 Yian Gu
Yian Gu Yuanyuan Ye
Yuanyuan Ye Hua Shu1
Hua Shu1 Ming Liu
Ming Liu Qing He
Qing He- 1Department of Endocrinology and Metabolism, Tianjin Medical University General Hospital, Tianjin, China
- 2Department of Endocrinology and Metabolism, Baodi District People’s Hospital, Tianjin, China
Background: Hereditary primary hyperparathyroidism (PHPT) accounts for 5-10% of all PHPT cases, necessitating genetic testing for diagnosis and management. Among these, hyperparathyroidism-jaw tumor syndrome (HPT-JT) is an autosomal dominant disorder caused by CDC73 mutations with variable clinical presentations and incomplete symptoms.
Case summary: The proband, diagnosed with PHPT, underwent parathyroidectomy at the age of 41 with pathological examination of parathyroid carcinoma (PC). Hereditary PHPT was initially suspected due to the early-onset PHPT and family history. Genetic testing identified a heterozygous CDC73 mutation, NM_024529.4: c. 687_688delAG (p. Arg229Serfs*37). Even in the absence of jaw tumors, the diagnosis of HPT-JT was confirmed based on the discovery of renal cysts. A secondary thyroidectomy was performed to reduce the risk of recurrence.
Conclusion: Genetic testing is strongly recommended in cases of early-onset PHPT, family history, jaw tumors, renal and uterine involvement, atypical parathyroid tumors, and PC. This testing provides valuable information for personalized management, and counseling is available for affected families.
Introduction
Hereditary primary hyperparathyroidism (PHPT) encompasses a spectrum of syndromes that require precise classification based on glandular involvement, familial presentation, and genetic testing. One of the rare familial etiologies is hyperparathyroidism-jaw tumor syndrome (HPT-JT), which is characterized by the combination of PHPT, ossifying fibroma of the jaw, and renal and uterine disorders (1). However, the exact genotype-phenotype relationship remains to be established (2).
This paper illuminates a case of HPT-JT within a family. It is noteworthy that despite the identification of a consistent CDC73 variant through genetic testing, the siblings presented distinct clinical phenotypes. The proband, diagnosed initially with parathyroid carcinoma (PC), underwent a second extended ipsilateral thyroidectomy to prevent recurrence. In summary, our case emphasizes the complexity of hereditary PHPT and its diverse clinical manifestations. It highlights the significance of genetic testing in guiding surgery, as it can provide essential information for personalized management.
Case presentation
Proband (II-3): A 41-year-old man was admitted to the hospital with a 15-year history of recurrent renal calculi and a 2-year history of a parathyroid mass. History: The patient had been experiencing recurrent renal calculi accompanied by severe abdominal pain since age 26. Importantly, he had no other symptoms such as bone pain, fractures, nausea, vomiting, thirst, or polyuria. Abdominal ultrasound revealed urolithiasis, leading to multiple extracorporeal lithotripsies. Two years ago, a parathyroid mass was incidentally discovered during a physical examination with parathyroid ultrasound, initially overlooked. Subsequently, he was referred to our hospital for a comprehensive evaluation, which encompassed the following laboratory findings: Hypercalcemia, hypophosphatemia, increased alkaline phosphatase (ALP), and elevated parathyroid hormone (PTH). Thyroid ultrasound showed a hypoechoic nodule, suspected of parathyroid origin, thus prompting his enrollment. Family history (Table 1, Figure 1): The patient’s father and sister both had parathyroid-related diseases. Physical examination revealed no evidence of thyroid goiter or renal tenderness on percussion. Admission investigations (Table 1): Several anomalies were noted, including hypercalciuria, vitamin D deficiency, increased bone turnover, and osteoporosis. Imaging studies (Figure 2), comprising parathyroid ultrasound, parathyroid ECT, and neck CT, identified a mass in the right inferior parathyroid gland. Skull radiographs indicated decreased bone mineral density without erosions or “salt and pepper” appearance. Abdominal CT disclosed bilateral renal cysts and calculi. No abnormalities were found in blood glucose, gastrin-17, adrenocortical function, plasma aldosterone-to-renin ratio, catecholamines and their metabolites, sex hormones, immunofixation electrophoresis, or pituitary MR. Surgical intervention involved the excision of the right inferior parathyroid gland. A brown mass measuring approximately 3.0 x 2.5 cm was observed, closely adherent to the adjacent thyroid tissue. Later pathology confirmed a parathyroid tumor characterized by a heterogeneous collection of neoplastic cells separated from the surrounding tissue by fibrous mesenchyme. Although there was evidence of peripheral invasion, no metastasis was observed in the nearby lymph nodes, seven of which were examined. Immunohistochemistry (Figure 3) revealed staining for PTH (+), CgA partial (+), CD vascular (+), Syn (-), and TTF-1 (-), with a Ki-67 index of approximately 8%.
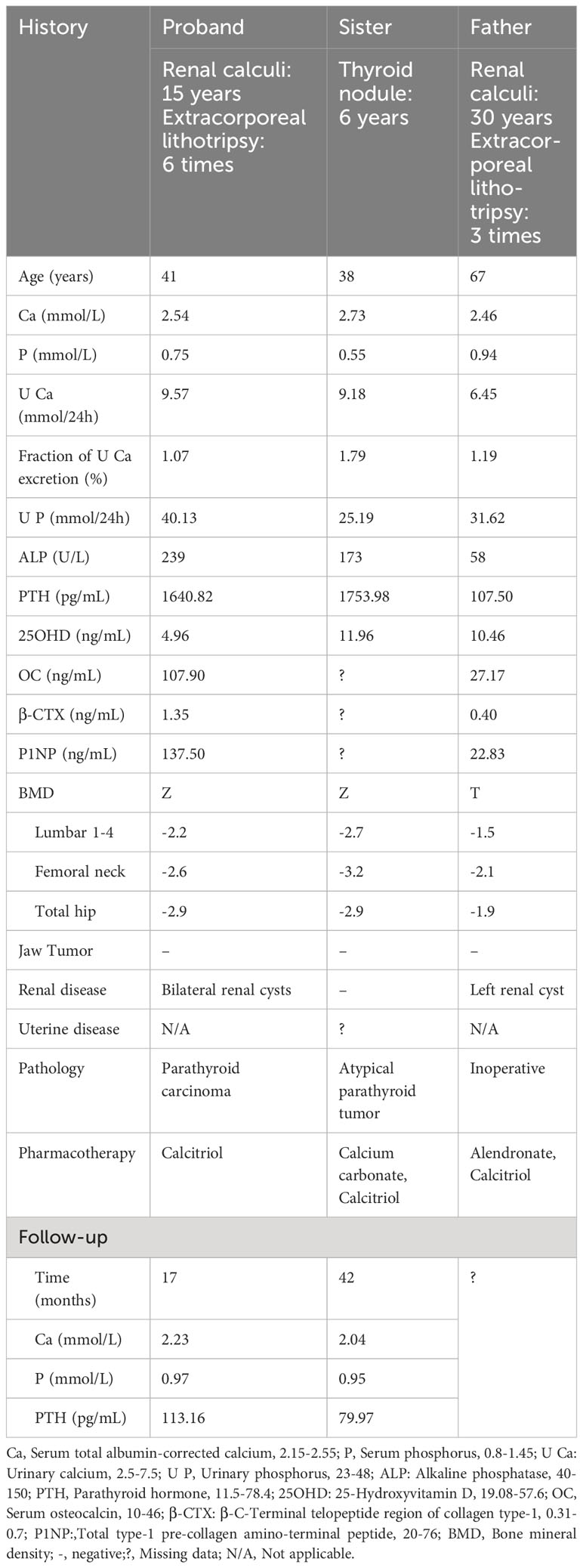
Table 1 Clinical data of a family with hyperparathyroidism-jaw tumor syndrome.
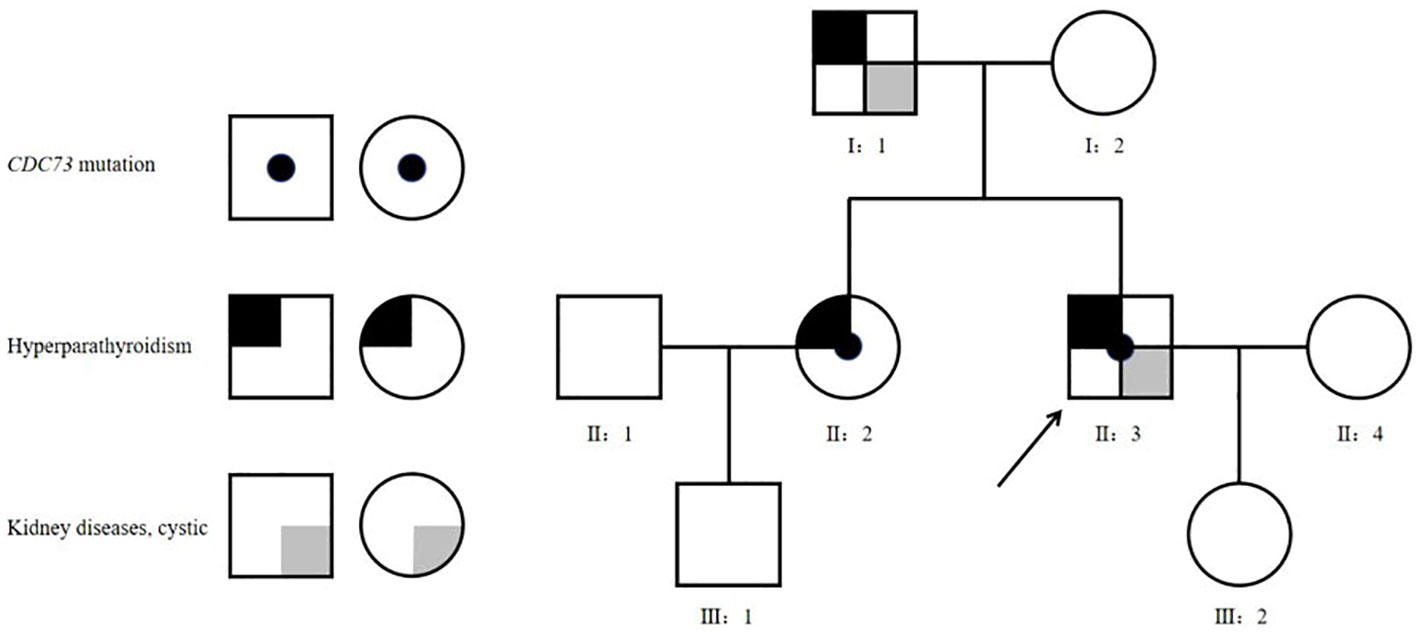
Figure 1 Family tree. Square - male, circle - female; arrow - the proband.
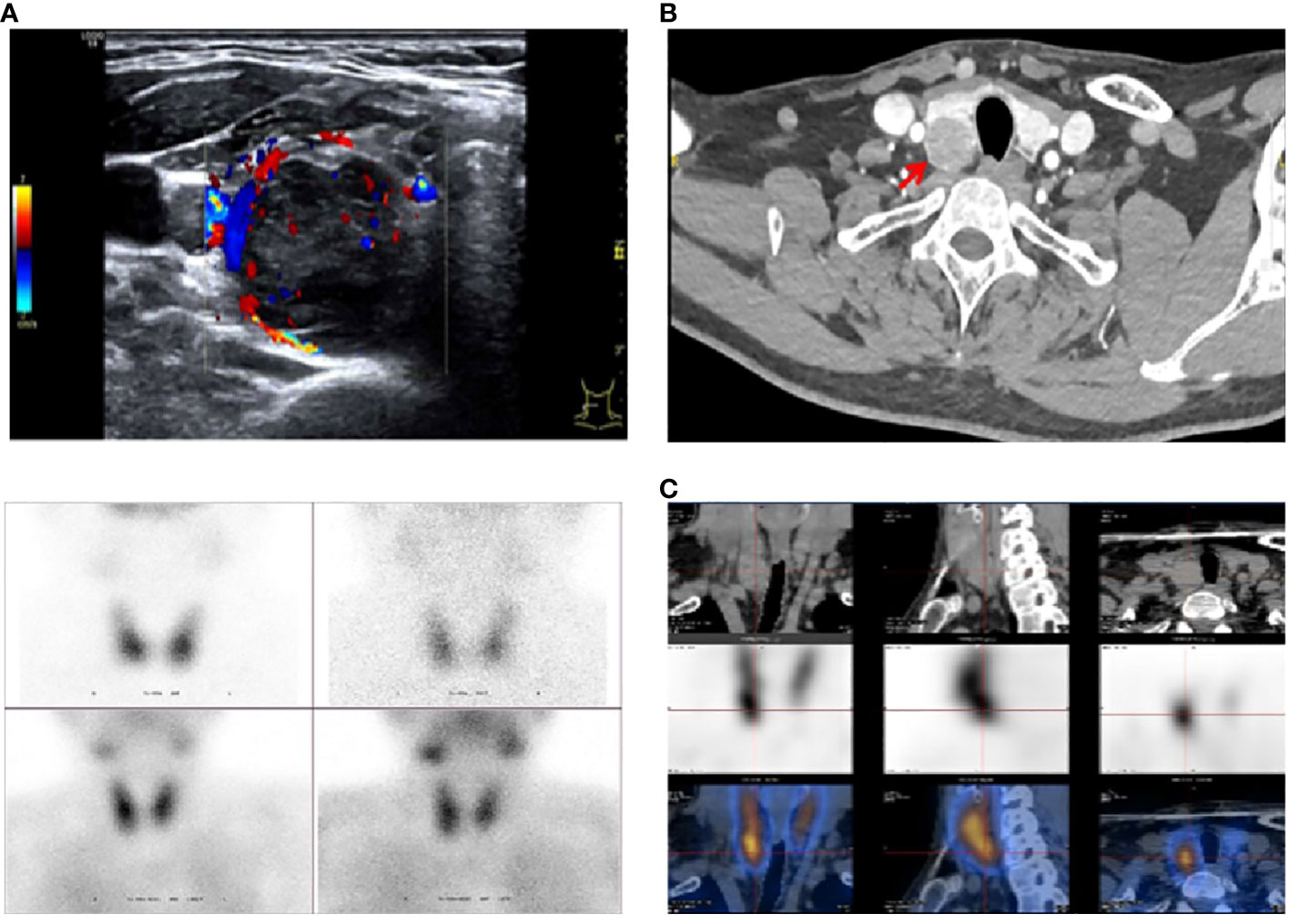
Figure 2 Imaging studies in the proband. (A) Parathyroid ultrasound: Dorsal aspect of the inferior pole of the suitable lobe: 3.1×2.4×1.9 cm. (B) Neck CT enhancement: Dorsal aspect of the inferior pole of the right lobe: Parathyroid origin? (C) Parathyroid ECT (99TcmO4¯MIBI): Right inferior parathyroid hyperfunctioning lesion: 2.2×2.3×2.9 cm.

Figure 3 Parathyroid histopathology in the proband. (A–D) in order of 20x (40x in the lower right corner) HE, PTH, CgA, Ki-67 index staining.
Following the histopathological examination of the PC, immediate reoperation of the right (ipsilateral) thyroid lobe, isthmus, and paratracheal lymph nodes was performed to minimize the risk of recurrence, adhering to guidelines and clinical experience. Considering the early-onset PHPT, family history, and the rare pathology, a peripheral blood sample was sent to HUADA Medical Laboratory for whole exome sequencing. The analysis revealed a heterozygous mutation identified as CDC73;NM_024529.4: c.687_688delAG (p. Arg229Serfs*37), classified and documented as a pathogenic variant in accordance with the American College of Medical Genetics and Genomics standards and guidelines (3). After the two surgical interventions, the patient developed hungry bone syndrome and received calcitriol treatment.
At the one-year regular follow-up, the patient maintained normal serum calcium levels, and thyroid ultrasound exhibited no irregularities at the operative site. However, a moderately hypoechoic nodule was detected in the left thyroid lobe, categorized as TI-RADS 4 with an abnormal aspect ratio, indicating a high-grade lesion. The patient opted for close follow-up.
The table shows the clinical data of two family members: the proband’s father (I-1) and sister (II-2). The sister underwent parathyroidectomy at the age of 38. Histopathologic findings included a right lower pole parathyroid adenoma (PA) with heterogeneous hyperplasia and several small satellite nodules with aggressive biological behavior. Additionally, left upper pole parathyroid hyperplasia (PH) with a cyst was identified. Immunohistochemistry showed: PTH (+), weak Bcl-2 (+), MDM-2 (-), Cyclin D1 (+), Galectin 3 (+), CK pan (+), Ki-67 (<2%+). These results supported the diagnosis of atypical parathyroid tumor (APT). At age 44, the patient’s sister, who resides in Canada, underwent genetic testing at Genetics & Genomics, Alberta Precision Laboratories. This analysis unveiled a genetic mutation consistent with the proband. With a 30-year history of recurrent renal calculi and a recent 4-month elevation in PTH levels, the father was admitted to our department at the age of 67. Laboratory results revealed fluctuating mild hypercalcemia. Thyroid ultrasound and parathyroid CT identified a small nodule in the inferior pole of the right lobe, raising suspicion of a parathyroid origin. However, parathyroid ECT did not exhibit typical highly functional PA images. Due to the uncertainty in localization, medical intervention was initiated. At present, the patient, aged 74, refuses genetic testing.
Discussion
PHPT is a relatively common endocrine disorder, third only to diabetes mellitus and thyroid disease in prevalence, affecting approximately 1 to 8.6 individuals per thousand. It has a higher incidence in the 45-75 age group and shows a gender bias, with women being more susceptible (4). Isolated PAs are responsible for over 80% of PHPT cases, while 0.5-5% are attributed to PCs (5). Around 5-10% of PHPT cases are hereditary. Hereditary PHPT typically manifests in late adolescence or early adulthood and follows an autosomal dominant inheritance pattern. Compared to sporadic PHPT, hereditary cases often involve multiple parathyroid glands and have a higher likelihood of recurrence. Common hereditary forms of PHPT are associated with specific genes, including the MEN1 gene in multiple endocrine neoplasia (MEN) 1, the RET gene in MEN 2, the CDKN1B gene in MEN 4, the CASR, GNA11, and AP2S1 genes in familial hypocalciuric hypercalcemia (FHH), the CASR gene in neonatal severe hyperparathyroidism, the CDC73 gene in HPT-JT, and the GCM2 gene in familial isolated hyperparathyroidism (FIHP) (2).
Distinguishing between FIHP and HPT-JT can be challenging as FIHP is considered either non-syndromic hereditary PHPT or an incomplete manifestation of syndromes such as MEN1, HPT-JT, and FHH associated with MEN1, CDC73, and CASR mutations (6). Latest literature also links GCM2 mutations to FIHP, necessitating further investigation (4).
HPT-JT, a rare familial form of hereditary PHPT with an incidence of less than one in a million, is attributed to mutations in the tumor suppressor gene CDC73 (formerly HRPT2), identified in 50-80% of cases (1, 2). The prevalence tends to rise with age, though onset may occur as early as age 7 (1, 5). Clinical manifestations are variable and incomplete. Unlike other hereditary forms of PHPT, HPT-JT is typically associated with involvement of a single parathyroid gland (7). PHPT is reported in 80-90% of HPT-JT cases, and there is a high incidence of PC, with a risk as high as 15-20% (8). In a national retrospective study of CDC73-associated PHPT in the Netherlands, eleven (12.4%) pathogenic germline mutations were identified in 89 patients with clinically heterogeneous PHPT. The mean age ( ± SD) at diagnosis was 32 ( ± 15) years with a range of 13-54 years (9). The early onset of clinical manifestations and the variable pathology in this family highlight the complexity of this disease. Despite its name, only about 30-40% of patients have maxilla and/or mandible fibro-osseous tumors, which are classified by the World Health Organization as slow-growing, painless, benign growths with a malignancy risk of less than 0.5% (10). They are primarily driven by genetic mutations rather than hyperparathyroidism (10, 11). Importantly, PHPT-induced fibrous dysplasia (osteoid osteoma) tends to resolve spontaneously after parathyroidectomy. Composed of fibroblastic mesenchyme and minerals, fibro-osseous tumors have distinct morphologic features that persist after parathyroidectomy (10). Treatment should be based on the size, location, and biological characteristics of the tumor, with postoperative surveillance recommended to prevent recurrence. Renal involvement constitutes 15% of HPT-JT cases, including cystic disease, hamartoma, and carcinoma, of which cystic disease is the most prevalent (12). These changes can manifest as small cysts or even polycystic kidney disease, either alone or in combination with rare tumors, requiring renal replacement therapy in severe cases (9, 12). Both the proband and his father were found to have simple renal cysts with normal renal function. Uterine disorders, such as leiomyoma, adenomyosis, endometrial hyperplasia, and sarcoma, are the second most common manifestations, affecting over half of women (6, 7). Although there is no information on the proband’s sister, regular follow-up is recommended. While other cancers have occasionally been reported in HPT-JT patients, the relationship to the syndrome remains uncertain. Vigilance is crucial for potential cancers, especially involving the thyroid, pancreas, colon, and testes (1, 2, 9). Following a year after surgery, the proband exhibited a TI-RADS 4 thyroid nodule, which carries a 9.1% risk of thyroid cancer (13). Fine-needle aspiration biopsy is advised if necessary. The differentiation of hereditary PHPT depends on the clinical presentation. However, the renal and uterine disorders are frequent manifestations that complicate the definitive diagnosis, especially when they coexist with PHPT. Furthermore, even in cases of isolated PHPT, HPT-JT should be considered. Therefore, genetic testing is required for diagnosis.
The CDC73 gene on chromosome 1q31.2 consists of 17 exons and encodes parafibromin, a 531-amino acid protein with antiproliferative properties (14). Parafibromin is expressed in various tissues, including parathyroid, adrenal, kidney, heart, and skeletal muscle. It primarily localizes within the nucleus, where it controls cell proliferation, apoptosis, and maintains chromosome stability as part of the human PAF1/RNA polymerase II-related complex (15). The pathogenesis of HPT-JT aligns with the two-hit hypothesis proposed by Knudson, in which a heterozygous germline mutation (inherited from a mutant parent or acquired during embryonic development in rare cases) is coupled with a somatic alteration (16). These lead to the loss of both alleles, resulting in the loss of heterozygosity within the tumor DNA. Consequently, the absence of parafibromin expression contributes to the development of neoplasia (15, 16). Genetic variations are distributed throughout the coding region and splice site, predominantly concentrated in exons 1, 2, and 7. Mutations in exons 3 and 4 are infrequent, while large intragenic deletions and partial intronic mutations are rare (8). The majority of CDC73 mutations, more than 75%, are frameshift and nonsense mutations (17). Although a clear genotype-phenotype relationship has not been formalized, recent research suggests that HPT-JT cases with frameshift mutations, nonsense mutations, and large intragenic deletions have an almost 7-fold higher risk of PC than missense mutations (8). In this family, a two-nucleotide deletion in exon seven of the CDC73 gene was identified, which was predicted to cause a frameshift and the premature termination codon, resulting in nonsense-mediated decay or a truncated protein. This exon variant has been identified in a Dutch family and a Chinese patient, but the clinical presentation differed between the index cases (9, 18).
Genetic testing plays a critical role in confirming hereditary PHPT, detecting associated complications, and providing essential guidance for surgical intervention. It is appropriate for individuals with early-onset PHPT, jaw tumors, renal and uterine disorders, APT, and PC. As other family members may be at risk of carrying variants, genetic testing of offspring may facilitate early tumor detection. In this family, a consistent mutation in the CDC73 gene was identified through national and international genetic testing. Unfortunately, other high-risk relatives did not undergo validation, missing an opportunity for early intervention and risk assessment.
PC is an extremely rare malignant endocrine tumor, comprising only 0.005% of all cancers (5). It usually presents as a sporadic tumor, but occasionally occurs in individuals with hereditary PHPT, leading to life-threatening hypercalcemia due to excessive production of PTH (5, 19). Preoperative serum calcium exceeding 3 mmol/L with PTH over three times the upper limit of normal and parathyroid lesions larger than 3 cm, referred to as the “>3+>3+>3 principle”, strongly suggests the possibility of PC (5, 8, 20). Nevertheless, histopathologic tissue biopsy remains the gold standard. The proband conforms to the above principle but also has severe 25-hydroxyvitamin D (25OHD) deficiency. Concerning secondary hyperparathyroidism, the expected upper limit of normal PTH (PTHmax, pg/mL) can be calculated using the formula , as shown in Jin’s diagnostic alignment diagram for PHPT with vitamin D deficiency (21). In this family, the level of PTH was significantly higher than PTHmax, which is consistent with a diagnosis of PHPT. It is noteworthy that avitaminosis D is prevalent in PHPT with potential mechanisms including increased conversion of 25OHD to 1,25-dihydroxyvitamin D and accelerated metabolism of 25OHD (21, 22). The patient underwent surgery without vitamin D supplementation due to strong motivation, family history of PHPT, kidney stones, and osteoporosis. Unfortunately, most cases of PC are diagnosed postoperatively. Fine-needle aspiration biopsy carries the risk of tumor dissemination and misdiagnosis because cytomorphology does not adequately differentiate between benign and malignant entities (5). Even experienced pathologists find it challenging to differentiate PC from APT. The diagnosis of PC relies on identifying vascular, lymphatic, perineural invasion, infiltration of adjacent structures, or histologic/cytologic metastasis (5, 19). However, APT may exhibit similar histologic features, such as adhesion to nearby structures, monotonous sheet-like or trabecular growth, fibrosis, necrosis, increased mitotic activity, cytologic atypia, and cellular extension into the capsule without complete penetration (19, 23). Immunohistochemistry can improve the accuracy of diagnosis, with positive staining for PTH and CgA often indicating parathyroid tissue (23). Additionally, a Ki-67 index greater than 5% suggests PC (5). The CDC73 mutation, resulting in the deletion of parafibromin, is found in approximately 70% of sporadic PC cases and only about 2% of PA cases, indicating a high risk of recurrence, metastasis, and mortality (23). Loss of parafibromin is emerging as a more promising predictor of adverse outcomes than mutation (5, 20). Therapeutically, surgery is a crucial prognostic determinant, with limited effects of radiotherapy and chemotherapy. Extensive surgery is advocated for PC, including complete resection of the primary tumor, ipsilateral thyroid lobe and isthmus, adjacent structures, and metastatic lymph nodes, to reduce the risk of persistence or recurrence (20, 24). Studies have shown that patients with PC who do not receive a complete resection at the initial operation may benefit from removal of the ipsilateral thyroid lobe and central group lymph nodes at reoperation within one month (24). The management of HPT-JT is based on the European Society of Endocrine Surgery guideline, which recommends selective resection of the abnormal parathyroid gland in cases of single involvement (1, 6). In instances where preoperative localization is negative or equivocal, bilateral neck exploration plus subtotal parathyroidectomy is preferred (6, 7, 25). Prophylactic parathyroidectomy increases the risk of postoperative hypoparathyroidism. A decline of more than 50% in intraoperative PTH indicates a successful procedure (23). In our case, both the proband and his sister experienced a significant decrease in PTH, demonstrating effective removal of the parathyroid mass causing PHPT. Furthermore, the overall survival rate for PC is 78-85% at 5 years and 49-70% at 10 years (5, 6).
Although there are no formal guidelines for the management of HPT-JT, individuals carrying the mutation should undergo lifelong follow-up. Surveillance should include regular monitoring of serum calcium, serum phosphorus, and PTH every six months to one year starting at 5-10 years of age. In addition, screening with parathyroid, renal, and gynecologic ultrasound, as well as jaw radiographs, should be conducted every five years beginning at 10 years of age (1, 2, 12). Genetic counseling is available to affected families.
In conclusion, this case report details the clinical features, pathological observations, and genotypic profiles within a family affected by HPT-JT, highlighting the variability in clinical manifestations and the tendency towards incomplete symptoms. Our primary objective is to raise awareness among clinicians about the significance of genetic testing and counseling in guiding surgical interventions and establishing routine follow-up.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The requirement of ethical approval was waived by the Medical Research Ethics Committee of Tianjin Medical University General Hospital for the studies involving humans. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
YG: Data curation, Writing – original draft, Writing – review & editing. YY: Data curation, Writing – review & editing. HS: Formal analysis, Writing – review & editing. LC: Writing – review & editing. YX: Writing – review & editing. FL: Writing – review & editing. TZ: Writing – review & editing. QH: Supervision, Writing – review & editing. ML: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Tianjin Key Medical Discipline (Specialty) Construction Project (grant number TJYXZDXK-030A), Major project of Tianjin Municipal Science and Technology Bureau (grant number 21ZXJBSY00060).
Acknowledgments
We thank all participants from the Department of Endocrinology and Metabolism, Tianjin Medical University General Hospital.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Torresan F, Iacobone M. Clinical features, treatment, and surveillance of hyperparathyroidism-jaw tumor syndrome: an up-to-date and review of the literature. Int J Endocrinol (2019) 2019:1761030. doi: 10.1155/2019/1761030
2. Newey PJ. Hereditary primary hyperparathyroidism. Endocrinol Metab Clin North Am (2021) 50(4):663–81. doi: 10.1016/j.ecl.2021.08.003
3. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
4. Minisola S, Arnold A, Belaya Z, Brandi ML, Clarke BL, Hannan FM, et al. Epidemiology, pathophysiology, and genetics of primary hyperparathyroidism. J Bone Miner Res (2022) 37(11):2315–29. doi: 10.1002/jbmr.4665
5. Rodrigo JP, Hernandez-Prera JC, Randolph GW, Zafereo ME, Hartl DM, Silver CE, et al. Parathyroid cancer: An update. Cancer Treat Rev (2020) 86:102012. doi: 10.1016/j.ctrv.2020.102012
6. Iacobone M, Carnaille B, Palazzo FF, Vriens M. Hereditary hyperparathyroidism–a consensus report of the European Society of Endocrine Surgeons (ESES). Langenbecks Arch Surg (2015) 400(8):867–86. doi: 10.1007/s00423-015-1342-7
7. Iacobone M, Camozzi V, Mian C, Pennelli G, Pagetta C, Casal Ide E, et al. Long-term outcomes of parathyroidectomy in hyperparathyroidism-jaw tumor syndrome: analysis of five families with CDC73 mutations. World J Surg (2020) 44(2):508–16. doi: 10.1007/s00268-019-05156-y
8. Li Y, Zhang J, Adikaram PR, Welch J, Guan B, Weinstein LS, et al. Genotype of CDC73 germline mutation determines risk of parathyroid cancer. Endocr Relat Cancer (2020) 27(9):483–94. doi: 10.1530/erc-20-0149
9. van der Tuin K, Tops CMJ, Adank MA, Cobben JM, Hamdy NAT, Jongmans MC, et al. CDC73-related disorders: clinical manifestations and case detection in primary hyperparathyroidism. J Clin Endocrinol Metab (2017) 102(12):4534–40. doi: 10.1210/jc.2017-01249
10. Ibrahem HM. Ossifying fibroma of the jaw bones in hyperparathyroidism-jaw tumor syndrome: Analysis of 24 cases retrieved from literatures. J Dent Sci (2020) 15(4):426–32. doi: 10.1016/j.jds.2019.12.007
11. Liu Y, Wang H, You M, Yang Z, Miao J, Shimizutani K, et al. Ossifying fibromas of the jaw bone: 20 cases. Dentomaxillofac Radiol (2010) 39(1):57–63. doi: 10.1259/dmfr/96330046
12. Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, Salenave S, et al. Frequent large germline HRPT2 deletions in a French National cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab (2013) 98(2):E403–8. doi: 10.1210/jc.2012-2789
13. Tessler FN, Middleton WD, Grant EG. Thyroid imaging reporting and data system (TI-RADS): A user's guide. Radiology (2018) 287(1):29–36. doi: 10.1148/radiol.2017171240
14. Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet (2002) 32(4):676–80. doi: 10.1038/ng1048
15. Jo JH, Chung TM, Youn H, Yoo JY. Cytoplasmic parafibromin/hCdc73 targets and destabilizes p53 mRNA to control p53-mediated apoptosis. Nat Commun (2014) 5:5433. doi: 10.1038/ncomms6433
16. Knudson A. Alfred Knudson and his two-hit hypothesis. (Interview by Ezzie Hutchinson). Lancet Oncol (2001) 2(10):642–5. doi: 10.1016/s1470-2045(01)00524-1
17. Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat (2010) 31(3):295–307. doi: 10.1002/humu.21188
18. Kong J, Wang O, Nie M, Shi J, Jiang Y, Li M, et al. CDC73 gene mutation and parafibromin expression status of parathyroid carcinoma in Chinese. Zhonghua Yi Xue Za Zhi (2013) 93(42):3364–8. doi: 10.3760/cma.j.issn.0376-2491.2013.42.010
19. Erickson LA, Mete O, Juhlin CC, Perren A, Gill AJ. Overview of the 2022 WHO classification of parathyroid tumors. Endocr Pathol (2022) 33(1):64–89. doi: 10.1007/s12022-022-09709-1
20. Marini F, Giusti F, Palmini G, Aurilia C, Donati S, Brandi ML. Parathyroid carcinoma: molecular therapeutic targets. Endocrine (2023) 81(3):409–18. doi: 10.1007/s12020-023-03376-w
21. Jin J, Mitchell J, Shin J, Berber E, Siperstein AE, Milas M. Calculating an individual maxPTH to aid diagnosis of normocalemic primary hyperparathyroidism. Surgery (2012) 152(6):1184–92. doi: 10.1016/j.surg.2012.08.013
22. Moosgaard B, Vestergaard P, Heickendorff L, Melsen F, Christiansen P, Mosekilde L. Vitamin D status, seasonal variations, parathyroid adenoma weight and bone mineral density in primary hyperparathyroidism. Clin Endocrinol (Oxf) (2005) 63(5):506–13. doi: 10.1111/j.1365-2265.2005.02371.x
23. Duan K, Gomez Hernandez K, Mete O. Clinicopathological correlates of hyperparathyroidism. J Clin Pathol (2015) 68(10):771–87. doi: 10.1136/jclinpath-2015-203186
24. Xue S, Chen H, Lv C, Shen X, Ding J, Liu J, et al. Preoperative diagnosis and prognosis in 40 Parathyroid Carcinoma Patients. Clin Endocrinol (Oxf) (2016) 85(1):29–36. doi: 10.1111/cen.13055
Keywords: hereditary primary hyperparathyroidism, hyperparathyroidism-jaw tumor syndrome, parathyroid carcinoma, CDC73 gene, parafibromin
Citation: Gu Y, Ye Y, Shu H, Chang L, Xie Y, Li F, Zhu T, Liu M and He Q (2024) A family case report of parathyroid carcinoma associated with CDC73 mutation in hyperparathyroidism-jaw tumor syndrome. Front. Endocrinol. 15:1330185. doi: 10.3389/fendo.2024.1330185
Received: 30 October 2023; Accepted: 08 January 2024;
Published: 29 January 2024.
Edited by:
Paraskevi Xekouki, University of Crete, GreeceReviewed by:
Dimitrios Kourkouliotis, University of Athens, GreeceJessica Costa-Guda, University of Connecticut, United States
Copyright © 2024 Gu, Ye, Shu, Chang, Xie, Li, Zhu, Liu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Liu, mingliu@tmu.edu.cn; Qing He, heqing202108@163.com
†These authors have contributed equally to this work and share first authorship