 Benjamin Chevalier1,2†
Benjamin Chevalier1,2† Hippolyte Dupuis1†
Hippolyte Dupuis1† Arnaud Jannin1,2Madleen Lemaitre1,2
Arnaud Jannin1,2Madleen Lemaitre1,2 Christine Do Cao1Catherine Cardot-Bauters1Stéphanie Espiard1,2,3Marie Christine Vantyghem1,2,3*
Christine Do Cao1Catherine Cardot-Bauters1Stéphanie Espiard1,2,3Marie Christine Vantyghem1,2,3*- 1Department of Endocrinology, Diabetology and Metabolism, Lille University Hospital, Lille, France
- 2University of Lille, Lille, France
- 3INSERM U1190, European Genomic Institute for Diabetes, Lille, France
Phakomatoses encompass a group of rare genetic diseases, such as von Hippel-Lindau syndrome (VHL), neurofibromatosis type 1 (NF1), tuberous sclerosis complex (TSC) and Cowden syndrome (CS). These disorders are due to molecular abnormalities on the RAS-PI3K-Akt-mTOR pathway for NF1, TSC and CS, and to hypoxia sensing for VHL. Phakomatoses share some phenotypic traits such as neurological, ophthalmological and cutaneous features. Patients with these diseases are also predisposed to developing multiple endocrine tissue tumors, e.g., pheochromocytomas/paragangliomas are frequent in VHL and NF1. All forms of phakomatoses except CS may be associated with digestive neuroendocrine tumors. More rarely, thyroid cancer and pituitary or parathyroid adenomas have been reported. These susceptibilities are noteworthy, because their occurrence rate, prognosis and management differ slightly from the sporadic forms. The aim of this review is to summarize current knowledge on endocrine glands tumors associated with VHL, NF1, TSC, and CS, especially neuroendocrine tumors and pheochromocytomas/paragangliomas. We particularly detail recent advances concerning prognosis and management, especially parenchyma-sparing surgery and medical targeted therapies such as mTOR, MEK and HIF-2 α inhibitors, which have shown truly encouraging results.
Introduction
Phakomatoses are a group of systemic diseases linked to ectodermal dysembryogenesis. The term comes from the Greek noun phakos (φακός, meaning “lentil” or “spot”) and the word termination -oma (for “tumor”), which refer to cutaneous birthmarks, i.e. hamartomas. Other cardinal features involve the central nervous system and the eyes (1). The most frequent forms of phakomatoses are:
- neurofibromatosis type 1 (NF1), also known as von Recklinghausen’s disease,
- von Hippel-Lindau disease (VHL),
- tuberous systemic complex (TSC),
- and Cowden syndrome (CS).
The prevalence is, however, less than 1/2,000 people. These rare diseases were clinically described in the 19th century by famous physicians such as the pathologist Friedrich Daniel von Recklinghausen, and the ophthalmologist Eugen von Hippel, both from Germany, Arvid Lindau, a Swedish pathologist and Désiré-Magloire Bourneville, a French neurologist. Although the hereditary nature of these syndromes was predicted early on, their molecular basis was only elucidated at the end of the 20th century by means of genetic developments and the characterization of the Ras-PI3K-Akt-mTOR pathway in NF1, TSC, and CS, as well as of the hypoxia signaling pathway in VHL.
Besides classical neurological, ophthalmological and cutaneous features, patients with phakomatoses are predisposed to developing tumors of the endocrine glands, with different spectrums for each disease (Figure 1).
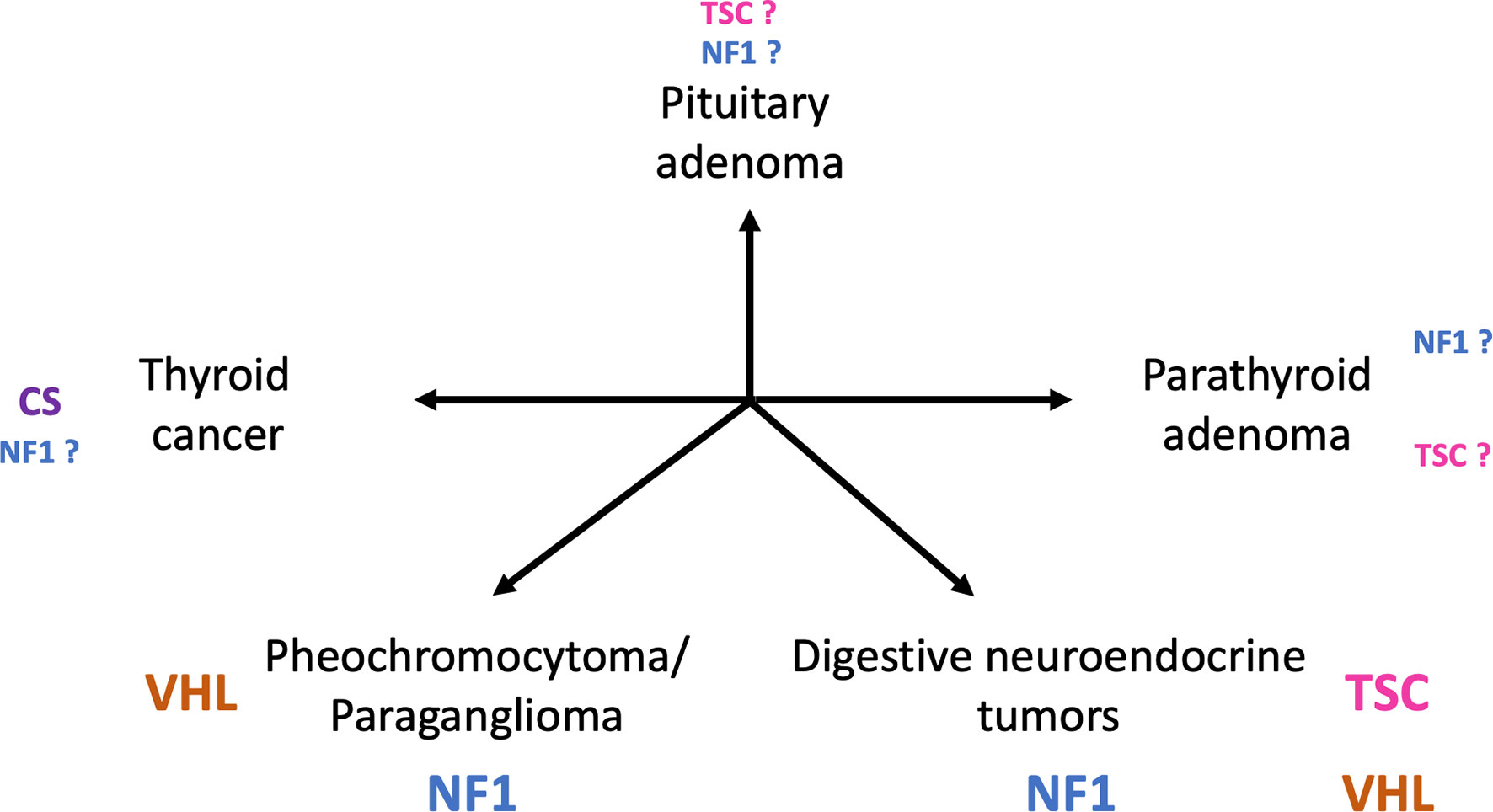
Figure 1 Main tumors of endocrine glands in patients diagnosed with phakomatoses. (TSC, tuberous sclerosis complex; VHL, von Hippel-Lindau disease; NF1, neurofibromatosis type 1; CS, Cowden syndrome).
After a brief review of the pathophysiology of common forms of phakomatoses, we describe the clinical features of endocrine tumors associated with them and focus on their specific features compared with sporadic counterparts; indeed, time of occurrence, clinical expression and prognosis can differ slightly. We then detail recent advances concerning the management of those tumors, especially focusing on parenchyma-sparing surgery in localized disease and pharmacological therapies targeting mTOR, MEK and HIF2-α in advanced/metastatic disease.
Pathophysiology and Molecular Biology of Phakomatoses
NF1, TSC and CS are caused by mutations on genes encoding for different components of the MAP kinase and Ras-PI3K-Akt-mTOR pathways (Figure 2). The PI3Kinase-Akt pathway is a classical signaling pathway involved in the regulation of metabolic processes, maintenance of the redox balance, and cell survival and growth (2). PI3Kinase is primarily activated by RAS proteins. This family of proteins (HRAS-NRAS-KRAS) also regulates other signaling pathways such as MAP kinase, which is involved in cell proliferation and survival (3). RAS proteins oscillate between active guanosine triphosphate (GTP)-bound and inactive guanosine diphosphate (GDP)-bound states, and deregulation has been observed in various diseases such as cancer or even psychiatric diseases (4).
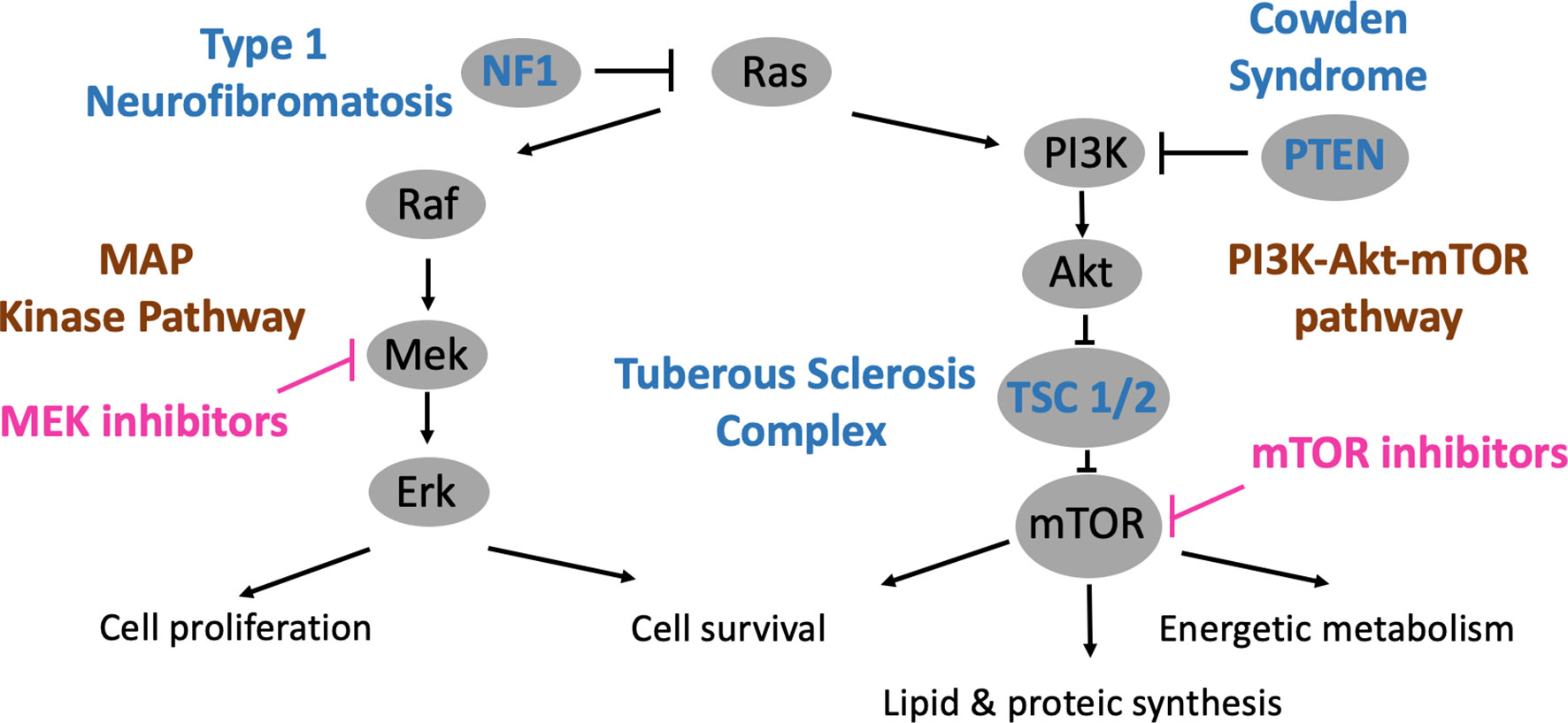
Figure 2 PI3K-Akt-mTOR and MAP kinase pathways, relations with phakomatoses (TSC, NF1 and CS) and therapeutic developments.
Downstream from PI3K and Akt is the mTOR pathway, which ultimately leads to the activation of mTOR complexes 1 and 2 (mTORC1 & 2). The mTOR pathway has been highly conserved during evolution and integrates several environmental cues, such as growth factors, amino acids or glucose, in order to guide cellular growth and fate (5). These multi-protein complexes regulate energy metabolism and lipid/protein synthesis and influence cell survival. In human diseases, mTOR deregulation is involved in cancer, diabetes and ageing (6).
Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1), an autosomal dominant disease, is caused by mutations of the NF1 gene located on chromosome 17q11.2, which encodes neurofibromin (7). This protein accelerates the conversion of active GTP-bound RAS to inactive GDP-RAS. Consistently, MAP kinase and PI3Kinase-Akt-mTOR are deregulated in NF1 (8, 9).
von Hippel-Lindau Disease
The pathophysiology of another type of phakomatoses, von Hippel-Lindau disease (VHL), is related to a different but major cellular dysfunction: oxygen sensing (10) (Figure 3). VHL is an autosomal dominant disease linked to mutations of the VHL gene, mapped on chromosome 3p25.3 (11). The VHL protein (pVHL) is part of a multiprotein complex that is also constituted by elongin B and C, Cullin 2 and RBX1, with the whole complex exhibiting E3 ubiquitin ligase properties (12, 13). Under normoxic conditions, hypoxia-inducible factors-α (HIF-α) are hydroxylated on proline residues by prolyl hydroxylase, which allows recognition by pVHL, then ubiquitylation of HIF-2 and subsequent degradation via the proteasome (Figure 3A). In hypoxic conditions, HIF-α cannot be hydroxylated and recognized by pVHL, and then tend to accumulate, dimerizing with HIF-1 β and activating the transcription of several genes involved in angiogenesis, erythropoiesis, metabolism, cell proliferation, migration and invasion. With VHL gene mutations, there is also no possibility of HIF-α degradation even in normoxic conditions, and this create a pseudo-hypoxic state with continuous activity of the HIF-α/HIF-1β heterodimer (Figure 3B). Consequently, VHL is crucial in the oxygen sensing process within the body. It also exhibits HIF-independent properties, including assembly and regulation of the extracellular matrix, microtubule stabilization, and regulation of apoptosis.
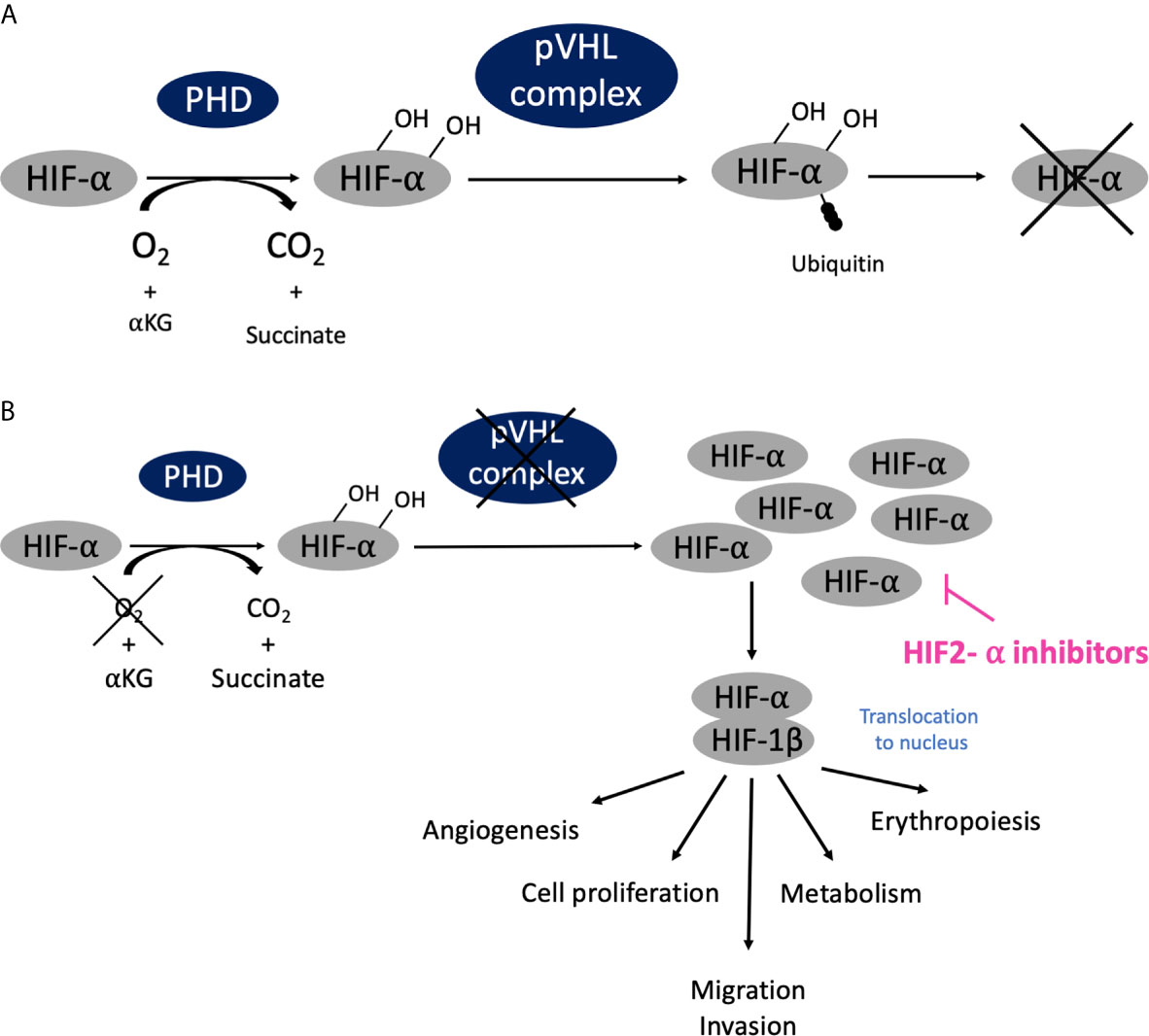
Figure 3 Oxygen sensing pathway, connection with phakomatoses (VHL) and therapeutic developments. (A) In normoxic conditions (normal oxygen concentration), HIF-α are hydroxylated on proline residues by prolyl hydroxylase, is then recognized by pVHL complex and ubiquitinated and subsequently degraded via the proteasome. (B) In hypoxic conditions (low oxygen concentration), HIF-α cannot the be recognized by pVHL and degraded; it accumulates, dimerizes with HIF-1 β, translocates into cellular nucleus and activates transcription of targeted genes. Similarly, when VHL is mutated there is also no possibility of HIF-α degradation even in normoxic conditions, and this creates a pseudo-hypoxic state with the HIF-α/HIF-1 β dimer constantly activated, leading to the development of tumor angiogenesis as compensation.
Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC), another autosomal dominant disorder, is related to mutations of the TSC1 or TSC2 genes, which are located on chromosomes 9q34 and 16p13.3 (14, 15). These genes encode for hamartin and tuberin, respectively, proteins that have the property of directly inhibiting the mTOR protein. Therefore, mutations on TSC1 or TSC2 lead to a hyperactive mTOR pathway.
Cowden Syndrome
Cowden syndrome (CS), also an autosomal dominant disease, is linked to mutations of the PTEN gene, mapped on chromosome 10q23.31 (16). PTEN has the ability to inactivate PI3Kinase, the first kinase that subsequently leads to activation of the mTOR pathway. Consistently, an overactive mTOR pathway is observed when PTEN is mutated.
Main Clinical Features of Phakomatoses
Neurofibromatosis Type 1
NF1 is the most common form of phakomatoses and affects 1 out of 3000–3500 births worldwide (17). The main symptoms of NF1 are cutaneous, ophthalmological and neurological (Table 1). NF1 predisposes to the development of multiple neoplasias including solid cancers, mainly malignant peripheral nerve sheath tumors, as well as malignant hemopathies (18). Endocrine tumors, mainly pheochromocytoma and digestive neuroendocrine tumors, are also observed. Primary hyperparathyroidism, pituitary adenomas and thyroid cancer have been reported but are indeed very rare and, to date, are not considered as classical phakomatoses-associated endocrine tumors (19).

Table 1 Neurofibromatosis type 1 (NF1) diagnostic criteria. Adapted from (18).
von Hippel-Lindau Disease
The classical clinical features of VHL disease usually include CNS and/or retinal hemangioblastoma, endolymphatic tumor, clear cell renal carcinoma, and a predisposition for developing cysts, mainly in the kidney and pancreas (Table 2). VHL disease occurs in about 1 per 36,000 births, with a penetrance at 65 years estimated at 90%. The disease is due to a de novo mutation in 20% of cases. Mosaic mutations can be identified in a minority (5%) of patients (20, 21). The two main endocrine tumors observed in VHL are pheochromocytoma/paraganglioma (PPGL) and pancreatic neuroendocrine tumors (pNETs). VHL disease is classified into two distinct phenotypes, which are based on the absence (type 1) or the presence (type 2) of PPGL. Type 1 VHL affects 80% of patients and is associated with large deletions or truncating mutations, whereas 20% of patients are in the type 2 group, which is associated with missense mutations. Sub-groups have also been defined within type 2 VHL: type 2a (low risk of renal cancer), type 2b (higher risk of renal cancer) and type 2c (PPGL only).

Table 2 von Hippel-Lindau disease diagnostic criteria. Adapted from (10).
Tuberous Sclerosis Complex
The classical phenotype of TSC associates cutaneous lesions, neurological features (cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas) and multiple retinal hamartomas (22) (Table 3). Its prevalence is estimated at 1/20,000 people. There is no clear genotype-phenotype correlation, but patients with TSC2 mutations show more severe disease than those with TSC1 mutations, although it does not involve endocrine tumors. The penetrance is high, with variable expression in the same family. Based on small studies, TSC may be associated with an increased risk of developing digestive neuroendocrine tumors, as well as pituitary and parathyroid adenomas (23).

Table 3 Tuberous sclerosis complex diagnostic criteria. Adapted from (22).
Cowden Syndrome
The incidence of CS is estimated to be 1/200,000 individuals. Patients with CS classically present with cutaneous features (facial papules, oral mucosal papillomatosis, palmoplantar keratoses), macrocephaly, and Lhermitte-Duclos disease (24) (Table 4). They also exhibit a predisposition for developing several types of cancer, the most prevalent being breast, endometrial, renal, colorectal and finally thyroid cancer. Thyroid cancer is typically differentiated, arise from follicular cells and occurs in 20 to 38% of CS patients, with a possible association with promoter or exon 1 mutations (25–28).

Table 4 Cowden syndrome diagnostic criteria. Adapted from (24).
Pheochromocytoma/Paraganglioma (PPGL) in Phakomatoses
PPGL arise from adrenal (pheochromocytoma) or extra-adrenal (paraganglioma) chromaffin cells associated with the paravertebral ganglia, and produce one or multiple catecholamines (adrenaline, noradrenaline, dopamine), which results in high blood pressure, palpitations, sweating, headaches, etc. (29, 30). These tumors are usually benign, and malignancy, characterized by distant metastasis, occurs in 8–10% of all PPGL patients: 10% for pheochromocytoma and ~25% for paraganglioma, the difference partially explained by SDH mutations more frequently encountered in paraganglioma (31, 32). Biochemical diagnosis is made with measurements of circulating or urinary catecholamine metabolites (Table 5) (33). Half of pheochromocytomas produce significant amounts of adrenaline and are diagnosed by an increase in metanephrines, with a linear relationship between catecholamine concentration and tumor size (34). The other half of pheochromocytomas and extra-adrenal paragangliomas are characterized by a predominant secretion of noradrenaline (35). Predominant production of dopamine is rare, preferentially encountered in head and neck paragangliomas or in malignant PPGLs (36). Some paragangliomas, especially localized in the head and neck, can also be non-secreting.
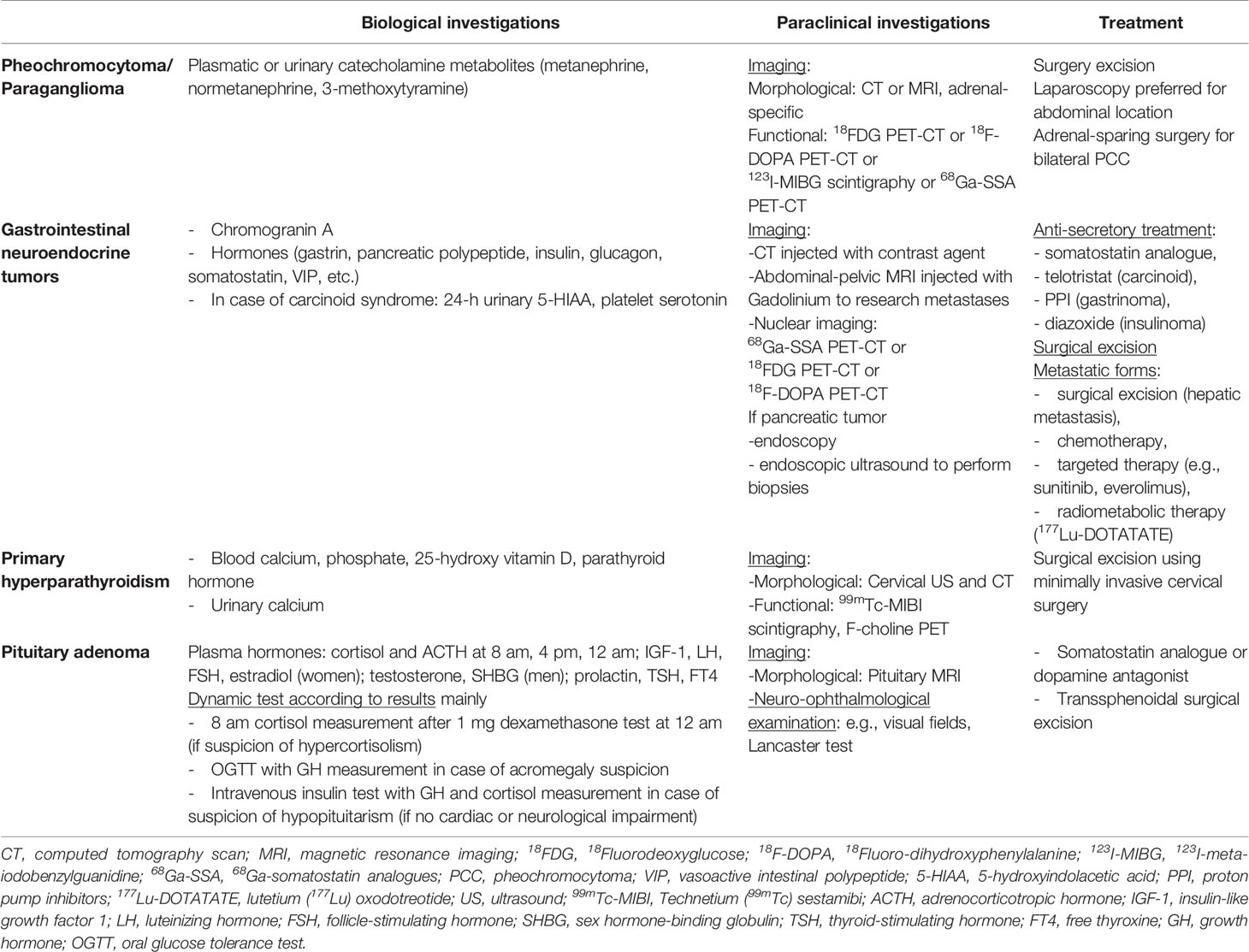
Table 5 General guidelines for diagnosis and treatment of tumors of endocrine glands.
NF1
NF1-associated PPGL prevalence varies from 2.9 to 14.6%, with no clear genotype-phenotype association with PPGL risk (37–42). Indeed, in NF1 patients diagnosed with PPGL, the mutational spectrum comprises both intragenic mutations and deletions, with mutations being preferentially located in the cysteine-rich region of the NF1 protein over the RAS-GAP domain (43). The median age at diagnosis is around 40–45 years, which is older than in other genetically-determined PPGLs (44). Most PPGLs are unilateral pheochromocytomas (75%) but bilateral tumors are not rare (up to 17% of cases) and are synchronous in 20 to 40% of cases (38). Paragangliomas are infrequent. In rare cases, mixed tumors can be observed with a ganglioneuroma/ganglioneuroblastoma contingent (45). Gangliocytic paragangliomas can also been observed in ~5% of NF1 patients (46, 47). The catecholamine secretion profile is adrenergic. PPGL tumor size in NF1 (median size 5.8 cm, range 0.8–20 cm) is roughly the same as in sporadic cases. Functional imaging, which is used to investigate bilateral pheochromocytomas and/or paragangliomas and/or metastatic extension, is based on 18F-DOPA PET-CT; second choices include 123I-MIBG scintigraphy or 68Ga-SSA PET-CT (48).
VHL
PPGL, which defines type 2 VHL, is observed in 20–30% of patients (49–51). The youngest patient diagnosed was 4 years old, and the median age at diagnosis is 25–30 years (52). VHL-associated PPGL are mostly pheochromocytomas; paragangliomas account for only 10 to 20% of these chromaffin tumors. The malignancy potential is lower than in sporadic cases, about 5% vs. 10–17% (53). One-third to one-half of patients with PPGLs have synchronous bilateral pheochromocytomas. The median tumor size is smaller than in the sporadic forms, about 30 mm, and this is possibly related to abdominal screening, which allows early diagnosis (49). Accordingly, the release of catecholamines and the prevalence of associated symptoms and hypertension are lower compared with the sporadic forms (16–55% and 8–46%). VHL-associated PPGL exhibit a specific secretion profile, which is almost exclusively noradrenergic. The absence of adrenaline secretion is due to the epigenetic silencing of phenylethanolamine N-methyltransferase, which catalyzes the production of adrenaline from noradrenaline (54). Functional imaging of PPGL can be performed with various radiopharmaceuticals, which are of particular interest because they can reveal multiple VHL-associated tumors. The recent European Nuclear Medicine Society guidelines prioritized these investigations and, as in NF1 patients, suggested 18F-DOPA PET-CT as first-line imaging for exploring potential bilateral pheochromocytomas and/or extra-adrenal paragangliomas (48, 55). If not available or feasible, 123I-MIBG scintigraphy or 68Ga-SSA PET-CT can be used, with the latter preferred since it also demonstrates great diagnostic performance with regard to neuroendocrine tumors (56).
Treatment of Phakomatoses-Associated PPGLs
Resection of benign PPGLs should be considered in order to limit cardiovascular complications and prevent unexpected death, which can be triggered with the administration of certain drugs (53, 57–60). Of note is that rare bilateral pheochromocytomas have led to adrenal-sparing surgery (61–63). This allows selective removal of the pheochromocytoma and leaves a sufficient amount of adrenal cortex tissue for maintaining corticosteroid independence and avoiding steroid dependence and its associated comorbidities, i.e. increased mortality risk especially in young patients, and deterioration of quality of life (64, 65). The risk is recurrence of the tumor or the occurrence of acute adrenal crisis in case of stress. Procedures can be performed by an open or preferentially laparoscopic approach, at least for lesions < 50 mm, while larger lesions should be removed by total adrenalectomy (66). Preoperative measures have to be taken, especially the introduction/optimization of anti-hypertensive drugs, in order to limit perioperative-associated morbidity and mortality (67).
Few data are currently available on adrenal-sparing surgery and NF1-associated PPGL, possibly because bilateral pheochromocytomas are present in a minority of patients. However, when performed, the procedure is safe and allows exogenous glucocorticoid-independence, with an estimated risk of recurrence between 0 and 10% (38, 68).
A greater number of studies have been dedicated to VHL-associated pheochromocytomas. One such study considered adrenal-sparing surgery in 26 VHL patients, none of whom developed metastatic pheochromocytoma; 11% exhibited local recurrence that could be treated with a second surgical procedure or active surveillance, 11% presented contralateral pheochromocytoma, and finally 11% became steroid-dependent (69). Similar results were published by an international consortium that compared morbidity and mortality among patients with bilateral pheochromocytomas undergoing total or cortical-sparing adrenal surgery. Of the 184 VHL patients included, 56% had bilateral synchronous pheochromocytomas. Cortical-sparing surgery was successful in 87% of procedures, with a 12% risk of ipsilateral recurrence (70). Sixty percent of VHL patients remained steroid-independent. Therefore, this procedure seems particularly recommended, when feasible, in VHL- and NF1-associated pheochromocytomas, and it can be performed in both adult and pediatric populations (71).
There are currently no specific recommendations for the management of rare cases of advanced/malignant VHL- or NF1-associated PPGL that currently do not differ from sporadic cases. Therapeutic options include locoregional therapies, radionuclide therapy and/or chemotherapy (72, 73). Targeted therapies such as sunitinib have been shown to induce a response in a VHL patient with malignant PPGL (74).
Screening and Follow-up of NF1 and VHL-Associated PPGLs
NF1
There are currently no recommendations for systematic screening of PPGL in NF1 patients in childhood, and experts suggest that investigations for PPGL must be done when there is an increase in heart rate and/or blood pressure (Figure 4) (75). Likewise, adult guidelines suggest that PPGL should be considered in hypertensive NF1 patients who are over 30 years old, pregnant and/or have catecholamine-related symptoms (76–79). Blood pressure should be evaluated at least annually, but systematic biochemical or morphological PPGL screening in asymptomatic patients with NF1 is not recommended under the current guidelines. Nevertheless, some studies suggest that only consider screening hypertensive patients would fail to recognize the majority of NF1-associated PPGL (80). Furthermore, prospective morphological and biochemical screening in a French series of 156 NF1 patients found a pheochromocytoma prevalence of 7.7% (12 patients), half (n = 6) of whom were secreting, and with only two of them symptomatic (81). This study and others showing the positive impact of genetic testing on the management and outcome of patients with paraganglioma-pheochromocytoma, and the poor prognosis of PPGL during pregnancy suggest that despite current recommendations, a screening for PPGL, at least before pregnancy, would improve the prognosis of these patients (82–85).
VHL
Current Danish and American guidelines as well as French recommendations suggest starting follow-up of VHL patients at a median age of 5 years old, with an annual measurement of plasma or urine metanephrine (Figure 4) (86–88). Monitoring of non-invasive imaging can be performed annually from the age of 8 years, initially by ultrasound used first alone, and then alternatively with MRI after the age of 16 years. French guidelines differ slightly and recommend starting abdominal ultrasound at the age of 5 years and the first MRI at 18 years. Functional imaging is useful only when pheochromocytoma is suspected, and it is currently not recommended to repeat it in asymptomatic patients. Particular attention should be paid to screening before pregnancy.
Digestive Neuroendocrine Neoplasms in PhakomatosEs
Neuroendocrine neoplasms are rare tumors that arise from neuroendocrine cells distributed throughout the entire body (89, 90). Those cells are derived from the embryonic gut (foregut, midgut, hindgut) and can be found in various locations, mainly in the digestive and thoracic regions, with various differentiation and hormonal secretion. Tumor aggressiveness can be assessed according to the WHO grade (grade 1 to 3) based on proliferation index values (Ki-67 and/or mitotic count) (91). Neuroendocrine neoplasms of the gastrointestinal tract comprise neuroendocrine tumors (NETs) and neuroendocrine carcinomas (NECs). Of note, the 2019 WHO classification has modified the cut-off used for the Ki-67 proliferative index to distinguish grade 1 from grade 2 NETs (Ki-67 above or below 3%), emphasized the distinction between grade 3 NETs (low-grade NETs with a high proliferative rate and Ki-67 above 20%) and NECs which are all high grades poorly differentiated aggressive neoplasms (92). Phakomatoses are mainly associated to NETs, which are well differentiated and progress slowly (most of them grade 1) in 85% of cases (93), even if they can be complicated with metastases. Even in metastatic conditions, the median overall survival is typically 5–10 years relative to the location and histological grade in well-differentiated NETs (94–96). In 15% of cases, a functional syndrome (FS) (hypoglycemia, recurrent gastric ulcers, necrolytic migrating erythema) is present, which is related to hormone production (e.g. insulin, gastrin, glucagon). About 4–5% of gastroenteropancreatic tumors arise in the context of an inherited tumor syndrome (mainly MEN1 but also phacomatosis), especially VHL, whereas the incidence of NET is lower in NF1 and TSC. Young age at diagnosis, multiple tumors in multiple organs, and familial history are clinically suggestive of the diagnosis. Neuroendocrine neoplasms in the context of phakomatoses are almost always well-differentiated neuroendocrine tumors. Except in VHL and NF1, tumors themselves do not show specific pathological features (97).
NF1
NF1-associated NETs are rare, and their prevalence is unknown. Most NF1-associated digestive NETs are located on the ampulla of Vater, followed by the duodenum and pancreas (98–100). The youngest published case was diagnosed at 23 years old and the median age at diagnosis is 48 years, which is 15 years younger than in the general population (94, 101). In a series of 58 NF1-associated NET cases, about 25% (14 cases) were marked with somatostatin antibodies on immunohistochemistry, but only 28% of the patients presented symptoms related to the hormone secretion, i.e. diabetes mellitus, diarrhea, gallstones, and less frequently dyspepsia and hypochlorhydria (102). Gastrinomas and insulinomas have also been observed (103, 104). In NF1, NETs are mostly well-differentiated, rarely multifocal, and can have specific pathological features such as a deceptive tubular/tubuloglandular appearance that can mimic adenocarcinoma. Metastases are present at diagnosis in 14% of reported patients. Specific survival data is not available yet. In a review of 76 published cases, 9% of patients died of NET progression; however, follow-up was short (median 31 months), and the grade as well as median overall survival were unknown. The standard differential diagnosis is gastrointestinal stromal tumors (GISTs), which can also be observed in NF1 patients (105). In this context, the tumors are preferentially located in the ileum/jejunum versus the stomach as in sporadic forms, and most NF1-associated GISTs have a favorable clinical course (106).
VHL
The VHL pancreatic lesions include solid NETs and cystic lesions. Cystic lesions (simple cysts and serous cystadenomas) are generally asymptomatic and do not require any treatment. They must be differentiated from other cystic tumors that have malignant potential, such as intraductal papillary mucin-producing tumors or mucinous cystic tumors. VHL pancreatic neuroendocrine tumors (pNETs) occur in 11–17% of cases and are frequently multifocal (40%) (107–109). In VHL patients, pNETs present with a characteristic microscopic appearance, with finely vacuolated cytoplasm and lipid-rich cells. Patients with missense mutations (type 2 VHL) rather than truncating mutations or large deletions (type 1 VHL) exhibit a higher prevalence of pNETs, with a hotspot on codons 161/67 in exon 3, which could be associated with a higher risk of metastases although the data are discordant (110, 111). Preferential association with PPGL is also discussed (107, 112). The mean age at diagnosis is 35–38 years; not surprisingly and because VHL does not seem to be involved in hormonal secretion, most patients are asymptomatic with nonfunctional tumors. However, ectopic secretion of ACTH with paraneoplastic Cushing syndrome is possible (113). The pathogenesis of VHL pNETs differ from that of MEN1 or sporadic pNETs notably because of:
- upregulation of genes related to hypoxia-inducible factor molecules, angiogenesis, epithelial mesenchymal transition and/or metastasis, cell cycle and growth factors and receptors (114),
- and hypomethylated CpGs, significantly more common in VHL-related versus sporadic and MEN1-related NETs (115).
The presence of local invasion or locoregional/distant metastasis varies between 7.5 and 12.8%; it reaches 20%, however, for the metastatic forms in the largest cohort, which is lower than in other genetic predisposition syndromes or in sporadic forms (112, 116–118). Metastasis is generally associated with pNETs larger than 28 mm, with a doubling time of 22 months vs. 126 months in non-metastatic lesions (111). Morphological investigations of VHL-associated pNETs do not differ from the management of sporadic forms. In the context of VHL, the differential diagnoses of pNETs include serous cystadenoma or metastatic renal cancer lesions (107). Simple cysts are also frequent (up to 75%), whereas cystic aspects of pNET are uncommon (119). Measurement of plasma chromogranin A can be of interest although there are several limitations of this assay, notably the false-positive results due to hypersecretion of gastrin, which is encountered, among other causes, in case of proton pump inhibitor use, or chronic kidney disease that can be associated with VHL (120). Nuclear medicine imaging recommends 68Ga-SSA PET-CT as a first-line investigation, or 18FDG PET-CT in more aggressive pNET cases (48). 18F-DOPA PET-CT is not useful in this context (55). The progression of pNET lesion size is not linear and may include periods of stability or even apparent decrease in size on imaging (121).
pNET-related mortality is not well documented in VHL patients. In two studies focusing on patients presenting with pNETs, tumor-associated mortality was estimated between 6.9 and 9.5%. However, death was associated with pNETs in 29 to 50% of VHL patients (108, 111). Ten-year overall survival is estimated at 50% in non-operated patients with a tumor size greater than 2.8 cm and rises to 94% in operated patients presenting with a pNET size less than 1.5 cm. No correlation was identified between VHL genotype and mortality.
TSC
The link between TSC and digestive NETs, especially pancreatic, is recognized because of the pathogenic role of the mTOR pathway in the development of NETs (122). However, the onset of those tumors in TSC remains low, with an estimated incidence of 1%, which can question the causal relation between these two conditions. Nevertheless, since the availability of medical treatment such as everolimus, their incidence could increase with the morphological follow-up of kidney lesions. Genetic data, available only in a small proportion of reported patients, revealed TSC2 mutations, most of them in or just upstream of the GTPase-activating protein (GAP) domain (exons 33–36) (123). TSC1 mutations can also be found, however to a lesser extent (124). The largest study focusing on pNETs in TSC patients (n = 18) found an average age of 26 years at diagnosis, significantly younger than in the sporadic cases; so, development in pediatric age or in young adults should be emphasized (123). pNETs were functional in 44% of patients, mostly due to insulin secretion resulting in hypoglycemia (125). The diagnosis can be challenging because of neurological features in TSC patients with seizures; the seizures may be attributed to neurological lesions, while they are indeed related to underrecognized insulinoma-related hypoglycemia, especially in those patients with intellectual disability. The mean pNET size of 5.1 cm was slightly higher than in sporadic cases, with a cystic aspect in one-third of patients without any multifocality, which is in contrast with other genetic NET predisposition syndromes (126). The proportion of synchronous metastasis was 13%, which is lower than in sporadic cases. The progression and specific associated mortality are unknown. Finally other locations such as rectal NET have also been described (127).
Treatment of Phakomatoses-Associated Digestive NETs
Treatment of NETs includes the management of tumor volume but also, when present, of the functional syndrome. The risk of resection must always be weighed with the risk of diabetes, especially in case of multiple tumors.
Functional Syndrome
Treatment of the functional syndrome does not differ from that of sporadic tumors, and we will only consider the most frequent secretions. The functional syndrome can be cured with surgical resection of the NET. However, while awaiting surgery and/or in advanced disease, medical treatment can be initiated in order to limit symptoms that may be life-threatening (128). In case of NF1-associated somatostatin-related symptoms, management is not well defined and can include treatment of diabetes, cholecystectomy, and pancreatic enzyme supplementation. Surprisingly, in a few cases the administration of somatostatin analogues can lead to clinical improvement (128–130), although careful monitoring is needed because hypoglycemia may worsen in some patients (131). Insulinoma-induced hypoglycemia can be reversed with small frequent meals and diazoxide. In malignant insulinoma, everolimus and sunitinib as well as pasireotide have been shown to improve glucose levels (132–134).
Antitumor Treatment of NETs
NF1
Treatment of NF1-associated NETs does not differ from the management of sporadic cases, whether or not they are metastatic. Treatment of localized NETs is based on tumor resection, which can be performed endoscopically or surgically according to NET location, size and pathological characteristics (128, 135, 136). In metastatic tumors, MEK inhibitors (selumetinib, trametinib) have shown great benefit in NF1 patients because they inhibit the MAP kinase pathway, which is overactivated due to NF1 mutation. Indeed, a decrease in the size of low-grade gliomas and plexiform neurofibromas has been reported (137–141). However, there is currently no data in NF1-associated NET patients.
VHL
Treatment of VHL-associated NETs does not differ from the management of sporadic cases. Given the relatively low risk of malignancy and the high frequency of asymptomatic forms, VHL-associated pNETs should not be removed if less than 15 mm in size and slowly progressing at the regular follow-up, with the rare exception of symptomatic forms (142). In case of non-distant metastatic forms, surgery should be considered for tumors with any of the following characteristics: 1) > 2 cm in the head of the pancreas, 2) > 3 cm in the body/tail of the pancreas, 3) doubling time < 500 days, or 4) in case of metastatic locoregional lymph nodes (143, 144). The recommended surgical procedures are enucleation or pancreaticoduodenectomy for pNETs in the pancreatic head relative to the position from the pancreatic duct (145). The risk of this last surgery is associated with a non-negligible morbidity-mortality rate of about 5%, which is much higher than for pancreatic body/tail pNETs in which enucleation or distal or central pancreatectomy should be performed if possible per laparoscopy. Consequently, total pancreatectomy should be discussed only in the very rare cases of symptomatic multifocal pancreatic tumors that cannot be safely enucleated. Post-pancreatectomy diabetes is a classical complication encountered at 10 years in up to 16% of patients after pancreaticoduodenectomy and in 35% after distal pancreatectomy; therefore, physicians and patients should be aware of this risk before validating surgical indication and educate patients to avoid weight gain (146, 147). Islet autotransplantation, now reimbursed through health insurance in France, could be an interesting option for limiting diabetes-associated morbidity; the procedure carries the risk of tumor occurrence in these genetically-determined diseases, although interesting results have been observed in pancreatic adenocarcinoma in an animal model (148, 149).
There is no difference in medical management between VHL-associated and sporadic metastatic pNETs, although antiangiogenic agents have been especially studied because of the specific deregulated angiogenesis mechanism of the disease (128). Nevertheless, there is not yet a direct comparison between VHL-associated and sporadic forms that evaluates the relative efficacy of these drugs. Sunitinib is approved for the management of pNETs and has shown stability in 5 out of 7 cases of VHL-associated pNETs (150, 151). Another study using multiple antiangiogenic treatments (sunitinib, sorafenib, axitinib, pazopanib) showed a partial response in 4 of the 15 VHL patients with cystic or solid pancreatic tumors (152). In a phase 2 trial, 53% of VHL pancreatic lesions responded to pazopanib (anti-VEGFR 1-3, anti-PDGFR α-β, c-KIT), but most of them were serous cystadenomas (153). Interestingly, pazopanib was also shown to reduce the size of renal lesions and hemangioblastomas.
Since the 2010s, specific HIF2-α inhibitors, which showed great benefits in sporadic clear cell renal cell carcinoma in phase1-2 clinical trials, have been undergoing evaluation alone or in combination with immunotherapy and/or antiangiogenic drugs (154). A phase 2 study using the HIF2-α inhibitor MK-6482 in 61 VHL patients with clear cell renal carcinoma and pNETs showed an objective response rate in 64% of pNET cases, with 4 complete responses. The data are not mature yet, but the 12-month progression-free survival rate was 98.3% (155). Therefore, HIF2-α inhibitors could offer promising prospects for VHL metastatic pNETs or even malignant PPGL.
TSC
The management does not differ from the sporadic forms, neither in localized or metastatic disease (128, 156). In advanced disease, mTOR inhibitors such as everolimus, which is used in all NET locations (as second-line treatment), could be of particular interest in TSC patients; however, no studies to date have demonstrated specific efficacy in those patients, except for one case with metastatic pNET and a TSC2 germline mutation that showed partial response shortly after the introduction of everolimus. Support for this hypothesis may be found in the results of the EXIST trials using everolimus in other features of TSC, with morphological responses observed in renal angiomyolipoma, subependymal giant cell astrocytomas and cutaneous nodules (157, 158).
Screening and Follow up of Phakomatoses-Associated Digestive NETs
NF1
Due to their rarity, there are currently no follow-up recommendations regarding digestive NETs in NF1 patients, and in particular no recommendations to perform and repeat abdominal imaging or endoscopy in asymptomatic patients (Figure 4) (76, 77). Nevertheless, visualization of the pancreas is suggested for morphological investigation of neurofibromas, pheochromocytomas, or GISTs.
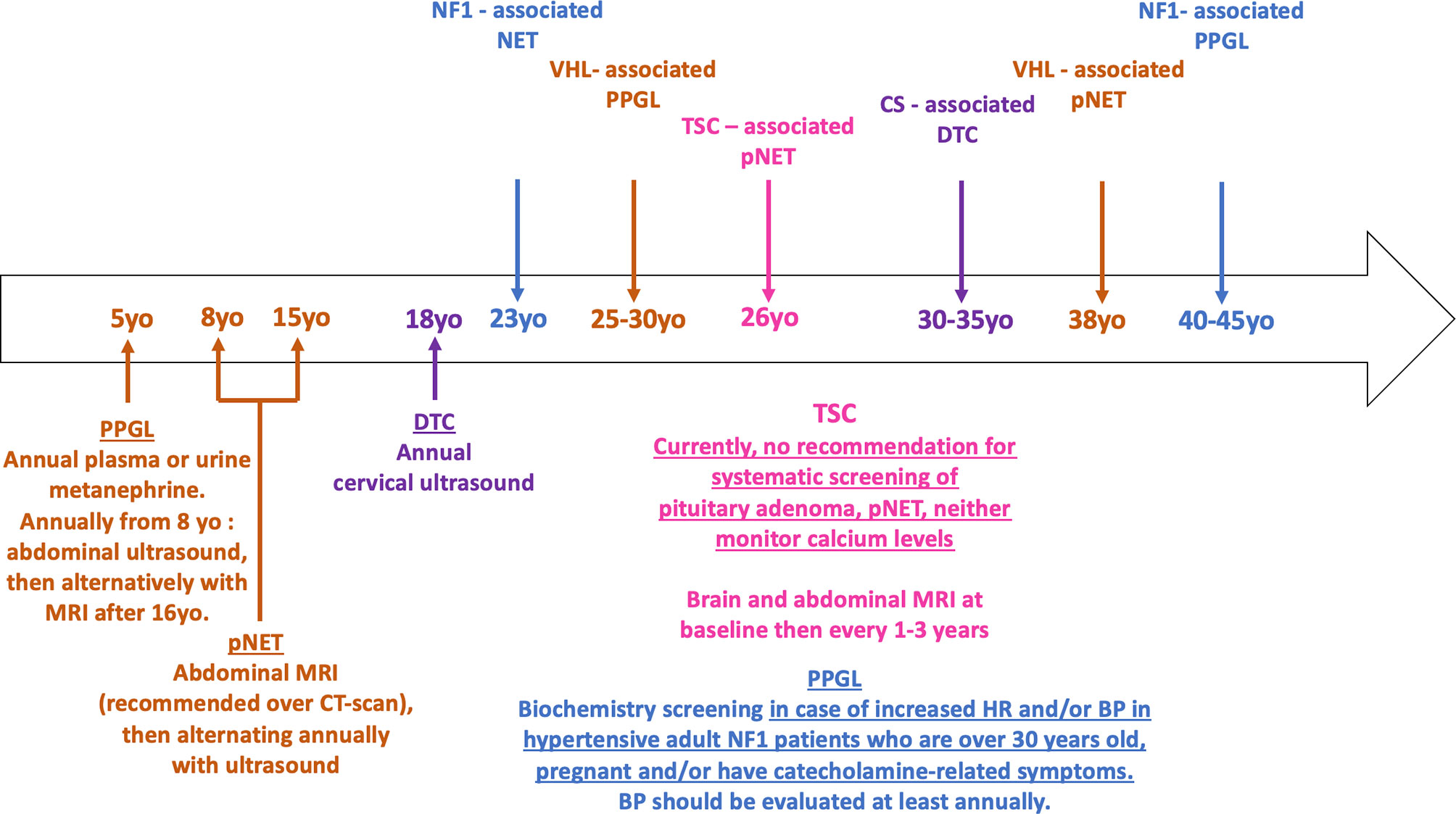
Figure 4 Timeline of phakomatoses-associated endocrine tumors and follow up strategy. The median age of onset of phakomatoses-associated endocrine tumors is shown in the arrow. The various follow up guidelines according to the syndromic presentation are shown below the arrow. Brown = VHL; purple = CS; fuchsia = TSC; blue = NF1. HR, heart rate; BP, blood pressure; pNET, pancreatic neuroendocrine tumor; PPGL, pheochromocytoma/paraganglioma; DTC, differentiated thyroid cancer.
VHL
In VHL patients, current surveillance guidelines regarding pNETs and more largely pancreatic lesions suggest first abdominal imaging between 8 and 15 years of age (Figure 4) (86, 87). MRI is recommended over CT scan to limit the consequences of repeated ionizing radiation exposure (159). Due to the non-functional characteristic of most pNETs, there is no indication for systematic biological follow-up.
TSC
Finally, there are currently no follow-up recommendations regarding digestive NETs in TSC patients; however, required abdominal imaging for the follow-up of renal angiomyolipoma enables simultaneous visualization of the pancreas and could detect non-functional pNETs at an early stage (Figure 4) (160).
Primary Hyperparathyroidism
NF1
A few cases of primary hyperparathyroidism occurring in NF1 patients have been published (161). The clinical presentation does not differ from that of sporadic cases, and, notably, the diagnosis is not made at a younger age in NF1 patients. Primary hyperparathyroidism is usually associated with a single adenoma, except for few cases of parathyroid carcinoma (162, 163) (Table 6). No recurrence has been observed after surgery. Therefore, a causal relationship between hyperparathyroidism and NF1 cannot be assumed.
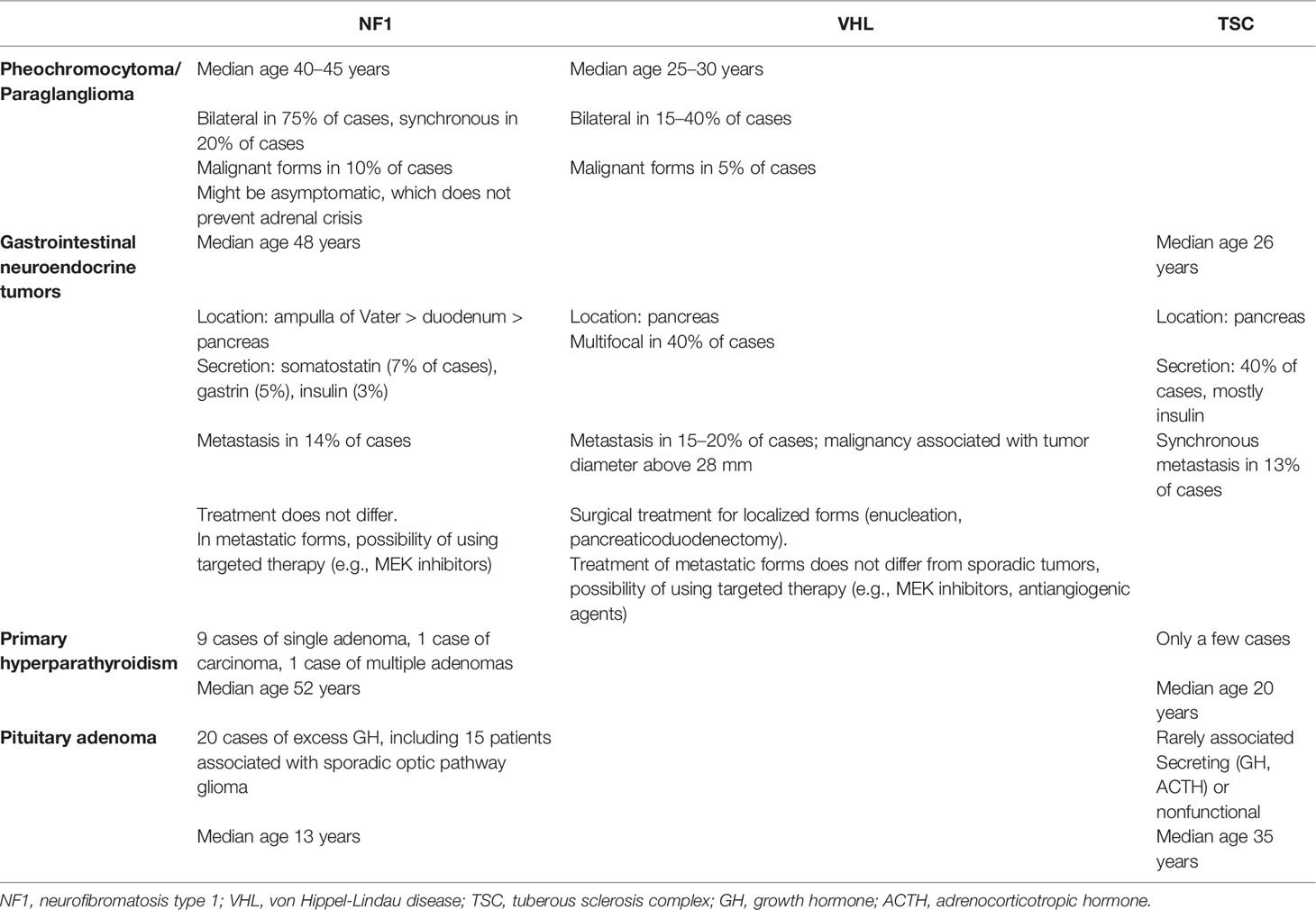
Table 6 Endocrine tumors and phakomatoses specificities.
TSC
A few cases of parathyroid adenomas have been described in young TSC patients. The occurrence before 20 years of age argues for a causal role of the TSC mutation (23, 164). However, recent advances in understanding the biology and pathogenesis of parathyroid adenomas do not seem to involve the mTOR pathway (165). Furthermore, primary hyperparathyroidism is considered the second most common endocrinopathy after diabetes mellitus (166), raising the hypothesis of a coincidental occurrence. However, TSC mutations could also lead to parathyroid adenoma development in an mTOR-independent pathway. In the literature, TSC patients with primary hyperparathyroidism presented symptoms related to hypercalcemia, with one parathyroid lesion at most.
Management
There are no specific recommendations for surgery and management of hyperparathyroidism in phakomatoses.
Screening and Follow up of Phakomatoses-Associated-Primary Hyperparathyroidism
There are currently no recommendations for monitoring calcium levels during the follow-up of NF1 or TSC patients (Figure 4).
Pituitary Adenomas
NF1
Pituitary adenomas are rarely reported in NF1 patients, although these genetic mutations are known to predispose patients to this condition (Table 6) (167–170). Excess growth hormone (GH) has been observed, notably in patients with central precocious puberty, but it seems to be associated with optic pathway tumors (OPT) rather than pituitary somatotroph adenomas (171, 172). Although the mechanism underlying excess GH in NF1 is still unknown, a loss of somatostatinergic inhibition from OPTs with dysregulation of GH secretion is suspected, particularly in tumors close to the hypothalamic and pituitary regions.
TSC
The association between TSC and pituitary adenomas is still debated. In preclinical models of TSC, such as in an Eker rat with TSC2 germline mutations, pituitary adenomas were observed in 40–60% of cases, and this was associated with premature death attributed to pituitary hemorrhage (173, 174). However, pituitary adenoma in human TSC patients have rarely been reported (23, 175). As TSC surveillance guidelines suggest repeating a brain MRI every 1 to 3 years because of the risk of astrocytoma, it is unlikely that pituitary adenomas are underdiagnosed (160). When diagnosed, TSC-associated pituitary adenomas are secreting (GH, ACTH) or non-functional tumors, and patients are diagnosed at a relatively young age, before 35 years, compared with sporadic cases.
Management
There is no rationale for managing phakomatoses and especially TSC-associated pituitary adenomas differently from sporadic cases. It should be noted that in in vitro situations, everolimus can lead to a significant decrease in cell viability in TSC; however, no human data are currently available (175).
Screening and Follow up of Phakomatoses-Associated-Pituitary Adenoma
There are currently no recommendations for screening of pituitary adenomas in asymptomatic NF1 or TSC patients (Figure 4).
Thyroid Tumors
Among cases of phakomatoses, thyroid cancer prevalence is increased in Cowden syndrome. Nevertheless, clinical cases have been reported in NF1.
NF1
The presence of thyroid disease in NF1 may be linked to different mechanisms including autoimmune thyroiditis, metastasis of another cancer, thyroid neurofibromas, and thyroid cancer. The latter is consistent with knowledge that somatic NF1 mutations have been identified in differentiated and anaplastic thyroid carcinoma (176, 177). In exceptional conditions, it can also be favored by GH secretion from pituitary somatotroph adenomas (168). A population-based study estimated the relative risk of NF1 patients developing thyroid cancer to be 4.9 (178). However, only a few cases have been reported in the literature, so a coincidental association cannot be excluded. Different types of thyroid tumors have been reported, such as well-differentiated papillary cancer or medullary thyroid cancer (179–182). Pathologists must be aware of the presence of NF1 because cancer should not be confused with rare intrathyroid neurofibroma (183, 184). Clinical presentation, notably age at diagnosis, does not seem to differ from the usual presentation. Differentiated thyroid cancer can be found through focal hypermetabolism on 18FDG PET-CT that is initially performed to distinguish between neurofibromas and malignant peripheral nerve sheath tumors (185, 186).
Cowden Syndrome
There is a clear molecular rationale linking Cowden syndrome (CS) and differentiated thyroid cancer because mutations resulting in PI3K/Akt pathway activation are known to be involved in thyroid carcinogenesis and cancer progression (176, 187). The diagnosis can be made during childhood or adolescence in 15% of patients, and the majority of CS patients are diagnosed with thyroid cancer before the age of 40 years. The main histological subtypes are papillary carcinoma (classical and follicular variants) and follicular carcinoma, with a potentially higher prevalence of the latter compared with sporadic forms (14–45% vs. 2%) (188–190). These cancers are often associated with specific pathological features such as multiple adenomatous nodules in the context of lymphocytic thyroiditis (190).
Management
Management of thyroid cancer in NF1 and CS does not differ from that of sporadic cases and includes surgery with or without complementary iodine treatment (191). There is no increased risk of lymph node extension, so systematic lymph node dissection is not recommended and depends on pre-operative ultrasound findings in individual patients. Since a significant proportion of patients with CS also develop nonmalignant thyroid diseases, some authors have suggested prophylactic thyroidectomy (192, 193). However, this procedure is not recommended given the good prognosis of CS-associated thyroid cancer, similar to that of sporadic cases. Recently, a pilot study investigating the benefits of sirolimus, an mTOR inhibitor, in patients with germline PTEN mutation demonstrated improvement in skin and gastrointestinal lesions, but there are no data focusing on thyroid cancer (194).
Screening and Follow up of Phakomatoses-Associated Thyroid Cancer
The European guidelines for CS follow-up suggest an systematic surveillance for thyroid cancer by cervical spine ultrasound (Figure 4) (195). An annual investigation starting at 18 years of age is proposed, although the levels of evidence supporting those modalities are moderate and some authors suggest beginning surveillance at 10 years of age (196). Indeed, systematic annual ultrasound monitoring could lead to overdiagnosis and excessive thyroidectomy, therefore follow up frequency could be modified based on the first cervical screening results.
There are currently no recommendations for screening of thyroid cancer in asymptomatic NF1 patients.
Conclusions
Phacomatoses are a group of rare diseases that can be associated with neoplasia of the endocrine glands. Clinicians must be aware of these features. The main tumors are PPGL and digestive, especially pancreatic neuroendocrine tumors. They can occur at a younger age compared with sporadic cases and are more frequently multiple. Usually benign, they can, however, be aggressive, and surveillance guidelines are available for detecting tumors at early stages and limiting associated morbidity and mortality, especially in VHL. Screening for catecholamine secretion in NF1 appears beneficial according to recent data, since these frequently asymptomatic but life-threatening tumors pose a high risk for cardiovascular morbidity and mortality and have a poor maternal-fetal prognosis in case of pregnancy. All secreting pheochromocytomas should be operated, but parenchyma-sparing surgery must be favored to avoid complete adrenal insufficiency in selected cases with bilateral pheochromocytomas. Management of pancreatic NET depends on the size, number, secretion, aggressiveness (Ki-67) and extra-pancreatic extension of the lesions. The treatment (including surveillance) must be discussed in order to limit surgical risk and post-pancreatectomy diabetes. In advanced/metastatic diseases, besides standard treatments, specific therapies that target the underlying genetic abnormality are under investigation. Importantly, those rare patients should be managed and followed by specialized and multidisciplinary teams and networks to weigh the benefit-risk ratio of each therapeutic strategy.
Author Contributions
BC and HD wrote the manuscript and are co first authors. AJ, ML, CD, CC-B, and SE contributed to the enrichment and the editing of the manuscript by their comments and remarks. MV supervised the work, wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ACTH, adrenocorticotropic hormone; CNS, central nervous system; CS, Cowden syndrome; CT, computerized tomography; 18FDG, 18fluorodeoxyglucose; 18F-DOPA, 18fluoro-dihydroxyphenylalanine; FS, functional syndrome; 68Ga-SSA, 68Ga-somatostatin analogues; GH, growth hormone; HIF, hypoxia-inducible factor; 123I-MIBG, 123I-metaiodobenzylguanidine; APK, mitogen-activated protein kinase; MEN1, multiple endocrine neoplasia type 1; MRI, magnetic resonance imaging; mTOR, mechanistic target of rapamycin; NET, neuroendocrine tumor; NF1, neurofibromatosis type 1; PET-CT, positron emission tomography-computerized tomography; PDGFR α-β, platelet-derived growth factor receptor α-β; PI3K, phosphoinositide 3-kinases; PPGL, pheochromocytoma/paraganglioma; TSC, tuberous sclerosis complex; VEGFR 1-3, vascular endothelial growth factor receptor 1-3; VHL, von Hippel-Lindau disease.
References
1. Huson SM, Korf BR. Chapter 121 - The Phakomatoses. In: Rimoin D, Pyeritz R, Korf B, editors. Emery and Rimoin’s Principles and Practice of Medical Genetics. Oxford: Academic Press (2008). p. 1–45. doi: 10.1016/B978-0-12-383834-6.00128-2
2. Hoxhaj G, Manning BD. The PI3K–AKT Network At the Interface of Oncogenic Signalling and Cancer Metabolism. Nat Rev Cancer (2020) 20:74–88. doi: 10.1038/s41568-019-0216-7
3. Gimple RC, Wang X. RAS: Striking At the Core of the Oncogenic Circuitry. Front Oncol (2019) 9:965. doi: 10.3389/fonc.2019.00965
4. Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell (2017) 170:17–33. doi: 10.1016/j.cell.2017.06.009
5. Saxton RA, Sabatini DM. TOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
6. Inoki K, Corradetti MN, Guan K-L. Dysregulation of the TSC-mTOR Pathway in Human Disease. Nat Genet (2005) 37:6. doi: 10.1038/ng1494
7. Gutmann DH, Wood DL, Collins FS. Identification of the Neurofibromatosis Type 1 Gene Product. Proc Natl Acad Sci (1991) 88:9658–62. doi: 10.1073/pnas.88.21.9658
8. Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic Analysis Reveals Hyperactivation of the Mammalian Target of Rapamycin Pathway in Neurofibromatosis 1–Associated Human and Mouse Brain Tumors. Cancer Res (2005) 65:2755–60. doi: 10.1158/0008-5472.CAN-04-4058
9. Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 Tumor Suppressor Critically Regulates TSC2 and Mtor. PNAS (2005) 102:8573–8. doi: 10.1073/pnas.0503224102
10. Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. Von Hippel-Lindau Disease. Lancet (2003) 361:2059–67. doi: 10.1016/S0140-6736(03)13643-4
11. Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the Von Hippel-Lindau Disease Tumor Suppressor Gene. Science (1993) 260:1317–20. doi: 10.1126/science.8493574
12. Gossage L, Eisen T, Maher ER. VHL, the Story of a Tumour Suppressor Gene. Nat Rev Cancer (2015) 15:55–64. doi: 10.1038/nrc3844
13. Richard S, Gardie B, Couvé S, Gad S. Von Hippel–Lindau: How a Rare Disease Illuminates Cancer Biology. Semin Cancer Biol (2013) 23:26–37. doi: 10.1016/j.semcancer.2012.05.005
14. The European Chromosome 16 Tuberous Sclerosis Consortium. Identification and Characterization of the Tuberous Sclerosis Gene on Chromosome 16. Cell (1993) 75:1305–15. doi: 10.1016/0092-8674(93)90618-Z
15. Kandt RS, Haines JL, Smith M, Northrup H, Gardner RJM, Short MP, et al. Linkage of an Important Gene Locus for Tuberous Sclerosis to a Chromosome 16 Marker for Polycystic Kidney Disease. Nat Genet (1992) 2:37–41. doi: 10.1038/ng0992-37
16. Liaw D, Marsh DJ, Li J, Dahia PLM, Wang SI, Zheng Z, et al. Germline Mutations of the PTEN Gene in Cowden Disease, an Inherited Breast and Thyroid Cancer Syndrome. Nat Genet (1997) 16:64–7. doi: 10.1038/ng0597-64
17. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis Type 1. Nat Rev Dis Primers (2017) 3:17004. doi: 10.1038/nrdp.2017.4
18. National Institutes of Health Consensus Development Conference, Neurofibromatosis: Conference Statement. Arch Neurol (1988) 45:575–8. doi: 10.1001/archneur.1988.00520290115023
19. Lodish MB, Stratakis CA. Endocrine Tumours in Neurofibromatosis Type 1, Tuberous Sclerosis and Related Syndromes. Best Pract Res Clin Endocrinol Metab (2010) 24:439–49. doi: 10.1016/j.beem.2010.02.002
20. Sgambati MT, Stolle C, Choyke PL, Walther MM, Zbar B, Linehan WM, et al. Mosaicism in Von Hippel–Lindau Disease: Lessons From Kindreds With Germline Mutations Identified in Offspring With Mosaic Parents. Am J Hum Genet (2000) 66:84–91. doi: 10.1086/302726
21. Coppin L, Plouvier P, Crépin M, Jourdain A-S, Ait Yahya E, Richard S, et al. Optimization of Next-Generation Sequencing Technologies for Von Hippel Lindau (VHL) Mosaic Mutation Detection and Development of Confirmation Methods. J Mol Diagn (2019) 21:462–70. doi: 10.1016/j.jmoldx.2019.01.005
22. Northrup H, Krueger DA, Northrup H, Krueger DA, Roberds S, Smith K, et al. Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol (2013) 49:243–54. doi: 10.1016/j.pediatrneurol.2013.08.001
23. Dworakowska D, Grossman AB. Are Neuroendocrine Tumours a Feature of Tuberous Sclerosis? A Systematic Review. Endocr Relat Cancer (2009) 16:45–58. doi: 10.1677/ERC-08-0142
24. Eng C. Will the Real Cowden Syndrome Please Stand Up: Revised Diagnostic Criteria. J Med Genet (2000) 37:828–30. doi: 10.1136/jmg.37.11.828
25. Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. Incidence and Clinical Characteristics of Thyroid Cancer in Prospective Series of Individuals With Cowden and Cowden-Like Syndrome Characterized by Germline PTEN, SDH, or KLLN Alterations. J Clin Endocrinol Metab (2011) 12:E2063–71. doi: 10.1210/jc.2011-1616
26. Son EJ, Nosé V. Familial Follicular Cell-Derived Thyroid Carcinoma. Front Endocrinol (2012) 3:3–61. doi: 10.3389/fendo.2012.00061
27. Bubien V, Bonnet F, Brouste V, Hoppe S, Barouk-Simonet E, David A, et al. High Cumulative Risks of Cancer in Patients With PTEN Hamartoma Tumour Syndrome. J Med Genet (2013) 50:255–63. doi: 10.1136/jmedgenet-2012-101339
28. Szabo Yamashita T, Baky FJ, McKenzie TJ, Thompson GB, Farley DR, Lyden ML, et al. Occurrence and Natural History of Thyroid Cancer in Patients With Cowden Syndrome. Eur Thyroid J (2020) 9:243–6. doi: 10.1159/000506422
29. Alrezk R, Suarez A, Tena I, Pacak K. Update of Pheochromocytoma Syndromes: Genetics, Biochemical Evaluation, and Imaging. Front Endocrinol (2018) 9:515. doi: 10.3389/fendo.2018.00515
30. Crona J, Taïeb D, Pacak K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr Rev (2017) 38:489–515. doi: 10.1210/er.2017-00062
31. Hamidi O, Young WF, Iñiguez-Ariza NM, Kittah NE, Gruber L, Bancos C, et al. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J Clin Endocrinol Metab (2017) 102:3296–305. doi: 10.1210/jc.2017-00992
32. Gimenez-Roqueplo A-P, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, et al. Mutations in the SDHB Gene Are Associated With Extra-adrenal and/or Malignant Phaeochromocytomas. Cancer Res (2003) 63:5615–21.
33. Eisenhofer G, Klink B, Richter S, Lenders JW, Robledo M. Metabologenomics of Phaeochromocytoma and Paraganglioma: An Integrated Approach for Personalised Biochemical and Genetic Testing. Clin Biochem Rev (2017) 38:69–100.
34. Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, et al. Pheochromocytoma Catecholamine Phenotypes and Prediction of Tumor Size and Location by Use of Plasma Free Metanephrines. Clin Chem (2005) 51:735–44. doi: 10.1373/clinchem.2004.045484
35. Kimura N, Miura Y, Nagatsu I, Nagura H. Catecholamine Synthesizing Enzymes in 70 Cases of Functioning and non-Functioning Phaeochromocytoma and Extra-Adrenal Paraganglioma. Vichows Arch A Pathol Anat (1992) 421:25–32. doi: 10.1007/BF01607135
36. Rao D, Peitzsch M, Prejbisz A, Hanus K, Fassnacht M, Beuschlein F, et al. Plasma Methoxytyramine: Clinical Utility With Metanephrines for Diagnosis of Pheochromocytoma and Paraganglioma. Eur J Endocrinol (2017) 177:103–13. doi: 10.1530/EJE-17-0077
37. Al-Sharefi A, Javaid U, Perros P, Ealing J, Truran P, Nag S, et al. Clinical Presentation and Outcomes of Phaeochromocytomas/Paragangliomas in Neurofibromatosis Type 1. Eur Endocrinol (2019) 15:95. doi: 10.17925/EE.2019.15.2.95
38. Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF, Bancos I. Pheochromocytoma and Paraganglioma in Patients With Neurofibromatosis Type 1. Clin Endocrinol (2017) 86:141–9. doi: 10.1111/cen.13163
39. Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. Von Recklingausen’s Disease and Pheochromocytomas. J Urol (2006) 162:1582–6. doi: 10.1016/S0022-5347(05)68171-2
40. Zografos GN, Vasiliadis GK, Zagouri F, Aggeli C, Korkolis D, Vogiaki S, et al. Pheochromocytoma Associated With Neurofibromatosis Type 1: Concepts and Current Trends. World J Surg Oncol (2010) 8:14. doi: 10.1186/1477-7819-8-14
41. Opocher G, Conton P, Schiavi F, Macino B, Mantero F. Pheochromocytoma in Von Hippel–Lindau Disease and Neurofibromatosis Type 1. Fam Cancer (2005) 4:13–6. doi: 10.1007/s10689-004-6128-y
42. Bausch B, Borozdin W, Neumann HPH. Clinical and Genetic Characteristics of Patients With Neurofibromatosis Type 1 and Pheochromocytoma. N Engl J Med (2006) 354:2729–31. doi: 10.1056/NEJMc066006
43. Bausch B, Borozdin W, Mautner VF, Hoffmann MM, Boehm D, Robledo M, et al. Germline NF1 Mutational Spectra and Loss-of-Heterozygosity Analyses in Patients With Pheochromocytoma and Neurofibromatosis Type 1. J Clin Endocrinol Metab (2007) 92:2784–92. doi: 10.1210/jc.2006-2833
44. Moramarco J, El Ghorayeb N, Dumas N, Nolet S, Boulanger L, Burnichon N, et al. Pheochromocytomas are Diagnosed Incidentally and At Older Age in Neurofibromatosis Type 1. Clin Endocrinol (2017) 86:332–9. doi: 10.1111/cen.13265
45. Kimura N, Miura Y, Miura K, Takahashi N, Osamura RY, Nagatsu I, et al. Adrenal and Retroperitoneal Mixed Neuroendocrine-Neural Tumors. Endocr Pathol (1991) 2:139–47. doi: 10.1007/BF02915454
46. Castoldi L, De Rai P, Marini A, Ferrero S, De Luca VM, Tiberio G. Neurofibromatosis-1 and Ampullary Gangliocytic Paraganglioma Causing Biliary and Pancreatic Obstruction. Int J Pancreatol (2001) 29:93–7. doi: 10.1385/IJGC:29:2:093
47. Okubo Y, Yoshioka E, Suzuki M, Washimi K, Kawachi K, Kameda Y, et al. Diagnosis, Pathological Findings, and Clinical Management of Gangliocytic Paraganglioma: A Systematic Review. Front Oncol (2018) 8:291. doi: 10.3389/fonc.2018.00291
48. Taïeb D, Hicks RJ, Hindié E, Guillet BA, Avram A, Ghedini P, et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for Radionuclide Imaging of Phaeochromocytoma and Paraganglioma. Eur J Nucl Med Mol Imaging (2019) 46:2112–37. doi: 10.1007/s00259-019-04398-1
49. Li SR, Nicholson KJ, Mccoy KL, Carty SE, Yip L. Clinical and Biochemical Features of Pheochromocytoma Characteristic of Von Hippel–Lindau Syndrome. World J Surg (2020) 44:570–7. doi: 10.1007/s00268-019-05299-y
50. Aufforth RD, Ramakant P, Sadowski SM, Mehta A, Trebska-McGowan K, Nilubol N, et al. Pheochromocytoma Screening Initiation and Frequency in Von Hippel-Lindau Syndrome. J Clin Endocrinol Metab (2015) 100:4498–504. doi: 10.1210/jc.2015-3045
51. Kittah NE, Gruber LM, Bancos I, Hamidi O, Tamhane S, Iñiguez-Ariza N, et al. Bilateral Pheochromocytoma: Clinical Characteristics, Treatment and Longitudinal Follow-Up. Clin Endocrinol (2020) 3:288–95. doi: 10.1111/cen.14222
52. Fagundes GFC, Petenuci J, Lourenco DM, Trarbach EB, Pereira MAA, Correa D’Eur JE, et al. New Insights Into Pheochromocytoma Surveillance of Young Patients With VHL Missense Mutations. J Endocr Soc (2019) 3:1682–92. doi: 10.1210/js.2019-00225
53. Lenders JWM, Duh Q-Y, Eisenhofer G, Gimenez-Roqueplo A-P, Grebe SKG, Murad MH, et al. Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2014) 99:1915–42. doi: 10.1210/jc.2014-1498
54. Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell (2013) 23:739–52. doi: 10.1016/j.ccr.2013.04.018
55. Weisbrod AB, Kitano M, Gesuwan K, Millo C, Herscovitch P, Nilubol N, et al. Clinical Utility of Functional Imaging With 18F-FDOPA in Von Hippel-Lindau Syndrome. J Clin Endocrinol Metab (2012) 97:E613–7. doi: 10.1210/jc.2011-2626
56. Shell J, Tirosh A, Millo C, Sadowski SM, Assadipour Y, Green P, et al. The Utility of 68Gallium-DOTATATE PET/CT in the Detection of Von Hippel-Lindau Disease Associated Tumors. Eur J Radiol (2019) 112:130–5. doi: 10.1016/j.ejrad.2018.11.023
57. Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, et al. Genetics, Diagnosis, Management and Future Directions of Research of Phaeochromocytoma and Paraganglioma: A Position Statement and Consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens (2020) 38:1443–56. doi: 10.1097/HJH.0000000000002438
58. Zelinka T, Petrák O, Turková H, Holaj R, Štrauch B, Kršek M, et al. High Incidence of Cardiovascular Complications in Pheochromocytoma. Horm Metab Res (2012) 44:379–84. doi: 10.1055/s-0032-1306294
59. Y-Hassan S, Falhammar H. Cardiovascular Manifestations and Complications of Pheochromocytomas and Paragangliomas. J Clin Med (2020) 9:2435. doi: 10.3390/jcm9082435
60. Eisenhofer G, Rivers G, Rosas AL, Quezado Z, Manger WM, Pacak K. Adverse Drug Reactions in Patients With Phaeochromocytoma. Drug Saf (2007) 30:1031–62. doi: 10.2165/00002018-200730110-00004
61. Rossitti HM, Söderkvist P, Gimm O. Extent of Surgery for Phaeochromocytomas in the Genomic Era. Br J Surg (2018) 105:e84–98. doi: 10.1002/bjs.10744
62. Neumann HPH, Bender BU, Reincke M, Eggstein S, Laubenberger J, Kirste G. Adrenal-Sparing Surgery for Phaeochromocytoma. Br J Surg (1999) 86:94–7. doi: 10.1046/j.1365-2168.1999.00974.x
63. Castinetti F, Taieb D, Henry JF, Walz M, Guerin C, Brue T, et al. MANAGEMENT OF ENDOCRINE DISEASE: Outcome of Adrenal Sparing Surgery in Heritable Pheochromocytoma. Eur J Endocrinol (2016) 174:R9–R18. doi: 10.1530/EJE-15-0549
64. Erichsen MM, Løvås K, Fougner KJ, Svartberg J, Hauge ER, Bollerslev J, et al. Normal Overall Mortality Rate in Addison’s Disease, But Young Patients are At Risk of Premature Death. Eur J Endocrinol (2009) 160:233–7. doi: 10.1530/EJE-08-0550
65. Tiemensma J, Andela CD, Kaptein AA, Romijn JA, van der Mast RC, Biermasz NR, et al. Psychological Morbidity and Impaired Quality of Life in Patients With Stable Treatment for Primary Adrenal Insufficiency: Cross-Sectional Study and Review of the Literature. Eur J Endocrinol (2014) 171:171–82. doi: 10.1530/EJE-14-0023
66. Brauckhoff M. Critical Size of Residual Adrenal Tissue and Recovery From Impaired Early Postoperative Adrenocortical Function After Subtotal Bilateral Adrenalectomy. Surgery (2003) 134:1020–7. doi: 10.1016/j.surg.2003.08.005
67. Fang F, Ding L, He Q, Liu M. Preoperative Management of Pheochromocytoma and Paraganglioma. Front Endocrinol (2020) 11:586795. doi: 10.3389/fendo.2020.586795
68. Ku YK, Sangla K, Tan YM, Williams DJ. A Case of Using Cortical Sparing Adrenalectomy to Manage Bilateral Phaeochromocytoma in Neurofibromatosis Type 1. Internal Med J (2010) 40:239–40. doi: 10.1111/j.1445-5994.2010.02181.x
69. Benhammou JN, Boris RS, Pacak K, Pinto PA, Linehan WM, Bratslavsky G. Functional and Oncologic Outcomes of Partial Adrenalectomy for Pheochromocytoma in Patients With Von Hippel-Lindau Syndrome After At Least 5 Years of Followup. J Urol (2010) 184:1855–9. doi: 10.1016/j.juro.2010.06.102
70. Neumann HPH, Tsoy U, Bancos I, Amodru V, Walz MK, Tirosh A, et al. Comparison of Pheochromocytoma-Specific Morbidity and Mortality Among Adults With Bilateral Pheochromocytomas Undergoing Total Adrenalectomy vs Cortical-Sparing Adrenalectomy. JAMA Netw Open (2019) 2:e198898. doi: 10.1001/jamanetworkopen.2019.8898
71. Volkin D, Yerram N, Ahmed F, Lankford D, Baccala A, Gupta GN, et al. Partial Adrenalectomy Minimizes the Need for Long-Term Hormone Replacement in Pediatric Patients With Pheochromocytoma and Von Hippel-Lindau Syndrome. J Pediatr Surg (2012) 47:2077–82. doi: 10.1016/j.jpedsurg.2012.07.003
72. Fassnacht M, Assie G, Baudin E, Eisenhofer G, de la Fouchardiere C, Haak HR, et al. Adrenocortical Carcinomas and Malignant Phaeochromocytomas: ESMO–EURACAN Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann Oncol (2020) 31:1476–90. doi: 10.1016/j.annonc.2020.08.2099
73. Otoukesh S, Cooper CJ, Lou W, Mojtahedzadeh M, Nasrazadani A, Wampler M, et al. Combination Chemotherapy Regimen in a Patient With Metastatic Malignant Pheochromocytoma and Neurofibromatosis Type 1. Am J Case Rep (2014) 15:123–7. doi: 10.12659/AJCR.890181
74. Jimenez C, Cabanillas ME, Santarpia L, Jonasch E, Kyle KL, Lano EA, et al. Use of the Tyrosine Kinase Inhibitor Sunitinib in a Patient With Von Hippel-Lindau Disease: Targeting Angiogenic Factors in Pheochromocytoma and Other Von Hippel-Lindau Disease-Related Tumors. J Clin Endocrinol Metab (2009) 94:386–91. doi: 10.1210/jc.2008-1972
75. Miller DT, Freedenberg D, Schorry E, Ullrich NJ, Viskochil D, Korf BR, et al. Health Supervision for Children With Neurofibromatosis Type 1. Pediatrics (2019) 143:e20190660. doi: 10.1542/peds.2019-0660
76. Stewart DR, Korf BR, Nathanson KL, Stevenson DA, Yohay K. Care of Adults With Neurofibromatosis Type 1: A Clinical Practice Resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med (2018) 20:671–82. doi: 10.1038/gim.2018.28
77. France Network NF, Bergqvist C, Servy A, Valeyrie-Allanore L, Ferkal S, Combemale P, et al. Neurofibromatosis 1 French National Guidelines Based on an Extensive Literature Review Since 1966. Orphanet J Rare Dis (2020) 15:37. doi: 10.1186/s13023-020-1310-3
78. Hirbe AC, Gutmann DH. Neurofibromatosis Type 1: A Multidisciplinary Approach to Care. Lancet Neurol (2014) 13:834–43. doi: 10.1016/S1474-4422(14)70063-8
79. Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, et al. Guidelines for the Diagnosis and Management of Individuals With Neurofibromatosis 1. J Med Genet (2006) 44:81–8. doi: 10.1136/jmg.2006.045906
80. Shinall MC, Solórzano CC. Pheochromocytoma in Neurofibromatosis Type 1: When Should it Be Suspected. Endocr Pract (2014) 20:792–6. doi: 10.4158/EP13417.OR
81. Képénékian L, Mognetti T, Lifante J-C, Giraudet A-L, Houzard C, Pinson S, et al. Interest of Systematic Screening of Pheochromocytoma in Patients With Neurofibromatosis Type 1. Eur J Endocrinol (2016) 175:335–44. doi: 10.1530/EJE-16-0233
82. Cornu E, Motiejunaite J, Belmihoub I, Vidal-Petiot E, Mirabel M, Amar L. Acute Stress Cardiomyopathy: Heart of Pheochromocytoma. Ann Endocrinol (2020) S0003–4266(20):30046–9. doi: 10.1016/j.ando.2020.03.011
83. Vermalle M, Tabarin A, Castinetti F. Hereditary Pheochromocytoma and Paraganglioma: Screening and Follow-Up Strategies in Asymptomatic Mutation Carriers. Ann Endocrinol (2018) 79:S10–21. doi: 10.1016/S0003-4266(18)31234-4
84. Buffet A, Ben Aim L, Leboulleux S, Drui D, Vezzosi D, Libé R, et al. Positive Impact of Genetic Test on the Management and Outcome of Patients With Paraganglioma and/or Pheochromocytoma. J Clin Endocrinol Metab (2019) 104:1109–18. doi: 10.1210/jc.2018-02411
85. Bancos I, Atkinson E, Eng C, Young WF, Neumann HPH, Yukina M, et al. Maternal and Fetal Outcomes in Phaeochromocytoma and Pregnancy: A Multicentre Retrospective Cohort Study and Systematic Review of Literature. Lancet Diabetes Endocrinol (2021) 9:13–21. doi: 10.1016/S2213-8587(20)30363-6
86. Rednam SP, Erez A, Druker H, Janeway KA, Kamihara J, Kohlmann WK, et al. Von Hippel–Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. Clin Cancer Res (2017) 23:e68–75. doi: 10.1158/1078-0432.CCR-17-0547
87. Binderup ML, Bisgaard ML, Harbud V, Møller HU, Gimsing S, Friis-Hansen L, et al. Danish vHL Coordination Group. Von Hippel-Lindau disease (vHL). National Clinical Guideline for Diagnosis and Surveillance in Denmark. 3rd edition. Dan Med J (2013) 60(12):B4763.
88. PREDIR National Reference Center For Rare Cancers. French Recommendations for Clinical Surveillance in Von Hippel Lindau (2018). Available at: http://www.vhlfrance.org/wp-content/uploads/2019/03/Fiche-de-recommandation-surveillance-clinique-VHL-France-du-28-ao%C3%BBt-2018.pdf.
89. Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic Neuroendocrine Tumours. Lancet Oncol (2008) 9:61–72. doi: 10.1016/S1470-2045(07)70410-2
90. Rindi G, Wiedenmann B. Neuroendocrine Neoplasia of the Gastrointestinal Tract Revisited: Towards Precision Medicine. Nat Rev Endocrinol (2020) 16:590–607. doi: 10.1038/s41574-020-0391-3
91. Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, et al. The 2019 WHO Classification of Tumours of the Digestive System. Histopathology (2020) 76:182–8. doi: 10.1111/his.13975
92. Gill AJ. Why did They Change That? Practical Implications of the Evolving Classification of Neuroendocrine Tumours of the Gastrointestinal Tract. Histopathology (2021) 78:162–70. doi: 10.1111/his.14172
93. Lombard-Bohas C, Mitry E, O’Toole D, Louvet C, Pillon D, Cadiot G, et al. Thirteen-Month Registration of Patients With Gastroenteropancreatic Endocrine Tumours in France. Neuroendocrinology (2009) 89:217–22. doi: 10.1159/000151562
94. Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol (2017) 3:1335. doi: 10.1001/jamaoncol.2017.0589
95. Pape U, Jann H, Müller-Nordhorn J, Bockelbrink A, Berndt U, Willich SN, et al. Prognostic Relevance of a Novel TNM Classification System for Upper Gastroenteropancreatic Neuroendocrine Tumors. Cancer (2008) 113:256–65. doi: 10.1002/cncr.23549
96. Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One Hundred Years After “Carcinoid”: Epidemiology of and Prognostic Factors for Neuroendocrine Tumors in 35,825 Cases in the United States. J Clin Oncol (2008) 26:3063–72. doi: 10.1200/JCO.2007.15.4377
97. Couvelard A, Scoazec J-Y. Inherited Tumor Syndromes of Gastroenteropancreatic and Thoracic Neuroendocrine Neoplasms. Ann Pathol (2020) 40:120–33. doi: 10.1016/j.annpat.2020.01.002
98. Relles D, Baek J, Witkiewicz A, Yeo CJ. Periampullary and Duodenal Neoplasms in Neurofibromatosis Type 1: Two Cases and an Updated 20-Year Review of the Literature Yielding 76 Cases. J Gastrointest Surg (2010) 14:1052–61. doi: 10.1007/s11605-009-1123-0
99. Tanaka S, Yamasaki S, Matsushita H, Ozawa Y, Kurosaki A, Takeuchi K, et al. Duodenal Somatostatinoma: A Case Report and Review of 31 Cases With Special Reference to the Relationship Between Tumor Size and Metastasis. Pathol Int (2000) 50:146–52. doi: 10.1046/j.1440-1827.2000.01016.x
100. Vanoli A, La Rosa S, Klersy C, Grillo F, Albarello L, Inzani F, et al. Four Neuroendocrine Tumor Types and Neuroendocrine Carcinoma of the Duodenum: Analysis of 203 Cases. Neuroendocrinology (2017) 104:112–25. doi: 10.1159/000444803
101. Samonakis DN, Quaglia A, Joshi NM, Tibballs JM, Nagree A, Triantos CK, et al. Obstructive Jaundice Secondary to Neuroendocrine Tumour in a Patient With Von Recklinghausen’s Disease. Eur J Gastroenterol Hepatol (2005) 17:1229–32. doi: 10.1097/00042737-200511000-00012
102. Sandru F, Carsote M, Valea A, Albu SE, Petca R-C, Dumitrascu MC. Somatostatinoma: Beyond Neurofibromatosis Type 1. Exp Ther Med (2020) 4:3383–8. doi: 10.3892/etm.2020.8965
103. Alshikho MJ, Noureldine SI, Talas JM, Nasimian A, Zazou S, Mobaed B, et al. Zollinger-Ellison Syndrome Associated With Von Recklinghausen Disease: Case Report and Literature Review. Am J Case Rep (2016) 17:398–405. doi: 10.12659/AJCR.898472
104. Rogers A, Wang LM, Karavitaki N, Grossman AB. Neurofibromatosis Type 1 and Pancreatic Islet Cell Tumours: An Association Which Should be Recognized. QJM (2015) 108:573–6. doi: 10.1093/qjmed/hcs203
105. Salvi PF, Lorenzon L, Caterino S, Antolino L, Antonelli MS, Balducci G. Gastrointestinal Stromal Tumors Associated With Neurofibromatosis 1: A Single Centre Experience and Systematic Review of the Literature Including 252 Cases. Int J Surg Oncol (2013) 2013:1–8. doi: 10.1155/2013/398570
106. Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, Nupponen N, Kindblom L-G. NF1-Associated Gastrointestinal Stromal Tumors Have Unique Clinical, Phenotypic, and Genotypic Characteristics. Am J Surg Pathol (2005) 29:1170–6. doi: 10.1097/01.pas.0000159775.77912.15
107. Hammel PR, Vilgrain V, Terris B, Penfornis A, Sauvanet A, Correas J, et al. Pancreatic Involvement in Von Hippel–Lindau Disease. Gastroenterology (2000) 119:1087–95. doi: 10.1053/gast.2000.18143
108. Corcos O, Couvelard A, Giraud S, Vullierme M-P, O’Toole D, Rebours V, et al. Endocrine Pancreatic Tumors in Von Hippel-Lindau Disease: Clinical, Histological, and Genetic Features. Pancreas (2008) 37:85–93. doi: 10.1097/MPA.0b013e31815f394a
109. Blansfield JA, Choyke L, Morita SY, Choyke PL, Pingpank JF, Alexander HR, et al. Clinical, Genetic and Radiographic Analysis of 108 Patients With Von Hippel-Lindau Disease (VHL) Manifested by Pancreatic Neuroendocrine Tumors (PNETs). Surgery (2007) 142:814–8.e2. doi: 10.1016/j.surg.2007.09.012
110. Tirosh A, El Lakis M, Green P, Nockel P, Patel D, Nilubol N, et al. In Silico VHL Gene Mutation Analysis and Prognosis of Pancreatic Neuroendocrine Tumors in Von Hippel–Lindau Disease. J Clin Endocrinol Metab (2018) 103:1631–8. doi: 10.1210/jc.2017-02434
111. Krauss T, Ferrara AM, Links TP, Wellner U, Bancos I, Kvachenyuk A, et al. Preventive Medicine of Von Hippel–Lindau Disease-Associated Pancreatic Neuroendocrine Tumors. Endocr Relat Cancer (2018) 25:783–93. doi: 10.1530/ERC-18-0100
112. Igarashi H, Ito T, Nishimori I, Tamura K, Yamasaki I, Tanaka M, et al. Pancreatic Involvement in Japanese Patients With Von Hippel-Lindau Disease: Results of a Nationwide Survey. J Gastroenterol (2014) 49:511–6. doi: 10.1007/s00535-013-0794-1
113. Zhang C, Jin J, Xie J, Ye L, Su T, Jiang L, et al. The Clinical Features and Molecular Mechanisms of ACTH-secreting Pancreatic Neuroendocrine Tumors. J Clin Endocrinol Metab (2020) 105:dgaa507. doi: 10.1210/clinem/dgaa507
114. Speisky D, Duces A, Bieche I, Rebours V, Hammel P, Sauvanet A, et al. Molecular Profiling of Pancreatic Neuroendocrine Tumors in Sporadic and Von Hippel-Lindau Patients. Clin Cancer Res (2012) 18:2838–49. doi: 10.1158/1078-0432.CCR-11-2759
115. Keutgen XM, Kumar S, Gara S, Boufraqech M, Agarwal S, Hruban RH, et al. Transcriptional Alterations in Hereditary and Sporadic Nonfunctioning Pancreatic Neuroendocrine Tumors According to Genotype. Cancer (2018) 124:636–47. doi: 10.1002/cncr.31057
116. Charlesworth M, Verbeke CS, Falk GA, Walsh M, Smith AM, Morris-Stiff G. Pancreatic Lesions in Von Hippel–Lindau Disease? A Systematic Review and Meta-synthesis of the Literature. J Gastrointest Surg (2012) 16:1422–8. doi: 10.1007/s11605-012-1847-0
117. Yamasaki I, Nishimori I, Ashida S, Kohsaki T, Onishi S, Shuin T. Clinical Characteristics of Pancreatic Neuroendocrine Tumors in Japanese Patients With Von Hippel-Lindau Disease. Pancreas (2006) 33:382–5. doi: 10.1097/01.mpa.0000240604.26312.e4
118. Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic Neuroendocrine Tumors (PNETs): Incidence, Prognosis and Recent Trend Toward Improved Survival. Ann Oncol (2008) 19:1727–33. doi: 10.1093/annonc/mdn351
119. Sharma A, Mukewar S, Vege SS. Clinical Profile of Pancreatic Cystic Lesions in Von Hippel-Lindau Disease: A Series of 48 Patients Seen At a Tertiary Institution. Pancreas (2017) 46:948–52. doi: 10.1097/MPA.0000000000000875
120. Oberg K, Couvelard A, Delle Fave G, Gross D, Grossman A, Jensen RT, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Biochemical Markers. Neuroendocrinology (2017) 105:201–11. doi: 10.1159/000472254
121. Weisbrod AB, Kitano M, Thomas F, Williams D, Gulati N, Gesuwan K, et al. Assessment of Tumor Growth in Pancreatic Neuroendocrine Tumors in Von Hippel Lindau Syndrome. J Am Coll Surg (2014) 218:163–9. doi: 10.1016/j.jamcollsurg.2013.10.025
122. Mafficini A, Scarpa A. Genetics and Epigenetics of Gastroenteropancreatic Neuroendocrine Neoplasms. Endocr Rev (2019) 40:506–36. doi: 10.1210/er.2018-00160
123. Larson A, Hedgire S, Deshpande V, Stemmer-Rachamimov A, Harisinghani M, Ferrone C, et al. Pancreatic Neuroendocrine Tumors in Patients With Tuberous Sclerosis Complex. Clin Genet (2012) 82:558–63. doi: 10.1111/j.1399-0004.2011.01805.x
124. Mehta S, Rusyn L, Ginsburg H, Hajdu C, Kohn B. Pancreatic Neuroendocrine Tumor in a Young Child With Tuberous Sclerosis Complex 1. J Endocr Soc (2019) 3:1201–6. doi: 10.1210/js.2019-00051
125. Borson-Chazot F, Cardot-Bauters C, Mirallie É, Pattou F. Insulinoma of Genetic Aetiology. Ann Endocrinol (2013) 74:200–2. doi: 10.1016/j.ando.2013.05.006
126. Crona J, Skogseid B. GEP- NETS UPDATE: Genetics of Neuroendocrine Tumors. Eur J Endocrinol (2016) 174:R275–90. doi: 10.1530/EJE-15-0972
127. Kolin DL, Duan K, Ngan B, Gerstle JT, Krzyzanowska MK, Somers GR, et al. Expanding the Spectrum of Colonic Manifestations in Tuberous Sclerosis: L-Cell Neuroendocrine Tumor Arising in the Background of Rectal Pecoma. Endocr Pathol (2018) 29:21–6. doi: 10.1007/s12022-017-9497-0
128. Falconi M, Eriksson B, Kaltsas G, Bartsch DK, Capdevila J, Caplin M, et al. Enets Consensus Guidelines Update for the Management of Patients With Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology (2016) 103:153–71. doi: 10.1159/000443171
129. Angeletti S, Corleto VD, Schillaci O, Marignani M, Annibale B, Moretti A, et al. Use of the Somatostatin Analogue Octreotide to Localise and Manage Somatostatin-Producing Tumours. Gut (1998) 42:792–4. doi: 10.1136/gut.42.6.792
130. Hofland J, Kaltsas G, de Herder WW. Advances in the Diagnosis and Management of Well-Differentiated Neuroendocrine Neoplasms. Endocr Rev (2020) 41:bnz004. doi: 10.1210/endrev/bnz004
131. Healy ML, Dawson SJ, Murray RML, Zalcberg J, Jefford M. Severe Hypoglycaemia After Long-Acting Octreotide in a Patient With an Unrecognized Malignant Insulinoma. Internal Med J (2007) 37:406–9. doi: 10.1111/j.1445-5994.2007.01371.x
132. Baudin E, Caron P, Lombard-Bohas C, Tabarin A, Mitry E, Reznick Y, et al. Malignant Insulinoma: Recommendations for Characterisation and Treatment. Ann Endocrinol (2013) 74:523–33. doi: 10.1016/j.ando.2013.07.001
133. Hendren NS, Panach K, Brown TJ, Peng L, Beg MS, Weissler J, et al. Pasireotide for the Treatment of Refractory Hypoglycaemia From Malignant Insulinoma. Clin Endocrinol (2018) 88:341–3. doi: 10.1111/cen.13503
134. Kulke MH. A Randomized, Open-Label, Phase 2 Study of Everolimus in Combination With Pasireotide LAR or Everolimus Alone in Advanced, Well-Differentiated, Progressive Pancreatic Neuroendocrine Tumors: COOPERATE-2 Trial. Ann Oncol (2017) 28:7. doi: 10.1093/annonc/mdx078
135. de Mestier L, Lepage C, Baudin E, Coriat R, Courbon F, Couvelard A, et al. Digestive Neuroendocrine Neoplasms (NEN): French Intergroup Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up (SNFGE, GTE, RENATEN, TENPATH, FFCD, GERCOR, UNICANCER, SFCD, SFED, SFRO, SFR). Dig Liver Dis (2020) 5:473–92. doi: 10.1016/j.dld.2020.02.011
136. Pavel M, Öberg K, Falconi M, Krenning EP, Sundin A, Perren A, et al. Gastroenteropancreatic Neuroendocrine Neoplasms: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann Oncol (2020) 31:844–60. doi: 10.1016/j.annonc.2020.03.304
137. Klesse LJ, Jordan JT, Radtke HB, Rosser T, Schorry E, Ullrich N, et al. The Use of MEK Inhibitors in Neurofibromatosis Type 1–Associated Tumors and Management of Toxicities. Oncol (2020) 7:1109–16. doi: 10.1634/theoncologist.2020-0069
138. Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N Engl J Med (2016) 375:2550–60. doi: 10.1056/NEJMoa1605943
139. Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A Phase I Trial of the MEK Inhibitor Selumetinib (AZD6244) in Pediatric Patients With Recurrent or Refractory Low-Grade Glioma: A Pediatric Brain Tumor Consortium (PBTC) Study. Neuro Oncol (2017) 19:1135–44. doi: 10.1093/neuonc/now282
140. Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in Paediatric Patients With BRAF-aberrant or Neurofibromatosis Type 1-Associated Recurrent, Refractory, or Progressive Low-Grade Glioma: A Multicentre, Phase 2 Trial. Lancet Oncol (2019) 20:1011–22. doi: 10.1016/S1470-2045(19)30277-3
141. Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in Children With Inoperable Plexiform Neurofibromas. N Engl J Med (2020) 382:1430–42. doi: 10.1056/NEJMoa1912735
142. de Mestier L, Gaujoux S, Cros J, Hentic O, Vullierme M-P, Couvelard A, et al. Long-Term Prognosis of Resected Pancreatic Neuroendocrine Tumors in Von Hippel-Lindau Disease Is Favorable and Not Influenced by Small Tumors Left in Place. Ann Surg (2015) 262:384–8. doi: 10.1097/SLA.0000000000000856
143. Libutti SK, Choyke PL, Bartlett DL, Vargas H, Walther M, Lubensky I, et al. Pancreatic Neuroendocrine Tumors Associated With Von Hippel Lindau Disease: Diagnostic and Management Recommendations. Surgery (1998) 124:1153–9. doi: 10.1067/msy.1998.91823
144. Keutgen XM, Hammel P, Choyke PL, Libutti SK, Jonasch E, Kebebew E. Evaluation and Management of Pancreatic Lesions in Patients With Von Hippel–Lindau Disease. Nat Rev Clin Oncol (2016) 13:537–49. doi: 10.1038/nrclinonc.2016.37
145. Howe JR, Merchant NB, Conrad C, Keutgen XM, Hallet J, Drebin JA, et al. The North American Neuroendocrine Tumor Society Consensus Paper on the Surgical Management of Pancreatic Neuroendocrine Tumors. Pancreas (2020) 49:1–33. doi: 10.1097/MPA.0000000000001454
146. Dai M, Xing C, Shi N, Wang S, Wu G, Liao Q, et al. Risk Factors for New-Onset Diabetes Mellitus After Distal Pancreatectomy. BMJ Open Diabetes Res Care (2020) 8:e001778. doi: 10.1136/bmjdrc-2020-001778
147. Maxwell DW, Jajja MR, Galindo RJ, Zhang C, Nadeem SO, Sweeney JF, et al. Post-Pancreatectomy Diabetes Index: A Validated Score Predicting Diabetes Development After Major Pancreatectomy. J Am Coll Surg (2020) 230:393–402.e3. doi: 10.1016/j.jamcollsurg.2019.12.016
148. Renaud F, Chetboun M, Thevenet J, Delalleau N, Gmyr V, Hubert T, et al. Safety of Islet Autotransplantation After Pancreatectomy for Adenocarcinoma. Transplantation (2019) 103:177–81. doi: 10.1097/TP.0000000000002419
149. Wojtusciszyn A, Branchereau J, Esposito L, Badet L, Buron F, Chetboun M, et al. Indications for Islet or Pancreatic Transplantation: Statement of the TREPID Working Group on Behalf of the Société Francophone Du Diabète (SFD), Société Francaise D’endocrinologie (SFE), Société Francophone De Transplantation (SFT) and Société Française De Néphrologie – Dialyse – Transplantation (SFNDT). Diabetes Metab (2019) 45:224–37. doi: 10.1016/j.diabet.2018.07.006
150. Raymond E, Dahan L, Raoul J-L, Bang Y-J, Borbath I, Lombard-Bohas C, et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N Engl J Med (2011) 364:501–13. doi: 10.1056/NEJMoa1003825
151. Jonasch E, McCutcheon IE, Waguespack SG, Wen S, Davis DW, Smith LA, et al. Pilot Trial of Sunitinib Therapy in Patients With Von Hippel–Lindau Disease. Ann Oncol (2011) 22:2661–6. doi: 10.1093/annonc/mdr011
152. Ma K, Hong B, Zhou J, Gong Y, Wang J, Liu S, et al. The Efficacy and Safety of Tyrosine Kinase Inhibitors for Von Hippel–Lindau Disease: A Retrospective Study of 32 Patients. Front Oncol (2019) 9:1122. doi: 10.3389/fonc.2019.01122
153. Jonasch E, McCutcheon IE, Gombos DS, Ahrar K, Perrier ND, Liu D, et al. Pazopanib in Patients With Von Hippel-Lindau Disease: A Single-Arm, Single-Centre, Phase 2 Trial. Lancet Oncol (2018) 19:1351–9. doi: 10.1016/S1470-2045(18)30487-X
154. Choueiri TK, Kaelin WG. Targeting the HIF2–VEGF Axis in Renal Cell Carcinoma. Nat Med (2020) 26:1519–30. doi: 10.1038/s41591-020-1093-z
155. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan V, Maughan BL, et al. Phase II Study of the Oral HIF-2α Inhibitor MK-6482 for Von Hippel-Lindau Disease–Associated Renal Cell Carcinoma. J Clin Oncol (2020) 38:5003–3. doi: 10.1200/JCO.2020.38.15_suppl.5003
156. Pavel M, O’’Toole D, Costa F, Capdevila J, Gross D, Kianmanesh R, et al. ENETS Consensus Guidelines Update for the Management of Distant Metastatic Disease of Intestinal, Pancreatic, Bronchial Neuroendocrine Neoplasms (NEN) and NEN of Unknown Primary Site. Neuroendocrinology (2016) 103:172–85. doi: 10.1159/000443167
157. Franz DN. Efficacy and Safety of Everolimus for Subependymal Giant Cell Astrocytomas Associated With Tuberous Sclerosis Complex (EXIST-1): A Multicentre, Randomised, Placebo-Controlled Phase 3 Trial. Lancet (2013) 381:8. doi: 10.1016/S0140-6736(12)61134-9
158. Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, et al. Everolimus for Angiomyolipoma Associated With Tuberous Sclerosis Complex or Sporadic Lymphangioleiomyomatosis (EXIST-2): A Multicentre, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet (2013) 381:817–24. doi: 10.1016/S0140-6736(12)61767-X
159. Tirosh A, Journy N, Folio LR, Lee C, Leite C, Yao J, et al. Cumulative Radiation Exposures From CT Screening and Surveillance Strategies for Von Hippel-Lindau–Associated Solid Pancreatic Tumors. Radiology (2019) 290:116–24. doi: 10.1148/radiol.2018180687
160. Amin S, Kingswood JC, Bolton PF, Elmslie F, Gale DP, Harland C, et al. The UK Guidelines for Management and Surveillance of Tuberous Sclerosis Complex. QJM Int J Med (2019) 112:171–82. doi: 10.1093/qjmed/hcy215
161. Kodama H, Iihara M, Okamoto T, Obara T. Water-Clear Cell Parathyroid Adenoma Causing Primary Hyperparathyroidism in a Patient With Neurofibromatosis Type 1: Report of a Case. Surg Today (2007) 37:884–7. doi: 10.1007/s00595-007-3484-x
162. Hayashibara N, Ogawa T, Tsuji E, Oya M, Fujii A. Parathyroid Carcinoma Coincident With Neurofibromatosis Type 1. J Clin Exp Oncol (2018) 5:6. doi: 10.4172/2324-9110.1000170
163. Demirjian AN, Grossman JM, Chan JL, Parangi S. Parathyroid Carcinoma and Neurofibromatosis. Surgery (2008) 144:827–9. doi: 10.1016/j.surg.2008.07.015
164. Shinzato Y, Ikehara Y. A Case of Tuberous Sclerosis Complex With Concomitant Primary Hyperparathyroidism Due to Parathyroid Adenoma: A Case Report. World J Surg Oncol (2015) 13:106. doi: 10.1186/s12957-015-0520-y
165. Brewer K, Costa-Guda J, Arnold A. Molecular Genetic Insights Into Sporadic Primary Hyperparathyroidism. Endocr Relat Cancer (2019) 2:R53–72. doi: 10.1530/ERC-18-0304
166. Clarke BL. Epidemiology of Primary Hyperparathyroidism. J Clin Densitom (2013) 16:6. doi: 10.1016/j.jocd.2012.11.009
167. Checa Garrido A, del Pozo Picó C. Acromegaly and Type 1 Neurofibromatosis. Is Association of Both Conditions Due to Chance? Endocrinol Nutr (2013) 60:144–5. doi: 10.1016/j.endoen.2012.01.014
168. Hozumi K, Fukuoka H, Odake Y, Takeuchi T, Uehara T, Sato T, et al. Acromegaly Caused by a Somatotroph Adenoma in Patient With Neurofibromatosis Type 1. Endocr J (2019) 66:853–7. doi: 10.1507/endocrj.EJ19-0035
169. Hannah-Shmouni F, Demidowich AP, Rowell J, Lodish M, Stratakis CA. Large Pituitary Gland With an Expanding Lesion in the Context of Neurofibromatosis 1. BMJ Case Rep (2017) 2017:bcr-2017-222411. doi: 10.1136/bcr-2017-222411
170. Lim CT, Korbonits MK. Update on the Clinicopathology of Pituitary Adenomas. Endocr Pract (2018) 24:473–88. doi: 10.4158/EP-2018-0034
171. Zacharin M. Precocious Puberty in Two Children With Neurofibromatosis Type I in the Absence of Optic Chiasmal Glioma. J Pediatr (1997) 130:155–7. doi: 10.1016/S0022-3476(97)70327-5
172. Cambiaso P, Galassi S, Palmiero M, Mastronuzzi A, Del Bufalo F, Capolino R, et al. Growth Hormone Excess in Children With Neurofibromatosis Type-1 and Optic Glioma. Am J Med Genet (2017) 173:2353–8. doi: 10.1002/ajmg.a.38308
173. Yeung RS, Katsetos CD, Klein-Szanto A. Subependymal Astrocytic Hamartomas in the Eker Rat Model of Tuberous Sclerosis. Am J Pathol (1997) 151:10.
174. Kenerson H, Dundon TA, Yeung RS. Effects of Rapamycin in the Eker Rat Model of Tuberous Sclerosis Complex. Pediatr Res (2005) 57:67–75. doi: 10.1203/01.PDR.0000147727.78571.07
175. Regazzo D, Gardiman MP, Theodoropoulou M, Scaroni C, Occhi G, Ceccato F. Silent Gonadotroph Pituitary Neuroendocrine Tumor in a Patient With Tuberous Sclerosis Complex: Evaluation of a Possible Molecular Link. Endocrinol Diabetes Metab Case Rep (2018) 2018. doi: 10.1530/EDM-18-0086
176. Agrawal N, Akbani R, Aksoy BA, Ally A, Arachchi H, Asa SL, et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell (2014) 159:676–90. doi: 10.1016/j.cell.2014.09.050
177. Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, et al. Characterization of the Mutational Landscape of Anaplastic Thyroid Cancer Via Whole-Exome Sequencing. Hum Mol Genet (2015) 24:2318–29. doi: 10.1093/hmg/ddu749
178. Seminog OO, Goldacre MJ. Risk of Benign Tumours of Nervous System, and of Malignant Neoplasms, in People With Neurofibromatosis: Population-Based Record-Linkage Study. Br J Cancer (2013) 108:193–8. doi: 10.1038/bjc.2012.535
179. Findakly D, Solsi A, Arslan W. A Novel Combination of Metachronous Primary Malignancies of the Thyroid and Breast in a Patient With Neurofibromatosis Type 1. Cureus (2020) 12. doi: 10.7759/cureus.7590
180. Hashiba T, Maruno M, Fujimoto Y, Suzuki T, Wada K, Isaka T, et al. Skull Metastasis From Papillary Thyroid Carcinoma Accompanied by Neurofibromatosis Type 1 and Pheochromocytoma: Report of a Case. Brain Tumor Pathol (2006) 23:97–100. doi: 10.1007/s10014-006-0203-z
181. Miyamoto K, Nakamura M, Suzuki K, Katsuki S, Kaki Y, Inoue G, et al. Diagnosis of Neurofibromatosis Type 1 After Rupture of Aneurysm and Consequent Fatal Hemothorax. Am J Emergency Med (2020) 38:1543.e3–1543.e5. doi: 10.1016/j.ajem.2020.04.004
182. Ercolino T, Lai R, Giachè V, Melchionda S, Carella M, Delitala A, et al. Patient Affected by Neurofibromatosis Type 1 and Thyroid C-cell Hyperplasia Harboring Pathogenic Germ-Line Mutations in Both NF1 and RET Genes. Gene (2014) 536:332–5. doi: 10.1016/j.gene.2013.12.003
183. Anagnostouli M, Piperingos G, Yapijakis C, Gourtzelidis P, Balafouta S, Zournas C, et al. Thyroid Gland Neurofibroma in a NF1 Patient: NF1 and Thyroid Gland. Acta Neurol Scand (2002) 106:58–61. doi: 10.1034/j.1600-0404.2002.01159.x
184. Koksal Y, Sahin M, Koksal H, Esen H, Sen M. Neurofibroma Adjacent to the Thyroid Gland and a Thyroid Papillary Carcinoma in a Patient With Neurofibromatosis Type 1: Report of a Case. Surg Today (2009) 39:884–7. doi: 10.1007/s00595-008-3946-9
185. van Lierop ZYGJ, Jentjens S, Anten MHME, Wierts R, Stumpel CT, Havekes B, et al. Thyroid Gland 18f-Fdg Uptake in Neurofibromatosis Type 1. Eur Thyroid J (2018) 7:155–61. doi: 10.1159/000488706
186. Nishida Y, Ikuta K, Ito S, Urakawa H, Sakai T, Koike H, et al. Limitations and Benefits of FDG-PET/CT in NF1 Patients With Nerve Sheath Tumors: A Cross-Sectional/Longitudinal Study. Cancer Sci (2021) 3:1114–22. doi: 10.1111/cas.14802
187. Robbins HL, Hague A. The PI3K/Akt Pathway in Tumors of Endocrine Tissues. Front Endocrinol (2016) 6:188. doi: 10.3389/fendo.2015.00188
188. Cameselle-Teijeiro J, Fachal C, Cabezas-Agrícola JM, Alfonsín-Barreiro N, Abdulkader I, Vega-Gliemmo A, et al. Thyroid Pathology Findings in Cowden Syndrome: A Clue for the Diagnosis of the PTEN Hamartoma Tumor Syndrome. Am J Clin Pathol (2015) 144:322–8. doi: 10.1309/AJCP84INGJUVTBME
189. Fagin JA, Wells SA. Biologic and Clinical Perspectives on Thyroid Cancer. N Engl J Med (2016) 375:1054–67. doi: 10.1056/NEJMra1501993
190. Laury AR, Bongiovanni M, Tille J-C, Kozakewich H, Nosé V. Thyroid Pathology in PTEN -Hamartoma Tumor Syndrome: Characteristic Findings of a Distinct Entity. Thyroid (2011) 21:135–44. doi: 10.1089/thy.2010.0226
191. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients With Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 26:1–133. doi: 10.1089/thy.2015.0020
192. Hall JE, Abdollahian DJ, Sinard RJ. Thyroid Disease Associated With Cowden Syndrome: A Meta-Analysis. Head Neck (2013) 35:1189–94. doi: 10.1002/hed.22971
193. Milas M, Mester J, Metzger R, Shin J, Mitchell J, Berber E, et al. Should Patients With Cowden Syndrome Undergo Prophylactic Thyroidectomy? Surgery (2012) 152:1201–10. doi: 10.1016/j.surg.2012.08.055
194. Komiya T, Blumenthal GM, DeChowdhury R, Fioravanti S, Ballas MS, Morris J, et al. A Pilot Study of Sirolimus in Subjects With Cowden Syndrome or Other Syndromes Characterized by Germline Mutations in PTEN. Oncologist (2019) 24:1510. doi: 10.1634/theoncologist.2019-0514
195. PHTS Guideline Development Group, The European Reference Network GENTURIS, Tischkowitz M, Colas C, Pouwels S, Hoogerbrugge N. Cancer Surveillance Guideline for Individuals With PTEN Hamartoma Tumour Syndrome. Eur J Hum Genet (2020) 28:1387–93. doi: 10.1038/s41431-020-0651-7
Keywords: neurofibromatosis type 1, von Hippel-Lindau, Cowden syndrome, tuberous sclerosis complex, pheochromocytoma, paraganglioma, digestive neuroendocrine tumors
Citation: Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, Espiard S and Vantyghem MC (2021) Phakomatoses and Endocrine Gland Tumors: Noteworthy and (Not so) Rare Associations. Front. Endocrinol. 12:678869. doi: 10.3389/fendo.2021.678869
Received: 10 March 2021; Accepted: 15 April 2021;
Published: 06 May 2021.
Edited by:
Antongiulio Faggiano, Sapienza University of Rome, ItalyReviewed by:
Hans Ghayee, University of Florida, United StatesJean-Yves Scoazec, Institut Gustave Roussy, France
Copyright © 2021 Chevalier, Dupuis, Jannin, Lemaitre, Do Cao, Cardot-Bauters, Espiard and Vantyghem. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marie Christine Vantyghem, mc-vantyghem@chru-lille.fr
†These authors have contributed equally to this work