 Rowan Hardy
Rowan Hardy Mark S. Cooper*
Mark S. Cooper*- Centre for Endocrinology, Diabetes and Metabolism, The Institute of Biomedical Research, The Medical School, University of Birmingham, Birmingham, UK
Glucocorticoids are a class of steroid hormones that are essential to life but cause serious harm in excess. The main clinical features of glucocorticoid excess are due to adverse effects on cells and tissues that arise from a common developmental precursor – the mesenchymal stromal cell (MSC; sometimes referred to as the mesenchymal stem cell). Interestingly glucocorticoids appear essential for the differentiation of cells and tissues that arise from MSCs. High levels of glucocorticoids are used in tissue engineering strategies to enhance the formation of tissues such as bone, cartilage, and muscle. This article discusses the paradox that glucocorticoids both enhance and impair MSC development and function. It will describe how endogenous glucocorticoids are likely to be important in these processes in vivo and will discuss the implications for therapies aimed at reducing the damage associated with the use of therapeutic glucocorticoids.
Introduction
Glucocorticoids are a class of steroid hormones that are essential to life but cause serious harm in excess. The main clinical features of glucocorticoid excess are due to adverse effects on cells and tissues that arise from a common developmental precursor – the mesenchymal stromal cell (MSC; sometimes referred to as the mesenchymal stem cell). Traditionally MSCs were most commonly derived from bone marrow but have also been identified in a range of other tissues (da Silva et al., 2006). Interestingly glucocorticoids appear essential for the differentiation of cells and tissues that arise from bone marrow derived MSCs. High levels of glucocorticoids are used in tissue engineering strategies to enhance the formation of tissues such as bone, cartilage, and muscle. This article discusses the paradox that glucocorticoids both enhance and impair MSC development and function. It will describe how endogenous glucocorticoids are likely to be important in these processes in vivo and will discuss the implications for therapies aimed at reducing the damage associated with the use of therapeutic glucocorticoids.
Cushing’s Syndrome
The term glucocorticoids comprises anti-inflammatory steroid hormones that are produced naturally by the adrenal cortex and related pharmaceuticals designed to treat inflammatory diseases. Prolonged exposure to high levels of glucocorticoids results in a range of clinical features referred to as Cushing’s syndrome (Fernandez-Rodriguez et al., 2009). These features were first noted by Harvey Cushing in rare patients who had excess production of cortisol (the main glucocorticoid made in the human adrenal gland – also referred to as hydrocortisone when administered as a pharmaceutical) as a result of an ACTH secreting pituitary tumor (Cushing, 1932). However, after the introduction of synthetic glucocorticoids in the 1950s to treat a range of inflammatory conditions the number of patients with features of excess glucocorticoid activity increased dramatically. The most prominent clinical features of Cushing’s syndrome in adults are muscle wasting and weakness, skin thinning (including easy bruising and the development of abdominal stretch marks called striae), osteoporosis and bone fracture, and redistribution of fat from the periphery to the center of the body (abdomen and face; Fernandez-Rodriguez et al., 2009). These features are thought to be due to the effects of high levels of glucocorticoids on muscle cells, dermal fibroblasts, osteoblasts, and adipocytes respectively. These cell types all share the common feature that they develop from MSCs (shown schematically in Figure 1). The main feature of Cushing’s syndrome in children is growth arrest, an effect attributed to the action of glucocorticoids on growth plate chondrocytes (Allen et al., 1994). Chondrocytes also arise from MSCs and, as such, all major cell types that arise from MSCs are implicated in the changes in body composition seen in patients with Cushing’s syndrome.
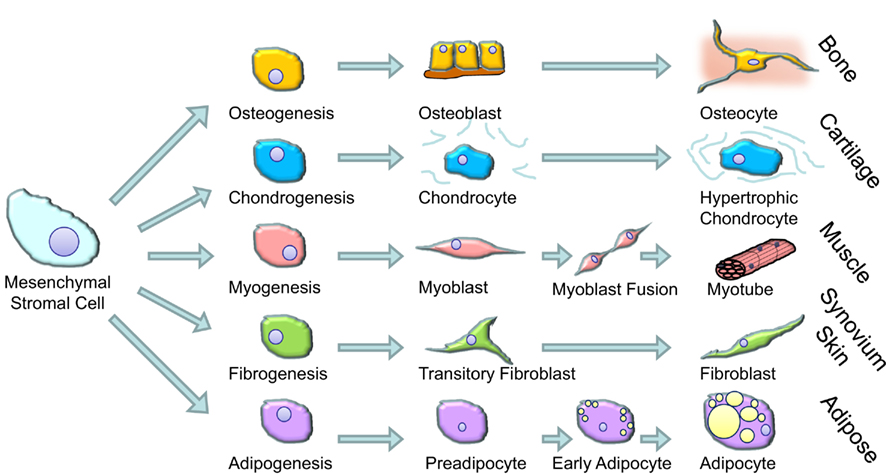
Figure 1. The cells and tissues that arise from bone marrow derived mesenchymal stromal cells.
Glucocorticoid-Induced Osteoporosis
The increased risk of fractures is frequently the most prominent problem associated with glucocorticoid excess. The scale of this problem is most clearly seen in patients who take oral glucocorticoids for the treatment of an underlying inflammatory disease (van Staa et al., 2000). The risk of hip fracture is approximately doubled in these individuals and the risk of hip fracture is increased up to fivefold in comparison to age matched individuals not treated with oral glucocorticoids. There is also evidence that subtle states of endogenous glucocorticoid overproduction, e.g., in the context of adrenal adenomas that produce “mild” increases in cortisol levels (subclinical hypercortisolemia) are associated with a substantial increase in fracture risk, particularly at the spine (Chiodini et al., 2009; Hardy and Cooper, 2010). The reason for the increase in fracture risk is probably due to effects on a variety of tissues. Glucocorticoid excess is associated with dramatic reductions in bone formation, an effect thought to be mediated primarily through direct actions of glucocorticoids on osteoblasts (Weinstein et al., 1998; Cooper, 2004). By contrast bone resorption is usually transiently increased leading to a degree of uncoupling of formation from resorption. The impairment of uncoupling is thought due to an increased production of RANKL and suppression of osteoprotegerin expression by osteoblasts (Hofbauer et al., 1999). Both of these actions would lead to continued stimulation of osteoclasts. Glucocorticoids also appear to reduce the quality of matrix produced by osteoblasts and increase the rate of osteocyte apoptosis (Weinstein et al., 1998; Lane et al., 2006). The combination of altered matrix production and “osteonecrosis” secondary to loss of osteocytes is likely to further reduce the resistance of bone to fracture.
Fracture risk with oral glucocorticoids appears to increase rapidly after starting glucocorticoids (van Staa et al., 2000). This rapid increase in risk occurs before significant amounts of bone tissue are lost suggesting the possible involvement of other mechanisms in this effect such as an increased falls risk. An obvious candidate for this is the effect of glucocorticoids on muscle. Glucocorticoid excess causes loss of muscle mass and weakness of muscle (myopathy) even when the effects on muscle mass are taken into account (Pereira and Freire de, 2011). This weakness appears to be especially apparent in the central/core muscles of the body resulting for example in difficulty rising from a chair. The impact of this muscle weakness on falls risk has not been evaluated formally but it is likely to be a major contributor to the increased risk of fracture in patients treated with glucocorticoids. The changes in fat distribution toward central fat accumulation would also be expected to alter an individual’s center of gravity and thus also contribute to a risk of falling. The effects of glucocorticoids on skin (thinning and easy bruising) would also negatively impact on the ability to withstand, and to resist damage, during a fall (Phillips and Weightman, 2007). As such glucocorticoid-induced osteoporosis and its consequences are likely to be dependent on effects on several tissues arising from MSCs.
The effects of glucocorticoids on MSCs and the cells that differentiate from MSC have been studied in vitro and in vivo. In general the effects of glucocorticoids have been broken down into the effects on the differentiation of MSCs to mature cells and the effects of glucocorticoids on mature, fully differentiated cells. The effects on MSC differentiation have primarily been explored in vitro whereas the effects on mature cells have been studied in vitro and in vivo. Limitations of these studies include the reliance on MSCs from a single source (usually bone marrow) for in vitro studies and the difficulty of examining fully differentiated cells in isolation in vivo since all tissues containing these cells are likely to contain some cells with MSC properties.
Effect of Glucocorticoids on the Differentiation of Mesenchymal Stromal Cells
Glucocorticoids are widely used in cell culture protocols to differentiate MSCs (e.g., those derived from bone marrow) into mature cells (Pittenger et al., 1999; outlined in Figure 2). Glucocorticoids increase the efficiency and yield of differentiated osteoblasts (Haynesworth et al., 1992; Shalhoub et al., 1992), myoblasts, adipocytes (Pittenger et al., 1999), and chondrocytes (Kato and Gospodarowicz, 1985; Mackay et al., 1998; Derfoul et al., 2006). The doses used in this setting is typically very high, e.g., 1 μM of dexamethasone (dexamethasone is approximately 25 times more potent than cortisol/hydrocortisone). How and why glucocorticoids are able to induce differentiation is not known. At a molecular level glucocorticoids bind to intracellular receptors found within the cytoplasm (glucocorticoid receptors, GR). When bound by GRs can migrate to the nucleus where they bind to specific sequences of DNA and influence gene transcription (Rhen and Cidlowski, 2005). Ligand bound GR can also influence gene transcription indirectly through interference with other signaling pathways either in the cytoplasm or the nucleus. This interference is particularly important for the anti-inflammatory effects of glucocorticoids which are primarily through interference with NF-κB and MAPK and AP-1 signaling pathways (Rhen and Cidlowski, 2005; Stahn and Buttgereit, 2008).
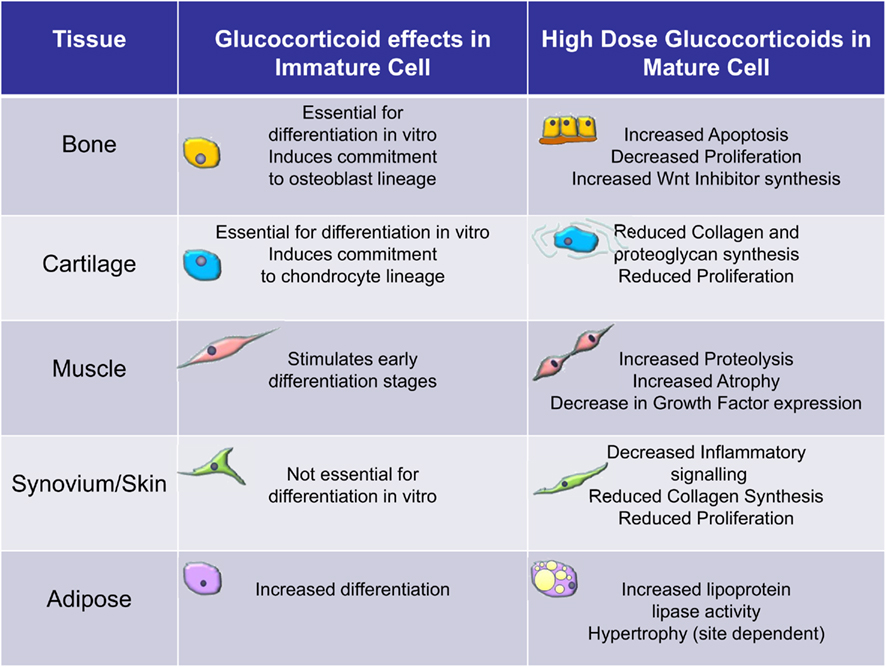
Figure 2. Overview of the effects of glucocorticoids on immature versus mature cells of the mesenchymal stromal cell lineage. These data are largely based on clinical observations and in vitro observations using bone marrow derived mesenchymal stromal cells.
The ability of glucocorticoids to stimulate differentiation appears to be generic to all cells that differentiate from bone marrow derived MSCs but the effect is more important to some cells than others. As an illustration of this it is generally considered that glucocorticoids are essential for the full differentiation of human MSCs to osteoblasts with the ability to mineralize matrix and the differentiation of chondrocytes (Eijken et al., 2005; Derfoul et al., 2006). It is also becoming clear that even within individual cell cultures, there may be differences in effect of glucocorticoids. A recent study explored the mechanisms underlying glucocorticoid stimulation of chondrocyte differentiation and found that selective apoptosis could be involved. Glucocorticoid treatment tended to favor the survival and proliferation of precursors with high potential for differentiation and proliferation whereas it induced apoptosis of cells with more limited proliferative capacity (Oshina et al., 2007). Whether such an effect occurs in vivo is unknown.
Effect of Glucocorticoids on Differentiated Cells Arising from Mesenchymal Stromal Cells
In contrast to the effects on immature cells glucocorticoids generally have a detrimental effect on mature cells that have differentiated from MSCs (Figure 2). In osteoblasts they can induce apoptosis and generally reduce the rate of proliferation (Weinstein et al., 1998; Rauch et al., 2010). At a molecular level these effects are thought to be due to alterations in various signaling pathways not least the wnt pathway (Yao et al., 2008). The canonical wnt signaling pathway is known to be anabolic for bone through a stimulation of bone formation. Glucocorticoids stimulate the expression of inhibitors of wnt signaling such as DKK1 and secreted frizzled related proteins (sFRPs; Wang et al., 2005, 2008). This results in reduced proliferation of osteoblasts. Glucocorticoids are thought to impact on muscle by increasing muscle proteolysis and by inducing atrophy of mature muscle cells. Signaling pathways affected by glucocorticoid excess include an inhibition of local growth factor production by myocytes and decreased expression of forkhead box class O transcription factors (FOXO) leading to increased expression of atrophy genes such as muscle RING-finger protein-1 (MuRf1) and atrogin-1/muscle atrophy F-box (MAFbx). Glucocorticoids impair the proliferation of growth plate chondrocytes (Chrysis et al., 2003). The downstream effectors of glucocorticoids are not known in this setting. However, in animals this effect appears partly reversible if glucocorticoid exposure is removed before closure of the growth plates at puberty (Schrier et al., 2006). However, in humans glucocorticoid treatment during childhood is associated with persistent decreases in final adult height (Lai et al., 2000). The mechanisms by which high levels of cortisol alter adipocytes are unclear. Changes in adipocyte function, e.g., lipoprotein lipase (LPL) activity, preadipocyte differentiation and survival, and altered adipokine secretion are all implicated (Paulsen et al., 2007). In Cushing’s syndrome, LPL activity is stimulated in retroperitoneal fat more than subcutaneous adipose tissue and this could explain the central fat accumulation (Burt et al., 2006). In human adipocytes cultured in vitro, glucocorticoids increase LPL mRNA expression and enzymatic activity (Fried et al., 1993). Pharmacological doses of glucocorticoids have been shown to stimulate lipolysis in vivo (Issekutz and Borkow, 1972; Tanaka et al., 1989). Glucocorticoids can also induce exaggerated adipocyte formation and hypertrophy (Rebuffe-Scrive et al., 1988; MacDougald and Lane, 1995). Fibroblasts are heterogeneous with phenotypic features often related to their tissue and site of origin (Parsonage et al., 2005). The effects of glucocorticoids on mature fibroblasts have been most extensively studied in skin (Schoepe et al., 2006). Dermal fibroblast proliferation and the ability to synthesize mature collagen are both substantially reduced by high doses of glucocorticoids (features thought to account for the clinical effects of glucocorticoids; Ponec et al., 1977). The molecular basis for these effects is unknown but likely to be complex given that fibroblasts isolated from different tissues can behave differently in response to the same treatment with glucocorticoids.
As illustrated in the examples above it appears that there is no single downstream mechanism by which glucocorticoids exert their negative actions on mature cells. This suggests that approaches to block actions of glucocorticoids downstream of the GR are unlikely to protect all tissues against the effects of glucocorticoid excess. Modulation of the effects of glucocorticoids on a broad range of cells and tissues that arise from MSCs would be more likely by the targeting of pre-receptor mechanisms as outlined below.
Role of Endogenous Glucocorticoids in Mesenchymal Stromal Cell Differentiation and Mature Cell Functioning in vivo
Although most attention has focused on the role of therapeutic glucocorticoids on MSC differentiation and function it is also likely that normal levels of endogenous glucocorticoids have an important role. Cortisol levels in the circulation are regulated by the hypothalamic-pituitary–adrenal (HPA) axis. Cortisol is secreted in a marked diurnal rhythm with high levels produced in the hours before waking and low levels being seen through the early part of the night. Cortisol levels are also raised dramatically in response to a range of stressors (Cooper and Stewart, 2003). As such glucocorticoid action at a tissue level could be regulated by changes in the level of cortisol within the circulation or by alteration of the sensitivity of the GR. Subtle changes in the HPA axis with a flattening out of the diurnal rhythm and a heightened nadir level of cortisol have been reported to occur with aging (Van Cauter et al., 1996; Hardy and Cooper, 2010). Correlations are also seen with peak cortisol responses on ACTH stimulation testing and the bone density and change in bone density over time at various sites (Reynolds et al., 2005). These changes correlate to some extent with bone loss and changes in body composition with age. However, the changes seen are not consistent between studies and results often apply only to one gender or the other or are discrepant between body regions. Further longitudinal studies are needed to address the role of differences in cortisol levels within the circulation in mediating changes in body composition.
Polymorphic variants of the GR which alter the sensitivity of the GR to cortisol have been described (Manenschijn et al., 2009). These are associated with subtle differences in body composition suggesting that subtle differences in glucocorticoid action at a tissue level could affect the development and/or health of muscle, bone, and fat tissue. These differences however appear subtle and only explain a small percentage of the variance in bone density (Huizenga et al., 1998).
Further insights into the role of the GR have come from experiments in which the GR has been deleted. Global deletion of the GR is not compatible with post-natal life due to a failure of lung-maturation. However, mice with global GR deletion appear to form bone, muscle, and adipose tissue indicating that there is no absolute requirement for GR signaling in the formation of these tissues (Rauch et al., 2010). More selective disruption of the GR has been used to dissect the role of GR signaling in various tissues. Deletion of the GR in osteoblasts results in mice with mildly reduced bone size but bone is otherwise relatively normal. Further understanding has come about through the study of mice with a mutation in the gene for the GR which prevents dimerization of the GR protein (Rauch et al., 2010). Dimerization is required for direct effects of GR on nuclear transcription but is not a requirement for the ability of GR to interfere with pro-inflammatory signaling pathways. Interestingly these dimerization deficient mice only have a mild bone phenotype, again with slightly reduced bone size. However the bone which is formed appears normal. Additionally, these animals remain sensitive to the effects of therapeutic glucocorticoids suggesting that the dominant pathway by which high doses of steroids affect bone is through interference with other signaling pathways. Overall, these data suggest that endogenous glucocorticoids have a mild anabolic action on bone size in vivo.
Similar GR deletion models have examined the role of endogenous glucocorticoids in muscle function. Targeted deletion of GR within muscle appears to have very little impact on muscle mass and function under normal circumstances. However, mice with GR deletion do not undergo muscle atrophy in response to stressful stimuli (Hu et al., 2009). These observations are supported by earlier work in which GR antagonists also provided protection against muscle atrophy in response to a range of stressors (Aboudrar et al., 1993; Tiao et al., 1996). This suggests that, at least under some circumstances, glucocorticoids are an essential endogenous regulator of muscle atrophy.
Findings from models in which chondrocytes, adipocytes, or fibroblasts have been targeted for GR gene deletion are not currently available but these should shed light on the importance of glucocorticoids in MSC differentiation and function in general.
More recently it has become appreciated that a major contributor to glucocorticoid action at a tissue level is the activity of glucocorticoid modifying enzymes. In particular the 11β-hydroxysteroid dehydrogenase enzymes (11β-HSD1) have been shown to regulate glucocorticoid action in a tissue specific fashion (Cooper and Stewart, 2009; Raza et al., 2010). These intracellular enzymes interconvert hormonally inactive glucocorticoids such as cortisone and dehydrocorticosterone with their active counterparts cortisol and corticosterone. They also metabolize the most commonly used oral glucocorticoids interchanging inactive prednisone with active prednisolone (Cooper et al., 2002). There are two 11β-HSD enzymes. The 11β-HSD1 is bidirectional but in vivo primarily activates glucocorticoids. 11β-HSD2 by contrast is a powerful inactivator of glucocorticoid and is found primarily in mineralocorticoid receptor (MR) expressing tissues. 11β-HSD2 inactivates cortisol to cortisone in these tissues which prevents cortisol from binding to the MR. This leaves aldosterone (which is not metabolized by these enzyme free to bind the MR; Stewart et al., 1987). 11β-HSD1 has been shown to be present in all cell types that arise from MSCs. Osteoblasts, myoblasts, adipocytes, chondrocytes, and fibroblasts all express the enzyme (Bujalska et al., 1997; Cooper et al., 2000; Hardy et al., 2006; Morgan et al., 2009). This suggests that these cells are able to regulate their exposure to glucocorticoids through modulating the metabolism of glucocorticoids within the cytoplasm before glucocorticoids have a chance to access the GR. Moreover the expression of 11β-HSD1 appears to be very dynamic across differentiation. In osteoblasts expression is relatively low in the earliest precursors but increases dramatically during differentiation before the activity falls back in mature cells (Eijken et al., 2005). In adipocytes 11β-HSD1 increases with differentiation and there is also a switch in the directionality of the enzyme favoring more glucocorticoid activation in mature adipocytes (Bujalska et al., 1999). 11β-HSD1 also increases dramatically across muscle differentiation (Morgan et al., 2009). The expression across differentiation has not been explored in fibroblasts or chondrocytes. In osteoblasts and adipocytes the functional importance of 11β-HSD1 expression has been demonstrated (Bujalska et al., 1999; Rabbitt et al., 2002). Differentiation of osteoblasts and adipocytes in the absence of glucocorticoids is impaired. However, this impairment can be corrected by the addition of inactive glucocorticoids. Addition of inactive glucocorticoids in the presence of an 11β-HSD1 inhibitor has no stimulatory effect on differentiation confirming the specificity of enzyme activity in these effects.
Although not expressed naturally in most cells of bone marrow derived MSC origin the 11β-HSD2 enzyme has been used to examine the effects of glucocorticoids on MSCs. Transgenic overexpression of 11β-HSD2 would be expected to confer resistance to glucocorticoid action. This has been demonstrated for osteoblasts in vitro (Rabbitt et al., 2002). Selective expression of 11β-HSD2 in osteoblasts in mice in vivo resulted in subtle abnormalities of bone structure and density (Sher et al., 2004, 2006). Bone density at the spine was reduced and the size of cortical bones was lower compared to wild type mice. These results again suggest that endogenous glucocorticoids have a mild anabolic effect on bone. Overexpression of 11β-HSD2 in osteoblasts also appeared to completely protect bone from the adverse effects of high doses of synthetic glucocorticoids (O’Brien et al., 2004). This data indicates that most of the effects of synthetic glucocorticoids on bone density and strength are mediated directly through effects on osteoblasts.
An interesting additional observation regarding 11β-HSD1 expression within stromal cells is the dramatic upregulation in activity that occurs in the presence of pro-inflammatory cytokines (Cooper et al., 2001; Tomlinson et al., 2001; Hardy et al., 2006, 2008; Kaur et al., 2010). The universal presence of this mechanism in cells arising from MSCs (in contrast to the lack of effect in lymphoid tissue or liver; Thieringer et al., 2001; Tomlinson et al., 2001) suggests that enhanced glucocorticoid action during stress is an important feature of the response of stromal cells to stress. How signals relating to differentiation are integrated with signals relating to stress and inflammation are currently unclear. A consequence of inflammation induced 11β-HSD1 expression is that cells expressing 11β-HSD1 in this setting will be disproportionately affected by endogenous and exogenous glucocorticoids. This effect could make patients with chronic inflammation, taking oral glucocorticoids, particularly at risk of the development of adverse effects in tissues arising from MSCs.
Newly Identified Mechanisms for Mesenchymal Stromal Cell Development in vivo
The vast majority of previous studies have focused on bone marrow derived MSCs as the model most likely to represent in vivo differentiation of cells of mesenchymal origin. It was also thought likely that these MSCs (or tissue resident cells with similar properties) were responsible for tissue repair, e.g., generation of osteoblasts as part of bone remodeling. However, a recent study has demonstrated that multipotent stem-like cells that are capable of forming bone and cartilage can be generated in vivo from vascular endothelial cells (Medici et al., 2010). This process of “endothelial to mesenchymal transition” appears to the be the mechanism by which abnormal bone is formed in fibrodysplasia ossificans progressiva (a condition characterized by the formation of heterotopic bone in muscle and soft tissues). It also appears to be operational in fracture repair. This new insight raises questions about the applicability to in vivo physiology of previous models that relied on bone marrow derived MSCs. Currently the extent to which endothelial to mesenchymal transition occurs in vivo and the sensitivity that these transiting cells have to glucocorticoids is unknown. Intriguingly, mature osteoblasts have been shown to regulate angiogenesis in bone through a glucocorticoid dependent pathway (Weinstein et al., 2010). This suggests that osteoblasts might be able to regulate bone formation through effects on the supply of new endothelial precursors. Vascular endothelial cells themselves are known to be very sensitive to glucocorticoids – endogenous glucocorticoid levels control vascular tone through effects on endothelial cells (Cooper and Stewart, 2003). Vascular endothelial cells also express high levels of 11β-HSD1 (Gong et al., 2008). This suggests that the adverse effects of high doses of glucocorticoids could be partly mediated through effects on the ability of these endothelial cells to transit into mesenchymal cells.
Implications for Therapy
Major advances have been made over the last decade in the prevention and protection against glucocorticoid-induced osteoporosis. Bisphosphonates have been proven to be an effective medication in maintaining bone density and reducing the risk of spine fractures in patients treated with oral glucocorticoids (Adachi et al., 1997, 2001; Cohen et al., 1999; Reid et al., 2009). Daily injection of the PTH analog teriparatide has been shown to be even more efficacious (Saag et al., 2007). However, the effectiveness of these medications in reducing non-vertebral fractures is still unclear. Additionally, these interventions do nothing to reverse the other changes in body composition brought about by glucocorticoid excess. The ideal treatment would strengthen bone whilst at the same time improving muscle strength, reducing falls and preventing the detrimental effects of glucocorticoids on skin, and adipose tissue redistribution. Given that the downstream pathways associated with the adverse effects of glucocorticoids vary considerably between tissues it is unlikely that any therapeutic approach to tackle these post-receptor mechanisms will address more than one component of Cushing’s syndrome. Attempts have been made over the last 50 years to generate glucocorticoids that specifically target the immune system without effects on body composition (Belvisi et al., 2001; Stahn and Buttgereit, 2008). However, despite considerable effort very little progress has been made in this area. The recent finding that bone tissue is affected by glucocorticoids through pathways normally associated with suppression of inflammation (Rauch et al., 2010) suggests that the previous view that the “good” effects (repression of pro-inflammatory pathways) could be dissected from the “bad” effects (stimulation of GR dimerization and direct regulation of nuclear gene transcription–translation) is overly simplistic. Additionally, some of the important anti-inflammatory effects of glucocorticoids appear to be through gene transactivation, e.g., the induction of dual specificity MAPK phosphatases (Abraham et al., 2006).
With this in mind a more fruitful approach would be the inhibition of pre-receptor glucocorticoid metabolism. Chemical inhibitors of the 11β-HSD1 enzyme have been developed (primarily to target obesity and insulin resistance in relation to endogenous glucocorticoids) and are now being used in clinical trials (Rosenstock et al., 2010). However, 11β-HSD1 appears to have an independent role in the immune system and global inhibition of enzyme activity could have detrimental consequences (Chapman et al., 2009; Cooper and Stewart, 2009). Additionally, a global approach would have no chance of dissecting out the positive and negative aspects of glucocorticoid action on MSCs. Currently the best hope for selectively protecting MSCs against the adverse effects of glucocorticoids would be to determine the molecular mechanisms underlying the induction of 11β-HSD1 during differentiation and during inflammation (Kaur et al., 2010). Selective inhibition of the exaggerated capacity of bone, muscle, adipose tissue and skin to generate glucocorticoids during chronic inflammation would be particularly attractive. The success of these approaches will of course depend on the nature of the specific pathways involved and the feasibility of inhibiting these systemically. The clinical application of these findings will also critically depend on the identification and characterization of the specific MSC subtypes involved in tissue repair in vivo.
Conclusion
Glucocorticoids appear to selectively impact on cells and tissues that arise from bone marrow derived MSCs in a negative way. This is in contrast to the positive, stimulatory effects that glucocorticoids have on immature cells. Therapeutic approaches to the problems caused by glucocorticoid excess will hopefully be able to address the adverse effects in tissues such as muscle and fat in addition to protecting bone tissue from bone loss and fracture. The most likely route to achieving this goal will be a focus on the regulation of pre-receptor glucocorticoid metabolism.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been funded by the Medical Research Council, UK, and Arthritis Research UK.
References
Aboudrar, S., Sempore, B., Koubi, H., Dechaud, H., and Desplanches, D. (1993). Effects of adrenalectomy or RU-486 on rat muscle fibers during hindlimb suspension. J. Appl. Physiol. 75, 2767–2773.
Abraham, S. M., Lawrence, T., Kleiman, A., Warden, P., Medghalchi, M., Tuckermann, J., Saklatvala, J., and Clark, A. R. (2006). Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 203, 1883–1889.
Adachi, J. D., Bensen, W. G., Brown, J., Hanley, D., Hodsman, A., Josse, R., Kendler, D. L., Lentle, B., Olszynski, W., Ste-Marie, L. G., Tenenhouse, A., and Chines, A. A. (1997). Intermittent etidronate therapy to prevent corticosteroid-induced osteoporosis. N. Engl. J. Med. 337, 382–387.
Adachi, J. D., Saag, K. G., Delmas, P. D., Liberman, U. A., Emkey, R. D., Seeman, E., Lane, N. E., Kaufman, J. M., Poubelle, P. E., Hawkins, F., Correa-Rotter, R., Menkes, C. J., Rodriguez-Portales, J. A., Schnitzer, T. J., Block, J. A., Wing, J., McIlwain, H. H., Westhovens, R., Brown, J., Melo-Gomes, J. A., Gruber, B. L., Yanover, M. J., Leite, M. O., Siminoski, K. G., Nevitt, M. C., Sharp, J. T., Malice, M. P., Dumortier, T., Czachur, M., Carofano, W., and Daifotis, A. (2001). Two-year effects of alendronate on bone mineral density and vertebral fracture in patients receiving glucocorticoids: a randomized, double-blind, placebo-controlled extension trial. Arthritis Rheum. 44, 202–211.
Allen, D. B., Mullen, M., and Mullen, B. (1994). A meta-analysis of the effect of oral and inhaled corticosteroids on growth. J. Allergy Clin. Immunol. 93, 967–976.
Belvisi, M. G., Wicks, S. L., Battram, C. H., Bottoms, S. E., Redford, J. E., Woodman, P., Brown, T. J., Webber, S. E., and Foster, M. L. (2001). Therapeutic benefit of a dissociated glucocorticoid and the relevance of in vitro separation of transrepression from transactivation activity. J. Immunol. 166, 1975–1982.
Bujalska, I. J., Kumar, S., Hewison, M., and Stewart, P. M. (1999). Differentiation of adipose stromal cells: the roles of glucocorticoids and 11β-hydroxysteroid dehydrogenase. Endocrinology 140, 3188–3196.
Bujalska, I. J., Kumar, S., and Stewart, P. M. (1997). Does central obesity reflect “Cushing’s disease of the omentum?” Lancet 349, 1210–1213.
Burt, M. G., Gibney, J., and Ho, K. K. (2006). Characterization of the metabolic phenotypes of Cushing’s syndrome and growth hormone deficiency: a study of body composition and energy metabolism. Clin. Endocrinol. (Oxf) 64, 436–443.
Chapman, K. E., Coutinho, A. E., Gray, M., Gilmour, J. S., Savill, J. S., and Seckl, J. R. (2009). The role and regulation of 11beta-hydroxysteroid dehydrogenase type 1 in the inflammatory response. Mol. Cell. Endocrinol. 301, 123–131.
Chiodini, I., Morelli, V., Masserini, B., Salcuni, A. S., Eller-Vainicher, C., Viti, R., Coletti, F., Guglielmi, G., Battista, C., Carnevale, V., Iorio, L., Beck-Peccoz, P., Arosio, M., Ambrosi, B., and Scillitani, A. (2009). Bone mineral density, prevalence of vertebral fractures, and bone quality in patients with adrenal incidentalomas with and without subclinical hypercortisolism: an Italian Italian multicenter study. J. Clin. Endocrinol. Metab. 94, 3207–3214.
Chrysis, D., Ritzen, E. M., and Savendahl, L. (2003). Growth retardation induced by dexamethasone is associated with increased apoptosis of the growth plate chondrocytes. J. Endocrinol. 176, 331–337.
Cohen, S., Levy, R. M., Keller, M., Boling, E., Emkey, R. D., Greenwald, M., Zizic, T. M., Wallach, S., Sewell, K. L., Lukert, B. P., Axelrod, D. W., and Chines, A. A. (1999). Risedronate therapy prevents corticosteroid-induced bone loss: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 42, 2309–2318.
Cooper, M. S., Bujalska, I., Rabbitt, E., Walker, E. A., Bland, R., Sheppard, M. C., Hewison, M., and Stewart, P. M. (2001). Modulation of 11β-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. J. Bone Miner. Res. 16, 1037–1044.
Cooper, M. S., Rabbitt, E. H., Goddard, P. E., Bartlett, W. A., Hewison, M., and Stewart, P. M. (2002). Osteoblastic 11β-hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. J. Bone Miner. Res. 17, 979–986.
Cooper, M. S., and Stewart, P. M. (2003). Corticosteroid insufficiency in acutely ill patients. N. Engl. J. Med. 348, 727–734.
Cooper, M. S., and Stewart, P. M. (2009). 11Beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. J. Clin. Endocrinol. Metab. 94, 4645–4654.
Cooper, M. S., Walker, E. A., Bland, R., Fraser, W. D., Hewison, M., and Stewart, P. M. (2000). Expression and functional consequences of 11β-hydroxysteroid dehydrogenase activity in human bone. Bone 27, 375–381.
Cushing, H. W. (1932). The basophil adenomas of the pituitary body and their clinical manifestations (pituitary basophilism). Bull. Johns Hopkins Hosp. 50, 137–195.
da Silva, M. L., Chagastelles, P. C., and Nardi, N. B. (2006). Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 119, 2204–2213.
Derfoul, A., Perkins, G. L., Hall, D. J., and Tuan, R. S. (2006). Glucocorticoids promote chondrogenic differentiation of adult human mesenchymal stem cells by enhancing expression of cartilage extracellular matrix genes. Stem Cells 24, 1487–1495.
Eijken, M., Hewison, M., Cooper, M. S., De Jong, F. H., Chiba, H., Stewart, P. M., Uitterlinden, A. G., Pols, H. A., and van Leeuwen, J. P. (2005). 11β-Hydroxysteroid dehydrogenase expression and glucocorticoid synthesis are directed by a molecular switch during osteoblast differentiation. Mol. Endocrinol. 19, 621–631.
Fernandez-Rodriguez, E., Stewart, P. M., and Cooper, M. S. (2009). The pituitary-adrenal axis and body composition. Pituitary 12, 105–115.
Fried, S. K., Russell, C. D., Grauso, N. L., and Brolin, R. E. (1993). Lipoprotein lipase regulation by insulin and glucocorticoid in subcutaneous and omental adipose tissues of obese women and men. J. Clin. Invest. 92, 2191–2198.
Gong, R., Morris, D. J., and Brem, A. S. (2008). Variable expression of 11beta Hydroxysteroid dehydrogenase (11beta-HSD) isoforms in vascular endothelial cells. Steroids 73, 1187–1196.
Hardy, R., Rabbitt, E. H., Filer, A., Emery, P., Hewison, M., Stewart, P. M., Gittoes, N. J., Buckley, C. D., Raza, K., and Cooper, M. S. (2008). Local and systemic glucocorticoid metabolism in inflammatory arthritis. Ann. Rheum. Dis. 67, 1204–1210.
Hardy, R. S., Filer, A., Cooper, M. S., Parsonage, G., Raza, K., Hardie, D. L., Rabbitt, E. H., Stewart, P. M., Buckley, C. D., and Hewison, M. (2006). Differential expression, function and response to inflammatory stimuli of 11β-hydroxysteroid dehydrogenase type 1 in human fibroblasts: a mechanism for tissue-specific regulation of inflammation. Arthritis Res. Ther. 8, R108.
Haynesworth, S. E., Goshima, J., Goldberg, V. M., and Caplan, A. I. (1992). Characterization of cells with osteogenic potential from human marrow. Bone 13, 81–88.
Hofbauer, L. C., Gori, F., Riggs, B. L., Lacey, D. L., Dunstan, C. R., Spelsberg, T. C., and Khosla, S. (1999). Stimulation of osteoprotegerin ligand and inhibition of osteoprotegerin production by glucocorticoids in human osteoblastic lineage cells: potential paracrine mechanisms of glucocorticoid-induced osteoporosis. Endocrinology 140, 4382–4389.
Hu, Z., Wang, H., Lee, I. H., Du, J., and Mitch, W. E. (2009). Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J. Clin. Invest. 119, 3059–3069.
Huizenga, N. A., Koper, J. W., De Lange, P., Pols, H. A., Stolk, R. P., Burger, H., Grobbee, D. E., Brinkmann, A. O., De Jong, F. H., and Lamberts, S. W. (1998). A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. J. Clin. Endocrinol. Metab. 83, 144–151.
Issekutz, B. Jr., and Borkow, I. (1972). Effect of catecholamines and dibutyryl-cyclic-AMP on glucose turnover, plasma free fatty acids, and insulin in dogs treated with methylprednisolone. Can. J. Physiol. Pharmacol. 50, 999–1006.
Kato, Y., and Gospodarowicz, D. (1985). Stimulation by glucocorticoid of the synthesis of cartilage-matrix proteoglycans produced by rabbit costal chondrocytes in vitro. J. Biol. Chem. 260, 2364–2373.
Kaur, K., Hardy, R., Ahasan, M. M., Eijken, M., van Leeuwen, J. P., Filer, A., Thomas, A. M., Raza, K., Buckley, C. D., Stewart, P. M., Rabbitt, E. H., Hewison, M., and Cooper, M. S. (2010). Synergistic induction of local glucocorticoid generation by inflammatory cytokines and glucocorticoids: implications for inflammation associated bone loss. Ann. Rheum. Dis. 69, 1185–1190.
Lai, H. C., FitzSimmons, S. C., Allen, D. B., Kosorok, M. R., Rosenstein, B. J., Campbell, P. W., and Farrell, P. M. (2000). Risk of persistent growth impairment after alternate-day prednisone treatment in children with cystic fibrosis. N. Engl. J. Med. 342, 851–859.
Lane, N. E., Yao, W., Balooch, M., Nalla, R. K., Balooch, G., Habelitz, S., Kinney, J. H., and Bonewald, L. F. (2006). Glucocorticoid-treated mice have localized changes in trabecular bone material properties and osteocyte lacunar size that are not observed in placebo-treated or estrogen-deficient mice. J. Bone Miner. Res. 21, 466–476.
MacDougald, O. A., and Lane, M. D. (1995). Transcriptional regulation of gene expression during adipocyte differentiation. Annu. Rev. Biochem. 64, 345–373.
Mackay, A. M., Beck, S. C., Murphy, J. M., Barry, F. P., Chichester, C. O., and Pittenger, M. F. (1998). Chondrogenic differentiation of cultured human mesenchymal stem cells from marrow. Tissue Eng. 4, 415–428.
Manenschijn, L., van den Akker, E. L., Lamberts, S. W., and van Rossum, E. F. (2009). Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann. N. Y. Acad. Sci. 1179, 179–198.
Medici, D., Shore, E. M., Lounev, V. Y., Kaplan, F. S., Kalluri, R., and Olsen, B. R. (2010). Conversion of vascular endothelial cells into multipotent stem-like cells. Nat. Med. 16, 1400–1406.
Morgan, S. A., Sherlock, M., Gathercole, L. L., Lavery, G. G., Lenaghan, C., Bujalska, I. J., Laber, D., Yu, A., Convey, G., Mayers, R., Hegyi, K., Sethi, J. K., Stewart, P. M., Smith, D. M., and Tomlinson, J. W. (2009). 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 58, 2506–2515.
O’Brien, C. A., Jia, D., Plotkin, L. I., Bellido, T., Powers, C. C., Stewart, S. A., Manolagas, S. C., and Weinstein, R. S. (2004). Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology 145, 1835–1841.
Oshina, H., Sotome, S., Yoshii, T., Torigoe, I., Sugata, Y., Maehara, H., Marukawa, E., Omura, K., and Shinomiya, K. (2007). Effects of continuous dexamethasone treatment on differentiation capabilities of bone marrow-derived mesenchymal cells. Bone 41, 575–583.
Parsonage, G., Filer, A. D., Haworth, O., Nash, G. B., Rainger, G. E., Salmon, M., and Buckley, C. D. (2005). A stromal address code defined by fibroblasts. Trends Immunol. 26, 150–156.
Paulsen, S. K., Pedersen, S. B., Fisker, S., and Richelsen, B. (2007). 11Beta-HSD type 1 expression in human adipose tissue: impact of gender, obesity, and fat localization. Obesity (Silver Spring) 15, 1954–1960.
Pereira, R. M., and Freire de, C. J. (2011). Glucocorticoid-induced myopathy. Joint Bone Spine 78, 41–44.
Phillips, P. J., and Weightman, W. (2007). Skin and Cushing syndrome. Aust. Fam. Physician 36, 545–547.
Pittenger, M. F., Mackay, A. M., Beck, S. C., Jaiswal, R. K., Douglas, R., Mosca, J. D., Moorman, M. A., Simonetti, D. W., Craig, S., and Marshak, D. R. (1999). Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147.
Ponec, M., de, H. C., Bachra, B. N., and Polano, M. K. (1977). Effects of glucocorticosteroids on primary human skin fibroblasts. I. Inhibition of the proliferation of cultured primary human skin and mouse L929 fibroblasts. Arch. Dermatol. Res. 259, 117–123.
Rabbitt, E., Lavery, G. G., Walker, E. A., Cooper, M. S., Stewart, P. M., and Hewison, M. (2002). Pre-receptor regulation of glucocorticoid action by 11β-hydroxysteroid dehydrogenase: a novel determinant of cell proliferation. FASEB J. 16, 36–44.
Rauch, A., Seitz, S., Baschant, U., Schilling, A. F., Illing, A., Stride, B., Kirilov, M., Mandic, V., Takacz, A., Schmidt-Ullrich, R., Ostermay, S., Schinke, T., Spanbroek, R., Zaiss, M. M., Angel, P. E., Lerner, U. H., David, J. P., Reichardt, H. M., Amling, M., Schutz, G., and Tuckermann, J. P. (2010). Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 11, 517–531.
Raza, K., Hardy, R., and Cooper, M. S. (2010). The 11beta-hydroxysteroid dehydrogenase enzymes – arbiters of the effects of glucocorticoids in synovium and bone. Rheumatology (Oxford) 49, 2016–2023.
Rebuffe-Scrive, M., Krotkiewski, M., Elfverson, J., and Bjorntorp, P. (1988). Muscle and adipose tissue morphology and metabolism in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 67, 1122–1128.
Reid, D. M., Devogelaer, J. P., Saag, K., Roux, C., Lau, C. S., Reginster, J. Y., Papanastasiou, P., Ferreira, A., Hartl, F., Fashola, T., Mesenbrink, P., and Sambrook, P. N. (2009). Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 373, 1253–1263.
Reynolds, R. M., Dennison, E. M., Walker, B. R., Syddall, H. E., Wood, P. J., Andrew, R., Phillips, D. I., and Cooper, C. (2005). Cortisol secretion and rate of bone loss in a population-based cohort of elderly men and women. Calcif. Tissue Int. 77, 134–138.
Rhen, T., and Cidlowski, J. A. (2005). Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N. Engl. J. Med. 353, 1711–1723.
Rosenstock, J., Banarer, S., Fonseca, V. A., Inzucchi, S. E., Sun, W., Yao, W., Hollis, G., Flores, R., Levy, R., Williams, W. V., Seckl, J. R., and Huber, R. (2010). The 11-beta-hydroxysteroid dehydrogenase type 1 inhibitor INCB13739 improves hyperglycemia in patients with type 2 diabetes inadequately controlled by metformin monotherapy. Diabetes Care 33, 1516–1522.
Saag, K. G., Shane, E., Boonen, S., Marin, F., Donley, D. W., Taylor, K. A., Dalsky, G. P., and Marcus, R. (2007). Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N. Engl. J. Med. 357, 2028–2039.
Schoepe, S., Schacke, H., May, E., and Asadullah, K. (2006). Glucocorticoid therapy-induced skin atrophy. Exp. Dermatol. 15, 406–420.
Schrier, L., Ferns, S. P., Barnes, K. M., Emons, J. A., Newman, E. I., Nilsson, O., and Baron, J. (2006). Depletion of resting zone chondrocytes during growth plate senescence. J. Endocrinol. 189, 27–36.
Shalhoub, V., Conlon, D., Tassinari, M., Quinn, C., Partridge, N., Stein, G. S., and Lian, J. B. (1992). Glucocorticoids promote development of the osteoblast phenotype by selectively modulating expression of cell growth and differentiation associated genes. J. Cell. Biochem. 50, 425–440.
Sher, L. B., Harrison, J. R., Adams, D. J., and Kream, B. E. (2006). Impaired cortical bone acquisition and osteoblast differentiation in mice with osteoblast-targeted disruption of glucocorticoid signaling. Calcif. Tissue Int. 79, 118–125.
Sher, L. B., Woitge, H. W., Adams, D. J., Gronowicz, G. A., Krozowski, Z., Harrison, J. R., and Kream, B. E. (2004). Transgenic expression of 11β-hydroxysteroid dehydrogenase type 2 in osteoblasts reveals an anabolic role for endogenous glucocorticoids in bone. Endocrinology 145, 922–929.
Stahn, C., and Buttgereit, F. (2008). Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 4, 525–533.
Stewart, P. M., Wallace, A. M., Valentino, R., Burt, D., Shackleton, C. H., and Edwards, C. R. (1987). Mineralocorticoid activity of liquorice: 11β-hydroxysteroid dehydrogenase deficiency comes of age. Lancet 2, 821–824.
Tanaka, H., Ichikawa, Y., Akama, H., and Homma, M. (1989). In vivo responsiveness to glucocorticoid correlated with glucocorticoid receptor content in peripheral blood leukocytes in normal humans. Acta Endocrinol. 121, 470–476.
Thieringer, R., Le Grand, C. B., Carbin, L., Cai, T. Q., Wong, B., Wright, S. D., and Hermanowski-Vosatka, A. (2001). 11 Beta-hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J. Immunol. 167, 30–35.
Tiao, G., Fagan, J., Roegner, V., Lieberman, M., Wang, J. J., Fischer, J. E., and Hasselgren, P. O. (1996). Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J. Clin. Invest. 97, 339–348.
Tomlinson, J. W., Moore, J., Cooper, M. S., Bujalska, I., Shahmanesh, M., Burt, C., Strain, A., Hewison, M., and Stewart, P. M. (2001). Regulation of expression of 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue: tissue-specific induction by cytokines. Endocrinology 142, 1982–1989.
Van Cauter, E., Leproult, R., and Kupfer, D. J. (1996). Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J. Clin. Endocrinol. Metab. 81, 2468–2473.
van Staa, T. P., Leufkens, H. G., Abenhaim, L., Zhang, B., and Cooper, C. (2000). Use of oral corticosteroids and risk of fractures. J. Bone Miner. Res. 15, 993–1000.
Wang, F. S., Ko, J. Y., Yeh, D. W., Ke, H. C., and Wu, H. L. (2008). Modulation of Dickkopf-1 attenuates glucocorticoid induction of osteoblast apoptosis, adipocytic differentiation, and bone mass loss. Endocrinology 149, 1793–1801.
Wang, F. S., Lin, C. L., Chen, Y. J., Wang, C. J., Yang, K. D., Huang, Y. T., Sun, Y. C., and Huang, H. C. (2005). Secreted frizzled-related protein 1 modulates glucocorticoid attenuation of osteogenic activities and bone mass. Endocrinology 146, 2415–2423.
Weinstein, R. S., Jilka, R. L., Parfitt, A. M., and Manolagas, S. C. (1998). Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Invest. 102, 274–282.
Weinstein, R. S., Wan, C., Liu, Q., Wang, Y., Almeida, M., O’Brien, C. A., Thostenson, J., Roberson, P. K., Boskey, A. L., Clemens, T. L., and Manolagas, S. C. (2010). Endogenous glucocorticoids decrease skeletal angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell 9, 147–161.
Yao, W., Cheng, Z., Pham, A., Busse, C., Zimmermann, E. A., Ritchie, R. O., and Lane, N. E. (2008). Glucocorticoid-induced bone loss in mice can be reversed by the actions of parathyroid hormone and risedronate on different pathways for bone formation and mineralization. Arthritis Rheum. 58, 3485–3497.
Keywords: glucocorticoids, osteoporosis, stromal, mesenchymal
Citation: Hardy R and Cooper MS (2011) Glucocorticoid-induced osteoporosis – a disorder of mesenchymal stromal cells? Front. Endocrin. 2:24. doi: 10.3389/fendo.2011.00024
Received: 10 June 2011; Paper pending published: 03 July 2011;
Accepted: 06 August 2011; Published online: 27 August 2011.
Edited by:
William Duncan Fraser, University of Liverpool/Royal Liverpool University Hospital, UKReviewed by:
Claire Elizabeth Clarkin, Kings College London, UKJan Josef Stepan, Charles University Faculty of Medicine Prague, Czech Republic
Copyright: © 2011 Hardy and Cooper. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Mark S. Cooper, The Institute of Biomedical Research, The Medical School, University of Birmingham, Birmingham, B15 2TH, UK. e-mail:bS5zLmNvb3BlckBiaGFtLmFjLnVr