 Jacob Bar-Tana
Jacob Bar-TanaIntroduction
The current paradigm of type 2 pre-diabetes/diabetes (T2D) maintains that glycemic control reflects the interplay between insulin production by beta-cells and the peripheral sensitivity/resistance to insulin. Insulin resistance implies failure of insulin to activate glucose uptake in muscle and adipose tissue and glycogen synthesis in liver and muscle, and to inhibit liver gluconeogenesis and adipose lipolysis. The pre-diabetes stage is considered to present progressive resistance to insulin, being offset by insulin hypersecretion by beta-cells, resulting in maintaining plasma glucose levels within the pre-diabetes limits. The overt diabetes stage that follows presents ‘exhaustion’ of beta cells, resulting in progressive hyperglycemia (1).
The close association between insulin secretion and insulin resistance leaves open the question which comes first (2, 3). Similarly, the association between insulin resistance and hyperinsulinemia in normoglycemic normolipemic off springs of T2D parents (4) is still undefined in terms of primary cause-effect. The classical paradigm maintains that insulin resistance is the primary defect of T2D, followed by ‘compensatory’ increase in beta cells insulin production (1, 5). An alternative paradigm maintains that insulin hypersecretion by beta-cells is the primary defect, resulting in hyperinsulinemia which drives peripheral insulin resistance (6). Each of the two paradigms appears to be substantiated by respective examples (2–6). However, the apparent cause-effect relationship outlined by each still remains unresolved in terms of molecular mode(s)-of-mediation. Thus, in spite of previous attempts (e.g., beta-trophin, irisin) no humoral and/or neuronal agents have yet been identified which may mediate between primary insulin resistance and increase in beta cells mass and function during the early normoglycemic normolipemic pre-diabetes stage of T2D. Also, the mode of suppression of the insulin transduction pathway in liver, muscle and adipose fat by primary hyperinsulinemia still remains unresolved. Moreover, plasma insulin levels are further determined by hepatic insulin clearance, which amounts to >50% of the insulin secreted by beta-cells (7). Indeed, hepatic insulin clearance is significantly inhibited in T2D patients, thereby synergizing with insulin hypersecretion by pre-diabetes beta cells (8, 9). Thus, the egg-chicken riddle of insulin resistance and secretion appears to be further complicated by the three-effector encounter of insulin resistance, secretion and clearance. Solving the concerned three-effector encounter may help in realizing the primary pathological driver and primary target for treatment of T2D.
The Three-Effector Encounter of T2D Is Driven by mTORC1
In lack of direct cause-effect relations between insulin resistance, secretion and clearance, the three effectors of T2D are proposed to be concomitantly driven by an upstream primary con-founder, namely, hyper activation of the mammalian target of rapamycin complex 1 (mTORC1) (Figure 1A). mTORC1 controls growth and metabolism by phosphorylating and/or affecting its downstream targets S6K1, 4EBP, CRTC2, lipin, ATF4, HIF1a, PPARg, PPARa, ULK1, TFEB, autophagy and others (10). Wildtype mTORC1 kinase activity may be hyper-activated by growth factors (e.g., insulin), energy/nutrients excess (e.g., glucose, leucine, arginine) and inflammation (e.g., NFkB/IKK), while being suppressed by metabolic stress (e.g., caloric, hypoxic, hyperosmotic, redox) (10, 11). Suppression of mTORC1 activity or its downstream targets is reported to ameliorate T2D phenotype in animal models (12, 13) and human (14–16), implying a putative upstream role of hyperactive mTORC1 in driving the three-effector encounter of T2D.
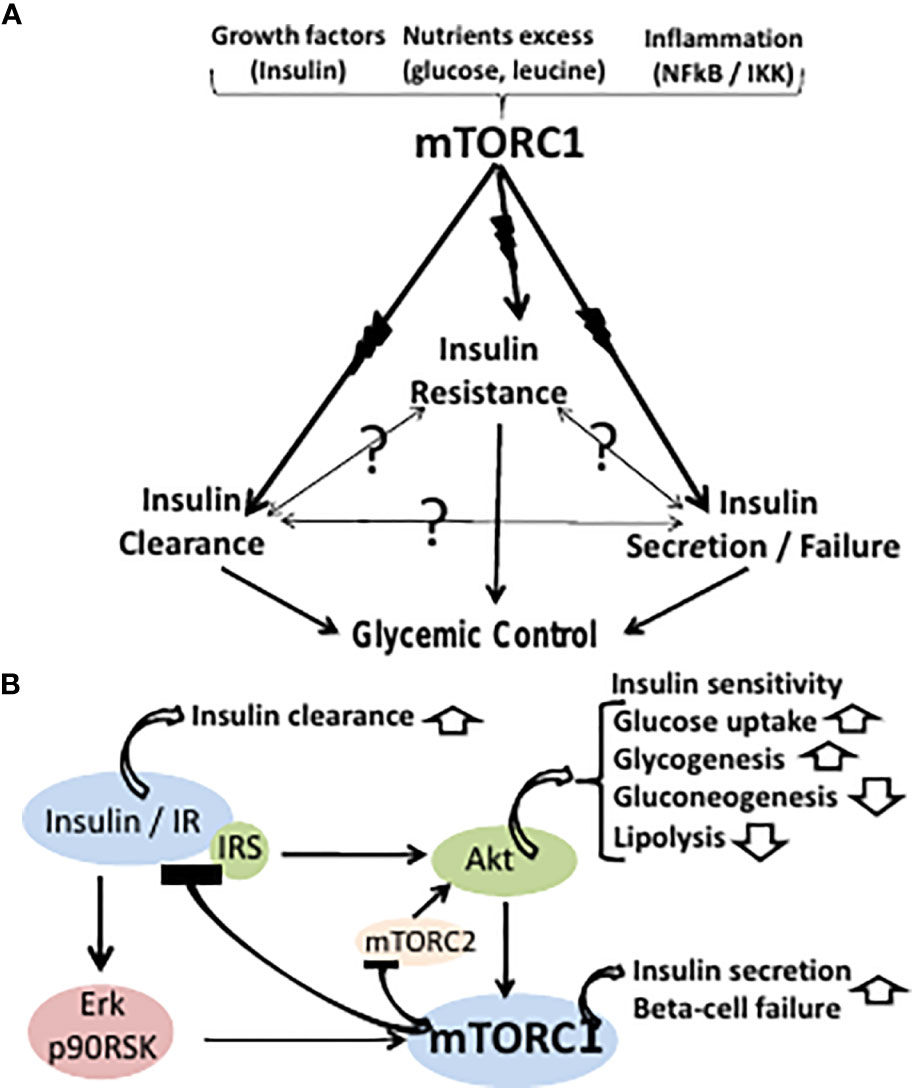
Figure 1 (A) The classical paradigm of T2D glycemic control maintains a cause-effect relationship between insulin resistance, secretion and clearance. However, the cause-effect paradigm is still unresolved (symbolized by question marks) in terms of molecular modes-of-mediation. The three effectors of T2D are proposed here to be concomitantly driven by an upstream primary hyperactive mTORC1. mTORC1 may be hyper-activated by growth factors (e.g., insulin), nutrients excess (e.g., glucose, leucine), or inflammation (e.g., NFkB/IKK), while being suppressed by metabolic stress. (B) Hyperactive mTORC1 may concomitantly drive peripheral insulin resistance, beta cells insulin hypersecretion and failure, and suppression of hepatic insulin clearance. Thus, hyperactive mTORC1 drives insulin resistance by disrupting the IR-Akt transduction pathway resulting in glycogenolysis, gluconeogenesis, inhibition of glucose uptake and adipose lipolysis. mTORC1 activation by the insulin-IR-Erk transduction pathway allows for sustained activation of mTORC1 by insulin upon disrupting the insulin-IR-Akt transduction pathway. Hyperactivation of beta cells mTORC1 serves as double-edged driver, allowing for insulin hypersecretion, while concomitantly promoting beta cells ER stress and apoptosis. Hepatic hyperactive mTORC1 suppresses hepatic insulin clearance.
Hyperactive mTORC1 drives insulin resistance by disrupting the insulin receptor (IR)-Akt transduction pathway in liver, muscle and adipose tissue, resulting in glycogenolysis, gluconeogenesis, inhibition of glucose uptake, unrestrained hyperglycemia and adipose lipolysis (17) (Figure 1B). Thus, phosphorylation of IRS1,2(Ser312, 636/639) by hyperactive mTORC1 and IRS1,2(Ser307, 1101) by hyperactive S6K1 result in suppressing the phosphorylation of IRS tyrosine(s) followed by IRS ubiquitination and degradation (18, 19). Also, phosphorylation and stabilization of GRB10 by hyperactive mTORC1 results in disrupting the IR/IRS transducer (20, 21). The IR-Akt transduction pathway is further disrupted by suppression of Akt(Ser473) phosphorylation by mTORC2, due to inhibition of mTORC2 kinase activity by hyperactive mTORC1/S6K1 (22, 23). Of note, disruption of the IR-Akt transduction pathway by hyperactive mTORC1 may still allow for sustained activation of mTORC1 by insulin, being mediated by the IR-Erk1,2 transduction pathway, implying a functional redundancy of the Akt and the Erk1,2 effectors in mediating mTORC1 activation by insulin (24) (Figure 1B).
mTORC1 drives beta-cells proliferation, cell size and insulin production (25). Hence, concomitant hyper activation of mTORC1 in liver, muscle, adipose fat and beta-cells may account for the close association between peripheral insulin resistance and insulin hypersecretion during the pre-diabetes stage of T2D (Figure 1B). However, hyper activation of beta cells mTORC1 serves as double-edged driver, allowing for beta cells high performance, while concomitantly promoting beta cells ER stress and apoptosis (26–28). Thus, chronic increased production of insulin and its islet amyloid polypeptide (IAPP) by-product may result in unfolded protein response (UPR), aimed at eliminating surplus by suppressing protein synthesis while increasing lysosomal autophagy and/or proteasome degradation. However, these degradation pathways are blocked by hyperactive mTORC1, resulting in apoptosis due to unresolved ER stress (29). Also, disruption of beta cells IR-Akt-FOXO1 pathway by hyperactive mTORC1 results in suppressing PDX1 and beta cells survival (30), while promoting alpha-cells glucagon expression (31) and glucagon-induced hepatic gluconeogenesis. The two concomitant contrasting aspects of beta-cells hyperactive mTORC1 may dynamically evolve during the clinical sequel of T2D, whereby the hyperplastic-hypertrophic pre-diabetes phase yields progressively to overt T2D beta-cells failure (1). Indeed, the decline in beta cells disposition index starts within the range of normal glucose tolerance and progressively deteriorates as patients progress to hyperinsulinemic prediabetes and then to overt T2D (1). Hence, the current two-stage paradigm of pre-diabetes/diabetes overlooks the inherent pathophysiological continuum of T2D.
Hepatic hyperactive mTORC1 suppresses hepatic insulin clearance, which depends on IR availability, and further depends on CEACAM1 phosphorylation by functional IR. Thus, hepatic insulin clearance is mediated by insulin binding to the IR followed by endocytosis, lysosomal insulin degradation and IR recycling to the cell membrane (32). Hyperactive mTORC1 phosphorylates and stabilizes GRB10(Ser476), resulting in its binding to IR, IR ubiquitination and degradation (33) (Figure 1B).
Hyperactive mTORC1 may further revoke a primary role of ‘lipotoxicity’ in driving the three effectors of T2D (34). Indeed, downstream mTORC1 targets [e.g., SREBP, CRTC2, Lipin and PPARalpha (10)] may drive the increase in long-chain fatty acyl-CoAs and the accumulation of diglycerides and ceramides in non-adipose tissues. Secondary ‘glucolipotoxicity’ may complement hyper-active mTORC1 in shaping the phenotype of overt T2D.
Discussion
Concomitant driving of the three-effectors of T2D by hyperactive mTORC1 turns redundant the question which effector comes first. However, mTORC1 activity is affected by genetic, epigenetic, ethnic and/or tissue-dependent factors which may determine its sensitivity to environmental and metabolic conditions. Hence, modulation of each of the three concerned effectors by hyperactive mTORC1 may be context-dependent, allowing for one effector to precede the others, thereby displaying an apparent cause-effect relationship between the three effectors (2–6).
Most importantly, the primary role played by hyperactive mTORC1 in driving the three-effector encounter of T2D implies that suppression of hyperactive mTORC1 may offer an all-in-one treatment for T2D. That is in contrast to targeting the concerned effectors individually by means of insulin sensitizers, potassium channel modulators, incretins, SGLT2 inhibitors, insulin degrading enzymes and other (35). Hyperactive mTORC1 may indeed be targeted by caloric/carbohydrate restriction (36, 37) and physical exercise (38). However, the compliance to behavioral modification is poor. Also, treatment of T2D patients with rapalogs is dubious since chronic treatment may result in inhibition of mTORC2, thereby suppressing the IR-Akt transduction pathway (39). By-passing that difficulty by intermittent rapalogs treatment (40) still remains to be verified under real life conditions.
Alternatively, hyperactive mTORC1 may be tamed by mitochondrial complex I inhibitors. Suppression of hyperactive mTORC1 due to inhibition of mitochondrial complex I is best exemplified by metformin (41–43) used as first-line therapy for T2D (44). The anti-diabetic efficacy of pioglitazone may similarly be ascribed to suppression of mTORC1 kinase activity due to inhibition of mitochondrial complex I (45, 46). Also, classical mitochondrial complex I inhibitors (e.g., rotenone) are reported to alleviate insulin resistance and beta-cell failure in T2D animal models (47). Suppression of hyperactive mTORC1 by mitochondrial complex I inhibitors may be ascribed to redox (NADH/NAD), energy (ATP/AMP), AMPK and/or oxidative (ROS) stress as function of the respective dose (11, 43). Since mTORC1 controls disease aspects of T2D beyond glycemic control (10, 17), suppression of mTORC1 kinase activity by metformin may further account for its pleiotropic effects in improving health- and life-span (48). The mitochondrial/mTORC1 connection may prompt a search for novel mitochondrial complex I inhibitors (49) which may tame hyperactive mTORC1 and the three-effector encounter of T2D.
Author Contributions
Composed and submitted by JB-T. The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. DeFronzo RA. Pathogenesis of Type 2 Diabetes Mellitus. In: Bonora E, DeFronzo R, editors. Diabetes Epidemiology, Genetics, Pathogenesis, Diagnosis, Prevention, and Treatment. Endocrinology. Springer International Publishing (2018).
2. Esser N, Utzschneider KM, Kahn SE. Early Beta Cell Dysfunction vs Insulin Hypersecretion as the Primary Event in the Pathogenesis of Dysglycaemia. Diabetologia (2020) 63:2007–21. doi: 10.1007/s00125-020-05245-x
3. Nolan CJ, Prentki M. Insulin Resistance and Insulin Hypersecretion in the Metabolic Syndrome and Type 2 Diabetes: Time for a Conceptual Framework Shift. Diabetes Vasc Dis Res (2019) 16:118–27. doi: 10.1177/1479164119827611
4. Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR. Slow Glucose Removal Rate and Hyperinsulinemia Precede the Development of Type II Diabetes in the Offspring of Diabetic Parents. Ann Intern Med (1990) 113:909–15. doi: 10.7326/0003-4819-113-12-909
5. DeFronzo RA, Tripathy D. Skeletal Muscle Insulin Resistance Is the Primary Defect in Type 2 Diabetes. Diabetes Care (2009) 32 Suppl 2:S157–63. doi: 10.2337/dc09-S302
6. Czech MP. Insulin Action and Resistance in Obesity and Type 2 Diabetes. Nat Med (2017) 23:804–14. doi: 10.1038/nm.4350
7. Kim SH, Reaven GM. Insulin Clearance: An Underappreciated Modulator of Plasma Insulin Concentration. J Investig Med (2016) 64:1162–5. doi: 10.1136/jim-2016-000149
8. Asare-Bediako I, Paszkiewicz RL, Kim SP, Woolcott OO, Kolka CM, Burch MA, et al. Variability of Directly Measured First-Pass Hepatic Insulin Extraction and Its Association With Insulin Sensitivity and Plasma Insulin. Diabetes (2018) 6:1495–503. doi: 10.2337/db17-1520
9. Gastaldelli A, Abdul Ghani M, DeFronzo RA. Adaptation of Insulin Clearance to Metabolic Demand Is a Key Determinant of Glucose Tolerance. Diabetes (2021) 70:377–85. doi: 10.2337/db19-1152
10. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
11. Demetriades C, Plescher M, Teleman AA. Lysosomal Recruitment of TSC2 Is a Universal Response to Cellular Stress. Nat Commun (2016) 7:10662. doi: 10.1038/ncomms10662
12. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, et al. Absence of S6K1 Protects Against Age- and Diet-Induced Obesity While Enhancing Insulin Sensitivity. Nature (2004) 431(7005):200–5. doi: 10.1038/nature02866
13. Khamzina L, Veilleux A, Bergeron S, Marette A. Increased Activation of the Mammalian Target of Rapamycin Pathway in Liver and Skeletal Muscle of Obese Rats: Possible Involvement in Obesity-Linked Insulin Resistance. Endocrinology (2005) 146:1473–81. doi: 10.1210/en.2004-0921
14. Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, et al. Overactivation of S6 Kinase 1 as a Cause of Human Insulin Resistance During Increased Amino Acid Availability. Diabetes (2005) 54:2674–84. doi: 10.2337/diabetes.54.9.2674
15. Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, et al. The Mammalian Target of Rapamycin Pathway Regulates Nutrient-Sensitive Glucose Uptake in Man. Diabetes (2007) 56:1600–7. doi: 10.2337/db06-1016
16. Yuan T, Rafizadeh S, Gorrepati KD, Lupse B, Oberholzer J, Maedler K, et al. Reciprocal Regulation of mTOR Complexes in Pancreatic Islets From Humans With Type 2 Diabetes. Diabetologia (2017) 60:668–78. doi: 10.1007/s00125-016-4188-9
17. Bar-Tana J. Type 2 Diabetes - Unmet Need, Unresolved Pathogenesis, Mtorc1-Centric Paradigm. Rev Endocr Metab Disord (2020) 21:613–29. doi: 10.1007/s11154-020-09545-w
18. Copps KD, White MF. Regulation of Insulin Sensitivity by Serine/Threonine Phosphorylation of Insulin Receptor Substrate Proteins IRS1 and IRS2. Diabetologia (2012) 55:2565–82. doi: 10.1007/s00125-012-2644-8
19. Yoneyama Y, Inamitsu T, Chida K, Iemura SI, Natsume T, Maeda T, et al. Serine Phosphorylation by Mtorc1 Promotes IRS-1 Degradation Through Scfβ-TRCP E3 Ubiquitin Ligase. Science (2018) 5:1–18. doi: 10.1016/j.isci.2018.06.006
20. Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, et al. The mTOR-Regulated Phosphoproteome Reveals a Mechanism of Mtorc1-Mediated Inhibition of Growth Factor Signaling. Science (2011) 332:1317–22. doi: 10.1126/science.1199498
21. Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villén J, et al. Phosphoproteomic Analysis Identifies Grb10 as an Mtorc1 Substrate That Negatively Regulates Insulin Signaling. Science (2011) 332:1322. doi: 10.1126/science.1199484
22. Liu P, Gan W, Inuzuka H, Lazorchak AS, Gao D, Arojo O, et al. Sin1 Phosphorylation Impairs Mtorc2 Complex Integrity and Inhibits Downstream Akt Signalling to Suppress Tumorigenesis. Nat Cell Biol (2013) 15:1340–50. doi: 10.1038/ncb2860
23. Julien LA, Carriere A, Moreau J, Roux PP. Mtorc1-Activated S6K1 Phosphorylates Rictor on Threonine 1135 and Regulates Mtorc2 Signaling. Mol Cell Biol (2010) 30:908–21. doi: 10.1128/MCB.00601-09
24. Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, et al. Insulin Resistance Differentially Affects the PI 3-Kinase- and MAP Kinase-Mediated Signaling in Human Muscle. J Clin Invest (2000) 105:311–20. doi: 10.1172/JCI7535
25. Blandino-Rosano M, Chen AY, Scheys JO, Alejandro EU, Gould AP, Taranukha T, et al. Mtorc1 Signaling and Regulation of Pancreatic β-Cell Mass. Cell Cycle (2012) 11:1892–902. doi: 10.4161/cc.20036
26. Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T, et al. Biphasic Response of Pancreatic Beta-Cell Mass to Ablation of Tuberous Sclerosis Complex 2 in Mice. Mol Cell Biol (2008) 28:2971–9. doi: 10.1128/MCB.01695-07
27. Bartolomé A, Kimura-Koyanagi M, Asahara S, Guillén C, Inoue H, Teruyama K, et al. Pancreatic β-Cell Failure Mediated by Mtorc1 Hyperactivity and Autophagic Impairment. Diabetes (2014) 63:2996–3008. doi: 10.2337/db13-0970
28. Ardestani A, Lupse B, Kido Y, Leibowitz G, Maedler K. Mtorc1 Signaling: A Double-Edged Sword in Diabetic β Cells. Cell Metab (2018) 27:314–31. doi: 10.1016/j.cmet.2017.11.004
29. Zhao J, Goldberg AL. Coordinate Regulation of Autophagy and the Ubiquitin Proteasome System by MTOR. Autophagy (2016) 12:1967–70. doi: 10.1080/15548627.2016.1205770
30. Jara MA, Werneck-De-Castro JP, Lubaczeuski C, Johnson JD, Bernal-Mizrachi E. Pancreatic and Duodenal Homeobox-1 (PDX1) Contributes to β-Cell Mass Expansion and Proliferation Induced by Akt/PKB Pathway. Islets (2020) 12:32–40. doi: 10.1080/19382014.2020.1762471
31. McKinnon CM, Ravier MA, Rutter GA. FoxO1 Is Required for the Regulation of Preproglucagon Gene Expression by Insulin in Pancreatic Alphatc1-9 Cells. J Biol Chem (2006) 281:39358–69. doi: 10.1074/jbc.M605022200
32. Najjar SM, Perdomo G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiol (Bethesda) (2019) 34:198–215. doi: 10.1152/physiol.00048.2018
33. Edick AM, Auclair O, Burgos SA. Role of Grb10 in Mtorc1-Dependent Regulation of Insulin Signaling and Action in Human Skeletal Muscle Cells. Am J Physiol Endocrinol Metab (2020) 318:E173–83. doi: 10.1152/ajpendo.00025.2019
34. Costes S, Bertrand G, Ravier MA. Mechanisms of Beta-Cell Apoptosis in Type 2 Diabetes-Prone Situations and Potential Protection by GLP-1-Based Therapies. Int J Mol Sci (2021) 22:5303. doi: 10.3390/ijms22105303
35. Artasensi A, Pedretti A, Vistoli G, Fumagalli L. Type 2 Diabetes Mellitus: A Review of Multi-Target Drugs. Molecules (2020) 25:1987. doi: 10.3390/molecules25081987
36. Margolis LM, Rivas DA, Berrone M, Ezzyat Y, Young AJ, McClung JP, et al. Prolonged Calorie Restriction Downregulates Skeletal Muscle Mtorc1 Signaling Independent of Dietary Protein Intake and Associated microRNA Expression. Front Physiol (2016) 7:445. doi: 10.3389/fphys.2016.00445
37. Kumar S, Behl T, Sachdeva M, Sehgal A, Kumari S, Kumar A, et al. Implicating the Effect of Ketogenic Diet as a Preventive Measure to Obesity and Diabetes Mellitus. Life Sci (2021) 264:118661. doi: 10.1016/j.lfs.2020.118661
38. Escobar KA, Cole NH, Mermier CM, VanDusseldorp TA. Autophagy and Aging: Maintaining the Proteome Through Exercise and Caloric Restriction. Aging Cell (2019) 18:e12876. doi: 10.1111/acel.12876
39. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-Induced Insulin Resistance Is Mediated by Mtorc2 Loss and Uncoupled From Longevity. Science (2012) 335(6076):1638–43. doi: 10.1126/science.1215135
40. Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, et al. Alternative Rapamycin Treatment Regimens Mitigate the Impact of Rapamycin on Glucose Homeostasis and the Immune System. Aging Cell (2016) 15:28–38. doi: 10.1111/acel.12405
41. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, Independent of AMPK, Induces mTOR Inhibition and Cell-Cycle Arrest Through REDD1. Cancer Res (2011) 71:4366–72. doi: 10.1158/0008-5472.CAN-10-1769
42. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife (2014) 3:e02242. doi: 10.7554/eLife.02242
43. Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, et al. Metformin Inhibits Hepatic Mtorc1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab (2017) 25:463–71. doi: 10.1016/j.cmet.2016.12.009
44. American Diabetes Association. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Medical Care in Diabetes-2021. Diabetes Care (2021) 44(Suppl 1):S111–24. doi: 10.2337/dc21-S009
45. García-Ruiz I, Solís-Muñoz P, Fernández-Moreira D, Muñoz-Yagüe T, Solís-Herruzo JA. Pioglitazone Leads to an Inactivation and Disassembly of Complex I of the Mitochondrial Respiratory Chain. BMC Biol (2013) 11:88. doi: 10.1186/1741-7007-11-88
46. San YZ, Liu Y, Zhang Y, Shi PP, Zhu YL. Peroxisome Proliferator-Activated Receptor-γ Agonist Inhibits the Mammalian Target of Rapamycin Signaling Pathway and has a Protective Effect in a Rat Model of Status Epilepticus. Mol Med Rep (2015) 12:1877–83. doi: 10.3892/mmr.2015.3641
47. Hou WL, Yin J, Alimujiang M, Yu XY, Ai LG, Bao YQ, et al. Inhibition of Mitochondrial Complex I Improves Glucose Metabolism Independently of AMPK Activation. J Cell Mol Med (2018) 22:1316–28. doi: 10.1111/jcmm.13432
48. Drzewoski J, Hanefeld M. The Current and Potential Therapeutic Use of Metformin-The Good Old Drug. Pharmaceut (Basel) (2021) 14:122. doi: 10.3390/ph14020122
Keywords: insulin resistance, insulin secretion, mTORC1 (mechanistic target of rapamycin complex 1), diabetes, metabolic syndrome and type II diabetes
Citation: Bar-Tana J (2021) Insulin Resistance, Secretion and Clearance –Taming the Three Effector Encounter of Type 2 Diabetes. Front. Endocrinol. 12:741114. doi: 10.3389/fendo.2021.741114
Received: 14 July 2021; Accepted: 14 September 2021;
Published: 29 September 2021.
Edited by:
Simone Baltrusch, University Medicine Rostock, GermanyReviewed by:
Christian Göbl, Medical University of Vienna, AustriaKathrin Maedler, University of Bremen, Germany
Copyright © 2021 Bar-Tana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacob Bar-Tana, amFjb2JiQGVrbWQuaHVqaS5hYy5pbA==