunits. Moreover, expression of the gene encoding glucose transporter subunit IIBC (VpaChn25_RS10110) was notably down-regulated (0.199-fold), which may be related with the repressed galactose, fructose, mannose and glycerol metabolisms, because glucose controls utilization of several other carbon sources including lactose, melibiose, maltose, and glycerol in E. coli (Holtman et al., 2001). Meanwhile, expression of about nine DEGs involved in TCA was slightly down-regulated (p<0.05). In addition, in the thiamine metabolism, expression of thiamine phosphorylase (VpaChn25_RS16405) was also down-regulated (p<0.05), which may result in decreased thiamine pyrophosphate, a cofactor for many essential enzymes in glucose and energy metabolisms (Rodionov et al., 2017). These data suggested inactive transport and utilization of the carbon sources as well as repressed energy production in the ΔVpaChn25_0724 mutant.
On the basis of the transcriptome data, major altered DGEs related with the phenotypic variations of the ΔVpaChn25_0724 mutant were also identified. For instance, bacterial polar flagellum is powered by a rotary motor and acts as semirigid helical propeller, which is attached via a flexible coupling, known as the hook, to the basal body. The latter consists of rings and rods that penetrate the membrane and peptidoglycan layers (Kim and McCarter, 2000). In this study, expression of the DEGs encoding flagellar basal body structure protein FliL (VpaChn25_RS22910), rod structure protein FlgD (VpaChn25_RS17140), and export and assembly structure protein FliR (VpaChn25_RS22850) were all significantly down-regulated in the ΔVpaChn25_0724 mutant (p<0.05). Recently, Takekawa et al. reported that FliL is a new stomatin-like protein that assists the Vibrio flagellar motor function (Takekawa et al., 2019). In this study, the FliR that belongs to a membrane-embedded part of flagellar export apparatus was greatly down-regulated in expression (0.094-fold). These data suggested a defective flagellar basal body in the ΔVpaChn25_0724 mutant that may have contributed to its affected swimming and biofilm formation.
T3SS was identified in V. parahaemolyticus CHN25 genome, which is necessary for bacterial survival in the environment (De Nisco et al., 2017; Matsuda et al., 2019). In this study, the genes encoding T3SS chaperones SycN (VpaChn25_RS08695) and YopN-like gatekeeper (VpaChn25_RS08705), YopR-like regulator (VpaChn25_RS08820), and export apparatus protein (VpaChn25_RS08740) were all greatly down-regulated in ΔVpaChn25_0724 (p<0.05). In Yersinia pestis, the secretion of toxic Yersinia outer proteins (Yops) is regulated by a YopN/SycN/YscB/TyeA complex (Joseph and Plano, 2013). It has been reported that YopN functions to prevent the secretion of Yops until T3SS apparatuses are in direct contact with a targeted eukaryotic cell and activated, which avoids the spurious loss of effector proteins to the extracellular environment (Plano and Schesser, 2013). Additionally, expression of the genes encoding OmpA (VpaChn25_RS22825) and OmpW (VpaChn25_RS16240) were significantly decreased (p<0.05). OmpA family proteins are heat-modifiable, surface-exposed, and porin proteins that are in high-copy number in the outer membrane of many gram-negative pathogenic bacteria. They are involved in bacterial adhesion, invasion or intracellular survival, as well as evasion of host defenses or stimulation of pro-inflammatory cytokine production (Confer and Ayalew, 2013). In this study, in contrast, the gene encoding T3SS chaperone CesT (VpaChn25_RS08785) was notably up-regulated in the ΔVpaChn25_0724 mutant (6.221-fold) (p<0.05). It has been reported that the enteropathogenic E. coli (EPEC) multicargo chaperone CesT interacts with at least ten effector proteins and contributes pathogenesis (Ramu et al., 2013).
The distinct transcriptome data also revealed strikingly down-regulated five key genes involved in the glycerolipid metabolism in the ΔVpaChn25_0724 mutant (0.034- to 0.041-fold), suggesting reduced important metabolites in the bacterial phospholipid biosynthesis, such as the G3P (Holtman et al., 2001) that plays a vital role in the regulation of membrane biogenesis. Meanwhile, a defective flagellar basal body in ΔVpaChn25_0724 was also revealed by the comparative transcriptomic analyses (see above). These results, coupled with the inactive transport and utilization of the carbon sources as well as repressed energy production in ΔVpaChn25_0724 may have significantly affected the cell membrane integrity of the mutant.
In addition, several key components in bacterial gene regulatory networks were greatly repressed in the ΔVpaChn25_0724 mutant, which can sense fluctuations under internal and external conditions (Brinkrolf et al., 2007). For instance, the gene (VpaChn25_RS04915) encoding a TetR/AcrR family transcriptional regulator was strikingly down-regulated (0.021-fold) (p<0.05). Regulators of this family are involved in a series of regulatory cascades, e.g., cell response to environmental insults, control of catabolic pathways, differentiation processes, and pathogenicity (Ramos et al., 2005). Another interesting observation was that expression of a regulator BetI (VpaChn25_RS18605) was also strikingly down-regulated in the ΔVpaChn25_0724 mutant (0.022-fold) (p<0.05), which negatively regulates betT and betIBA genes that govern glycine betaine (GB) biosynthesis from choline in E. coli (Rkenes et al., 1996). The down-regulated BetI possibly in turn activated the target genes in ΔVpaChn25_0724, which perhaps led to increased amount of GB to maintain the integrity of cell membranes against the damaging effects, as in other stress responses to excessive salt, cold, heat and freezing in bacteria (Sun et al., 2014).
Taken together, the results in this study facilitate better understanding of biological function of prophage-related genes remaining in V. parahaemolyticus, and meet the increasing need for novel diagnosis candidates of the leading seafood-borne pathogen worldwide.
Data Availability Statement
A complete list of the DEGs is available in the NCBI SRA database (http://www.ncbi.nlm.nih.gov/sra/) under the accession number SRP258529.
Author Contributions
LY, YW, PY, SR, ZZ, YJ, JY, XP, and LC participated in the design and or discussion of the study. LY, YW, and ZZ carried out the major experiments. PY analyzed the data. SR supervised this study. JY helped the bacteria-host intestinal epithelial cell interaction experiments. LY, YW, ZZ, and LC wrote the manuscript. XP, and LC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Science and Technology Commission of Shanghai Municipality (No. 17050502200) and the National Natural Science Foundation of China (No. 31671946).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge Professor Dominique Schneider, for kindly providing us the plasmid pDS132 for construction of the gene deletion mutant, and Professor Weicheng Bei for the E. coli β2155 λpir for conjugation experiments in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.595709/full#supplementary-material
Supplementary Figure 1 | Construction of the ΔVpaChn25_0724, and ΔVpaChn25_0724-com mutants by agarose gel electrophoresis analysis. (A) amplicons of upstream (Lane 1) and downstream (Lane 2) sequences of the VpaChn25_0724 gene. (B) amplicons franking the upstream and downstream sequence of VpaChn25_0724. (C) amplicons of the upstream and downstream sequence of VpaChn25_0724 in the recombinant pDS132+VpaChn25_0724 plasmids. Lines 1–6, positive recombinant plasmids; Line 7, blank control; Line 8, positive control. (D) amplicons of transformants with the VpaChn25_0724-up-F and VpaChn25_0724-down-R primers. Lines 1–5, positive strain of the first change; Lines 6–7, positive control; Lines 8 to 9, blank control. (E) amplicons of the tlh gene. Lines 1–5, positive exconjugants; Line 6, positive control; Line 7, blank control. (F) amplicons of exconjugants with the VpaChn25_0724-up-ex-F and VpaChn25_0724-down-ex-R primers. Lines 1–22, selected exconjugants strains; Line 23, blank control; Line 24, genomic DAN control. (G) Amplicons of the tlh gene from ΔVpaChn25_0724 mutants. Lines 1–5, positive ΔVpaChn25_0724 mutants; Line 6, positive control; Line 7, blank control. (H) Amplicons of the VpaChn25_0724 gene. Lines 1–2, positive bands; Line 3, blank control. (I) double digestion of the recombinant pMMB207+VpaChn25_0724 plasmid. (J) amplicons of VpaChn25_0724 from positive transformants. (K) amplicons of tlh from ΔVpaChn25_0724-com mutants. M, 100 bp DNA Ladder.
Supplementary Figure 2 | Activities of the malate dehydrogenase and citrate synthase encoded by the DEGs Vpachn25_RS01720 and Vpachn25_RS04440 in V. parahaemolyticus CHN25 (WT), ΔVpaChn25_0724, and ΔVpaChn25_0724-com strains. A. Malate dehydrogenase. B. Citrate synthase.
Supplementary Table S1 | Expression of representative DEGs in ΔVpaChn25_0724 mutant by RT-qPCR analysis.
References
Bonete M. J., Ferrer J., Pire C., Penades M., Ruiz J. L. (2000). 2-Hydroxyacid dehydrogenase from Haloferax mediterranei, a D-isomer-specific member of the 2-hydroxyacid dehydrogenase family. Biochimie 82 (12), 1143–1150. doi: 10.1016/s0300-9084(00)01193-7
Brinkrolf K., Brune I., Tauch A. (2007). The transcriptional regulatory network of the amino acid producer Corynebacterium glutamicum. J. Biotechnol. 129 (2), 191–211. doi: 10.1016/j.jbiotec.2006.12.013
Castillo D., Kauffman K., Hussain F., Kalatzis P., Rorbo N., Polz M. F., et al. (2018). Widespread distribution of prophage-encoded virulence factors in marine Vibrio communities. Sci. Rep. 8 (1), 9973. doi: 10.1038/s41598-018-28326-9
Collado S., Oulego P., Alonso S., Diaz M. (2017). Flow cytometric characterization of bacterial abundance and physiological status in a nitrifying-denitrifying activated sludge system treating landfill leachate. Environ. Sci. Pollut. Res. Int. 24 (26), 21262–21271. doi: 10.1007/s11356-017-9596-y
Confer A. W., Ayalew S. (2013). The OmpA family of proteins: roles in bacterial pathogenesis and immunity. Vet. Microbiol. 163 (3-4), 207–222. doi: 10.1016/j.vetmic.2012.08.019
De Nisco N. J., Kanchwala M., Li P., Fernandez J., Xing C., Orth K. (2017). The cytotoxic type 3 secretion system 1 of Vibrio rewires host gene expression to subvert cell death and activate cell survival pathways. Sci. Signal. 10 (479), eaal4501. doi: 10.1126/scisignal.aal4501
Desai S. K., Kenney L. J. (2019). Switching lifestyles is an in vivo adaptive strategy of bacterial pathogens. Front. Cell. Infect. Microbiol. 9:421. doi: 10.3389/fcimb.2019.00421
Echazarreta M. A., Kepple J. L., Yen L. H., Chen Y., Klose K. E. A. (2018). Critical Region in the FlaA Flagellin Facilitates Filament Formation of the Vibrio cholerae Flagellum. J. Bacteriol. 200 (15), e00029–e00018. doi: 10.1128/jb.00029-18
Feiner R., Argov T., Rabinovich L., Sigal N., Borovok I., Herskovits A. A. (2015). A new perspective on lysogeny: prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 13 (10), 641–650. doi: 10.1038/nrmicro3527
Flores-Kim J., Darwin A. J. (2016). Interactions between the cytoplasmic domains of PspB and PspC silence the Yersinia enterocolitica phage shock protein response. J. Bacteriol. 198 (24), 3367–3378. doi: 10.1128/jb.00655-16
Fujino T., Okuno Y., Nakada D., Aoyama A., Fukai K., Mukai T., et al. (1953). On the bacteriological examination of Shirasu food poisoning. Med. J. Osaka Univ. 4, 299–304.
Gallegos M. T., Schleif R., Bairoch A., Hofmann K., Ramos J. L. (1997). Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61 (4), 393–410. doi: 10.1128/.61.4.393-410.1997
Ghenem L., Elhadi N., Alzahrani F., Nishibuchi M. (2017). Vibrio parahaemolyticus: a review on distribution, pathogenesis, virulence determinants and epidemiology. Saudi J. Med. Med. Sci. 5 (2), 93–103. doi: 10.4103/sjmms.sjmms_30_17
Gomez-Gil B., Soto-Rodriguez S., Lozano R., Betancourt-Lozano M. (2014). Draft genome sequence of Vibrio parahaemolyticus strain M0605, which causes severe mortalities of shrimps in Mexico. Genome Announc. 2 (2), e00055–e00014. doi: 10.1128/genomeA.00055-14
Guo D., Yang Z., Zheng X., Kang S., Yang Z., Xu Y., et al. (2019). Thymoquinone inhibits biofilm formation and attachment-invasion in host cells of Vibrio parahaemolyticus. Foodborne Pathog. Dis. 16 (10), 671–678. doi: 10.1089/fpd.2018.2591
Harrison E., Brockhurst M. A. (2017). Ecological and evolutionary benefits of temperate phage: what does or doesn’t kill you makes you stronger. Bioessays 39, 1700112. doi: 10.1002/bies.201700112
He Y., Wang H., Chen L. (2015). Comparative secretomics reveals novel virulence-associated factors of Vibrio parahaemolyticus. Front. Microbiol. 6:707. doi: 10.3389/fmicb.2015.00707
Holtman C. K., Pawlyk A. C., Meadow N. D., Pettigrew D. W. (2001). Reverse genetics of Escherichia coli glycerol kinase allosteric regulation and glucose control of glycerol utilization in vivo. J. Bacteriol. 183 (11), 3336–3344. doi: 10.1128/JB.183.11.3336–3344
Howard-Varona C., Hargreaves K. R., Abedon S. T., Sullivan M. B. (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11 (7), 1511–1520. doi: 10.1038/ismej.2017.16
Huang H. H., Chen W. C., Lin C. W., Lin Y. T., Ning H. C., Chang Y. C., et al. (2017). Relationship of the CreBC two-component regulatory system and inner membrane protein CreD with swimming motility in Stenotrophomonas maltophilia. PLoS One 12 (4), e0174704. doi: 10.1371/journal.pone.0174704
Jensen R. V., Depasquale S. M., Harbolick E. A., Hong T., Kernell A. L., Kruchko D. H., et al. (2013). Complete genome sequence of prepandemic Vibrio parahaemolyticus BB22OP. Genome Announc. 1 (1), e00002–e00012. doi: 10.1128/genomeA.00002-12
Jiang Y., Chu Y., Xie G., Li F., Wang L., Huang J., et al. (2019). Antimicrobial resistance, virulence and genetic relationship of Vibrio parahaemolyticus in seafood from coasts of Bohai Sea and Yellow Sea, China. Int. J. Food. Microbiol. 290, 116–124. doi: 10.1016/j.ijfoodmicro.2018.10.005
Joseph S. S., Plano G. V. (2013). The SycN/YscB chaperone-binding domain of YopN is required for the calcium-dependent regulation of Yop secretion by Yersinia pestis. Front. Cell. Infect. Microbiol. 3, 1. doi: 10.3389/fcimb.2013.00001
Kalburge S. S., Polson S. W., Boyd Crotty K., Katz L., Turnsek M., Tarr C. L., et al. (2014). Complete genome sequence of Vibrio parahaemolyticus environmental strain UCM-V493. Genome Announc. 2 (2), e00159–e00114. doi: 10.1128/genomeA.00159-14
Kim Y. K., McCarter L. L. (2000). Analysis of the polar flagellar gene system of Vibrio parahaemolyticus. J. Bacteriol. 182 (13), 3693–3704. doi: 10.1128/jb.182.13.3693-3704.2000
Kim H. W., Hong Y. J., Jo J. II, Ha S. D., Kim S. H., Lee H. J., et al. (2017). Raw ready-to-eat seafood safety: microbiological quality of the various seafood species available in fishery, hyper and online markets. Lett. Appl. Microbiol. 64 (1), 27–34. doi: 10.1111/lam.12688
Kim G., Azmi L., Jang S., Jung T., Hebert H., Roe A. J., et al. (2019). Aldehyde-alcohol dehydrogenase forms a high-order spirosome architecture critical for its activity. Nat. Commun. 10 (1), 4527. doi: 10.1038/s41467-019-12427-8
Lan S. F., Huang C. H., Chang C. H., Liao W. C., Lin I. H., Jian W. N., et al. (2009). Characterization of a new plasmid-like prophage in a pandemic Vibrio parahaemolyticus O3:K6 strain. Appl. Environ. Microbiol. 75 (9), 2659–2667. doi: 10.1128/aem.02483-08
Li W., Wen L., Li C., Chen R., Ye Z., Zhao J., et al. (2016). Contribution of the outer membrane protein OmpW in Escherichia coli to complement resistance from binding to factor H. Microb. Pathog. 98, 57–62. doi: 10.1016/j.micpath.2016.06.024
Li L., Meng H., Gu D., Li Y., Jia M. (2019). Molecular mechanisms of Vibrio parahaemolyticus pathogenesis. Microbiol. Res. 222, 43–51. doi: 10.1016/j.micres.2019.03.003
Loyola D. E., Navarro C., Uribe P., Garcia K., Mella C., Diaz D., et al. (2015). Genome diversification within a clonal population of pandemic Vibrio parahaemolyticus seems to depend on the life circumstances of each individual bacteria. BMC Genomics 16, 176. doi: 10.1186/s12864-015-1385-8
Matsuda S., Okada R., Tandhavanant S., Hiyoshi H., Gotoh K., Iida T., et al. (2019). Export of a Vibrio parahaemolyticus toxin by the Sec and type III secretion machineries in tandem. Nat. Microbiol. 4 (5), 781–788. doi: 10.1038/s41564-019-0368-y
Morgelin M. (2017). Negative Staining and transmission electron microscopy of bacterial surface structures. Methods Mol. Biol. 1535, 211–217. doi: 10.1007/978-1-4939-6673-8_13
O’Boyle N., Boyd A. (2013). Manipulation of intestinal epithelial cell function by the cell contact-dependent type III secretion systems of Vibrio parahaemolyticus. Front. Cell. Infect. Microbiol. 3:114. doi: 10.3389/fcimb.2013.00114
Pelletier C., Bouley C., Cayuela C., Bouttier S., Bourlioux P., Bellon-Fontaine M. N. (1997). Cell surface characteristics of Lactobacillus casei subsp. casei, Lactobacillus paracasei subsp. paracasei, and Lactobacillus rhamnosus strains. Appl. Environ. Microbiol. 63 (5), 1725–1731. doi: 10.1128/AEM.63.5.1725-1731.1997
Penades J. R., Chen J., Quiles-Puchalt N., Carpena N., Novick R. P. (2015). Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 23, 171–178. doi: 10.1016/j.mib.2014.11.019
Plano G. V., Schesser K. (2013). The Yersinia pestis type III secretion system: expression, assembly and role in the evasion of host defenses. Immunol. Res. 57 (1-3), 237–245. doi: 10.1007/s12026-013-8454-3
Postma P., Lengeler J., Jacobson G. (1993). Phosphoenolpyruvate: carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 57 (3), 543–594. doi: 10.1128/MMBR.57.3.543-594.1993
Raghunath P. (2014). Roles of thermostable direct hemolysin (TDH) and TDH- related hemolysin (TRH) in Vibrioparahaemolyticus. Front. Microbiol. 5:805. doi: 10.3389/fmicb.2014.00805
Ramos J. L., Martínez-Bueno M., Molina-Henares A. J., Terán W., Watanabe K., Zhang X., et al. (2005). The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69 (2), 326–356. doi: 10.1128/MMBR.69.2.326-356.2005
Ramu T., Prasad M. E., Connors E., Mishra A., Thomassin J. L., Leblanc J., et al. (2013). A novel C-terminal region within the multicargo type III secretion chaperone CesT contributes to effector secretion. J. Bacteriol. 195 (4), 740–756. doi: 10.1128/jb.01967-12
Rkenes T., Lamark T., Strøm A. R. (1996). DNA-binding properties of the BetI repressor protein of Escherichia coli: The inducer choline stimulates BetI-DNA complex formation. J. Bacteriol. 178 (6), 1663–1670. doi: 10.1128/jb.178.6.1663-1670.1996
Rodionov D. A., Leyn S. A., Li X., Rodionova I. A. (2017). A novel transcriptional regulator related to thiamine phosphate synthase controls thiamine metabolism genes in archaea. J. Bacteriol. 199 (4), e00743–e00716. doi: 10.1128/jb.00743-16
Song Y., Yu P., Li B., Pan Y., Zhang X., Cong J., et al. (2013). The mosaic accessory gene structures of the SXT/R391-like integrative and conjugative elements derived from Vibrio spp. isolated from aquatic products and environment in the Yangtze River estuary, China. BMC Microbiol. 13:214. doi: 10.1186/1471-2180-13-214
Sun X., Liu T., Peng X., Chen L. (2014). Insights into Vibrio parahaemolyticus CHN25 response to artificial gastric fluid stress by transcriptomic analysis. Int. J. Mol. Sci. 15 (12), 22539–22562. doi: 10.3390/ijms151222539
Takekawa N., Isumi M., Terashima H., Zhu S., Nishino Y., Sakuma M., et al. (2019). Structure of Vibrio FliL, a new stomatin-like protein that assists the bacterial flagellar motor function. MBio 10 (2), e00292–e00219. doi: 10.1128/mBio.00292-19
Tan L., Zhao F., Han Q., Zhao A., Malakar P. K., Liu H., et al. (2018). High correlation between structure development and chemical variation during biofilm formation by Vibrio parahaemolyticus. Front. Microbiol. 9:1881. doi: 10.3389/fmicb.2018.01881
Tsai C. C., Hung Y. H., Chou L. C. (2018). Evaluation of lactic acid bacteria on the inhibition of Vibrio parahaemolyticus infection and its application to food systems. Molecules 23 (5):1238. doi: 10.3390/molecules23051238
Voss D., Montville T. J. (2014). 1,6-Diphenyl-1,3,5-hexatrine as a reporter of inner spore membrane fluidity in Bacillus subtilis and Alicyclobacillus acidoterrestris. J. Microbiol. Methods 96, 101–103. doi: 10.1016/j.mimet.2013.11.009
Wang L., Ling Y., Jiang H., Qiu Y., Qiu J., Chen H., et al. (2013). AphA is required for biofilm formation, motility, and virulence in pandemic Vibrio parahaemolyticus. Int. J. Food. Microbiol. 160 (3), 245–251. doi: 10.1016/j.ijfoodmicro.2012.11.004
Whiteley M., Diggle S. P., Greenberg E. P. (2017). Progress in and promise of bacterial quorum sensing research. Nature 551 (7680), 313–320. doi: 10.1038/nature24624
Wong H. C., Wang T. Y., Yang C. W., Tang C. T., Ying C., Wang C. H., et al. (2019). Characterization of a lytic vibriophage VP06 of Vibrio parahaemolyticus. Res. Microbiol. 170 (1), 13–23. doi: 10.1016/j.resmic.2018.07.003
Xu F., Feng X., Sui X., Lin H., Han Y. (2017). Inactivation mechanism of Vibrio parahaemolyticus via supercritical carbon dioxide treatment. Food. Res. Int. 100 (Pt 2), 282–288. doi: 10.1016/j.foodres.2017.08.038
Yang A., Tang W. S., Si T., Tang J. X. (2017). Influence of physical effects on the swarming motility of Pseudomonas aeruginosa. Biophys. J. 112 (7), 1462–1471. doi: 10.1016/j.bpj.2017.02.019
Yildiz F. H., Visick K. L. (2009). Vibrio biofilms: so much the same yet so different. Trends Microbiol. 17 (3), 109–118. doi: 10.1016/j.tim.2008.12.004
Zabala B., Hammerl J. A., Espejo R. T., Hertwig S. (2009). The linear plasmid prophage Vp58.5 of Vibrio parahaemolyticus is closely related to the integrating phage VHML and constitutes a new incompatibility group of telomere phages. J. Virol. 83 (18), 9313–9320. doi: 10.1128/jvi.00672-09
Zhu C., Sun B., Liu T., Zheng H., Gu W., He W., et al. (2017). Genomic and transcriptomic analyses reveal distinct biological functions for cold shock proteins (VpaCspA and VpaCspD) in Vibrio parahaemolyticus CHN25 during low-temperature survival. BMC Genomics 18 (1), 436. doi: 10.1186/s12864-017-3784-5
Keywords: Vibrio parahaemolyticus, prophage, virulence, gene knockout, secretome, transcriptome
Citation: Yang L, Wang Y, Yu P, Ren S, Zhu Z, Jin Y, Yan J, Peng X and Chen L (2020) Prophage-Related Gene VpaChn25_0724 Contributes to Cell Membrane Integrity and Growth of Vibrio parahaemolyticus CHN25. Front. Cell. Infect. Microbiol. 10:595709. doi: 10.3389/fcimb.2020.595709
Received: 17 August 2020; Accepted: 06 November 2020;
Published: 09 December 2020.
Edited by:
Lixing Huang, Jimei University, ChinaReviewed by:
Yanni Zhao, Shaanxi University of Science and Technology, ChinaXiaojun Zhang, Yangzhou University, China
Copyright © 2020 Yang, Wang, Yu, Ren, Zhu, Jin, Yan, Peng and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lanming Chen, lmchen@shou.edu.cn
†These authors have contributed equally to this work