 Christine Bellanné-Chantelot1,2
Christine Bellanné-Chantelot1,2 Matthieu Mosca1,3,4
Matthieu Mosca1,3,4 Caroline Marty1,3,4
Caroline Marty1,3,4 Rémi Favier1,5
Rémi Favier1,5 William Vainchenker1,3,4
William Vainchenker1,3,4 Isabelle Plo1,3,4*
Isabelle Plo1,3,4*
- 1INSERM UMR1170, Gustave Roussy, Villejuif, France
- 2Department of Genetics, Assistance Publique-Hôpitaux de Paris (AP-HP) Hôpitaux Universitaires Pitié Salpêtrière—Charles Foix, UPMC Univ Paris 06, Paris, France
- 3Université Paris-Saclay, UMR1170, Gustave Roussy, Villejuif, France
- 4Gustave Roussy, UMR1170, Villejuif, France
- 5Assistance Publique-Hôpitaux de Paris (AP-HP), Service d’Hématologie biologique, Centre de Référence des Pathologies Plaquettaires (CRPP), Hôpital Armand Trousseau, Paris, France
The molecular basis of hereditary thrombocytosis is germline mutations affecting the thrombopoietin (TPO)/TPO receptor (MPL)/JAK2 signaling axis. Here, we report one family presenting two cases with a mild thrombocytosis. By sequencing JAK2 and MPL coding exons, we identified a germline MPL R102P heterozygous mutation in the proband and his daughter. Concomitantly, we detected high TPO levels in the serum of these two patients. The mutation was not found in three other unaffected cases from the family except in another proband’s daughter who did not present thrombocytosis but had a high TPO level. The MPL R102P mutation was first described in congenital amegakaryocytic thrombocytopenia in a homozygous state with a loss-of-function activity. It was previously shown that MPL R102P was blocked in the endoplasmic reticulum without being able to translocate to the plasma membrane. Thus, this case report identifies for the first time that MPL R102P mutation can differently impact megakaryopoiesis: thrombocytosis or thrombocytopenia depending on the presence of the heterozygous or homozygous state, respectively. The paradoxical effect associated with heterozygous MPL R102P may be due to subnormal cell-surface expression of wild-type MPL in platelets inducing a defective TPO clearance. As a consequence, increased TPO levels may activate megakaryocyte progenitors that express a lower, but still sufficient level of MPL for the induction of proliferation.
Background
Hereditary thrombocytosis is characterized by an increase in platelets above 450 × 109/L due to germline genetic abnormalities. These mutations affect the thrombopoietin (TPO)/TPO receptor (MPL)/JAK2 axis that is essential for megakaryopoiesis. First, heterozygous mutations in the THPO gene located in the 5′-untranslated region or in splice donor sites lead to an increased mRNA translation and synthesis of TPO (1–3). Second, several heterozygous JAK2 mutations were discovered that are located not only in the pseudokinase but also in the kinase domains such as JAK2 H608N, JAK2 R564Q, JAK2 S755R, JAK2 R938Q, and JAK2 R867Q (4–7). These mutants harbor low constitutive kinase activity that is dependent on the presence of MPL (5). Third, MPL mutations were identified affecting different residues of the receptor and involving different mechanisms. The heterozygous gain-of-function mutation MPL S505N was identified in the transmembrane domain of MPL and was found to induce its dimerization and activation (8). Interestingly, loss-of-function mutations have also been identified including the homozygous MPL P106L in Saoudian and Koweitian families (9, 10). Studies have shown that MPL P106L presents a defective trafficking to the cell membrane leading to a decrease in TPO clearance by platelets, which results in high circulating TPO levels that induce a proliferative response to TPO in immature cells (9, 10). The MPL K39N polymorphism was also discovered in 7% of African Americans and homozygous cases present a thrombocytosis associated with an abnormal MPL maturation (11).
Case Presentation
We have identified two members of a family displaying mild thrombocytosis diagnosed at an early age (21 and 41 years old) with platelet counts around 600 × 109/L. Four other members of the family were included in the study and the pedigree was registered in the National collection of myeloproliferative neoplasms (MPNs) (DC 2009-957). All participants gave their written informed consent in accordance to the Declaration of Helsinki and the study approved by Ethical committee of Ile de France (no. IDRCB 2008-A00658-47). Clinical and biological annotations were recorded in an Access database approved by the French computer commission (CNIL #815419).
All exons of MPL and JAK2 genes were sequenced in the proband’s DNA extracted from total blood cells (indicated by an arrow in Figure 1) using the BigDye Terminator chemistry (Life Technologies) on an ABI3730 Genetic Analyser (Applied Biosystem). We found a heterozygous MPL R102P mutation that had been previously reported in congenital amegakaryocytic thrombocytopenia (CAMT) but was previously found to be in a homozygous or compound heterozygous state leading to a complete defect in MPL function (12–14). This MPL R102P mutation was also identified in the other affected case demonstrating that this event was germline. TPO levels were measured by ELISA in the two affected subjects and were found elevated (119 and 260 pg/mL) compared to the normal range (<30 pg/mL). The mutation was not found in the other non-affected members of the family except in the second 23-year-old proband’s daughter. This case had a normal but rather high level of 397 × 109/L platelets and a high TPO level of 131 pg/mL. However, this asymptomatic carrier has not reached the father’s age at diagnosis and thus will be carefully followed in clinics. These results suggest an incomplete penetrance of the MPL mutation depending on age.
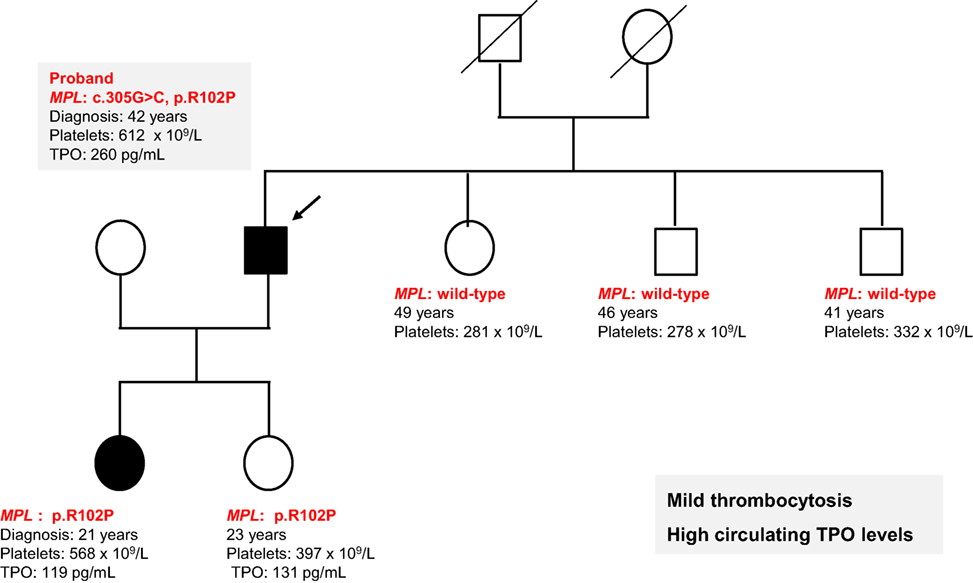
Figure 1. Heterozygous MPL R102P mutation induces a mild thrombocytosis with high thrombopoietin (TPO) levels. Pedigree from a family harboring a mild thrombocytosis with platelet counts around 600 × 109/L. The proband indicated by an arrow was diagnosed at 42 years old and one of her daughter was 21 years old. MPL was sequenced and a heterozygous MPL R102P mutation was identified. TPO levels were found elevated 119 and 260 pg/mL. Another daughter presented a high TPO level 131 pg/mL but without disease.
Discussion
MPL is a homodimeric type 1 receptor that controls megakaryopoiesis after TPO binding. It is expressed at high levels in hematopoietic stem cells, megakaryocytic (MK) progenitors, megakaryocytes, and platelets, while the TPO levels are controlled in large part by the platelet mass due to internalization of TPO/MPL/JAK2 complexes.
Many mutations affecting MPL induce deregulation of megakaryopoiesis. Although some are gain-of-function, such as MPL W515K/L and S505N, leading to MPNs or hereditary thrombocytosis (8, 15), others such as MPL R102P and F104S are loss-of-function leading to CAMT (12–14). The MPL R102P receptor function was previously investigated and studies have shown that it is not addressed to the cell-surface and induces no proliferation of Ba/F3 cell lines (14). This loss of function was caused by the presence of an abnormal glycosylated receptor resulting from its blockade in the endoplasmic reticulum (ER) (12–14). In CAMT, MPL mutations are recessive events (homozygous or compound heterozygous state) generally located throughout the MPL gene, implying that no functional MPL receptors are present at the cell surface.
This case report identifies for the first time that MPL R102P mutation can differently impact megakaryopoiesis depending on heterozygous and homozygous states (Figure 2). Indeed, based on previous studies showing that MPL R102P remains in the ER, we propose that in the heterozygous state, only the wild-type MPL form is addressed to the cell surface leading to subnormal expression in platelets and a consequent deficit in TPO clearance. Therefore, heterozygous MPL R102P results in an increased TPO levels in the serum, which is able to induce the proliferation of MK progenitors expressing sufficient MPL. Altogether this phenomenon leads to thrombocytosis by a decreased TPO clearance, as has also been demonstrated in transgenic mice with a low expression of MPL in megakaryocytes and platelets (16, 17).
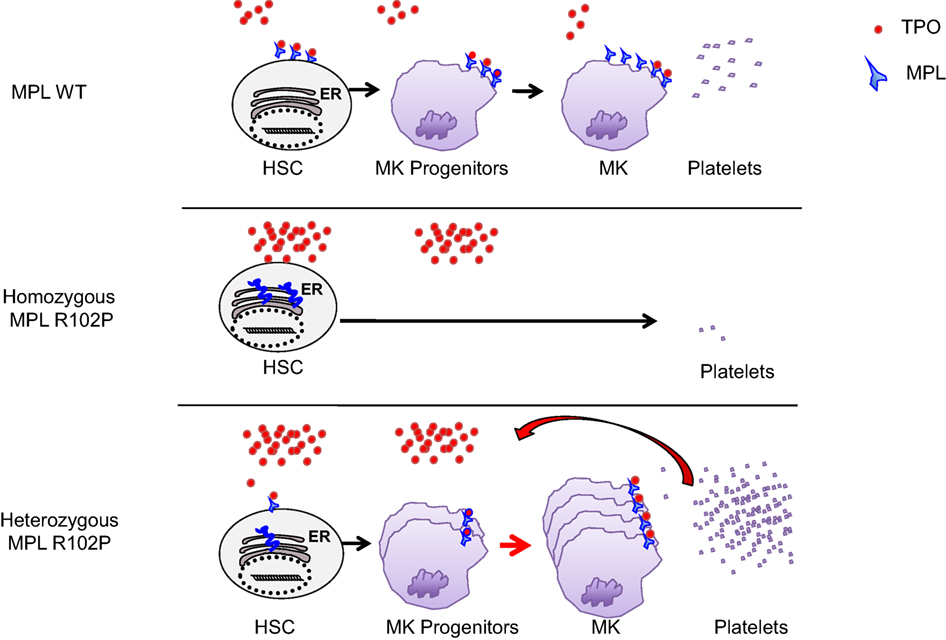
Figure 2. Model of MPL R102P function. In wild-type, MPL is increasingly upregulated in megakaryocytic progenitors (MK progenitors) and platelets. The level of thrombopoietin (TPO) is controlled by the platelet mass due to the internalization and degradation of TPO/MPL/JAK2 complexes. In homozygous MPL R102P situation, MPL cannot signal to induce megakaryopoiesis and platelets production even if the TPO level is high since it is blocked in the endoplasmic reticulum (ER). In MPL R102P heterozygous condition, only wild-type MPL is expressed at the cell surface leading to subnormal expression in platelets and defective TPO clearance. Therefore, the TPO levels increased and TPO is able to favor the proliferation of MK progenitors through the wild-type MPL, leading to a thrombocytosis.
Concluding Remarks
We identified one family of hereditary thrombocytosis with three members harboring a germline MPL R102P heterozygous mutation with high TPO levels in the serum but with incomplete penetrance of the phenotype. These results contrast with the presence of the same MPL R102P mutation at homozygous state in CAMT.
Ethics Statement
Four other members of the family were included in the study, and the pedigree was registered in the National collection of myeloproliferative neoplasms (MPNs) (DC 2009-957). All participants gave their written informed consent in accordance to the Declaration of Helsinki and the study approved by Ethical committee of Ile de France (No. IDRCB 2008-A00658-47). Clinical and biological annotations were recorded in an Access database approved by the French computer commission (CNIL #815419).
Author Contributions
C-BC, IP, and WV designed the work and wrote the manuscript. MM and RF performed experiments. CM performed experiments and contributed to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from l’Agence Nationale de la Recherche (ANR-13-JVSV1-GERMPN-01), the Laurette Fugain foundation, the GIS-Institute for rare diseases for high throughput sequencing (AO9102LS), the Association de recherche sur la moelle osseuse (ARMO), the Association pour la Recherche contre le Cancer (ARC) (Fondation ARC libre 2012), and the regional PHRC AOR07014. The program “Investissements d’avenir” is founding the Labex GR-Ex (IP, WV). MM is a recipient from La ligue Nationale contre le cancer.
References
1. Ghilardi N, Skoda RC. A single-base deletion in the thrombopoietin (TPO) gene causes familial essential thrombocythemia through a mechanism of more efficient translation of TPO mRNA. Blood (1999) 94:1480–2.
2. Kondo T, Okabe M, Sanada M, Kurosawa M, Suzuki S, Kobayashi M, et al. Familial essential thrombocythemia associated with one-base deletion in the 5’-untranslated region of the thrombopoietin gene. Blood (1998) 92:1091–6.
3. Wiestner A, Schlemper RJ, van der Maas AP, Skoda RC. An activating splice donor mutation in the thrombopoietin gene causes hereditary thrombocythaemia. Nat Genet (1998) 18:49–52. doi:10.1038/ng0198-49
4. Etheridge SL, Cosgrove ME, Sangkhae V, Corbo LM, Roh ME, Seeliger MA, et al. A novel activating, germline JAK2 mutation, JAK2R564Q, causes familial essential thrombocytosis. Blood (2014) 123:1059–68. doi:10.1182/blood-2012-12-473777
5. Marty C, Saint-Martin C, Pecquet C, Grosjean S, Saliba J, Mouton C, et al. Germ-line JAK2 mutations in the kinase domain are responsible for hereditary thrombocytosis and are resistant to JAK2 and HSP90 inhibitors. Blood (2014) 123:1372–83. doi:10.1182/blood-2013-05-504555
6. Mead AJ, Rugless MJ, Jacobsen SE, Schuh A. Germline JAK2 mutation in a family with hereditary thrombocytosis. N Engl J Med (2012) 366:967–9. doi:10.1056/NEJMc1200349
7. Rumi E, Harutyunyan AS, Casetti I, Pietra D, Nivarthi H, Moriggl R, et al. A novel germline JAK2 mutation in familial myeloproliferative neoplasms. Am J Hematol (2014) 89:117–8. doi:10.1002/ajh.23614
8. Ding J, Komatsu H, Wakita A, Kato-Uranishi M, Ito M, Satoh A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-MPL gene, which encodes for the receptor for thrombopoietin. Blood (2004) 103:4198–200. doi:10.1182/blood-2003-10-3471
9. El-Harith el HA, Roesl C, Ballmaier M, Germeshausen M, Frye-Boukhriss H, von Neuhoff N, et al. Familial thrombocytosis caused by the novel germ-line mutation p.Pro106Leu in the MPL gene. Br J Haematol (2009) 144:185–94. doi:10.1111/j.1365-2141.2008.07430.x
10. Favale F, Messaoudi K, Varghese LN, Boukour S, Pecquet C, Gryshkova V, et al. An incomplete trafficking defect to the cell-surface leads to paradoxical thrombocytosis for human and murine MPL P106L. Blood (2016) 128:3146–58. doi:10.1182/blood-2016-06-722058
11. Moliterno AR, Williams DM, Gutierrez-Alamillo LI, Salvatori R, Ingersoll RG, Spivak JL. Mpl Baltimore: a thrombopoietin receptor polymorphism associated with thrombocytosis. Proc Natl Acad Sci U S A (2004) 101:11444–7. doi:10.1073/pnas.0404241101
12. Tijssen MR, di Summa F, van den Oudenrijn S, Zwaginga JJ, van der Schoot CE, Voermans C, et al. Functional analysis of single amino-acid mutations in the thrombopoietin-receptor Mpl underlying congenital amegakaryocytic thrombocytopenia. Br J Haematol (2008) 141:808–13. doi:10.1111/j.1365-2141.2008.07139.x
13. van den Oudenrijn S, Bruin M, Folman CC, Peters M, Faulkner LB, de Haas M, et al. Mutations in the thrombopoietin receptor, Mpl, in children with congenital amegakaryocytic thrombocytopenia. Br J Haematol (2000) 110:441–8. doi:10.1046/j.1365-2141.2000.02175.x
14. Varghese LN, Zhang JG, Young SN, Willson TA, Alexander WS, Nicola NA, et al. Functional characterization of c-Mpl ectodomain mutations that underlie congenital amegakaryocytic thrombocytopenia. Growth Factors (2014) 32:18–26. doi:10.3109/08977194.2013.874347
15. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med (2006) 3:e270. doi:10.1371/journal.pmed.0030270
16. Lannutti BJ, Epp A, Roy J, Chen J, Josephson NC. Incomplete restoration of Mpl expression in the mpl-/- mouse produces partial correction of the stem cell-repopulating defect and paradoxical thrombocytosis. Blood (2009) 113:1778–85. doi:10.1182/blood-2007-11-124859
Keywords: thrombopoietin, JAK2, MPL R102P, thrombocytosis, thrombocytopenia, CAMT
Citation: Bellanné-Chantelot C, Mosca M, Marty C, Favier R, Vainchenker W and Plo I (2017) Identification of MPL R102P Mutation in Hereditary Thrombocytosis. Front. Endocrinol. 8:235. doi: 10.3389/fendo.2017.00235
Received: 09 August 2017; Accepted: 28 August 2017;
Published: 20 September 2017
Edited by:
Pierre De Meyts, de Duve Institute, BelgiumReviewed by:
Jean-Baptiste Demoulin, Catholic University of Louvain, BelgiumJames M. Murphy, Walter and Eliza Hall Institute of Medical Research, Australia
Copyright: © 2017 Bellanné-Chantelot, Mosca, Marty, Favier, Vainchenker and Plo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Isabelle Plo, isabelle.plo@gustaveroussy.fr